

Abstract
Hemophilia A gene therapy has been hampered by immune responses to vector-associated antigens and by neutralizing antibodies or inhibitors to the factor VIII (FVIII) protein; these ‘inhibitors’ more commonly effect hemophilia A patients than those with hemophilia B. A gene replacement strategy beginning in the neonatal period may avoid the development of these immune responses and lead to prolonged expression with correction of phenotype thereby avoiding long-term consequences. Serotype rh10 AAV was developed splitting the FVIII coding sequence into heavy and light chains with the chicken β-actin promoter/CMV enhancer for dual recombinant AAV vector delivery. Coinjection of virions of each FVIII chain intravenously to mice on the second day of life was performed. Mice express sustained FVIII antigen levels of ≥5% to 22 months of life without the development of antibodies to FVIII. Phenotypic correction was manifest in all AAV-FVIII-treated mice as demonstrated by functional assay and reduction in bleeding time. This study demonstrates the use of AAV in a gene replacement strategy in neonatal mice that establishes both long-term phenotypic correction of hemophilia A and lack of antibody development to FVIII in this disease model where AAV is administered shortly after birth. These studies support consideration of gene replacement therapy for diseases that are diagnosed in utero or in the early neonatal period.
Keywords: Neonate, Hemophilia A, Gene Therapy, AAV, Immune Response
Introduction
Neonatal gene therapy is a promising strategy for treating multiple congenital diseases that can be diagnosed shortly after birth. A number of reports suggest that there may be advantages including 1) therapeutic gene expression may allow for avoidance of the development of irreversible pathology; 2) with earlier vector administration, there is greater vector to cell ratio allowing for less vector; and 3) stem and progenitor cells, likely less accessible later in life, may be more easily transduced. In addition, transduction and expression of genes prior to the maturation of immunity may enable induction of operational immune tolerance and prolonged transgene expression. A number of animal models have been treated in the neonatal period with encouraging results, including Crigler-Najjar syndrome1, mucopolysaccharidosis type VII2-4, Pompe disease5, methylmalonic acidemia6-7, hemophilia B8-9 and spinal muscular atrophy.10 However, in some models in immunocompetent adult animals, delivery and expression of genes has resulted in immune responses to the gene product and/or to vector-associated antigens that have limited or abrogated expression.
Hemophilia A is an inherited disease of coagulation due to a deficiency of functional factor VIII characterized by spontaneous and prolonged bleeding in joints, muscles, and internal organs.11 Patients with this disease require invasive, lifelong management and can become untreatable due to immune responses to factor VIII protein (FVIII); neutralizing antibodies or ‘inhibitors’ can complicate therapy in up 25% of patients. Gene therapy is a compelling goal for the treatment of hemophilia as the therapeutic window is wide and amounts as little as 1-2% of normal physiologic levels are therapeutic and result in substantial improvement in the clinical phenotype.12 However, hemophilia A requires expression of FVIII to be persistent for gene transfer to be considered an effective therapy.
The success in canine studies was not successfully translated in the human clinical trial with AAV2 for gene therapy of hemophilia B;13 multiple pieces of evidence suggest that the failure of this trial was due to the development of an anamnestic immune response to the AAV serotype 2 capsid proteins, not factor IX (FIX) with the specific immune-mediated destruction of vector-transduced cells. A recent and ongoing clinical trial14-15 enrolled severe hemophilia B patients without histories of inhibitor development to receive AAV8 with a liver-specific promoter for the expression of FIX. Two patients, one in a lower-dose cohort and one in a medium-dose cohort achieved levels of FIX of 2-4%14; while two patients in the highest dose cohort did have brief periods of elevated hepatic transaminases weeks after administration suggestive of an immune response, they have both maintained FIX expression. This most recent clinical trial suggests that in selected non-inhibitor prone hemophilia B patients the barrier to achieving sustained expression of therapeutic amounts of protein may have been reached. While our approach with neonatal transgene expression in hemophilia A may avert complications in the perinatal period, such as intracranial hemorrhage in hemophilic neonates, it may also allow for avoidance of a humoral immune response to the factor VIII protein. Earlier studies of neonatal murine gene transfer for hemophilia A with adeno-associated virus (AAV) serotype 216 and retroviral vectors (RV)17 did demonstrate prolonged FVIII protein expression in some animals (22 weeks with AAV [n=8] and up to 14 months with RV [n=2]); however these were complicated by the development of inhibitory antibodies in many of the animals.
Recombinant adeno-associated viral vectors (rAAV) have specific advantages as gene transfer vehicles. These include 1) long-term gene expression with minimal toxicity following administration in adult mammals18, 2) a broad host cell range with the ability to infect dividing and growth-arrested cells, and 3) the availability of procedures for generating high-titer, helper virus-free preparations.19 Furthermore the identification of additional AAV serotypes that demonstrate higher levels of transduction in adult mammalian tissues including the liver have been described.20-21 While not completely understood, the mechanisms underlying these differences in biological performance have been attributed to differences in cell entry, trafficking, viral uncoating and genome processing.22-24 One of the more recently described serotypes is rh10, a clade E primate AAV25 with favorable biological properties as a vector for neonatal liver-directed gene transfer, including higher levels of transgene expression and vector copy number persistence during the period of rapid neonatal growth.20 With no known pathogenicity of wild type AAV, the features of rAAV vectors in general, and with the properties of the most recently described serotypes, suggest that they may be well-suited for neonatal gene transfer and thus require a thorough evaluation in appropriate animal models.
In these studies we demonstrate that initiating therapy for hemophilia A shortly after birth using recombinant AAV serotype rh10 expressing factor VIII results in in vivo production of biologically active protein initially at supraphysiological levels, then declining to relatively stable therapeutic levels; this results in an improvement of the bleeding phenotype by tail clip and a functional FVIII assay (Coatest). This persistent expression is life-long in the murine model of hemophilia A after co-injection of rAAV vectors, one expressing the heavy chain of FVIII and the other expressing the FVIII light chain. Importantly, no antibodies develop to factor VIII protein subsequent to vector administration or with protein challenge in the presence of adjuvant.
Results
Tolerability of virus administration
Matings of FVB/n hemophilic males (XHY) and hemophilic females (XHXH) were setup to produce offspring that were all affected. Previously published data demonstrate that these mice develop antibodies to human factor VIII (hFVIII) in adult animals when injected with hFVIII.26 C57Bl/6 mice were purchased for reporter gene (i.e. luciferase) studies. On the second day of life, mice were intravenously administered either 1) pharmaceutical saline (negative controls, n=12) or AAVrh10 (n=54). Of the AAVrh10-injected groups, mice received either AAVrh10-chicken β-actin promoter/CMV enhancer (CBA)-Luciferase (n=20) or AAV rh10 serotypes expressing both the FVIII light chain (LC) and the FVIII heavy chains (HC) (n=34) each under control of the CBA promoter (Figure 1).
Figure 1.

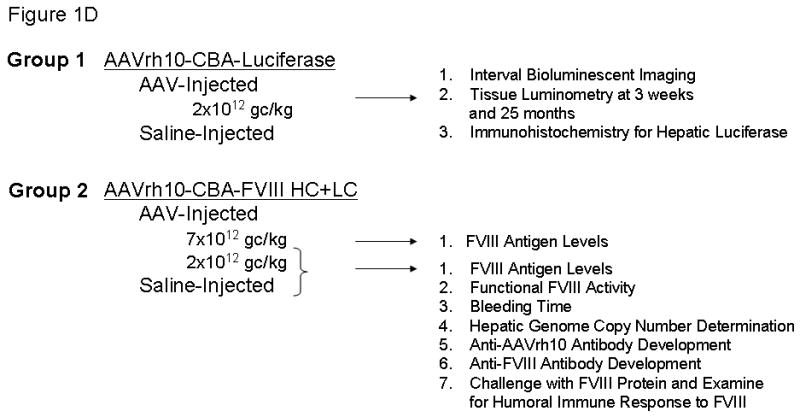
Schematic of the gene structures of AAVrh10 vectors. The vectors encode A) luciferase, B) human FVIII heavy chain cDNA (base pairs 1-2292), and C) human FVIII light chain cDNA (base pairs 1-57 and 4744-7053). Vector was administration was performed on the second day of life. (CBA=chicken β-actin promoter/CMV enhancer, hgH pA=human growth hormone polyadenylation signal, ITR=AAV inverted terminal repeat, ss=signal sequence. Letters represent domains of the factor VIII cDNA and * indicates incomplete domain.)
Wild type C57Bl/6 mice were administered pharmaceutical saline (negative controls) (n=3) or 2.0×1012 gc/kg AAVrh10 expressing firefly luciferase (n=20). Affected hemophilia A neonatal mice received either 2.0×1012 genome copies/kilogram (gc/kg) of AAVrh10 carrying each of FVIII-heavy chain (HC) and FVIII-light chain (LC) (referred to as moderate dose) (n=26) or 7×1012 gc/kg of AAVrh10 carrying each of FVIII-HC and FVIII-LC or saline (referred to as high dose) (n=8). Hemophilia A mice were followed longitudinally except for a subset euthanized at 6 months of life after receiving 2×1012 gc/kg of AAVrh10 FVIII-HC and FVIII-LC on day 2 of life (n=4).
All of the animals having received AAVrh10 expressing factor VIII and AAVrh10 expressing luciferase appeared well during the neonatal and juvenile periods and did not demonstrate any evidence of growth retardation compared to pharmaceutical saline-injected controls. ALT levels of mice having received 2.0×1012 gc/kg of each of FVIII-HC and FVIII-LC at 30 days of age (n=5 per group) were similar to those of controls (49.7±4.0 vs. 49.2±19.6 IU/L, respectively [p=ns]).
Luciferase gene expression is long-lived after neonatal administration
Bioluminescent imaging (BLI) was performed of mice having received the neonatal injection of 2.0×1012 gc/kg AAVrh10-CBA-Luciferase to examine for the distribution and longevity of expression of the reporter gene (Figure 2A, B, C). Mice were imaged from 2 days after injection to 96 weeks of life, the length of the study (n=6-8 mice at each time point), to generate a time course plot allowing for analysis of the level of expression. Mice were imaged from the lateral aspect beginning 72 hours after vector administration (5th day of life) and from the ventral surface beginning on day 9; photon diffusion patterns were acquired. Subsequent images were obtained on weeks 2 through 6, 8, 12, 26, 52, 78, and 96. Expression was detected at the earliest time point and this was the peak as detected by BLI in the course of these studies.
Figure 2.
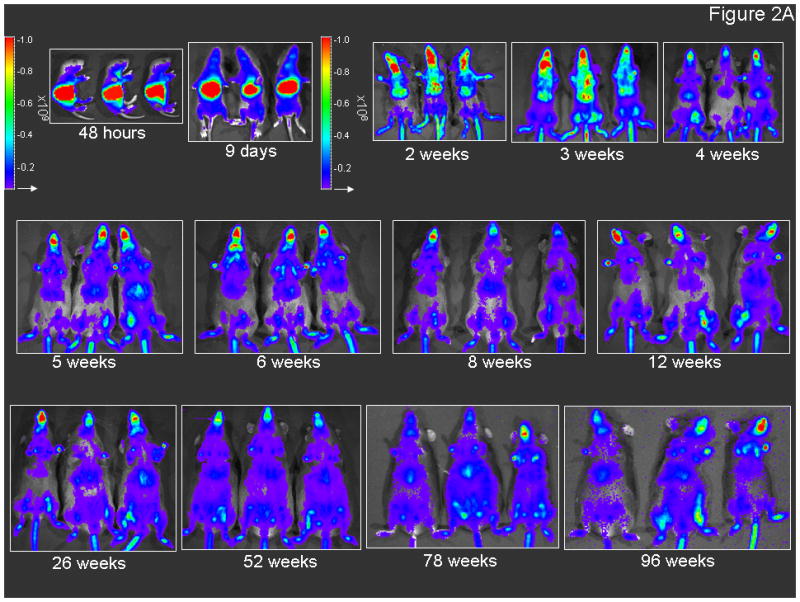
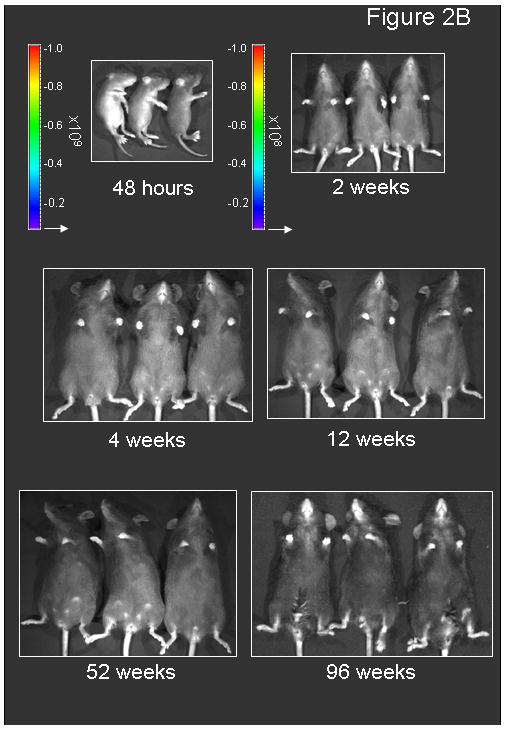
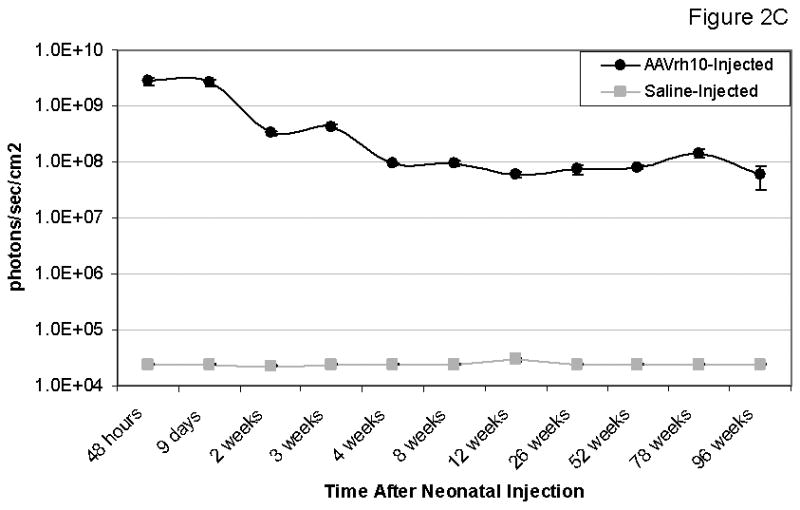
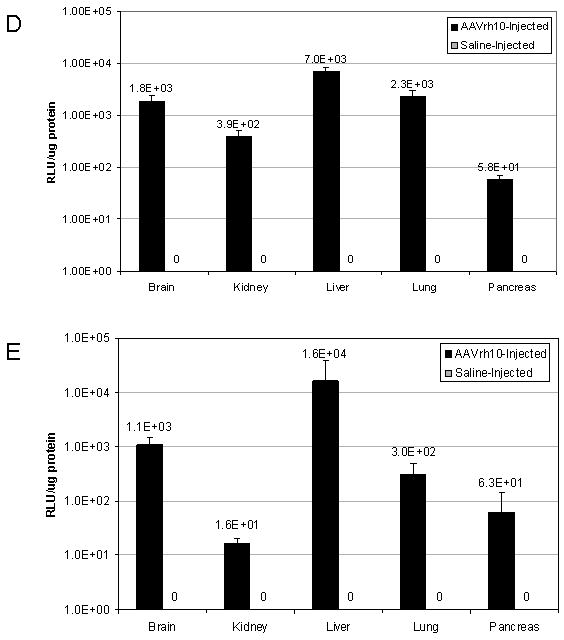
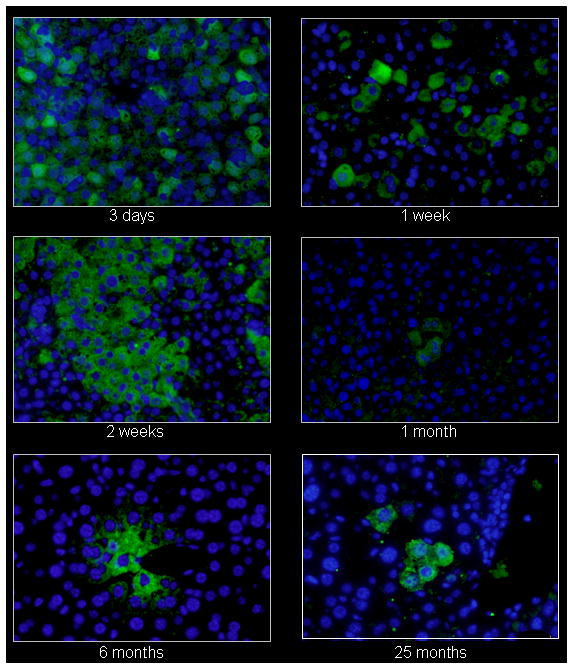
Persistence of luciferase expression after vector administration to neonatal mice. In vivo imaging of firefly luciferase after intravenous injection of A) 2.0×1012 gc/kg of AAV or B) saline on the 2nd day of life demonstrates photon diffusion patterns. The images are shown at 2 days (48 hours after injection), 9 days, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 8 weeks, 12 weeks, 26 weeks, 52 weeks, 78 weeks and 96 weeks after vector administration and at 48 hours, 2 weeks, 4 weeks, 12 weeks, 52 weeks, and 96 weeks after saline administration. Total flux in photons/second was acquired. For all time points except at 48 hours, images were acquired with the mice in the ventral position. Images at 48 hours and 9 days and from 2 to 96 weeks were set with the same references such that side-by-side comparison can be made. Note changes in the legend of the pseudocolor scale per group between these two groups. C) BLI was followed longitudinally in animals as was luminometry in selected tissues at D) 1 month and E) 25 months after vector administration. F) Representative histological liver sections examined at indicated times after intravenous injection of two-day-old mice receiving 2×1012 gc/mouse of AAVrh10-CBA-luciferase (640× magnification). (n=6-8 animals per group at each time point for A, B and C, and n=5 animals per group at for D and E.) (Error bars represent mean + SD.) (AAV=adeno-associated virus)
The time course of expression (Figure 2C) demonstrates the greatest period of decline occurs over the first 2 weeks during this period of rapid growth. This decline is substantial (graphical data demonstrates a negative slope and images show a substantive loss of red and yellow [higher intensity] areas and more predominant appearance of blue and purple [lower intensity] or areas of no expression); however, by 4 weeks whole animal BLI time course demonstrates stability throughout the length of these studies.
Furthermore we examined the expression of luciferase by tissue luminometry to 25 months of life (Figures 2D, E). Comparing expression in multiple tissues at 1 month of life (D) demonstrates that the highest expression was in the liver in moderate dose AAVrh10-CBA-luciferase-injected animals; all other tissues demonstrated lower levels of expression. At 25 months of life (length of study), similar levels of expression with the same general organ distribution was demonstrated (E). While some bioluminescent images do demonstrate an increased intensity in the craniofacial region of some mice, this is due to the intravascular injection with local expression at the site of injection as this intensity of expression was not found in the brain; during the injection some injectate does enter the local tissues due to the elevated intravascular pressure. Luminometry of individual tissues demonstrates the liver was the highest expressing organ of the transgene-encoded protein; animals receiving saline only on the second day of life demonstrated no luciferase expression or bioluminescence as expected.
Immunohistochemistry for luciferase expression in hepatocytes was performed to further examine the decline in expression after neonatal administration of vector (Figure 2F). Using an FITC-conjugated secondary to detect the anti-luciferase antibody, widespread hepatic expression of luciferase is demonstrated by fluoroscein positive cells in the liver by one week after administration. Subsequent images demonstrate a decline in the number of luciferase-expressing hepatocytes. Examination of hepatocytes at 6 and 25 months demonstrates low but stable expression in groups of clustered cells.
Factor VIII expression in the hemophilia murine model
A similar time course of expression was detected with factor VIII antigen expression. Mice receiving 2.0×1012 genome copies/kg of each AAVrh10 FVIII-HC+LC demonstrated supranormal levels of hFVIII antigen of 230.2 ± 79.1 ng/ml at 1 week of life (Figure 3A). The level declined to 20.2 ± 10.2 ng/ml by 1 month of life. From 2 until 22 months of age (length of study), FVIII antigen levels ranged from 4.2 ± 1.3 ng/ml to 10.9 ± 5.2 ng/ml. At 22 months, the average factor VIII antigen level was 8.6 ± 5.6 ng/ml. Mice receiving 7.0×1012 genome copies/kg of each AAVrh10 FVIII-HC+LC also demonstrated supranormal levels of hFVIII antigen of 470.6 ± 87.1 ng/ml of normal at 1 week of life (Figure 3B). The level declined to 122.3 ± 56.6 ng/ml by 1 month and from 2 until 20 months of age, FVIII antigen levels ranged from 42.9 ±13.0 ng/ml to 10.5 ± 2.4 ng/ml (length of study). At 20 months, the average FVIII antigen level was 19.9 ± 10.9 ng/ml.
Figure 3.
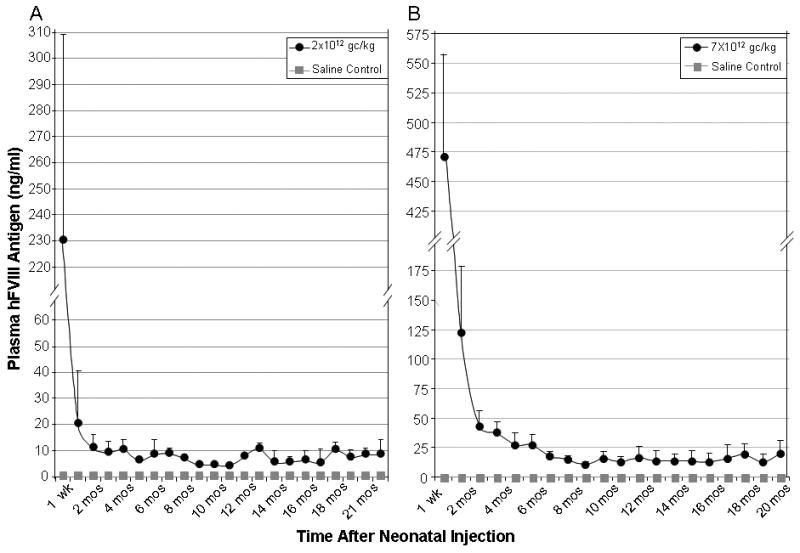
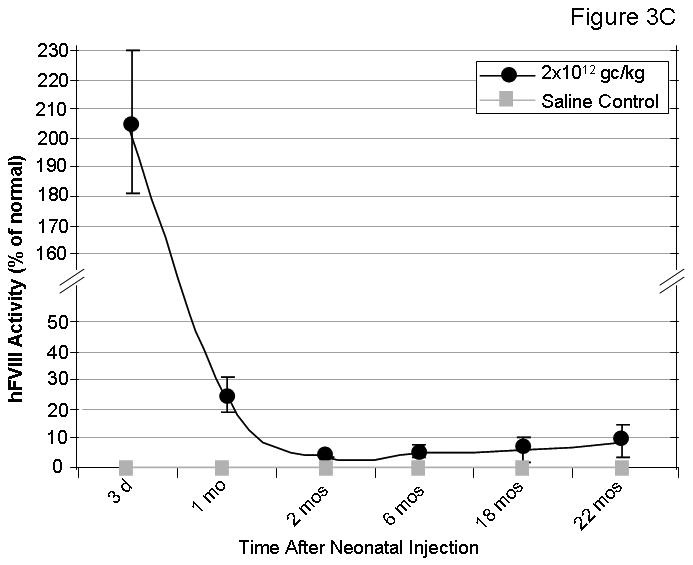
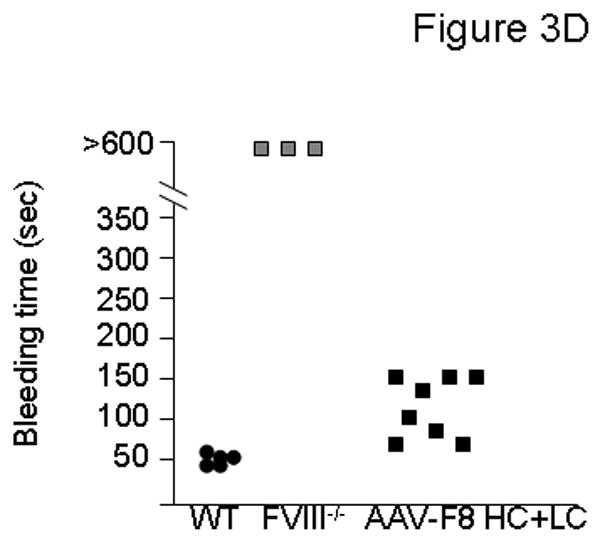
Factor VIII levels and function were assessed in hemophilic FVIII knockout mice following AAVrh10-FVIII HC+LC administration. Hemophilia A mice were injected intravenously with saline, 2.0×1012 gc/kg, or 7.0×1012 gc/kg of each AAVrh10-CBA-FVIII expressing HC and LC on the second day of life. Blood was collected at regular intervals. Examination of hFVIII antigen in plasma by ELISA after A) 2.0×1012 gc/kg or B) 7.0×1012 gc/kg of AAVrh10-CBA-FVIII HC+LC were administered (n=4-12 animals at each time point) (Error bars represent mean + SD); C) Examination of functional hFVIII in plasma (by Coatest) (n=5-7 animals at each time point) after administration of 2.0×1012 gc/kg of AAVrh10-CBA-FVIII HC+LC (Error bars represent mean + SD); D) Bleeding times were assessed in adult mice (n=3-8 animals per group) after administration of 2.0×1012 gc/kg of AAVrh10-CBA-FVIII HC+LC. (WT=wild type, FVIII-/-=factor VIII deficient [hemophilic], AAV=neonatal AAV injected, HC=heavy chain, LC=light chain)
Mice receiving 2.0×1012 gc/kg of each hFVIII chain were examined for functional factor VIII activity. In order to assess the FVIII function, 1) a chromogenic assay based on FVIII activity was performed and 2) bleeding time was analyzed by tail clip assay 8 months after neonatal FVIII knockout mice received AAVrh10 expressing FVIII-HC and FVIII-LC. Mice demonstrated supraphysiological levels of FVIII activity 3 days after vector administration (205.6 ± 24.6%) which stabilized at 3.6 ± 0.1 to 8.9 ± 5.6% from 2 to 22 months of life (n=5-7 mice per time point) (Figure 3C). With the tail clip bleeding time assay, saline-injected wild type mice (n=5) were examined for comparison (range 44-56 seconds). The bleeding time in all AAVrh10 expressing FVIII-HC+LC-injected mice (n=8) (range 85-168 seconds) shortened from that of saline-administered knockout mice (n=3) (>600 seconds) (Figure 3D), also demonstrating that functional factor VIII is produced and that partial correction of the phenotype is present.
In a subset of animals that received 2.0×1012 gc/kg of AAVrh10 FVIII-HC+LC as neonates, examination of the quantity of heavy chain and light chain along with the chromogenic assay was performed in the same animals to assess if there was an imbalance between the secretion of the 2 chains. Utilizing samples from 6 month old animals collected at the same time (n=4), light chain was expressed 6.9 times higher than that of the heavy chain (compared to standardized plasma, heavy chain was 1.8 ± 0.2% and light chain was 12.4 ± 0.7%). FVIII activity by Coatest at the same time point with the same animals was 2.6 ± 0.2%.
Genome copy number declines with animal growth
Genome copy number per hepatocyte was examined after injection with the viral vector. Livers were removed and DNA prepared for nucleic acid analysis. Viral DNA was quantitated by real-time polymerase chain reaction to determine the total copy number per genomic DNA. Three days after injection there were 69.6 ± 34.7 copies per hepatocyte (1.04×104 ± 5.21 ×103 gc/ng liver DNA). Subsequently, mice were randomly selected at pre-determined time points after injection of AAVrh10 expressing FVIII-HC+LC to examine vector copy number stability. Vector copy number declined from day 5 to 153.9 ± 57.6 gc/ng liver DNA at 1 month, 84.9 ± 37.1 gc/ng liver DNA at 2 months with 30.3 ± 19.4 gc/ng liver DNA at 1½ years of life (n=3-6 mice per time point) (Figure 4).
Figure 4.
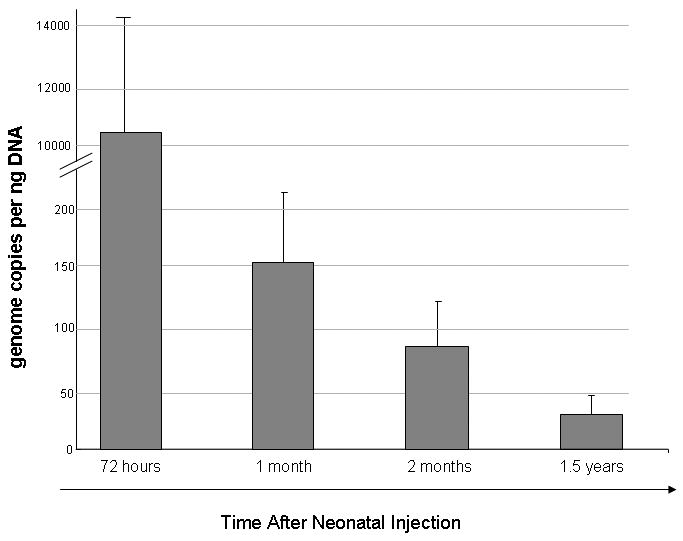
Viral copy number in liver was measured at selected time points by quantitative PCR. Hemophilia A mice were injected intravenously with 2.0×1012 gc/kg of each AAVrh10-CBA-FVIII expressing HC and LC on the second day of life. Genome copy number kinetics 72 hours after injection followed by 1 month, 2 months, and 1.5 years were examined (n ≥ 3 animals at each time point). (Error bars represent mean + SD)
Antibodies to AAV and FVIII do not develop after neonatal injection
Antibodies to vector-associated proteins and hFVIII were examined after neonatal injection of AAV. As adults, mice injected with 2.0×1012 gc/kg AAVrh10-FVIII-HC+LC as neonates were bled at 1 month, 3 months, 6, months, 9 months, and 1 year with plasma examined for antibodies against hFVIII by ELISA. Antibodies to FVIII were not present (fig. 5A [n=8]) in any of the mice tested as they continued to demonstrate expression of FVIII. Plasma was also examined for antibodies to rh10 AAV capsid proteins. All mice demonstrated no antibodies against vector-associated antigens (Figure 5B [n=6]). Mice injected with 7.0×1012 gc/kg AAVrh10-FVIII-HC+LC as neonates similarly did not demonstrate antibodies to FVIII or vector-associated antigens when tested (data not shown).
Figure 5.
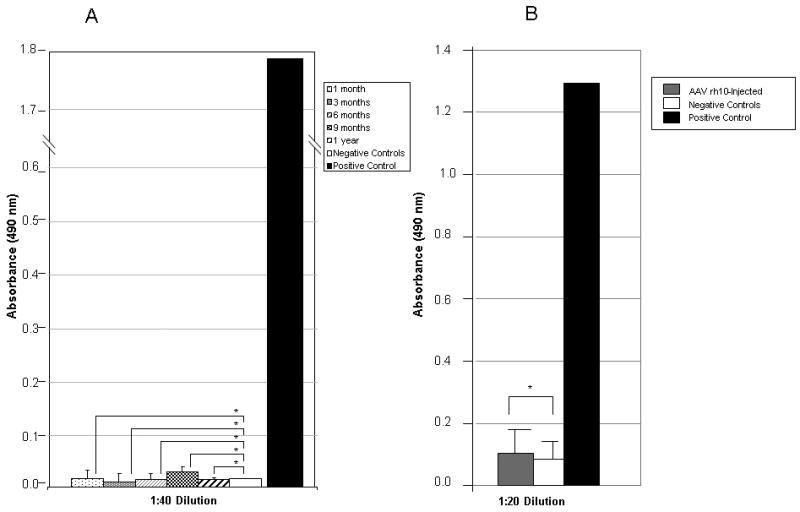
Humoral immune response to hFVIII and AAVrh10 capsid in hemophilic mice injected on the second day of life. Neonatal mice received intravenously either saline or AAVrh10-FVIII expressing HC and LC. A) At 1, 3, 6, 9, and 12 months mice were bled and plasma was analyzed for antibodies to FVIII (n = 8 animals per group). B) At 1 month mice were bled and plasma was analyzed for antibodies to rh10 capsid protein. (n=6 animals per group) (* p=ns). (Error bars represent mean + SD)
Antibodies do not develop to recombinant FVIII after challenge with adjuvant
As corrected mice (having received 2.0×1012 gc/kg AAVrh10-FVIII-HC+LC as neonates) demonstrate long-term expression of factor VIII at ≥5%, we sought to examine if these animals were immunologically unresponsive to exogenously-administered hFVIII; control mice, that had been administered saline on the 2nd day of life, were included. Mice were administered by intraperitoneal route 1 unit of B-domain-deleted hFVIII as an immunogen mixed with alum (an adjuvant) at 8 weeks of life (n=3); antibody development to hFVIII was examined. Control adult animals (injected as neonates with saline) had development of anti-hFVIII antibodies when plasma was analyzed 16 days after protein challenge (n=3) (Figure 6); however and most importantly, none of the neonatally-injected AAVrh10-hFVIII HC+LC animals demonstrated development of antibodies to hFVIII indicating a long-term unresponsiveness to FVIII.
Figure 6.
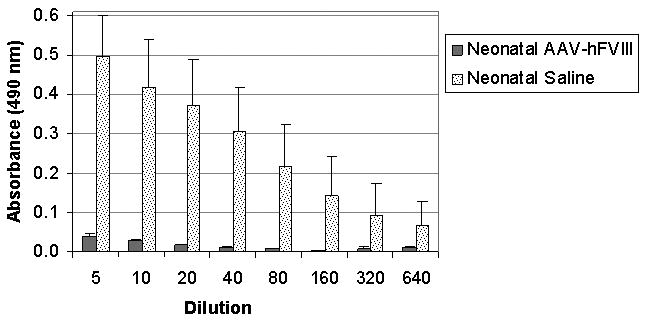
Humoral immune response to hFVIII in hemophilic mice challenged with hFVIII as adults. At 16 days after alum-hFVIII injection, mice were bled and plasma analyzed for antibodies to hFVIII in both control (saline-injected as neonates) and neonatal AAVrh10-hFVIII-HC+LC-injected mice. (n≥3 animals per group.) (Error bars represent mean + SD)
Discussion
Multiple animal studies suggest that gene replacement strategies are a promising approach for the treatment of inherited monogenic diseases of the liver such as hemophilia.27-30 The goal of gene therapy for hemophilia A has been to safely and stably achieve levels of FVIII activity of at least 1% to 2% of normal to convert severe hemophilia to a milder form of the disease;27 levels this low have previously been shown to be efficacious using protein replacement therapy.31 The low levels of FVIII necessary for therapeutic correction, the wide therapeutic window, and lack of need for tight regulation of gene expression make hemophilia A a first target for developing such gene therapy strategies. Long-term AAV-mediated expression of FVIII has been observed in large animal models without an immune response to FVIII.28-30 Furthermore, in one study continuous expression of FVIII by AAV eradicated pre-existing inhibitory antibodies to FVIII in dogs.32 However, published clinical trials have not realized the success obtained in these canine models due to both pre-existing and subsequent development of immune responses to AAV capsid when these vectors have been administered to humans.13
The current study examined the neonatal treatment of hemophilia A by administration of AAV serotype rh10 with FVIII expressed by the split chain approach to overcome the limited vector capacity. The main findings of this study were 1) early high-level transduction is possible with AAVrh10 for neonatal gene transfer; 2) moderate doses of vector are tolerated when administered in early postnatal life; 3) a rapid decline in vector number and gene expression during the first month after administration is detected; 4) phenotypic improvement of hemophilia A is possible; and 5) an immune response to subsequent hFVIII administration is lacking.
In mature animals at any specific time, the number of hepatocytes in cycle is estimated to be 1 in 10,000 to 1 in 20,000;33 hepatocyte lifespan has been estimated to be 180-400 days.34-35 This stability of adult hepatocytes allows for long-term AAV-mediated transgene expression that results in stable persistence of transcriptional active vector genomes that remain episomal or are in an integrated form.36-37 To date, few studies have examined the administration and persistence of episomal viral gene therapy vectors in the context of in vivo cellular proliferation associated with growth and maturation of tissues;20, 38-39 however, like neonatal tissues it has been demonstrated in adult mice that loss of episomal genomes, the primary source of rAAV-mediated gene expression,36 does occur with cell division40-41 and proliferation after hepatic resection.36, 42-43
Daily postnatal weight gain in mice peaks at the time of weaning, up to 0.9 grams per day by day 20 postpartum, with subsequent decline afterwards.44 The thoracic (heart, lung) and abdominal organs (liver, pancreas, GI tract) and brain reach their adult size by day 35 of life.20 During this time of rapid animal growth and organ size maturation, loss of AAV-mediated vector genomes occurs.20 Tissues undergoing active cell cycling during neonatal life like hepatocytes (unlike the adult liver) result in a substantive loss of vector after administration with stability only achieved when adult size is attained.20, 38 However, postmitotic tissues (e.g. brain, skeletal muscle, and heart) have lower rates of vector genome loss during this time.20 The decline in replication-incompetent vector number is only partly explained by organ growth with vector dilution; as we20 and others38 have previously demonstrated this substantial early vector loss after administration is suggestive of active degradation rather than being solely dilutional. While the mechanism is not known we hypothesize that the unintegrated genomes in the nucleus are not linked to chromatin and thus with cellular division and breakdown of the nuclear envelope during prophase, episomes may move to the cytoplasm and then are degraded. We believe that this is similar to what occurs with cytosolic plasmid DNA: Naked plasmid DNA entering the cytoplasm is unstable, is subjected to cytosolic nucleases, and is rapidly turned over.45 In addition, the factor VIII level, as a secreted protein, is further affected by its dilution in the expanding blood volume with animal growth. Thus, the rapid cellular proliferation, active genome degradation, and expanding volume of distribution in the early neonatal period make the potential for efficacious therapeutic gene replacement strategies with episomal vectors more challenging. While integrating vectors may be able to overcome limitations of vector loss that are likely to occur in humans as detected in mice, concerns of oncogenesis and insertional mutagenesis remain.46-47
In the studies conducted herein we were able to obtain long-term persistent expression of the reporter gene luciferase and expression and function of factor VIII with AAV rh10-mediated gene delivery. Supraphysiologic levels of FVIII were obtained shortly after vector administration, declining to stable levels of ≥5% that persisted to 22 months (length of study) when 2.0×1012 gc/kg of AAVrh10 FVIII-HC+LC or ≥19% at 20 months when 7.0×1012 gc/kg were administered neonatally (length of study). Chain-specific ELISA confirmed that more light chain is produced compared to heavy chain as demonstrated by others.16, 48-49 An improvement in bleeding phenotype was observed with tail clipping in this clinically representative model of hemophilia A. Other studies by our group have demonstrated a similar longevity of expression, alteration of bleeding phenotype, and lack of immune response in the murine model of hemophilia A using a helper-dependent adenoviral vector expressing FVIII.50
Luciferase expression was widespread and high-level shortly after administration with a similar decline detected by bioluminescent imaging when compared to imaging shortly after administration; tissue studies with luciferase demonstrate that expression is maintained in multiple organs long-term. While some bioluminescent images demonstrate higher levels of expressions in the craniofacial region, organ luminometry does not demonstrate these levels of expression as this appears to be from expression in the overlying soft tissues at the site of injection secondary to unavoidable viral extravasation due to the elevated venous pressure of injection during the time of vector administration.
Vector persistence in these studies has demonstrated that genome copies endure for greater than 1 year with relative stability making continual expression of FVIII possible; in addition, persistence of gene expression has been demonstrated in multiple studies to be important in both induction and maintenance of immune tolerance.51-53 Expression in hepatocytes appears to be maintained long-term in small clusters of cells, also previously demonstrated by others with AAV8.38-39 Re-administration of serotype rh10 AAV appears to be possible to subsequently augment FVIII expression as the animals mature as a humoral immune response did not develop to this AAV serotype after early postnatal administration; these studies, while preliminary and limited, demonstrate with subsequent vector administration (2×1012 gc/kg at 3 weeks of age) human FVIII antigen in plasma increased on average to 70 ng/mL when examined at 3 months of age (data not shown). In humans, however, it is unclear if this will be possible due to 1) the stage of development of immune ontogeny at birth and 2) to the seroprevalence of anti-AAV antibodies in the human population. While the prevalence of neutralizing antibodies to AAVrh10 is not known, seroprevalence to AAV2 has been reported to be as high as 30%54-72%55 of the population. Neutralizing antibodies to more novel serotypes are lower (e.g. AAV7 ∼15% in US54; AAV8 16%54-19%55 in US, France, respectively; and AAV9 33.5%55 in France). In a study limited to AAV2, 5, and 6, children (age <18 years) had a lower prevalence of neutralizing antibodies than adults and serotypes 5 and 6 were lower than that of AAV2. Taken together, these results support the development of more novel serotypes of AAV directed to future clinical applications in this area.
Unlike hemophilia B where it is estimated that the development of inhibitory or neutralizing antibodies occurs in 3% of those afflicted,12 hemophilia A is a disease where 20-25% develop inhibitors56 and may be a more appropriate model for examining the ability of the neonatal immune system to avoid this pathologic consequence. Antibodies to FVIII can complicate treatment of acute bleeding episodes in human patients as they neutralize the activity of the infused clotting factor making it difficult to establish hemostasis effectively and despite years of investigation, it is not possible to predict which patients will develop inhibitory antibodies. The ability to induce anergy or tolerance to FVIII would be advantageous in preventing inhibitor formation, thus surmounting the major complication for this disease and would allow for subsequent administration of gene therapy vectors or factor infusions if necessary without the worry of immune consequences.
Avoidance of humoral immune responses to FVIII in these murine studies was possible by both initiating expression of the transgene before immune ontogeny was complete and maintaining expression continuously. In contrast to human development, mice are born at an immunologically immature stage with few B cells and almost no T cells in the peripheral lymphoid tissues and they do not develop alloreactivity until birth.57 While likely strain dependent, it has been suggested that the neonatal murine response to some antigens may not be fully developed until they are 4-5 days old;58 mammals with gestational periods of 30 days or longer generally have immunocompetence at birth.44 While B cell tolerance to selected antigens has been established in humans up to 2 years of life,59 humans are born with a considerably more mature immune system capable of generating effective T and B cell responses with populated peripheral lymphoid tissues; human fetal thymocytes first respond to phytohemagglutinin at 14 weeks of gestation.60 While the studies described herein demonstrate lack of an immune response after neonatal administration of AAV in mice, further investigation is necessary in appropriate large animal models with an immune ontogeny similar to that of humans.
Materials and Methods
Generation of Recombinant Adeno-Associated Viral Vectors
The DNA sequences encoding the heavy and light chains of human FVIII were constructed as described61 and cloned into expression cassettes in AAV vectors with subsequent pseudotyping to serotype rh10. The factor VIII cDNA was obtained by reverse transcriptase PCR from RNA prepared from human liver as previously described.62 In brief, poly(A) RNA was primed with an oligo (dT) primer, and first-strand synthesis was catalyzed by SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA) at 42°C. The cDNA was then amplified with two specific oligonucleotides and Pfu DNA polymerase (Stratagene, La Jolla, CA). Sequencing was performed to confirm integrity by comparing with GENBANK. The luciferase cDNA was obtained by excising from pGL3-enhancer plasmid (Promega, Madison, WI) at NheI and XbaI.
The chick β-actin/CMV enhancer (CAG)-luciferase plasmid was derived by first excising the hCMV promoter at MluI and SacII from pAAV-hrGFP (Stratagene, La Jolla, CA). The CAG promoter was excised at AccI and ApaI from pturboCre (provided by M. Jiang, UCLA). The CAG promoter was inserted and ligated by blunt-blunt cloning procedures to create the intermediate plasmid CAG-hrGFP. The GFP cDNA was then excised at XhoI and EcoRI; in its place, the luciferase cDNA was inserted by blunt-blunt methods. Correct orientation was determined by restriction digestion.
The cloning plasmid CAG-hrGFP was used to create both the CAG-FVIII heavy chain and CAG-FVIII light chain plasmids. The heavy chain construct encodes the A1 and A2 domains and 5 amino acids from the amino terminus of the B domain. The light chain vector encodes 85 amino acids of the carboxyl terminal B domain in addition to the complete A3, C1, and C2 domains. The CAG-hrGFP plasmid was digested at restriction sites BsteII and EcoRI to remove the hrGFP cDNA as well as the hGH PA tail. The heavy and light chains of factor VIII and their accompanying hGH polyA tail were amplified by PCR using Platinum Taq Hi-Fidelity DNA Polymerase (Invitrogen, Carlsbad, CA) and sequence specific primers. The PCR products were blunted and cloned into the intermediate cloning plasmid. Firefly luciferase cDNA was excised from pGL2-enhancer plasmid (Promega, Madison, WI), blunted, and subcloned into the sites with orientation determined by restriction digestion.
Mice and Animal Procedures
Factor VIII exon 16 knockout hemophilia A mice on the FVB/n background (FVIII-deficient knockout mice) were used in all the experiments described in this study. FVIII-deficient mice were purchased from Jackson Laboratories (Bar Harbor, ME). The characteristics of the exon-16 strains of hemophilic mice have been previously reported63 were backcrossed with FVB/n mice (Charles River, Wilmington, MA) through ten cycles prior to these studies. The immune characteristics of these mice to human FVIII has been previously described.26 All mice were housed under specific pathogen-free conditions; food and water were provided ad libitum. Genotyping was performed as previously described.62 All mice were kept according to the National Institutes of Health guidelines and all experimental procedures were conducted in accordance with guidelines for the care and use of research animals at our institution. Newborn pups on the second day of life and from XHXH and XHY matings were injected with 2.0×1012 gc/kg of AAVrh10 FVIII-LC and 2.0×1012 gc/kg FVIII-HC or 7.0×1012 gc/kg of AAVrh10 FVIII-LC and 7.0×1012 gc/kg FVIII-HC diluted in pharmaceutical grade saline by the superficial facial vein. The injections were performed in a total volume of 50 μl. Confirmatory PCR genotyping was performed on all pups. Similarly C57Bl/6 mice (purchased from Charles River, Wilmington, MA) were bred and pups were administered 2.0×1012 gc/kg of AAVrh10-CBA-Luciferase. Males and females were equally represented throughout the study. After the vector injection, scheduled blood sampling was taken from retro-orbital plexus. Plasma was frozen immediately and stored at -80°C until analysis.
Alum (Pierce Biotechnologies, Rockford, IL) was mixed with 1 unit of B-domain-deleted hFVIII (Wyeth, NY, NY) in normal saline and mixed for 30 minutes before injecting IP into 8-week-old mice. Plasma was collected 16 days later and frozen at minus 80°C until ready for analysis.
Bioluminescent Imaging
At predetermined times after AAV administration, mice were placed on a warming table and administered luciferase substrate (D-luciferin, Caliper, Hopkinton, MA, at 150 mg/kg IP) and imaged using the bioluminescence optical imager (IVIS 200; Xenogen, Alameda, CA) as previously described.20 At each time-point a region of interest was generated surrounding each animal (excluding the tail) in order to quantify the total photons emitted by luciferase activity.
Immunohistochemistry
For detection of luciferase, liver sections (5 μm) were deparafinized and rehydrated followed by a heat-mediated antigen retrieval step performed using sodium citrate buffer (10mM, pH 6.0). Tissue was permeabilized using 0.2% Triton X100. Samples were then blocked for 20 minutes with protein buffer (Dako, Carpinteria, CA [Cat# X0909]) at room temperature followed by incubation with the firefly luciferase antibody (Abcam, Cambridge, MA [Cat# ab81823]) at a 1/100 dilution overnight at 4°C. Bound primary antibody was detected with a FITC conjugated donkey anti-goat IgG secondary antibody (Abcam, Cat# ab6881) at a 1/100 dilution for 1 hour at room temperature. After washing three times with 1X TBS, tissue was counter stained with DAPI. (Vector Laboratories, Burlingame, CA [Cat# H-1500]). Images were captured using an Olympus IX71 fluorescent microscope using cellSens software (Olympus, Center Valley, PA).
Assays for Luciferase Activity
Mice were euthanized by isoflurane overdose. Tissues were removed and placed in flat bottom 2.0 ml small capped tubes (USA Scientific, Ocala, FL) with cell culture lysis reagent (Promega, Madison, WI [E153A]) and placed on ice. Tissues were homogenized with a hand-held homogenizer (Omni International, Marietta, GA). Specimens were centrifuged at 13,200 rpm for 5 minutes and then placed at 4°C. A luminometer (Berthold Detection Systems, Oak Ridge, TN) was used to measure total light emission according to the manufacturer's instructions. All samples were measured for 10 seconds. Luciferase levels were determined in duplicate and recorded as relative light units (RLU).
A Lowry-based protein assay was performed (Bio-Rad, Hercules, CA) and specimens were analyzed on a microplate reader (BioTek, Winooski, VT) at 750 nm. Samples were performed in duplicate and compared with protein standards. Mean and standard error of the mean (SEM) was calculated. Luciferase levels by luminometry were normalized after protein concentrations were determined.
Assays for measuring human factor VIII antigen and activity level
hFVIII antigen levels were examined by enzyme-linked immunosorbent assay (ELISA) using a polyclonal antibody to human FVIII light chain and heavy chain that are species-specific and do not cross-react with murine FVIII. Plasma used for this assay of human FVIII was collected at regular intervals from retroorbital bleeding using heparinized tubes. Factor VIII antigen was quantitated by following the manufacturer's instructions (Enzyme Research Laboratories, South Bend, Indiana) using 20 uls plasma performed in duplicate. FVIII standards were generated using normal human pooled plasma (Sigma-Aldrich, St. Louis, MO). The background antigen of hFVIII in control animals was negligible. An ELISA to quantitate light and heavy chain antigen levels, based on the technique of Sabatino64 and personal communication with D. Sabatino, was established using antibodies specific for human FVIII. A monoclonal antibody (1:500 diluted in coating buffer) to the light chain (ESH-2, American Diagnostica, Stamford, CT) or heavy chain (6345, Abcam, Cambridge, MA) (both kindly provided by Denise Sabatino, University of Pennsylvania) was used to capture the protein. After blocking with 5% milk a rabbit anti-hFVIII polyclonal antibody conjugated to horseradish peroxidase as a secondary (Enzyme Research Laboratory, South Bend Indiana) was added. After washing OPD substrate was added and after 10 minutes the reaction was stopped and the plate was read at 450 nm. The standard curve was generated using serial dilution of normal human pooled plasma as above.
FVIII activity was quantitated by a chromogenic assay (COATEST SP FVIII, Chromogenix, Instrumental Laboratory Company, Lexington, MA) using citrated plasma collected at selected time points following the manufacturer's instructions for microplate method. FVIII levels were calculated from a standard curve generated using a series of dilutions of normal human pooled plasma (Hemosil Calibration Plasma, Instrumental Laboratory Company, Lexington, MA).
Phenotypic correction was assessed by tail transection bleeding time as described.65 The mouse tail was transected at 5 mm from the tip and it was immediately immersed into 37°C saline. The bleeding time was determined as the time from the tail transection to the moment the blood flow stopped for more than 60 seconds. Bleeding time beyond 600 seconds was considered as cut-off time for the purpose of analysis.
Examination of immune responses to AAV and human factor VIII after viral injection
Immune response to viral capsid protein was measured by anti-AAVrh10 ELISA. A positive control for anti-AAVrh10 antibody was generated by intraperitoneally administering a naïve 8-week-old adult mouse with 5×1010 gc of AAVrh10-luciferase mixed with Alum (Pierce, Rockford, IL). Blood (>200 uls) was collected 4 weeks later and serum was removed and stored at -80°C until needed. Ninety-six well immunoplates (NUNC, Rochester, NY) were coated with 1 × 109 viral particles/well of AAVrh10-luciferase that were prepared in 75 μl of 50mM carbonate buffer (pH 9.6). To inactivate the virus, the plates were exposed to UV light for 30 minutes. Following an overnight incubation at 4°C and washing with 1X PBS, 200 μl of blocking reagent (1X PBS with 5% FCS) was added to each well and incubated at 37°C for 2 hours. Meanwhile, two-fold dilutions of plasma samples from experimental (neonatally-injected with AAVrh10 FVIII-LC and FVIII-HC) and control groups, beginning from 1:40, were prepared in blocking buffer. After the plates were washed, 100 μl of diluted plasma was added in duplicate and subsequently incubated for 2 hours at 37°C. Fifty microliters of 1:1000 dilution of HRP-conjugated goat-anti-mouse secondary antibody (BD Pharmingen, Franklin, NJ) was added and incubated for 1 hour at 37°C. The plates were washed as above and incubated with 50 μl of OPD substrate for up to 6 minutes. Color development was stopped by adding 50 μl of 2.5 M sulfuric acid and was measured for absorbance at 492 nm; background was defined as the absorbance measured in the absence of primary and secondary antibody.
Total antibodies against hFVIII were detected with an antigen-specific ELISA technique using purified commercial factor VIII protein concentrate. The anti-hFVIII ELISA was similarly performed to ELISA described above except the sample volume was 50ul per well. The immunoplates were initially coated with 0.122 IU/well of purified human recombinant FVIII (1 U=100 ng hFVIII protein) (Xyntha, Wyeth, NY, NY). The background levels of anti-hFVIII antibodies measured in control hemophilia A mice were negligible. A mouse anti-human FVIII antibody was generated by injecting an adult mouse intravenously with recombinant human FVIII and was used as a positive control for these studies. Results were presented as average ± standard deviation.
Determination of serum alanine aminotransferase (ALT)
Blood was collected from mice at 30 days of life and analyzed as described (IDEXX Veterinary Services Inc., West Sacramento, CA); 65 uls per animal was subjected to analysis. The results were pooled and mean and standard deviation (SD) calculated.
Statistical Analysis
Statistical calculations were performed using the SPSS version 17.0 statistical software package (Chicago, IL). Data are presented as mean ± SD unless stated otherwise. Groups were compared by T test and a p value of less than 0.05 was considered statistically significant.
Acknowledgments
This work was supported by grants from the National Institutes of Health (5K08HD057555-03,4, 3KO8HD057555-03S1, and 1R01NS071076-01A1) and from the Stein-Oppenheimer Foundation to G.S.L. The authors thank Tamara Horwich MD MS (UCLA) for her assistance with the statistical analysis; Denise Sabatino PhD (University of Pennsylvania) for discussion of FVIII antigen, activity, and light and heavy chain determination, and for providing antibodies that are no longer commercially available; Robin Rosenblatt, Fides Lay, Eun K. Lee, and Luciano Castaneda for their technical assistance.
Footnotes
Conflict of Interest: The authors declare no competing financial interests in relation to the work described.
References
- 1.Takahashi M, Ilan Y, Chowdhury NR, Guida J, Horwitz M, Chowdhury JR. Long term correction of bilirubin-UDP-glucuronosyltransferase deficiency in Gunn rats by administration of a recombinant adenovirus during the neonatal period. J Biol Chem. 1996;271(43):26536–42. doi: 10.1074/jbc.271.43.26536. [DOI] [PubMed] [Google Scholar]
- 2.Xu L, Haskins ME, Melniczek JR, Gao C, Weil MA, O'Malley TM, et al. Transduction of hepatocytes after neonatal delivery of a Moloney murine leukemia virus based retroviral vector results in long-term expression of beta-glucuronidase in mucopolysaccharidosis VII dogs. Mol Ther. 2002;5(2):141–53. doi: 10.1006/mthe.2002.0527. [DOI] [PubMed] [Google Scholar]
- 3.Xu L, Mango RL, Sands MS, Haskins ME, Ellinwood NM, Ponder KP. Evaluation of pathological manifestations of disease in mucopolysaccharidosis VII mice after neonatal hepatic gene therapy. Mol Ther. 2002;6(6):745–58. doi: 10.1006/mthe.2002.0809. [DOI] [PubMed] [Google Scholar]
- 4.Ponder KP, Melniczek JR, Xu L, Weil MA, O'Malley TM, O'Donnell PA, et al. Therapeutic neonatal hepatic gene therapy in mucopolysaccharidosis VII dogs. Proc Natl Acad Sci U S A. 2002;99(20):13102–7. doi: 10.1073/pnas.192353499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mah C, Cresawn KO, Fraites TJ, Jr, Pacak CA, Lewis MA, Zolotukhin I, et al. Sustained correction of glycogen storage disease type II using adeno-associated virus serotype 1 vectors. Gene Ther. 2005;12(18):1405–9. doi: 10.1038/sj.gt.3302550. [DOI] [PubMed] [Google Scholar]
- 6.Chandler RJ, Venditti CP. Long-term rescue of a lethal murine model of methylmalonic acidemia using adeno-associated viral gene therapy. Mol Ther. 2010;18(1):11–6. doi: 10.1038/mt.2009.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carrillo-Carrasco N, Chandler RJ, Chandrasekaran S, Venditti CP. Liver-directed recombinant adeno-associated viral gene delivery rescues a lethal mouse model of methylmalonic acidemia and provides long-term phenotypic correction. Hum Gene Ther. 2010;21(9):1147–54. doi: 10.1089/hum.2010.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sabatino DE, Mackenzie TC, Peranteau W, Edmonson S, Campagnoli C, Liu YL, et al. Persistent expression of hF.IX After tolerance induction by in utero or neonatal administration of AAV-1-F.IX in hemophilia B mice. Mol Ther. 2007;15(9):1677–85. doi: 10.1038/sj.mt.6300219. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J, Xu L, Haskins ME, Parker Ponder K. Neonatal gene transfer with a retroviral vector results in tolerance to human factor IX in mice and dogs. Blood. 2004;103(1):143–51. doi: 10.1182/blood-2003-06-2181. [DOI] [PubMed] [Google Scholar]
- 10.Foust KD, Wang X, McGovern VL, Braun L, Bevan AK, Haidet AM, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol. 2010;28(3):271–4. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Kaufman RJ. Advances toward gene therapy for hemophilia at the millennium. Hum Gene Ther. 1999;10(13):2091–107. doi: 10.1089/10430349950017095. [DOI] [PubMed] [Google Scholar]
- 12.Kay MA, High K. Gene therapy for the hemophilias. Proc Natl Acad Sci U S A. 1999;96(18):9973–5. doi: 10.1073/pnas.96.18.9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12(3):342–7. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 14.Ponder KP. Hemophilia gene therapy: a Holy Grail found. Mol Ther. 2011;19(3):427–8. doi: 10.1038/mt.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nathwani AC, Tuddenham E, Rosales C, McIntosh J, Riddel A, Rustagi PK, et al. Early clinical trial results following administration of a low dose of a novel self complementary adeno-associated viral vector encoding human Factor IX in two subjects with severe hemophilia B. Blood. 2010;116:114. [Google Scholar]
- 16.Mah C, Sarkar R, Zolotukhin I, Schleissing M, Xiao X, Kazazian HH, et al. Dual vectors expressing murine factor VIII result in sustained correction of hemophilia A mice. Hum Gene Ther. 2003;14(2):143–52. doi: 10.1089/104303403321070838. [DOI] [PubMed] [Google Scholar]
- 17.VandenDriessche T, Vanslembrouck V, Goovaerts I, Zwinnen H, Vanderhaeghen ML, Collen D, et al. Long-term expression of human coagulation factor VIII and correction of hemophilia A after in vivo retroviral gene transfer in factor VIII-deficient mice. Proc Natl Acad Sci U S A. 1999;96(18):10379–84. doi: 10.1073/pnas.96.18.10379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiao X, Li J, Samulski RJ. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J Virol. 1996;70(11):8098–108. doi: 10.1128/jvi.70.11.8098-8108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsushita T, Elliger S, Elliger C, Podsakoff G, Villarreal L, Kurtzman GJ, et al. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther. 1998;5(7):938–45. doi: 10.1038/sj.gt.3300680. [DOI] [PubMed] [Google Scholar]
- 20.Hu C, Busuttil RW, Lipshutz GS. RH10 provides superior transgene expression in mice when compared with natural AAV serotypes for neonatal gene therapy. J Gene Med. 2010;12(9):766–78. doi: 10.1002/jgm.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A. 2002;99(18):11854–9. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanlioglu S, Monick MM, Luleci G, Hunninghake GW, Engelhardt JF. Rate limiting steps of AAV transduction and implications for human gene therapy. Curr Gene Ther. 2001;1(2):137–47. doi: 10.2174/1566523013348788. [DOI] [PubMed] [Google Scholar]
- 23.Thomas CE, Storm TA, Huang Z, Kay MA. Rapid uncoating of vector genomes is the key to efficient liver transduction with pseudotyped adeno-associated virus vectors. J Virol. 2004;78(6):3110–22. doi: 10.1128/JVI.78.6.3110-3122.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Xie J, Lu H, Chen L, Hauck B, Samulski RJ, et al. Existence of transient functional double-stranded DNA intermediates during recombinant AAV transduction. Proc Natl Acad Sci U S A. 2007;104(32):13104–9. doi: 10.1073/pnas.0702778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L, Wang H, Bell P, McCarter RJ, He J, Calcedo R, et al. Systematic evaluation of AAV vectors for liver directed gene transfer in murine models. Mol Ther. 2010;18(1):118–25. doi: 10.1038/mt.2009.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qian J, Borovok M, Bi L, Kazazian HH, Jr, Hoyer LW. Inhibitor antibody development and T cell response to human factor VIII in murine hemophilia A. Thromb Haemost. 1999;81(2):240–4. [PubMed] [Google Scholar]
- 27.Scallan CD, Lillicrap D, Jiang H, Qian X, Patarroyo-White SL, Parker AE, et al. Sustained phenotypic correction of canine hemophilia A using an adeno-associated viral vector. Blood. 2003;102(6):2031–7. doi: 10.1182/blood-2003-01-0292. [DOI] [PubMed] [Google Scholar]
- 28.Jiang H, Lillicrap D, Patarroyo-White S, Liu T, Qian X, Scallan CD, et al. Multiyear therapeutic benefit of AAV serotypes 2, 6, and 8 delivering factor VIII to hemophilia A mice and dogs. Blood. 2006;108(1):107–15. doi: 10.1182/blood-2005-12-5115. [DOI] [PubMed] [Google Scholar]
- 29.Sarkar R, Mucci M, Addya S, Tetreault R, Bellinger DA, Nichols TC, et al. Long-term efficacy of adeno-associated virus serotypes 8 and 9 in hemophilia a dogs and mice. Hum Gene Ther. 2006;17(4):427–39. doi: 10.1089/hum.2006.17.427. [DOI] [PubMed] [Google Scholar]
- 30.Sabatino DE, Lange AM, Altynova ES, Sarkar R, Zhou S, Merricks EP, et al. Efficacy and Safety of Long-term Prophylaxis in Severe Hemophilia A Dogs Following Liver Gene Therapy Using AAV Vectors. Mol Ther. 2010 doi: 10.1038/mt.2010.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lofqvist T, Nilsson IM, Berntorp E, Pettersson H. Haemophilia prophylaxis in young patients--a long-term follow-up. J Intern Med. 1997;241(5):395–400. doi: 10.1046/j.1365-2796.1997.130135000.x. [DOI] [PubMed] [Google Scholar]
- 32.Finn JD, Ozelo MC, Sabatino DE, Franck HW, Merricks EP, Crudele JM, et al. Eradication of neutralizing antibodies to factor VIII in canine hemophilia A after liver gene therapy. Blood. 2010;116(26):5842–8. doi: 10.1182/blood-2010-06-288001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fausto N, Webber EM, editors. The Liver: Biology and Pathobiology. Raven; New York: 1994. [Google Scholar]
- 34.Fausto N, Campbell JS. The role of hepatocytes and oval cells in liver regeneration and repopulation. Mech Dev. 2003;120(1):117–30. doi: 10.1016/s0925-4773(02)00338-6. [DOI] [PubMed] [Google Scholar]
- 35.Magami Y, Azuma T, Inokuchi H, Kokuno S, Moriyasu F, Kawai K, et al. Cell proliferation and renewal of normal hepatocytes and bile duct cells in adult mouse liver. Liver. 2002;22(5):419–25. doi: 10.1034/j.1600-0676.2002.01702.x. [DOI] [PubMed] [Google Scholar]
- 36.Nakai H, Yant SR, Storm TA, Fuess S, Meuse L, Kay MA. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J Virol. 2001;75(15):6969–76. doi: 10.1128/JVI.75.15.6969-6976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miao CH, Snyder RO, Schowalter DB, Patijn GA, Donahue B, Winther B, et al. The kinetics of rAAV integration in the liver. Nat Genet. 1998;19(1):13–5. doi: 10.1038/ng0598-13. [DOI] [PubMed] [Google Scholar]
- 38.Cunningham SC, Dane AP, Spinoulas A, Logan GJ, Alexander IE. Gene delivery to the juvenile mouse liver using AAV2/8 vectors. Mol Ther. 2008;16(6):1081–8. doi: 10.1038/mt.2008.72. [DOI] [PubMed] [Google Scholar]
- 39.Wang L, Bell P, Lin J, Calcedo R, Tarantal AF, Wilson JM. AAV8-mediated Hepatic Gene Transfer in Infant Rhesus Monkeys (Macaca mulatta) Molecular Therapy. 2011 doi: 10.1038/mt.2011.151. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malik P, McQuiston SA, Yu XJ, Pepper KA, Krall WJ, Podsakoff GM, et al. Recombinant adeno-associated virus mediates a high level of gene transfer but less efficient integration in the K562 human hematopoietic cell line. J Virol. 1997;71(3):1776–83. doi: 10.1128/jvi.71.3.1776-1783.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flotte TR. Adeno-associated virus-based gene therapy for inherited disorders. Pediatr Res. 2005;58(6):1143–7. doi: 10.1203/01.pdr.0000189226.03684.fe. [DOI] [PubMed] [Google Scholar]
- 42.Grimm D, Pandey K, Nakai H, Storm TA, Kay MA. Liver transduction with recombinant adeno-associated virus is primarily restricted by capsid serotype not vector genotype. J Virol. 2006;80(1):426–39. doi: 10.1128/JVI.80.1.426-439.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Conlon TJ, Cossette T, Erger K, Choi YK, Clarke T, Scott-Jorgensen M, et al. Efficient hepatic delivery and expression from a recombinant adeno-associated virus 8 pseudotyped alpha1-antitrypsin vector. Mol Ther. 2005;12(5):867–75. doi: 10.1016/j.ymthe.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 44.Solomon JB. Foetal and neonatal immunology. In: Neuberger A, Tatum EL, editors. Frontiers of Biology. Vol. 20. North-Holland Publishing Company; Amsterdam/London: 1971. pp. 353–354. [Google Scholar]
- 45.Lechardeur D, Sohn KJ, Haardt M, Joshi PB, Monck M, Graham RW, et al. Metabolic instability of plasmid DNA in the cytosol: a potential barrier to gene transfer. Gene Ther. 1999;6(4):482–97. doi: 10.1038/sj.gt.3300867. [DOI] [PubMed] [Google Scholar]
- 46.Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118(9):3132–42. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302(5644):415–9. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 48.Scallan CD, Liu T, Parker AE, Patarroyo-White SL, Chen H, Jiang H, et al. Phenotypic correction of a mouse model of hemophilia A using AAV2 vectors encoding the heavy and light chains of FVIII. Blood. 2003;102(12):3919–26. doi: 10.1182/blood-2003-01-0222. [DOI] [PubMed] [Google Scholar]
- 49.Sabatino DE, Lange AM, Altynova ES, Sarkar R, Zhou S, Merricks EP, et al. Efficacy and safety of long-term prophylaxis in severe hemophilia A dogs following liver gene therapy using AAV vectors. Mol Ther. 2011;19(3):442–9. doi: 10.1038/mt.2010.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu C, Cela RG, Suzuki M, Lee B, Lipshutz GS. Neonatal helper-dependent adenoviral vector gene therapy mediates correction of hemophilia A and tolerance to human factor VIII. Proc Natl Acad Sci U S A. 2011;108(5):2082–7. doi: 10.1073/pnas.1015571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mingozzi F, Liu YL, Dobrzynski E, Kaufhold A, Liu JH, Wang Y, et al. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest. 2003;111(9):1347–56. doi: 10.1172/JCI16887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jamieson BD, Butler LD, Ahmed R. Effective clearance of a persistent viral infection requires cooperation between virus-specific Lyt2+ T cells and nonspecific bone marrow-derived cells. J Virol. 1987;61(12):3930–7. doi: 10.1128/jvi.61.12.3930-3937.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jamieson BD, Ahmed R. T-cell tolerance: exposure to virus in utero does not cause a permanent deletion of specific T cells. Proc Natl Acad Sci U S A. 1988;85(7):2265–8. doi: 10.1073/pnas.85.7.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis. 2009;199(3):381–90. doi: 10.1086/595830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21(6):704–12. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 56.Lollar P. Pathogenic antibodies to coagulation factors. Part one: factor VIII and factor IX. J Thromb Haemost. 2004;2(7):1082–95. doi: 10.1111/j.1538-7836.2004.00802.x. [DOI] [PubMed] [Google Scholar]
- 57.Fadel S, Sarzotti M. Cellular immune responses in neonates. Int Rev Immunol. 2000;19(2-3):173–93. doi: 10.3109/08830180009088504. [DOI] [PubMed] [Google Scholar]
- 58.Evans MM, Williamson WG, Irvine WJ. The appearance of immunological competence at an early age in New Zealand black mice. Clin Exp Immunol. 1968;3(5):375–83. [PMC free article] [PubMed] [Google Scholar]
- 59.Fan X, Ang A, Pollock-Barziv SM, Dipchand AI, Ruiz P, Wilson G, et al. Donor-specific B-cell tolerance after ABO-incompatible infant heart transplantation. Nat Med. 2004;10(11):1227–33. doi: 10.1038/nm1126. [DOI] [PubMed] [Google Scholar]
- 60.Kay HE, Doe J, Hockley A. Response of human foetal thymocytes to phytohaemagglutinin (PHA) Immunology. 1970;18(3):393–6. [PMC free article] [PubMed] [Google Scholar]
- 61.Yonemura H, Sugawara K, Nakashima K, Nakahara Y, Hamamoto T, Mimaki I, et al. Efficient production of recombinant human factor VIII by co-expression of the heavy and light chains. Protein Eng. 1993;6(6):669–74. doi: 10.1093/protein/6.6.669. [DOI] [PubMed] [Google Scholar]
- 62.Nguyen AT, Dow AC, Kupiec-Weglinski J, Busuttil RW, Lipshutz GS. Evaluation of gene promoters for liver expression by hydrodynamic gene transfer. J Surg Res. 2008;148(1):60–6. doi: 10.1016/j.jss.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bi L, Sarkar R, Naas T, Lawler AM, Pain J, Shumaker SL, et al. Further characterization of factor VIII-deficient mice created by gene targeting: RNA and protein studies. Blood. 1996;88(9):3446–50. [PubMed] [Google Scholar]
- 64.Sabatino DE, Freguia CF, Toso R, Santos A, Merricks EP, Kazazian HH, Jr, et al. Recombinant canine B-domain-deleted FVIII exhibits high specific activity and is safe in the canine hemophilia A model. Blood. 2009;114(20):4562–5. doi: 10.1182/blood-2009-05-220327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dejana E, Callioni A, Quintana A, de Gaetano G. Bleeding time in laboratory animals. II - A comparison of different assay conditions in rats. Thromb Res. 1979;15(1-2):191–7. doi: 10.1016/0049-3848(79)90064-1. [DOI] [PubMed] [Google Scholar]