

Abstract
Signaling by target of rapamycin (mTOR in mammals) has been shown to modulate lifespan in several model organisms ranging from yeast to mice. In mice, reduced mTOR signaling by chronic rapamycin treatment leads to lifespan extension, raising the possibility that rapamycin and its analogs may benefit the aging brain and serve as effective treatments of age-related neurodegenerative diseases. Here, we review mTOR signaling and how neurons utilize mTOR to regulate brain function, including regulation of feeding, synaptic plasticity and memory formation. Additionally, we discuss recent findings that evaluate the mechanisms by which reduced mTOR activity might benefit the aging brain in normal and pathological states. We will focus on recent studies investigating mTOR and Alzheimer s disease, Parkinson s disease, and polyglutamine expansion syndromes such as Huntington s disease.
Keywords: rapamycin, TOR, neurodegeneration, Alzheimer's disease, Huntington's Disease Parkinson's disease
Part I: regulation and function of mTORC1
1.1 Introduction
Aging is a major risk factor for neurodegenerative disease. Studies in model organisms ranging from yeast to mice have shown that lifespan can be extended by reducing TOR activity (Stanfel et al, 2009), raising the possibility that interventions that reduce TOR signaling might delay or reduce the pathology of age-related neurodegenerative disorders. However, recent studies have also shown that TOR activity contributes to several functions of the adult brain; activation of TOR contributes to processes such as synaptic plasticity and metabolic regulation. Thus, the roles of TOR activity in normal brain functions should be considered when evaluating the therapeutic potential of reduced TOR activity. Here, we will discuss cellular and neuronal regulation of TOR, the role of TOR activity in synaptic plasticity, memory formation, and feeding behavior. We will then discuss how TOR activity may be altered in neurodegenerative diseases, and review studies examining the potential benefits of reducing TOR activity in several models of neurodegeneration.
1.2 Rapamycin selectively inhibits mTORC1
TOR genes were first described in yeast as the pharmacological targets of the microbicide rapamycin (Heitman et al., 1991). TORs were subsequently described other invertebrate and vertebrate organisms. TOR is a member of the phosphoinositol kinase related kinase (PIKK) family of kinases, although TOR phosphorylates proteins on serine and threonine residues. In yeast, nematodes, flies, and mammals, TOR exists in two functionally distinct complexes (TORC1 and TORC2.) TORC2 contains specific proteins including rictor, is involved in cyctoskeletal remodeling (Cybulski and Hall, 2009) and is required for activation of Akt (Sarbassov et al., 2005). TORC1 also contains specific proteins including raptor, and is involved in translation, energy regulation, autophagy, and cell growth. TORC1 is the primary target of inhibition by rapamycin and will be the subject of this review. Rapamycin is produced by the bacterium Streptomyces hygroscopicus. The drug gets its name from its habitat: soil samples from Rapa Nui (Easter Island; Sehgal, 2003). Rapamycin selectively inhibits TORC1 activity by forming a trimolecular complex with mTOR and FKBP12. Rapamycin and its analogs have proved to be a useful tool in elucidating the functions of mTORC1. Of note, increasing evidence suggests that chronic rapamycin exposures results in TORC2 inhibition likely through indirect mechanisms (Sarbassov et al., 2006).
Rapamycin treatment has been shown to extend lifespan in a wide range of models, including yeast, C. elegans, D. melanogaster, and mice (Vellai et al, 2003; Kapahi et al, 2004; Kaeberlein et al, 2005; Harrison et al, 2009). These findings have generated interest in rapamycin and its derivatives as potential treatments for age-related diseases, including neurodegenerative syndromes. However, mTORC1 activity also regulates aspects of normal brain function. Thus, the potential benefit of TOR inhibition as a treatment for neurodegerative disease must be considered along with its potential for disrupting normal brain function. TORC1 plays a role in brain development, as reviewed previously (Jaworski and Sheng, 2006; Swiech et al., 2008). Here, we will discuss the role of TOR signaling in adult brain functioning.
1.3 Regulation of mTORC1 in mammalian cells
In mammalian cells, enhanced mTORC1 function is mediated through activation of receptors for growth factors, hormones, and cytokines. For example, binding of insulin to the insulin receptor recruits insulin receptor substrate (IRS) and the regulatory subunit of type I phosphoinositol 3-kinase (PI3K). These events activate the catalytic subunit of PI3K, generating the production of phosphatidylinolitol (3,4,5)-triphosphate (PIP3). PIP3 recruits PDK1 and Akt to the membrane, allowing PDK1 to phosphorylate Akt. This event allows for a second phosphorylation of Akt by mTORC2. This dual phosphorylation stabilizes and activates Akt s kinase activity, which in turn promotes cel l growth and survival through its numerous targets (Scheid and Woodgett, 2001). In addition, Akt removes inhibitory constraint of mTORC1 signaling by phosphorylating TSC2 and PRAS40 (Inoki et al., 2002; Potter et al., 2002; Vander Haar et al., 2007). TSC2 (and TSC1) act as GTP activating proteins (GAPs) to inhibit Rheb, a kinase which activates mTORC1. Phophorylation of TSC2 by akt inhibits the GAP activity. PRAS40 binds and prevents mTORC1 activity and phosphorylation of PRAS40 by Akt releases it from the complex (Vander Haar et al., 2007). In addition to PI3K activation, insulin receptor activation can trigger ERK/MAPK signaling through ras activation. Ras activates raf, and raf phosphorylates and activates MEK, which in turn activates ERK1 and ERK2. ERK1/2 can phosphorylate and inhibit TSC2, and so, along with akt signaling, MAPK can remove inhibition of mTORC1 activity.
In addition to extracellular stimuli, mTORC1 is responsive to internal cellular signals. For example, when cellular energy is low, AMPK can activate TSC2 and drive inhibition of mTORC1. ATP is the main source of cellular energy, and as energy is expended, levels of AMP accumulate. Increasing levels of AMP allow for the activation of AMPK by LKB1 (Hardie, 2008), while glycogen, a cellular energy reserve, can bind and inhibit AMPK. Activated AMPK can phosphoryate and promote the GAP activity of TSC2, thereby reducing TORC1 activity. Furthermore, AMPK can phosphorylate raptor, removing it from the TORC1 complex and disabling TORC1 activity. Through AMPK, mTORC1 activity responds to the cell s current and short-term energy availability.
The activity of mTORC1 is also sensitive to nutrient availability. Increasing the cellular concentration of amino acids activates mTORC1, although the mechanism by which it does so has not been fully described (Wang and Proud, 2009). Activity of mTORC1 is responsive the levels of essential amino acids, such as leucine. Amino acid stimulation of mTORC1 occurs downstream of the TSC1/2, since mTORC1 activity still responds to amino acid levels in cells lacking TSC2. Activation of mTORC1 by amino acids is thought to require MAP4K3, a map kinase kinase (Findlay et al., 2007), and the Rag GTPases (Sancak et al., 2008).
1.5 Actions of mTORC1 in mammalian cells
Perhaps the best characterized action of the mTORC1 pathway is its regulation of translation initiation. Mammalian TORC1 stimulates translational activity by phosphorylating two targets, 4E-BP and S6K. Phosphorylation of 4E-BP by mTORC1 releases 4E-BP from eiF4E. This allows eiF4E to associate with eiF-4G leading to the formation of the eiF-4F complex, which facilitates the loading of ribosomes onto mRNA. Mammals have 3 isoforms of 4E-BP; 4E-BP2 is the main 4E-BP in the mammalian brain (Banko et al., 2005). Phosphoryation of the S6Ks by mTOR also promotes translation initiation. The S6Ks take their name for their ability to phosphorylate the ribosomal protein S6. While S6 phosphorylation correlates with translational activity, there is controversy as to what contribution S6 phosphorylation makes to modify translational activity (Ruvinsky and Meyuhas, 2006). S6K promotes initiation by phosphorylation of eIF4B and regulates the elongation phase of translation through phosphoryation of eEF2 kinase. Since eEF2 kinase down regulates eEF2, the actions of S6K1 promote eEF2 s role in elongation.
One consequence of mTORC1 activation is enhanced ribosome biogenesis and levels of translation machinery. While mTORC1 promotes CAP-dependent translation, the 5 TOP mRNAs are preferentially sensitive to mTORC1 activity and include messages that encode ribosomal proteins and translational machinery such as elongation factors (Ruvinsky and Meyuhas, 2006). Thus, translational initiation activity regulated by mTORC1 can increase the cell s capacity to generate new proteins. Additionally, mTORC1 activity promotes ribosome biogenesis, and rapamycin inhibits transcription of ribosomal RNA genes, ribosomal protein genes, and transfer RNA genes.
Another activity of mTOR is the suppression of macroautophagy (autophagy). Autophagy degrades material; cyctosolic material and organelles are engulfed in a double membrane bound vesicle and shuttled to the lysozome. By degrading existing proteins, one result of autophagy is to increase available amino acids for the synthesis of new proteins. Another important contribution is the turnover of damaged molecules and organelles. Thus, from a more holistic perspective, mTORC1 can be thought of as regulating the balance between protein synthesis and turnover.
1.6 mTORC1 and lifespan
Dietary restriction is an intervention known to increase lifespan in model organisms, including yeast, nematodes, flies, and mice (Stanfel et al., 2009). Dietary restriction has recently been demonstrated to extend lifespan in non-human primates (Colman et al., 2009), suggesting that similar interventions may benefit human lifespan. Dietary restriction has been shown to reduce TORC1 activity, and genetic or pharmacological reduction of TOR activity is sufficient to extend lifespan (Stanfel et al., 2009). Furthermore, dietary restriction fails to extend lifespan when TOR activity is reduced in yeast, nematodes, or flies (Hansen et al., 2008; Kaeberlein et al., 2005; Kapahi et al., 2004). Thus, data from experiments in these models suggest that dietary restriction may modulate lifespan through suppression of TOR activity, or that rapamycin and dietary restriction benefit lifespan through overlapping mechanisms.
New evidence demonstrates that pharmacological disruption of mTORC1 is sufficient to extend lifespan in mice as well. The NIA s Aging Intervention Testing Program has recently identified mTOR as a pharmacological target to increase longevity in mice (Harrison et al., 2009). The most surprising aspect of this study was that rapamycin treatment was effective at increasing lifespan even though treatment began at 600 days of age, which the authors point out is the equivalent of 60 years in humans. Additional evidence that the mTOR pathway is important for lifespan in mammals is the recent observation that mice lacking S6K1 have increased lifespan (Selman et al., 2009). Since S6K1 is activated upon phosphorylation by mTOR, this observation further demonstrates the importance of this pathway. These mice have also been shown to have resistance to diet-induced obesity (Um et al., 2004), a trait associated with longevity in mice.
Part II neuronal mTORC1
2.1 Regulation of mTORC1 in neurons
In neurons, increased mTORC1 activity can result from several stimuli, including growth factors such as brain derived neurotrophic factor (BDNF), cytokines such as leptin, influx of calcium, and neurotransmitters through activation of G protien coupled receptors (GPCRS) or ionotropic receptors (Swiech et al., 2008).
One important trigger for activation of mTORC1 is influx of calcium through membrane channels. Calcium influx can result from increased neural activity, for example, through the opening of voltage gated calcium channels or NMDA receptors. Calcium/calmodulin can activate ras, which in turn can stimulate ERK/MAPK and PI3K. Calcium/calmodulin may also directly activate regulatory PI3K. Furthermore, calcium/calmodulin can stimulate calcium-stimulated adenyly cyclases, generating the secondary messenger cAMP. In neurons, cAMP can trigger ERK/MAPK signaling through activation of PKA and EPAC. Finally, stimulation of Gq, and Gi coupled GCPRs can all lead to stimulation of mTORC1. Beta/gamma subunits of G proteins coupled to GPCRs can activate PI3K. So while Gs stimulation has the potential to stimulate mTORC1 in a cAMP and ERK/MAPK dependent manner, Gi can inhibit cAMP production and still activate mTORC1. Thus, the crosstalk between cAMP, ERK/MAPK, and PI3K pathways allow for mTORC1 to be activated by a range of stimuli (Figure 1).
Figure 1.
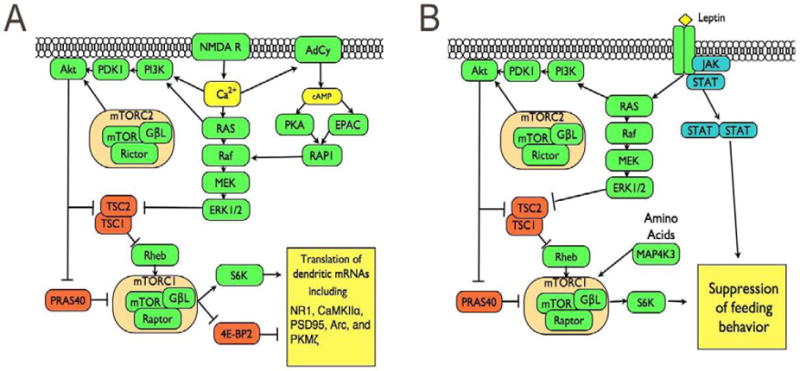
Activation of mTORC1 in neurons. A) mTORC1 activation by LTP-inducing stimulus. Tetanic stimualation results in depolarization and opening of NMDA receptor channels. The resulting influx of calcium triggers activation of PI3K, ERK/MAPK, and cAMP/PKA pathways. Resulting activation of ERK1/2 and Akt inhibit TSC1/2, removing constraint on Rheb kinase. Akt activation also serves to remove inhibition of the mTORC1 complex by PRAS40. Removal of these constraints allow for mTORC1-depend phosphorylation of S6K and 4E-BP2, which promotes translation, including for mRNAs with known functions in synaptic function. B) mTORC1 activation in the hypothamalus can suppress feeding behavior. Leptin activates JAK/STAT and PI3K signaling pathways. As above, PI3K activity can remove inhibition of mTORC1 activity. In addition, aminio acids can increase mTORC1 activity in part through the inhibition of TSC2. Activation of mTORC1 suppresses feeding behavior in an S6 kinase dependent manner.
Recently, Hoeffer et al (2008) identified a role for FKBP12 in the regulation of mTORC1 activity. A traditional FKBP12 knockout mouse is inviable, so a conditional knockout was generated that lacked FKBP12 specifically in the hippocampus. Removal of FKBP12 increased the association of raptor to mTOR without altering the total levels of either protein. This increased mTOR/raptor association was accompanied by elevated mTORC1 activity, suggesting an endogenous role of FKBP12 for opposing mTORC1 activity by limiting mTORC1 complex formation.
2.3 mTORC1 and Synaptic Plasticity
Learning and memory are thought to be mediated by altering the strength or number of connections (synapses) between neurons. Given the complexity of the brain, our understanding of the mechanisms by which neurons alter their connectivity has benefited greatly from in vitro tissue preparations that model synaptic plasticity. A large body of work has tried to enumerate the mechanisms by which physiological or chemical stimulation can increase (potentiation) or decrease (depression) the electrophysiological response of neurons. Two well-modeled forms of synaptic plasticity include long-term potentiation (LTP) and long-term depression (LTD).
De novo protein synthesis has a well defined role in LTP and LTD. For both processes, there exists an early phase and a late phase with distinct molecular requirements. For example, in rodent hippocampal tissue preparations, induction of multiple trains of tetanic stimulation to the Schaffer Collateral (a connection between the CA3 and CA1 regions of the hippocampus) results in an elevated response to stimulation that can last for hours. Requirements for the induction of early-phase LTP include the passage of calcium through NMDA receptors and activation of Calcium/calmodulin depent protien kinase II (CamKII). Extension beyond the early-phase of LTP requires activation of cAMP and ERK/MAPK signaling, CREB-mediated transcription, and de novo protein synthesis (Xia and Storm, 2005).
The mTORC1 pathway likely contributes to synaptic plasticity by regulating protein synthesis. mTORC1 is required for late-phase LTP, since application of rapamycin specifically attenuates late-phase LTP in Schaffer-collateral preparations (Tang et al, 2002). Furthermore, stimuli that induce LTP activate mTOR in a PI3K and ERK/MAPK dependent manner (Gobert et al., 2008; Kelleher et al., 2004; Tsokas et al., 2005; Tsokas et al., 2007). The induction of LTP leads to mTORC1-dependent translation, including translation of 5 TOP containing mRNAs (Tsokas et al., 2005). Since 5 TOP messages include ribosomal proteins and elongation factors, mTORC1 activation may prime further translation events in the neuron. Additional mRNAs that are translated in response to mTORC1 activation have well characterized roles in synaptic function and plasticity, and include NR1, CamKII alpha, PSD95, Arc, and PKM zeta (Gong et al., 2006; Kelly et al., 2007; Lee et al., 2005; Schratt et al., 2004).
2.4 mTORC1 and localized protein synthesis
If the brain is thought to store and encode information by adjusting specific connections, then a challenge to understanding molecular mechanisms of learning and memory is identifying how neurons might specifically modulate some synapses while leaving others unaltered. A potential mechanism has been termed synaptic tagging, where by a local, persistent protein modification might serve as a marker for where long-term synaptic alterations must occur. Additionally, localized protein synthesis might support such as function, whereby proteins required for plasticity are synthesized in the vicinity of the activated synapse.
Mammalian TORC1 can facilitate local protein synthesis. Dendrites contain numerous mRNAs encoding proteins influencing synaptic function (Bramham and Wells, 2007). Dendritic localization is likely not a result of passive diffusion from the soma since localization of mRNA in neurons can depend on sequences in the 3 untranslated region of mRNA (Miller et al., 2002). Furthermore, synaptic activity recruits ribosomes from the dendritic shaft to spines (Ostroff et al., 2002), thus mobilizing the ribosome to the synapse. Polysome analysis in cultured neurons has demonstrated that stimulation of mTORC1 by BDNF increases translational activity in synapatosomes (Schratt et al., 2004). BDNF increased the amount of mRNA bound to ribosomes at the synapse. This increase was not due to increased levels of total mRNA by BDNF, since neurons were co-treated with actinomysin D, an inhibitor of general transcriptional activity. While rapamycin reduced translation initiation, translation was not completely suppressed. Furthermore, rapamycin only disrupted translation of a subset of messages, suggesting that other mechanisms contribute to localized protein synthesis.
Additional evidence suggests a role for mTORC1-dependent synaptic translation stimulated at the synapse. Protocols have been developed to induce LTP in preparations where dendrites are separated from the soma and nucleus (Cracco et al., 2005; Vickers et al., 2005). In these studies, general protein synthesis inhibitors or rapamycin prevented LTP induction in these preps, illustrating how mTORC1-mediated protein synthesis in active dendrites can be sufficient to modulate long lasting synaptic alterations. Further suggesting the ability of mTORC1 to selectively activate regions of the neuron is the observation that calcium stimulation results in localized pockets of mTORC1 activity as opposed to evenly dispersed activation throughout the dendrite (Cammalleri et al., 2003).
Activation of mTOR can directly act at synapses to promote synthesis of proteins necessary to facilitate plasticity, but mTOR may also modulate neural activity by suppressing translation of specific messages. Kv1.1 is a potassium channel whose expression is induced near in the dendrites during periods of low neuronal activity. Functional Kv1.1 lowers neuronal activity. When cultured neurons are treated with rapamycin, Kv1.1 is synthesized in dendrites. Furthermore, calcium stimulation of mTORC1 activity reduces levels of Kv1.1. This observation suggests that mTORC1 supports neuronal activity both by the selective promotion and suppression of translation of mRNAs (Raab-Graham et al., 2006).
2.5 mTORC1 and memory
Protein synthesis is a well established requirement for memory formation. Rodent models have demonstrated that de novo protein synthesis is required to stabilize a short-term memory into a long-term memory (Abel and Lattal, 2001). Rapamycin has been shown to disrupt this process in several behavior paradigms. For example, cued fear conditioning is a memory task where rodents form an association between an auditory tone and a food shock. Formation of long-term fear memory was impaired when rapamycin was injected bilaterally into the amygdala during training (Parsons et al., 2006). Rapamycin injection disrupted memory performance 24 hours, but not 3 hours, after training (Hoeffer et al., 2008), demonstrating that the animal could form the fear association and its accompanying short-term memory. Thus, rapamycin specifically impaired the consolidation of the memory to long-term storage. Rapamycin has been shown to disrupt consolidation of memories that depend on other brain regions, including hippocampus-dependent spatial memory (Dash et al., 2006), auditory cortex-dependent memory (Schicknick et al., 2008; Tischmeyer et al., 2003), gustatory cortex-dependent memory for taste aversion (Belelovsky et al., 2009), and prefrontal cortex-dependent trace fear memory (Sui et al., 2008).
Enhancing neuronal mTORC1 activity may improve memory function to an extent. Injection of glucose into the hippocampus during training improved long-term spatial memory in an mTOR-dependent manner (Dash et al., 2006). In an aforementioned model, removal of FKBP12 in mouse hippocampal neurons increases the complex formation of mTORC1 and its activity. Enhancing mTORC1 in this manner improved memory on tests for hippocampus-dependent memory (Hoeffer et al., 2008). However, these mice also displayed increased tendency for behavioral perseverance and repetition, characteristics associated with animal models of neurological disorders such as obsessive compulsive disorder and schizophrenia.
Alternatively, increasing mTORC1 activity can disrupt memory processing. This has been a proposed mechanism for memory loss caused by cannabinoids. In mice activation of CB1 receptors, the main endocannabinoid receptors in the brain, by THC disrupts formation of hippocampus-dependent memory (Puighermanal et al., 2009). CB1 receptor activation by THC on inhibitory GABAergic neurons increased hippocampal mTORC1 activity by increasing excitatory neurotransmission in the hippocampus. Injection of low dose of rapamycin along with THC removed memory impairment. Another important example of memory deficits associated with overactive mTORC1 are human patients and animal models of Tuberous Sclerosis. This condition stems from mutations in TSC1 or 2 that cause reduced TSC1/2 and enhanced mTORC1 activity (Ehninger et al., 2009). In a transgenic mouse model of tuberous sclerosis, rapamycin treatment rescued memory deficits (Ehninger et al., 2008) suggesting that memory impairments are due, at least in part, to abnormal mTOR signaling in adult neurons as opposed to developmental effects of the disease. Thus, while mTORC1 activation is required for memory formation, unregulated mTORC1 signaling disrupts memory function.
The requirement of mTORC1 activation for synaptic plasticity and the consolidation of long-term memories should must be taken into account when evaluating the potential of rapamycin to treat disease or extend lifespan. For example, while mice lacking S6K1 are resistant to obesity and are long-lived, they display impaired memory function (Antion et al., 2008). Furthermore, rapamycin can impair memory performance in several learning paradigms. Prolonged treatment with rapamycin can lead to inhibition of TORC2, a concern since PI3K signaling supports memory function (Chen et al., 2005; Lin et al., 2001). Thus, cognitive function should be taken into account when designing rapamycin treatments. However, recent observations suggest that doses of rapamycin that attenuate pathology in disease models may be below the threshold to disrupt memory formation (Caccamo et al., 2010; Spilman et al., 2010). Additionally, reports of human heart transplant patients taking Everolimus, an mTORC1 inhibitor, as an immunosuppressant did not experience cognitive impairment due to the drug (Lang et al., 2009). Since interventions do not necessarily effect different memory functions equally (Mizuno and Giese, 2005), further work is required to evaluate if beneficial levels of rapamycin leave memory function intact. Still, these data suggest a strong possibility that inhibitors of mTORC1 may benefit health without detrimental cognitive side-effects; specifically, there may be therapeutic dose ranges which provide the benefits and avoid detrimental consequences.
2.6 Hypothalamic mTORC1 regulation of feeding behavior
Given how mTORC1 can respond to extracellular and intracellular signals regarding nutrient and energy status, it should not be surprising that the brain utilizes mTORC1 signaling to regulate food intake. The arcuate nucleus and paraventricular nucleus of the hypothalamus in rats have a high level of phosphorylated S6K1, a marker for mTORC1 activity (Cota et al., 2006). When rats are fasted, this signal decreases, demonstrating that this activity reflects feeding status. Injecting the amino acid leucine intracerebroventricularly (ICV) raised phospho-S6K1 levels in the arcuate nucleus and suppressed feeding behavior in fasted rats. Infusion of rapamycin along with leucine removed this supression. Furthermore, leptin, a circulating cyctokine that suppresses feeding, was shown to stimulate mTORC1 activity in the hypothalamus. As with leucine, suppression of feeding by leptin required mTORC1 activity (Cota et al., 2006). Thus, hypothalamic mTORC1 activity can coordinate multiple signals to regulate feeding behavior.
A subsequent study demonstrated that hypothalamic mTORC1 regulates feeding behavior, at least in part, through S6K1 (Blouet et al., 2008). Viral delivery of dominant negative (DN) S6K1 into the hypothalamus increased feeding behavior of rats. Conversely, constitutively active (CA) S6K1 suppressed feeding behavior. Rats with hypothalamic CA S6K1 were resistant to weight gain when fed a high fat diet for 2 weeks. Interestingly, while S6K1 KO are resistant to high fat diet induced obesity, S6K1 KO mice display increased feeding behavior, as measured by food intake normalized to body weight (Um et al., 2004).
These data might suggest that increasing hypothalamic mTORC1 activity could be a strategy for treatment of metabolic disorders. However, this notion is contradicted by a study which chronically activated mTORC1 activity in POMC neurons of the hypothalamus by conditional knock out of TSC1 (Mori et al., 2009). POMC neurons are found in the arcuate nuclease of the hypothalamus and are activated by leptin to repress feeding behavior. POMC TSC1 KO mice developed leptin resistance, increased feeding behavior and obesity. Furthermore, these phenotypes were reversible by treatment with rapamycin, suggesting that the metabolic phenotypes observed in POMC TSC1 KO were not due to developmental alterations, but persistent mTORC1 overactivity. Since overfeeding and excess nutrient intake result in elevated hypothalamic mTORC1 activity, this mouse may model changes in the brain during the development of metabolic disorders. Together, studies examining hypothalamic mTOR illustrate how acute mTOR activity responds to sufficient nutrient levels and signals cessation of feeding behavior, and how chronic elevation of mTORC1 activity can contribute to complications associated with metabolic disorders and homeostatic imbalance.
2.7 Other potential brain-specific functions of mTORC1
The ability of the mTORC1 pathway to rapidly and selectively translate mRNAs allows neurons to utilize this pathway to produce long-term changes to various signals. This suggests that mTORC1 likely contributes to other brain functions. For example, the suprachiasmatic nucleus (SCN) is the master regulator of circadian rhythm, and can adjust the circadian cycle in response to environmental cues. Recent work has demonstrated that mTORC1-mediated translation in the SCN is required for circadian entrainment (Cao et al., 2010). Since many functions of the brain require neurons to swiftly alter properties in response to stimuli, more functional roles of neuronal mTOR signaling will likely be identified.
Part III mTORC1 and the aging brain
3.1 Normal Brain Aging
Initial studies on aging and brain morphology beginning in the 1950 s reported an age-associated loss of neurons. When these studies were corrected for technical issues such as tissue processing and screening for healthy as opposed to diseased individuals, it appeared that changes in the aging brain are much more subtle, (reviewed in Burke and Barnes, 2006). Significant neuron loss is not seen in non-diseased aged individuals for most brain regions, although there are exceptions; a notable one is the substantia nigra. In humans, the number of dopaminergic neurons in this region decreases steadily with age (McGeer et al., 1977; Rudow et al., 2008). In a similar vein, morphological changes of neurons that accompany aging are subtle and vary by neuron type and region of the brain. Normal aging reduces the number of dendritiic spines, dendritic arborization is increased in certain brain regions, while remaining unchanged in others. As is the case with other cells in the body, damage accumulates in neurons as aging progresses. Aged neurons are more sensitive to oxidative stress, and accumulate increased levels of damaged proteins, phospholipids, and nucleic acids (Mattson et al., 2002).
Work with animal models also demonstrates changes in the aged brain. Tissue taken from aged rodents shows altered plasticity. Aged neurons have slower cyctosolic calcium clearance and higher resting concentration of cyctosolic calcium. Normal aging is accompanied by reduced memory function, and signaling pathways that support memory function are altered in the aged brain (Thibault et al., 2007; Toescu, 2005). Reductions in cAMP levels, adenylyl cyclase activity, and CREB mediated transcription have all been reported in aged rodent brain (Garelick et al., 2009; Mouravlev et al., 2006). Learning events fail to trigger CREB activitation in neurons of aged animals to the same degree that is seen in younger animals (Mouravlev et al., 2006). Another change associated with aging is reduced adult neurogenesis. Neurogenesis occurs in the adult brain but is limited to the dentate gyrus region of the hippocampus and the subventricular zone. There progenitor cells continually divide and giving rise to cells that can develop into functional neurons. This process continues through adult life but is reduced in normal aging.
3.2 mTORC1 and normal brain aging
As rapamycin has only recently been established to extend lifespan in mice, not much is known about its effects on age-related phenotypes pertaining to the mammalian brain at doses associated with delayed aging. It also remains to seen if the function of neuronal mTOR changes throughout age. However, given the potential overlap between rapamycin and dietary restriction as interventions to extend lifespan, results from dietary restriction studies may predict potential benefits of rapamycin treatment for the aged brain. Since dietary restriction and rapamycin treatment may extend life-span through overlapping mechanisms, effects of dietary restriction on the aging brain could generate hypotheses predicting the outcome of rapamycin treatment.
Dietary restriction has been shown to reduce phenotypes associated with advanced age in the brain (Mattson et al., 2002). For example, this reduced feeding regimen leads to lower levels of oxidative stress in aged neurons. Dietary restriction increases neurogenesis in the dentate gyrus of the hippocampus in aged mice (Lee et al, 2000). Importantly, dietary restriction can reduce pathology in models of age related neurodegeneration, including models of Alzheimer s, Parkinson s, and poly-glutamine diseases. Another benefit of dietary restriction could be memory improvement in aged rodents (reviewed by Mattson et al, 2002).
3.3 mTORC1 as a target for treating neurodegeneration
There are different hypothetical mechanisms by which rapamycin treatment might benefit patients with age-related neurodegenerative disorders. First, rapamycin treatment extends lifespan; if rapamycin can delay the aging process, it may delay or prevent neurodegenaration by preserving a young phenotype of the brain. Alternatively, mTOR inhibition might induce stress response pathways or altar metabolism in neurons allowing them to be better equipped to survive the neurotoxic insults involved with neurodegenerative diseases. Lastly, mTOR activity may be altered in these diseases, and this alteration may be intrinsically linked to pathogenesis. These potential mechanisms need not be mutually exclusive, and will be discussed in the examples below.
3.4 Alzheimer s disease
Alzheimer s disease is the most common neurodegenerative disorder and affects about 10% of the population over the age of 70. Alzheimer s disease is the most common form of dementia in the elderly (Mattson et al., 2002). As the disease progresses, Alzheimer s patients lose memory, language, and can exhibit emotiona l disturbances. Patients in late phase of Alzheimer's are completely dependent on others for care.
Alzheimer s diseased brains are characterized by the presence of amyloid plaques and tau tangles. While in most cases the cause of Alzhiemer s disease is unknown, a small fraction of Alzheimer s cases are triggered by mutations in the genes coding for amyloid precursor protein (APP) gene or in Presinilin. In both cases, these mutations result in processing of APP leading to a higher level of Aβ342, a cleavage product of APP (LaFerla et al., 2007). Additionally, mutations in the gene that encodes tau, a main component of tau tangles, lead to inherited forms of neurodegeneration that contain neurofibulary tangles but not Aβ plaques, such as frontotemporal dementias (Ballatore et al., 2007). The fact that mutated tau can lead to tangle formation and neurodegeneration in diseases that do not feature amyloid beta plaques, and the appearance of amyloid plaques before tau tangles in affected human brains suggest that abnormal tau processing may be downstream of amyloid beta accumulation in Alzheimer s disease pathology (Hardy and Higgins, 1992).
Studies examining human tissue report increased activation of the mTOR pathway in affected brain areas in Alzheimer s diseased brains compared to non-diseased control tissue (Li et al., 2005). However, this increase in mTOR activation is accompanied by a reduction in translational activity (Langstrom et al., 1989), a decrease in ribosomal material, and an increase in oxidative-damaged ribosomal RNA (Ding et al., 2005). These changes in mTORC1 activity and translation were limited to brain regions affected with Alzheimer s pathology. Yet while analysis suggests that overal l translation is decreased, levels of certain proteins can accumulate, including tau. Interestingly, activation of mTORC1 can promote translation of tau (An et al., 2003; Li et al., 2005).
There is a lack of consensus of how mTORC1 might become activated in Alzheimer s disease and how it may contribute to pathogenesis. Cell culture models have been mixed examining the roles of Aβ, with some models reporting that Aβ decreases mTORC1 signaling, and others reporting an increase (reviewed by Swiech et al, 2008). Studies examining tau are also conflicting. As stated before, mTOR activity can increase levels of tau protein production, suggesting that activation of mTORC1 may be upstream of tau pathology. Conversely, in a drosophila model of tauopathy, overexpression of wild-type or mutant tau induced mTORC1 activity, neural cell cycle activation, and neurodegeneration (Khurana et al., 2006). This feature recapitulated an aspect of Alzhimer s; abnormal expression of cell cycle markers and tetraploidy have been observed in neurons form autopsied Alzhiemer s brains (Husseman et al., 2000; Yang et al., 2001). Rapamycin treatment to flies expressing mutant tau prevented the abnormal cell cycle activation and neuron death (Khurana et al., 2006). This suggests that elevated mTORC1 may drive neurodegeneration in Alzheimer s disease.
In addition to rapamycin s ability to ameliorate pathology in the fly model, two recent reports demonstrate that rapamycin can benefit mouse models of Alzheimer s disease. The triple transgenic mouse model of Alzheimer s disease contains mutations in PS1, APP, and tau (Oddo et al., 2003). While mouse models of Alzheimer s disease don t display the widespread loss of neurons seen in human cases, the triple transgenic mice experience progressive neuron loss and develop extracellular Aβ plaques and tau tangles. These mice model also develop impaired hippocampus-dependent memory. Another mouse model of Alzheimer s, PDAPP, has mutant APP that leads to the generation of amyloid beta plaques and memory impairments (Games et al., 1995). In both models,the rapamycin feeding regimen shown to extend life span in mice (Harrison et al., 2009) reduced pathology (Caccamo et al., 2010; Spilman et al., 2010). Both studies demonstrated that rapamycin treatment increased markers of autophagy in neurons, and reduced levels of Aβ and plaques. Tangle formation was reduced in the triple transgenic mouse. In both studies, rapamycin feeding rescued memory impairments in the diseased mice. Importantly, rapamycin feeding did not impair memory in non-transgenic control mice, suggesting that levels of rapamycin that induce autophagy and improve pathology in Alzheimer s models do not disrupt mTORC1 activity that is required for memory function in these paradigms.
3.5 Parkinson s disease
Parkinson s disease is the second most common neurodegenerative disorder, and affects up to 1 % of the elderly population. Parkinson s disease is characterized by a loss of dopamine (DA) producing neurons in the substantia nigra (SN) and intraneuronal inclusions termed Lewy bodies. The symptoms of Parkinson s disease include a difficulty in starting or continuing movements termed bradykinesia, rigidity, and involuntary tremors. Approximately 5-10% of Parkinson s disease cases are inherited. The familial forms of Parkinson s stem from autosomal dominant mutations in SCNA and LRRK2, and from autosomal recessive mutations in PARKIN, PINK1, and DJ1. SCNA encodes alpha synuclein, a major component of Lewy bodies. Additionally, chemicals that are selectively toxic to DA neurons have been identified, and include 6-OHDA, MPTP, and the pesticides rotenone and paraquat (Betarbet et al., 2002).
Studies suggest there may be a complex relationship between pathology observed in Parkinson s disease and mTORC1 activity. REDD1, a protein that inhibits TOR activity, is increased in the substantia nigra of MPTP treated mice and in human Parkinson s patients (Malagelada et al., 2006). When Parkinson s disease was then modeled in PC12 cells, a catecholaminergic cell line that shares sensitivity to toxins associated with Parkinson s, REDD1 could be induced by 6-OHDA treatment, a drug that induces dopaminergic cell death. Treatment with siRNA against REDD1 blunted the toxicity of 6-OHDA to PC12 cells and increased mTORC1 activity in these cells. Furthermore, increasing the activity of mTORC1 by silencing TSC2 also reduced 6-OHDA toxicity, suggesting that increasing mTORC1 activity could be protective (Malagelada et al., 2006).
However, a follow up study from the same group demonstrated that rapamycin could also reduce 6-OHDA toxicity in PC12 cells (Malagelada et al., 2010). Importantly, rapamycin treatment increased survival of DA neurons in the SN in mice treated with MPTP. Further investigation with the PC12 cell model demonstrated REDD1 was translated in an mTORC1 dependent manner. Additionally, REDD1 disrupted mTORC2-mediated activation of akt, and activation of this kinase has been shown to reduce apoptosis in models of Parkinson s associated cell death (Hsuan et al., 2006). Taken together, these studies suggest a complicated relationship between mTORC1 and Parkinson s pathology. While decreasing mTORC1 may have protective effects in diseased cells, decreasing mTORC1 could be beneficial at the expense of other pro-survival pathways. For example, activation of S6K1 has been shown to be anti-apoptotic under some conditions (Harada et al, 2001).
Other data has suggested that rapamycin might benefit Parkinson s patients. Alpha synuclein is observed in Lewy bodies, and mutations in alpha synuclein are associated with inherited forms of Parkinson s disease. Alpha synuclein can be degraded by the ubiquitin proteosome pathway and by autophapgy (Webb et al., 2003). When autophagy is inhibited in cell culture, A53T alpha synuclien levels increased more than wild-type alpha synuclein, suggesting that pathological mutations of alpha synuclein, which are more aggregate prone than wild-type, depend on autophagy for clearance (Webb et al, 2003). Furthermore, rapamycin facilitated clearance of both wild-type and mutant alpha synuclein. In an SH-SY5Y cell culture model, cells treated with rotenone had reduced apoptosis when co-treated with rapamycin (Pan et al., 2009). The rapamycin-conferred protection was reduced when cells were treated with siRNA against ATG5, a requirement for autophagy induction. These results suggest that rapamycin may be beneficial in several models of Parkinson s disease through the induction of autophagy.
Another mechanism by which rapamycin may benefit Parkinson s disease could be through reducing levels of translational initiation. In the Pink1 and Park2 mutant Drosophila models of PD, rapamycin treatment rescued aspects of the fly pathology, including muscle degeneration and DA neuron death (Tain et al., 2009). Since mTORC1 reduces 4E-BP activity, the drosophila 4E-BP, Thor, was over-expressed to mimic rapamycin s effects of 4E-BP. 4E-BP over-expression also reduced pathology in the Pink1 and Park2 mutant drosophila, suggesting that rapamycin s effects were mediated through reduction of translation initiation. Furthermore, the benefits of 4E-BP over-expression did not require ATG5, suggesting that reduced translation initiation is beneficial independently from autophagy. Both rapamycin and 4E-BP overexpression increased levels of Drosophila glutathion-S-transferase, an enzyme whose activity can suppress Park2 toxicity. Thus, work in Drosophila suggests that reducing translational activity can up-regulate expression of proteins that protect against cellular stress. Taken together, these studies suggest that rapamycin may have therapeutic value for the treatment of PD through several of its actions.
Interestingly, a recent report suggests that rapamycin might be useful to alleviate side effects of L-DOPA, a common treatment for Parkinson s (Santini et al., 2009). L-DOPA is a dopamine precursor, and administration of L-DOPA increases the production of DA in the remaining substantia nigra neurons, compensating for the loss of DA neurons and improving motor ability. However, extended treatment can lead to L-DOPA induced dyskinesia. Mice with unilateral SN 6-OHDA lesions were treated with L-DOPA, an animal model of L-DOPA induced dyskinesia. L-DOPA activated ERK/MAPK and mTOR signaling in a subset of neurons in the striatum, a region that regulates motor behavior and receives projections from the substantia nigra. In these neurons, GABAergic medium spiny neurons containing D1 receptors, L-DOPA had previously been shown to altar synaptic plasticity (Picconi et al., 2003). Co-administration of rapamycin along with L-DOPA prevented the increase of mTORC1 activity in D1 containing neurons. More importantly, rapamycin reduced episodes of dyskinesia in mice treated with L-DOPA. Together, these results suggest that rapamycin might benefit both symptoms and pathology in patients with Parkinson s disease.
3.6 Huntington s Disease
Huntington s disease and related poly-glutamine expansion disease are heritable. CAG expansion occurs within the genes, and when an allele has expanded beyond a specific threshold (above 34 repeats in Huntington s disease), the allele produces neurodegeneration. The number of repeats can determine the severity of the disease; longer repeats result in more severe disease with earlier onset. Huntington s is the most common poly-glutamine repeat disease, and symptoms include involuntary movements and progressive cognitive disfunction (Walker, 2007). Poly-glutamine expanded proteins aggregate and form inclusion bodies.
In animal models as well as in diseased human tissue, markers of mTORC1 activity are decreased in affected cells. This could be due to the fact that mTOR is found in inclusions bodies in human tissue and well as in tissue from mouse models (Ravikumar et al., 2004), suggesting that incorporation into inclusion bodies sequesters mTOR and precludes its activity. The reduced mTOR activity is accompanied by an upregulation of autophagy. This may be a protective mechanism for the cell; increasing mTOR activity in a cell culture model of Huntington s reduces autophagy and increases toxicity of mutant huntingtin (Ravikumar et al., 2004).
The huntingtin protein can be degraded by the ubiquitin proteosome pathway or by autophagy. As with other proteins implicated in neurodegenerative diseases, degradation of the aggregate prone expanded species relies more on autophagy than the non-expanded one (Ravikumar et al., 2002).
Rapamycin treatment reduces polyglutamine neurotoxicity in several disease models. Ross/Borchelt mice, mice expressing huntingtin with 82 glutamine repeats were treated with rapamycin with a regimen that began before disease phenotypes appeared. This strategy induced autophagy in neurons and reduced the number of inclusions in tissue. More importantly, rapamycin treatment improved motor deficits associated with the disease (Ravikumar et al., 2004). Rapamycin also reduces toxicity in drosphila models of polyglutamine diseases (Berger et al., 2006; Pandey et al., 2007; Ravikumar et al., 2004). In drosophila models, rescue by rapamycin was blunted when levels of atg1 (Berger et al., 2006) or atg12 (Pandey et al., 2007), factors critical for autophagy, were knocked down. However, work in a cell culture model of Huntington s suggested that reduction of translation initiation can also contribute to the protective effects of rapamycin (King et al., 2008). These studies suggest that rapamycin benefits models of polyglutamine expansion diseases through the clearance and reduction of pathogenic protein species.
Part IV Conclusions
Since many neurodegenerative diseases are associated with accumulation of misfolded or aggregate prone proteins, rapamycin may be particularly relevant to neuronal health. Rapamycin both slows translation of new proteins and induces autophagy. Slowing down the rate of protein synthesis may make the cell more effective and clearing and removing misfolded or damaged proteins. Additionally, slowing protein synthesis may be a cue for neurons to upregulate machinery to cope with damaged protiens, oxidative damage, and other stressors. Beyond slowing the production of proteins, the induction of autophagy by rapamycin can clear damaged organelles or proteins that are resistant to degradation by the proteosome. Furthermore, brain specific removal of ATG5 or ATG7 (Hara et al., 2006; Komatsu et al., 2006), requirements of autophagy, are sufficient to induce neurodegeneration in mice, suggesting that autophagy is required for normal maintenance in neurons. Thus, the duel actions of rapamycin on protein turnover may support neuronal well-being by slowing production and increasing removal of damaged material.
In healthy neurons, the mTORC1 signaling pathway can respond to a variety of stimuli and trigger appropriate, long-lasting responses. The mTORC1 pathway is involved in synaptic plasticity, in part by coordinating the timing and location for the synthesis of new proteins. mTOR activity is used by the brain to monitor nutrient status, regulate feeding and consolidate long-term memories. However important the role of mTORC1 activity in the healthy brain, evidence is mounting that the diseased brain may benefit from reducing mTOR activity. Treatment with rapamycin benefits a range of models for Alzheimer s disease, Parkinson s disease, and poly-glutamine expansion diseases. Further characterizing mechanisms by which rapamycin and its analogs reduce toxicity should yield promising potential therapies for these diseases: treatments that may alleviate pathology while sparing mTOR s ability to support normal brain function.
Acknowledgments
M.G.G. is supported by NIA training grant T32 AG 00057. Studies of mTOR and aging in the Kennedy lab are supported by NIA grants AG033373-03 and AG035336-02.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Abel T, Lattal KM. Molecular mechanisms of memory acquisition, consolidation and retrieval. Curr Opin Neurobiol. 2001;11:180–7. doi: 10.1016/s0959-4388(00)00194-x. [DOI] [PubMed] [Google Scholar]
- An WL, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K, Iqbal IG, Winblad B, Pei JJ. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in disease. Am J Pathol. 2003;163:591–607. doi: 10.1016/S0002-9440(10)63687-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antion MD, Hou L, Wong H, Hoeffer CA, Klann E. mGluR-dependent long-term depression is associated with increased phosphorylation of S6 and synthesis of elongation factor 1A but remains expressed in S6K-deficient mice. Mol Cell Biol. 2008;28:2996–3007. doi: 10.1128/MCB.00201-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, Klann E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci. 2005;25:9581–90. doi: 10.1523/JNEUROSCI.2423-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–72. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Belelovsky K, Kaphzan H, Elkobi A, Rosenblum K. Biphasic activation of the mTOR pathway in the gustatory cortex is correlated with and necessary for taste learning. J Neurosci. 2009;29:7424–31. doi: 10.1523/JNEUROSCI.3809-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, Schmitt I, Wullner U, Evert BO, O'Kane CJ, Rubinsztein DC. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–42. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, Greenamyre JT. Animal models of Parkinson's disease. Bioessays. 2002;24:308–18. doi: 10.1002/bies.10067. [DOI] [PubMed] [Google Scholar]
- Blouet C, Ono H, Schwartz GJ. Mediobasal hypothalamic p70 S6 kinase 1 modulates the control of energy homeostasis. Cell Metab. 2008;8:459–67. doi: 10.1016/j.cmet.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Wells DG. Dendritic mRNA: transport, translation and function. Nat Rev Neurosci. 2007;8:776–89. doi: 10.1038/nrn2150. [DOI] [PubMed] [Google Scholar]
- Burke SN, Barnes CA. Neural plasticity in the ageing brain. Nat Rev Neurosci. 2006;7:30–40. doi: 10.1038/nrn1809. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–20. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammalleri M, Lutjens R, Berton F, King AR, Simpson C, Francesconi W, Sanna PP. Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1. Proc Natl Acad Sci U S A. 2003;100:14368–73. doi: 10.1073/pnas.2336098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R, Li A, Cho HY, Lee B, Obrietan K. Mammalian target of rapamycin signaling modulates photic entrainment of the suprachiasmatic circadian clock. J Neurosci. 2010;30:6302–14. doi: 10.1523/JNEUROSCI.5482-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Garelick MG, Wang H, Lil V, Athos J, Storm DR. PI3 kinase signaling is required for retrieval and extinction of contextual memory. Nat Neurosci. 2005;8:925–31. doi: 10.1038/nn1482. [DOI] [PubMed] [Google Scholar]
- Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–4. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312:927–30. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- Cracco JB, Serrano P, Moskowitz SI, Bergold PJ, Sacktor TC. Protein synthesis-dependent LTP in isolated dendrites of CA1 pyramidal cells. Hippocampus. 2005;15:551–6. doi: 10.1002/hipo.20078. [DOI] [PubMed] [Google Scholar]
- Cybulski N, Hall MN. TOR complex 2: a signaling pathway of its own. Trends Biochem Sci. 2009;34:620–7. doi: 10.1016/j.tibs.2009.09.004. [DOI] [PubMed] [Google Scholar]
- Dash PK, Orsi SA, Moore AN. Spatial memory formation and memory-enhancing effect of glucose involves activation of the tuberous sclerosis complex-Mammalian target of rapamycin pathway. J Neurosci. 2006;26:8048–56. doi: 10.1523/JNEUROSCI.0671-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Markesbery WR, Chen Q, Li F, Keller JN. Ribosome dysfunction is an early event in Alzheimer's disease. J Neurosci. 2005;25:9171–5. doi: 10.1523/JNEUROSCI.3040-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat Med. 2008;14:843–8. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, de Vries PJ, Silva AJ. From mTOR to cognition: molecular and cellular mechanisms of cognitive impairments in tuberous sclerosis. J Intellect Disabil Res. 2009;53:838–51. doi: 10.1111/j.1365-2788.2009.01208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay GM, Yan L, Procter J, Mieulet V, Lamb RF. A MAP4 kinase related to Ste20 is a nutrient-sensitive regulator of mTOR signalling. Biochem J. 2007;403:13–20. doi: 10.1042/BJ20061881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–7. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Garelick MG, Chan GC, DiRocco DP, Storm DR. Overexpression of type I adenylyl cyclase in the forebrain impairs spatial memory in aged but not young mice. J Neurosci. 2009;29:10835–42. doi: 10.1523/JNEUROSCI.0553-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobert D, Topolnik L, Azzi M, Huang L, Badeaux F, Desgroseillers L, Sossin WS, Lacaille JC. Forskolin induction of late-LTP and up-regulation of 5′ TOP mRNAs translation via mTOR, ERK, and PI3K in hippocampal pyramidal cells. J Neurochem. 2008;106:1160–74. doi: 10.1111/j.1471-4159.2008.05470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong R, Park CS, Abbassi NR, Tang SJ. Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity-dependent dendritic protein synthesis in hippocampal neurons. J Biol Chem. 2006;281:18802–15. doi: 10.1074/jbc.M512524200. [DOI] [PubMed] [Google Scholar]
- Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008;4:e24. doi: 10.1371/journal.pgen.0040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad Sci U S A. 2001;98:9666–9670. doi: 10.1073/pnas.171301998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. AMPK: a key regulator of energy balance in the single cell and the whole organism. Int J Obes (Lond) 2008;32(4):S7–12. doi: 10.1038/ijo.2008.116. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–5. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905–9. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- Hoeffer CA, Tang W, Wong H, Santillan A, Patterson RJ, Martinez LA, Tejada-Simon MV, Paylor R, Hamilton SL, Klann E. Removal of FKBP12 enhances mTOR-Raptor interactions, LTP, memory, and perseverative/repetitive behavior. Neuron. 2008;60:832–45. doi: 10.1016/j.neuron.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsuan SL, Klintworth HM, Xia Z. Basic fibroblast growth factor protects against rotenone-induced dopaminergic cell death through activation of extracellular signal-regulated kinases 1/2 and phosphatidylinositol-3 kinase pathways. J Neurosci. 2006;26:4481–91. doi: 10.1523/JNEUROSCI.4922-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husseman JW, Nochlin D, Vincent I. Mitotic activation: a convergent mechanism for a cohort of neurodegenerative diseases. Neurobiol Aging. 2000;21:815–28. doi: 10.1016/s0197-4580(00)00221-9. [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–57. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Sheng M. The growing role of mTOR in neuronal development and plasticity. Mol Neurobiol. 2006;34:205–19. doi: 10.1385/MN:34:3:205. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, Powers RW, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–6. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–90. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004;116:467–79. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Kelly MT, Crary JF, Sacktor TC. Regulation of protein kinase Mzeta synthesis by multiple kinases in long-term potentiation. J Neurosci. 2007;27:3439–44. doi: 10.1523/JNEUROSCI.5612-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana V, Lu Y, Steinhilb ML, Oldham S, Shulman JM, Feany MB. TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr Biol. 2006;16:230–41. doi: 10.1016/j.cub.2005.12.042. [DOI] [PubMed] [Google Scholar]
- King MA, Hands S, Hafiz F, Mizushima N, Tolkovsky AM, Wyttenbach A. Rapamycin inhibits polyglutamine aggregation independently of autophagy by reducing protein synthesis. Mol Pharmacol. 2008;73:1052–63. doi: 10.1124/mol.107.043398. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- Lang UE, Heger J, Willbring M, Domula M, Matschke K, Tugtekin SM. Immunosuppression using the mammalian target of rapamycin (mTOR) inhibitor everolimus: pilot study shows significant cognitive and affective improvement. Transplant Proc. 2009;41:4285–8. doi: 10.1016/j.transproceed.2009.08.050. [DOI] [PubMed] [Google Scholar]
- Langstrom NS, Anderson JP, Lindroos HG, Winblad B, Wallace WC. Alzheimer's disease-associated reduction of polysomal mRNA translation. Brain Res Mol Brain Res. 1989;5:259–69. doi: 10.1016/0169-328x(89)90060-0. [DOI] [PubMed] [Google Scholar]
- Lee J, Duan W, Long JM, Ingram DK, Mattson MP. Dietary restriction increases the number of newly generated neural cells, and induces BDNF expression, in the dentate gyrus of rats. J Mol Neurosci. 2000;15:99–108. doi: 10.1385/JMN:15:2:99. [DOI] [PubMed] [Google Scholar]
- Lee CC, Huang CC, Wu MY, Hsu KS. Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J Biol Chem. 2005;280:18543–50. doi: 10.1074/jbc.M414112200. [DOI] [PubMed] [Google Scholar]
- Li X, Alafuzoff I, Soininen H, Winblad B, Pei JJ. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer's disease brain. FEBS J. 2005;272:4211–20. doi: 10.1111/j.1742-4658.2005.04833.x. [DOI] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Lu KT, Leu TH, Chang WC, Gean PW. A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron. 2001;31:841–51. doi: 10.1016/s0896-6273(01)00433-0. [DOI] [PubMed] [Google Scholar]
- Malagelada C, Ryu EJ, Biswas SC, Jackson-Lewis V, Greene LA. RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson's disease by a mechanism involving mammalian target of rapamycin inactivation. J Neurosci. 2006;26:9996–10005. doi: 10.1523/JNEUROSCI.3292-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson's disease. J Neurosci. 2010;30:1166–75. doi: 10.1523/JNEUROSCI.3944-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Chan SL, Duan W. Modification of brain aging and neurodegenerative disorders by genes, diet, and behavior. Physiol Rev. 2002;82:637–72. doi: 10.1152/physrev.00004.2002. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG, Suzuki JS. Aging and extrapyramidal function. Arch Neurol. 1977;34:33–5. doi: 10.1001/archneur.1977.00500130053010. [DOI] [PubMed] [Google Scholar]
- Miller S, Yasuda M, Coats JK, Jones Y, Martone ME, Mayford M. Disruption of dendritic translation of CaMKIIalpha impairs stabilization of synaptic plasticity and memory consolidation. Neuron. 2002;36:507–19. doi: 10.1016/s0896-6273(02)00978-9. [DOI] [PubMed] [Google Scholar]
- Mizuno K, Giese KP. Hippocampus-dependent memory formation: do memory type-specific mechanisms exist? J Pharmacol Sci. 2005;98:191–7. doi: 10.1254/jphs.crj05005x. [DOI] [PubMed] [Google Scholar]
- Mori H, Inoki K, Munzberg H, Opland D, Faouzi M, Villanueva EC, Ikenoue T, Kwiatkowski D, MacDougald OA, Myers MG, Jr, Guan KL. Critical role for hypothalamic mTOR activity in energy balance. Cell Metab. 2009;9:362–74. doi: 10.1016/j.cmet.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouravlev A, Dunning J, Young D, During MJ. Somatic gene transfer of cAMP response element-binding protein attenuates memory impairment in aging rats. Proc Natl Acad Sci U S A. 2006;103:4705–10. doi: 10.1073/pnas.0506137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Ostroff LE, Fiala JC, Allwardt B, Harris KM. Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron. 2002;35:535–45. doi: 10.1016/s0896-6273(02)00785-7. [DOI] [PubMed] [Google Scholar]
- Pan T, Rawal P, Wu Y, Xie W, Jankovic J, Le W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience. 2009;164:541–51. doi: 10.1016/j.neuroscience.2009.08.014. [DOI] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–63. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Parsons RG, Gafford GM, Helmstetter FJ. Translational control via the mammalian target of rapamycin pathway is critical for the formation and stability of long-term fear memory in amygdala neurons. J Neurosci. 2006;26:12977–83. doi: 10.1523/JNEUROSCI.4209-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picconi B, Centonze D, Hakansson K, Bernardi G, Greengard P, Fisone G, Cenci MA, Calabresi P. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci. 2003;6:501–6. doi: 10.1038/nn1040. [DOI] [PubMed] [Google Scholar]
- Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4:658–65. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- Puighermanal E, Marsicano G, Busquets-Garcia A, Lutz B, Maldonado R, Ozaita A. Cannabinoid modulation of hippocampal long-term memory is mediated by mTOR signaling. Nat Neurosci. 2009;12:1152–8. doi: 10.1038/nn.2369. [DOI] [PubMed] [Google Scholar]
- Raab-Graham KF, Haddick PC, Jan YN, Jan LY. Activity- and mTOR-dependent suppression of Kv1.1 channel mRNA translation in dendrites. Science. 2006;314:144–8. doi: 10.1126/science.1131693. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–17. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Rudow G, O'Brien R, Savonenko AV, Resnick SM, Zonderman AB, Pletnikova O, Marsh L, Dawson TM, Crain BJ, West MJ, Troncoso JC. Morphometry of the human substantia nigra in ageing and Parkinson's disease. Acta Neuropathol. 2008;115:461–70. doi: 10.1007/s00401-008-0352-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvinsky I, Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci. 2006;31:342–8. doi: 10.1016/j.tibs.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Heiman M, Greengard P, Valjent E, Fisone G. Inhibition of mTOR signaling in Parkinson's disease prevents L-DOPA-induced dyskinesia. Sci Signal. 2009;2:ra36. doi: 10.1126/scisignal.2000308. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–68. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Woodgett JR. PKB/AKT: functional insights from genetic models. Nat Rev Mol Cell Biol. 2001;2:760–8. doi: 10.1038/35096067. [DOI] [PubMed] [Google Scholar]
- Schicknick H, Schott BH, Budinger E, Smalla KH, Riedel A, Seidenbecher CI, Scheich H, Gundelfinger ED, Tischmeyer W. Dopaminergic modulation of auditory cortex-dependent memory consolidation through mTOR. Cereb Cortex. 2008;18:2646–58. doi: 10.1093/cercor/bhn026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schratt GM, Nigh EA, Chen WG, Hu L, Greenberg ME. BDNF regulates the translation of a select group of mRNAs by a mammalian target of rapamycin-phosphatidylinositol 3-kinase-dependent pathway during neuronal development. J Neurosci. 2004;24:7366–77. doi: 10.1523/JNEUROSCI.1739-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal SN. Sirolimus: its discovery, biological properties, and mechanism of action. Transplant Proc. 2003;35:7S–14S. doi: 10.1016/s0041-1345(03)00211-2. [DOI] [PubMed] [Google Scholar]
- Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, Gems D, Withers DJ. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–4. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfel MN, Shamieh LS, Kaeberlein M, Kennedy BK. The TOR pathway comes of age. Biochim Biophys Acta. 2009;1790:1067–74. doi: 10.1016/j.bbagen.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui L, Wang J, Li BM. Role of the phosphoinositide 3-kinase-Akt-mammalian target of the rapamycin signaling pathway in long-term potentiation and trace fear conditioning memory in rat medial prefrontal cortex. Learn Mem. 2008;15:762–76. doi: 10.1101/lm.1067808. [DOI] [PubMed] [Google Scholar]
- Swiech L, Perycz M, Malik A, Jaworski J. Role of mTOR in physiology and pathology of the nervous system. Biochim Biophys Acta. 2008;1784:116–32. doi: 10.1016/j.bbapap.2007.08.015. [DOI] [PubMed] [Google Scholar]
- Tain LS, Mortiboys H, Tao RN, Ziviani E, Bandmann O, Whitworth AJ. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat Neurosci. 2009;12:1129–35. doi: 10.1038/nn.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:467–72. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault O, Gant JC, Landfield PW. Expansion of the calcium hypothesis of brain aging and Alzheimer's disease: minding the store. Aging Cell. 2007;6:307–17. doi: 10.1111/j.1474-9726.2007.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischmeyer W, Schicknick H, Kraus M, Seidenbecher CI, Staak S, Scheich H, Gundelfinger ED. Rapamycin-sensitive signalling in long-term consolidation of auditory cortex-dependent memory. Eur J Neurosci. 2003;18:942–50. doi: 10.1046/j.1460-9568.2003.02820.x. [DOI] [PubMed] [Google Scholar]
- Toescu EC. Normal brain ageing: models and mechanisms. Philos Trans R Soc Lond B Biol Sci. 2005;360:2347–54. doi: 10.1098/rstb.2005.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokas P, Grace EA, Chan P, Ma T, Sealfon SC, Iyengar R, Landau EM, Blitzer RD. Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J Neurosci. 2005;25:5833–43. doi: 10.1523/JNEUROSCI.0599-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokas P, Ma T, Iyengar R, Landau EM, Blitzer RD. Mitogen-activated protein kinase upregulates the dendritic translation machinery in long-term potentiation by controlling the mammalian target of rapamycin pathway. J Neurosci. 2007;27:5885–94. doi: 10.1523/JNEUROSCI.4548-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–23. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–5. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426:620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- Vickers CA, Dickson KS, Wyllie DJ. Induction and maintenance of late-phase long-term potentiation in isolated dendrites of rat hippocampal CA1 pyramidal neurones. J Physiol. 2005;568:803–13. doi: 10.1113/jphysiol.2005.092924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker FO. Huntington's disease. Lancet. 2007;369:218–28. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- Wang X, Proud CG. Nutrient control of TORC1, a cell-cycle regulator. Trends Cell Biol. 2009;19:260–7. doi: 10.1016/j.tcb.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–13. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- Xia Z, Storm DR. The role of calmodulin as a signal integrator for synaptic plasticity. Nat Rev Neurosci. 2005;6:267–76. doi: 10.1038/nrn1647. [DOI] [PubMed] [Google Scholar]
- Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci. 2001;21:2661–8. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]