

Abstract
The formation of amyloid-like fibrils is a hallmark of several neurodegenerative diseases. How the assembly of amyloid-like fibrils contributes to cell death is a major unresolved question in the field. The budding yeast Saccharomyces cerevisiae is a powerful model organism to study basic mechanisms for how cellular pathways regulate amyloid assembly and proteotoxicity. For example, studies of the amyloidogenic yeast prion [RNQ+] have revealed novel roles by which molecular chaperones protect cells from the accumulation of cytotoxic protein species. In budding yeast there are a variety of cellular assays that can be employed to analyze the assembly of amyloid-like aggregates and mechanistically dissect how cellular pathways influence proteotoxicity. In this review, we describe several assays that are routinely used to investigate aggregation and toxicity of the [RNQ+] prion in yeast.
Keywords: amyloid, Rnq1, prion, molecular chaperone, yeast, proteotoxicity
1. Introduction
An extensive group of neurodegenerative disorders are characterized by the formation of amyloid-like fibrils, including Alzheimer’s disease, Huntington’s disease, and Creutzfeldt-Jakob disease (1). Amyloid-like fibrils are composed of β-sheet-rich conformers of a non-native protein that are assembled in a unique structure called a cross-β spine (2). This structure confers several unique properties that distinguish amyloid-like fibrils from amorphous, or disordered aggregates including resistance to proteases and insolubility in ionic detergents (3). While the accumulation of amyloid deposits is a diagnostic biomarker of amyloidosis, the connection between the assembly of amyloid-like fibrils and neuronal cell death is currently unclear (4–6). Furthermore, specific neuronal subpopulations are selectively susceptible to misfolding and aggregation of particular disease proteins suggesting there are complex cellular pathways that influence amyloid assembly and proteotoxicity. Our understanding of this process is still rudimentary and many questions remain to be answered.
There are numerous model organisms available to study amyloid-linked diseases. While every model organism has unique benefits and limitations, studies in the budding yeast Saccharomyces cerevisiae have provided substantial insight on mechanisms underlying the assembly of amyloid-like fibrils and proteotoxicity (7). Several proteins in S. cerevisiae known collectively as yeast prions can exist in a beta-rich conformation that possesses a high propensity to assemble into amyloid-like fibrils inside the cell (8). Molecular chaperones shear these amyloid-like assemblies to generate a steady pool of prion seeds that convert the native form of the protein into the beta-rich prion conformation and propagate the prion state (9). While studies on yeast prion biogenesis have centered on the yeast prions [PSI+] and [URE3], the prion [RNQ+]/[PIN+] has recently emerged as a powerful tool in the study of amyloid-linked toxicity in yeast. The [RNQ+] prion is formed by the yeast protein Rnq1 (10, 11). Like other proteins that propagate as prions in budding yeast, Rnq1 possesses a domain enriched in glutamine/asparagines residues that is sufficient to assemble into amyloid-like fibrils and propagate as a prion (12–14). Interestingly, the induction of other prions in yeast requires Rnq1 to be in the [RNQ+] state. Rnq1 has no other known function in the cell outside of this role in prion induction although yeast strains isolated in the wild are predominantly in a [RNQ+] state (15) suggesting there may be an important, yet undiscovered function for this prion (16). In addition, expression of the expanded-polyglutamine form of huntingtin is selectively toxic to yeast when Rnq1 is the [RNQ+] prion conformation (17). Thus, the [RNQ+] prion influences aggregation and toxicity of other glutamine-enriched proteins in yeast although the mechanism underlying this relationship is unclear.
Importantly, overexpression of Rnq1 is toxic to yeast specifically in the presence of the [RNQ+] prion (18). Toxicity was correlated with the accumulation of a soluble, non-amyloid-like Rnq1 protein species and chaperone-dependent [RNQ+] assembly protected cells from Rnq1-induced toxicity. Therefore, aberrations in [RNQ+] prion biogenesis can result in the accumulation of proteotoxic species and further investigation into this pathway will likely yield additional discoveries on how amyloid assembly pathways contribute to cell death. This review will describe a variety of assays to analyze Rnq1-induced toxicity and methods for characterizing amyloid-like aggregates in yeast.
2. Basic methods for culturing yeast
Methods for culturing and manipulating yeast are well-established and readily available (19). As such, this section will focus primarily on central guidelines for growing yeast cultures to study amyloid assembly and toxicity. All experiments described herein were performed at 30°C although the specific experimental temperature may vary depending upon the optimal growth conditions for the yeast strain background. Plasmids expressing aggregation-prone proteins can easily be transformed into yeast and maintained under selection by utilizing auxotrophic markers (ex. amino acids). Individual colonies are used to inoculate liquid cultures that can be expanded into liter-size volumes as long as cultures are properly aerated and nutrient-rich. The ability to collect large quantities of cells is one of the distinct advantages of using budding yeast over most other model systems. The most important consideration when culturing yeast is to maintain the cells in log-phase growth by consistently diluting cells in fresh media. However, yeast cells will undergo physiological changes similar to aging after repetitive diluting or indefinite time in stationary phase (20). As a result, exogenous plasmids should be freshly transformed to improve reproducibility between experiments.
One additional advantage to working with budding yeast is the availability of diverse promoters (both inducible and constitutive) that provide extensive flexibility in the time and level of protein expression. Inducible promoters are particularly important when studying proteotoxicity because the expression of a toxic protein needs to be tightly controlled during an experiment. One of the more common promoters for this kind of analysis is the GAL1 promoter (21). As described below, this promoter is often used to examine how high level expression of a disease protein impacts cell viability by spotting assays or growth curves. There are several advantages to using the GAL1 promoter for this kind of analysis. Expression from this promoter is repressed in the presence of glucose permitting very robust control of protein expression. To facilitate rapid expression from the GAL1 promoter, yeast cells are initially grown in media containing raffinose as the carbon source because raffinose does not induce or repress expression from the GAL1 promoter. Addition of galactose to the media rapidly induces protein expression within 30 minutes. One drawback is that expression from the GAL1 promoter cannot be titrated by varying the levels of galactose. Furthermore, some yeast strains are unable to unable to metabolize galactose and are thus inviable when galactose is the sole carbon source.
In contrast to the GAL1 promoter, the CUP1 promoter can be tightly controlled by varying levels of copper sulfate added to the media. Expressing a toxic protein such as Rnq1 from the CUP1 promoter can inhibit cell growth (22); however, caution should be exercised if levels of copper sulfate cause growth defects independent of protein expression. This promoter is also used to constitutively express a protein of choice using low levels of copper sulfate (ex. 50μM or lower). Long-term, low level expression of an aggregation-prone protein such as Rnq1 allows the exogenous protein to reach steady state equilibrium with cellular pathways that regulate assembly of amyloid-like aggregates. As such, Rnq1 aggregation can be studied under conditions when this protein is not toxic to yeast and independent of changes in cell physiology that might accompany cytotoxicity. Constitutive, high level expression can also be achieved using promoters from housekeeping enzymes such as glyceraldehyde-3-phosphate dehydrogenase (GPD) or alcohol dehydrogenase (ADH). These promoters are powerful tools to analyze how overexpression of various cellular factors affects amyloid assembly and toxicity.
Expression constructs can also be placed in different plasmid backbones with unique elements that control the plasmid copy number within the cell (19). For example, episomal (2μ) vectors vary from 10–40 copies per cell and thus provide high levels of expression yet also display extensive variation from cell to cell. These plasmids are generally used in growth assays (ex. high copy suppressor screens) where cell to cell variation is not visible in measurements of population growth. Centromeric (CEN) plasmids are maintained at approximately 1–3 plasmids per cell and thus display more consistency in expression from cell to cell, yet also result in lower expression levels. To maintain consistent expression from cell to cell, expression constructs can also be integrated into the yeast genome. Ultimately, the decision to use one plasmid backbone may depend upon the specific protein of choice and the experimental context.
In the context of studying prion biogenesis, budding yeast offer one unique advantage over other model systems. Yeast strains can be “cured” of the prion state. For example, Rnq1 is soluble and does not assemble into amyloid-like aggregates in strains that are cured of the [RNQ+] prion state; now designated [rnq−]. The cured strain can be compared back to the [RNQ+] strain to determine how cellular factors such as molecular chaperones interact with the soluble versus amyloidogenic conformer of the same protein. Yeast strains can be cured of the prion state by two methods. First, if the protein determinant is non-essential such as Rnq1 then deleting the RNQ1 gene will eventually cure yeast of the [RNQ+] prion state as the remaining Rnq1 protein is diluted over successive generations. If the Rnq1 protein is subsequently expressed in this strain from an exogenous plasmid, it will not assemble into amyloid-like aggregates because pre-existing [RNQ+] prion seeds are not present in the cell. An alternative and more common method is to treat cells with low doses of guanidine hydrochloride. This compound inhibits a molecular chaperone called Hsp104 that shears prion fibrils to generate new prion seeds and is required to propagate yeast prions (23, 24). Two to three passages on solid media containing 3mM guanidine hydrochloride is typically sufficient to cure yeast of the prion state. One drawback to this method is that it is not selective for any specific prion. Both methods permanently cure yeast of the prion state because spontaneous prion induction is a highly unfavorable event (25).
3. Growth and viability assays
3.1. Spotting Assay
The most basic tool for studying amyloid toxicity in yeast is to monitor cell growth under conditions where an amyloidogenic protein is expressed compared to conditions when this protein either is not expressed or does not assemble into amyloid-like fibrils (ex. a cured prion strain). This method is readily applicable to high through-put screening of genetic factors that influence toxicity. Defects in cell growth can most easily be assessed by spotting yeast cells on solid agar media and comparing growth between strain expressing a toxic protein and a control strain. As shown in Figure 1, expressing Rnq1 from the GAL1 promoter is toxic to yeast in a [RNQ+] dependent-manner (18). To perform this assay, single colonies from freshly transformed yeast are used to inoculate a liquid culture and when yeast cells have reach mid-log phase, equal concentrations of cells (~0.5 OD600) are serially diluted onto solid selective media containing 2% galactose as the carbon source and incubated for 2–4 days at 30°C depending upon the strain. As a control, strains are simultaneously spotted on selective media containing glucose to show that changes in growth rates are dependent upon expression of the aggregation-prone protein.
Figure 1.

Expression of amyloid-forming proteins is toxic to yeast. Yeast cells (strain BY4741-[RNQ+] or [rnq−]) were transformed with an empty pRS416 vector or pRS416(GAL1-RNQ1). Liquid cultures were inoculated with freshly transformed yeast and incubated for one day at 30°C. Equal quantities of cells in mid-log phase (0.5 OD600) were serially diluted (1:5) onto selective media containing glucose or galactose and incubated for approximately 3 days at 30°C and photographed.
3.2 Growth Curve
If differences in growth are too slight to detect by the spotting assay then toxicity can be quantitatively assessed by measuring growth curves in liquid media. The procedure begins as described above, except liquid cultures are induced with galactose at a very low density (OD600 0.01–0.05) and OD600 measured approximately every 2 hours until strains reach stationary phase. Compared to the spotting assay, measuring growth curves is more labor intensive however this assay can determine at what point expression of a proteotoxic protein such as Rnq1 affects cell growth. Furthermore, resolving whether a defect in cell growth reflect cell cycle arrest or cell death might lead to a better understanding of the mechanism underlying proteotoxicity in this model system. To determine whether yeast cells have entered cell cycle arrest or cell death, an aliquot is removed from liquid cultures at varying timepoints post-expression and plated on selective media with 2% glucose to repress the GAL1 promoter. If cell viability is indeed reduced upon protein expression, then yeast cells will not grow on media with glucose while yeast cells would be expected to reenter the cell cycle when the proteotoxic insult is removed.
4. Morphological analysis of protein aggregation
4.1. Fluorescence microscopy
A rapid method to analyze protein aggregation is fluorescently-tagging a protein of interest and visualizing intracellular localization via fluorescence microscopy. For example, Rnq1-GFP forms punctuate structures in [RNQ+] cells while is predominantly diffuse in [rnq−] cells (Figure 2). If tagged constructs are expressed from inducible promoters, then a time-course can be performed to determine the kinetics of aggregation. To freeze cells at specific timepoints, cells in mid-log phase are treated with 0.1M phosphate buffer (pH6.8) and 3.7% formaldehyde then incubated at 25°C for 30 minutes to 1 hour. Cells are then washed once in phosphate buffer (pH7.4) and resuspended in phosphate buffer (pH 7.4) plus 1.2M sorbitol. Cells can be stored in this solution for several days at 4°C and further processed for immunofluorescent detection of other proteins or stained with commercially available dyes to identify organelles. Alternatively, fluorophore-conjugated proteins can be visualized by live cell imaging to characterize the dynamics of protein aggregation on the order of seconds to minutes.
Figure 2.
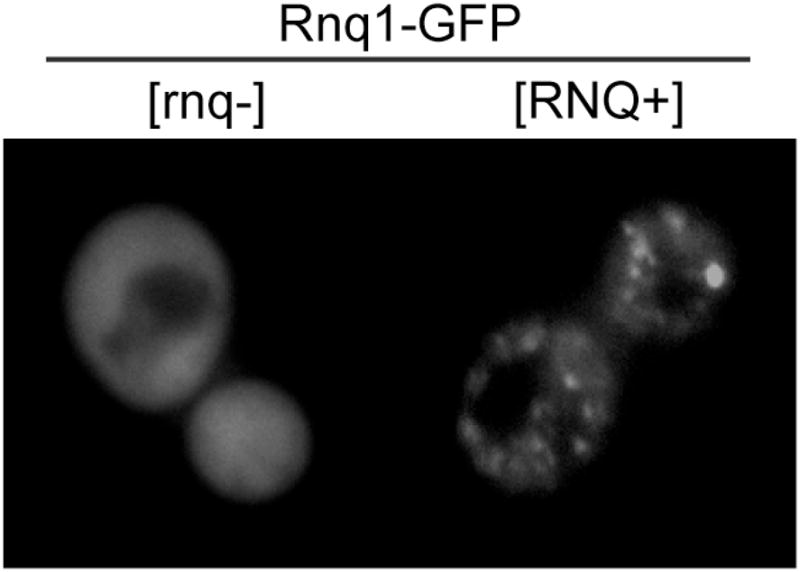
Visualization of protein aggregates by fluorescence microscopy. (A) BY4741 cells ([RNQ+] or [rnq−]) in mid-log phase expressing Rnq1-GFP from the CUP1 promoter were induced with 50uM CuSO4 for 2 hours and live cells visualized with a FITC filter set.
There are several considerations when assessing protein aggregation by fluorescence microscopy. First, the appearance of foci within the cell does not imply the protein of interest is aggregating, yet may represent localization to a specific subcellular compartment or protein complex. Indeed, recent studies have suggested that misfolded and aggregated proteins are transported to distinct subcellular compartments (26), Thus, subcellular localization of a protein of interest should be compared with a panel of known organelle markers (ex nuclei, vacuole, etc). Furthermore, intracellular foci might be disordered, amorphous aggregates rather than amyloid-like assemblies. Additional analysis using the methods described below need to be performed to determine whether intracellular foci are indeed amyloid-like fibrils.
4.2. Thioflavin T Staining of Amyloid-like Fibrils in Yeast
One of the classic, defining characteristics of an amyloid-like fibril is recognition by amyloid-indicator dyes such as thioflavin T (27). Similar to amyloid fibrils formed in human diseases, intracellular yeast prion assemblies are also recognized by thioflavin T (18, 28, 29). To stain intracellular, amyloid-like particles with thioflavin T, an aggregation-prone protein such as Rnq1 is induced for an extended period of time (at least four hours) until it is predominantly assembled into single, distinct foci. Cells are fixed as described above, briefly permeabilized with phosphate buffer (pH 7.5) plus a weak non-ionic detergent such as 0.1% Triton-X, and treated with 0.001% thioflavin T for approximately 10 minutes at room temperature. The cells should be washed at least 3–4 times with phosphate buffer. Thioflavin T has excitation and emission wavelengths of 450nm and 482nm respectively when bound to amyloid-like fibrils and can be visualized in cells under standard FITC filter range. As a control [RNQ+] cells transformed with an empty vector can be used to determine the level of background staining and show thioflavin-t positive foci are dependent upon Rnq1 overexpression. If background ThT staining is very high, then reducing the concentration of ThT or increasing the number of washes should alleviate this problem.
5. Biochemical analysis of amyloid-like aggregates
The structure of amyloid-like aggregates renders these assemblies insoluble in ionic detergents such as sodium dodecyl sulfate (SDS). There are several assays that exploit this unique property to biochemically distinguish large, SDS-insoluble assemblies from unassembled, SDS-soluble protein species in yeast cell extracts. As mentioned above, maintaining cells in mid-log phase is critical when performing these experiments. Additionally, if the aggregate-prone protein is expressed from an inducible promoter, a time-course experiment should be performed to characterize assembly kinetics. Once the basic dynamics of protein assembly into SDS-resistant aggregates is established, then these assays can be performed under conditions where select cellular factors are overexpressed or deleted to determine how specific cellular pathways influence the assembly of amyloid-like aggregates.
5.1. Differential high speed centrifugation
The most simple approach to characterizing the formation of amyloid-like aggregates is to separate SDS-insoluble aggregates by high speed centrifugation. This technique allows direct comparison between soluble and insoluble pools of an amyloid-forming protein. However, this technique is relatively insensitive to slight changes in the size of amyloid-like oligomers or fibrils. As shown in Figure 3A, excess Rnq1-YFP partitions predominantly in the SDS-insoluble fraction yet a pool also remains in the SDS-soluble supernatant. In contrast, excess Rnq1-YFP resolves exclusively in the SDS-soluble fraction in [rnq−] cells (18).
Figure 3.

Biochemical analysis of SDS-insoluble, amyoid-like aggregates. (A) Separation of SDS-insoluble Rnq1-GFP aggregates by high-speed differential centrifugation. BY4741 cells ([RNQ+] or [rnq−] transformed with pGAL-RNQ1YFP were induced for 2 hours with 2% galactose. Cells were processed as described in the main text and equal volumes of total, supernatant, and pellet fractions were analyzed by SDS-PAGE and western immunoblotting for GFP (Roche) and Pgk1 (Invitrogen). (B) SDD-AGE analysis of Rnq1-GFP SDS-insoluble aggregates. BY4741 cells ([RNQ+] or [rnq−] transformed with pCUP-RNQ1GFP were induced with 50uM CuSO4 and samples were collected at 0hr, 1hr, 2hr, and 4hr post-induction. Cells were processed as described in the main text and the PVDF membrane analyzed by western immunoblotting for GFP. Lysates were also analyzed by SDS-PAGE and western immunobloting for GFP (lower panel).
To analyze solubility of Rnq1, cells expressing Rnq1 are first lysed by glass bead disruption under moderately denaturing conditions (150mM NaCl, Hepes pH7.4, 1mM EDTA, 1mM DTT, 1% Triton-X, 0.2% SDS, 1mM PMSF, protease inhibitor cocktail). Depending upon the amyloidogenic protein, the concentration of SDS can be increased to 2% to increase the stringency of this assay. Cell lysates are first processed by a short centrifugation step (650×g for 3min at 4°C) to remove unbroken cells. After the spin, the supernatant is subsequently spun at 100,000 × g for 30min to 1 hour in 1.5mL Beckman polyallomer centrifuge tubes. The speed and time may vary depending upon the specific protein but these conditions appear sufficient for a majority of endogenous Rnq1 to resolve in the insoluble pellet in [RNQ+] cells. An aliquot from the lysate is saved prior to the spin represent the input (or Total) fraction. After the spin, an aliquot from the supernatant is saved and the pellet is resupended in lysis buffer (in a volume equivalent to the original volume used for the spin). Each fraction (total, supernatant, pellet) is mixed with a equal volume of 2X sample buffer (125mM Tris-HCl pH6.8, 4% SDS, 4mM EDTA, and 20% glycerol, 8% beta-mercaptoethanol) and samples are analyzed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE).
5.2. Semi-denaturing detergent agarose gel electrophoresis (SDD-AGE)
SDS-insoluble aggregates can also be separated from SDS-soluble protein species in an agarose gel. In contrast to methods that rely on SDS-PAGE, this method can resolve changes in the size of SDS-insoluble oligomers. For example, Kryndushkin et al demonstrated that inhibiting the chaperone Hsp104 results in a transient increase in the size of SDS -insoluble oligomers and subsequent conversion into the SDS-soluble pool (30). This experiment provided crucial evidence that Hsp104 shears prion fibrils to generate new prion seeds in order to propagate the prion state. In Figure 3B, Rnq1-GFP was induced from a copper promoter and lysates from various timepoints resolved by SDD-AGE. As Rnq1-GFP accumulates, a SDS-insoluble pool appears first then a SDS-soluble pool appears at the latest time point. However, Rnq1-GFP is entirely soluble in [rnq−] cells. Compared to differential high speed centrifugation, this technique is relatively more labor intensive, though it can provide significant insight into the relative ratio of soluble vs insoluble pools as well as changes in the size of SDS-insoluble oligomers. To perform this assay, yeast cells are lysed by glass bead disruption moderately denaturing conditions (150mM NaCl, 50mM Hepes pH 7.4, 1mM EDTA, 1mM DTT, 1% Triton X, 0.2% SDS, 1mM PMSF, yeast protease inhibitor cocktail). Yeast lysates are cleared by centrifugation (650 × g, 3min, 4°C). The supernatant is saved and protein concentrations normalized between samples (higher concentrations are ideal >2.0mg/mL). Normalized lysates are added to an equal volume of 2X SDD-AGE buffer (1XTAE buffer [0.8mM Tris-Acetate, 0.02mM EDTA], 4% SDS, 10% glycerol 8% beta-mercaptoethanol ). Lysates are incubated at 37°Cfor 10 minutes and at least 100μg loaded into a 1.5% agarose gel (made with 1xTAE buffer + 0.1%SDS). Lysates are run at ~70 volts for approximately 2–3 hours. The gel is transferred onto a PVDF membrane at 24 volts for 1.5 hours in standard Tris-based buffer (alternatively you can transfer for 8 hours at 12 volts). The transfer step generates a large amount of heat so the buffer and transfer apparatus should be chilled at 4°C prior to use. The transfer step is inefficient and requires practice and some optimization. The PVDF membrane is analyzed by western immunoblotting for the protein of interest using standard methods.
5.3. Filter Trap
A filter trap assay is another common and rapid method for analyzing the formation of large, SDS-insoluble aggregates. Initially, this technique was used to analyze aggregation of the polyglutamine-expanded form of huntingtin from cell and tissue extracts (31). However, this assay can also be applied to the analysis of large, SDS-insoluble aggregates formed by yeast prions (32, 33). In contrast to the assays described above only the large, SDS-insoluble pool is observed and cannot be directly compared to the SDS-soluble pool that is lost through the cellulose acetate membrane. However, this technique is applicable to large scale analysis, for example, assessing changes in SDS-insoluble aggregate formation under a variety of environmental conditions or genetic backgrounds.
To perform this assay, yeast lysates are generated as described above for SDD-AGE analysis. After protein concentrations are standardized between samples, lysates are applied to a cellulose acetate membrane (0.2μM pore size -Beckman) that is previously equilibrated with lysis buffer and assembled in a slot blot apparatus. The samples are allowed to flow through the membrane by gravity. While still in the slot blot apparatus, the membrane is washed with lysis buffer containing 2% SDS. Once the membrane is dry, it is subsequently processed by western immunoblotting for the protein of interest. Samples should also be spotted onto a nitrocellulose membrane or analyzed by SDS-PAGE to analyze total protein levels. If there is a high level of background, then the concentration of SDS can be increased or the samples heated at various temperatures prior to application on the membrane. Alternatively, the samples can be titrated onto the membrane to identify the linear range of the signal. If there is a poor signal, then the amount of SDS can be reduced or the protein induction time increased to accumulate more of the SDS-insoluble aggregate pool.
5.4. Size-exclusion chromatography
The assays described above separate amyloid-like particles into a SDS-soluble and SDS-insoluble protein species. However, there are times when more detailed resolution is required to characterize assembly intermediates that are solubilized by the presence of SDS. In addition, protein-protein interactions are typically disrupted by the presence of SDS and lost in these assays. To bypass this complication, amyloid-like assemblies can be resolved by size exclusion chromatography. We have previously used this technique to distinguish the ratio of assembled, high molecular weight pools of Rnq1 from unassembled, low molecular protein species (22, 32, 33). Resolving yeast cell extracts by size-exclusion chromatography requires a large quantity of yeast (>100 OD600) because cell extracts are diluted over the column volume which can exceed 100mL depending upon the specific column. Cell growth conditions are the same was described above with a particular emphasis on maintaining cells in mid-log phase and proper aeration throughout the experiment especially as the culture volume is expanded to liter-size to collect enough yeast cells for this experiment. Cells are lysed as described above by glass bead disruption in non-denaturing lysis buffer (150mM NaCl, 50mM Hepes pH 7.4, 1mM EDTA, 0.1% Triton X, plus 1mM PMSF and protease inhibitor cocktail). Specific components in this buffer can be modified depending upon the protein-protein interactions that are under investigation. For example, if a specific protein-protein interaction is ATP-dependent, then EDTA should be omitted and ATP included in the buffer. Cell lysates are precleared twice by centrifugation (650 × g, 3min, 4°C) to ensure cell debris is completely removed from the lysate. Inject about l–5mg cell lysate into size-exclusion column (pre-equilibrated with cold lysis buffer) and collect fractions spanning the void volume to the fractions eluting with the molecular weight of the monomer. The void volume is typically identified by the elution volume of blue dextran and the included volume of the column calibrated by a series of known molecular weight markers. Fractions can be assessed for proteins of interest by SDS-PAGE and western immunoblotting.
If lysates are separated on the appropriate resin then the relative ratio of assembled to unassembled protein can be directly compared. One important consideration is that this approach does not distinguish SDS-insoluble, amyloid-like particles from disordered aggregates or large protein complexes because denaturing buffers are not always compatible with resins in size-exclusion columns. To overcome this restriction, elution fractions can be applied to the assays described above to identify fractions that contain SDS-insoluble aggregates. Furthermore, the elution profile of Rnq1 in a [RNQ+] strain can be compared back to the elution profile in a [rnq−] strain when these proteins do not assemble into amyloid-like conformers (17, 18, 32).
In summary, these biochemical assays offer distinct methods for analyzing the assembly of amyloid-like aggregates. Differential centrifugation and filter trap are the least labor techniques yet do not reveal insight on aggregate size and are most insensitive to subtle changes in the levels of SDS-insoluble aggregates. In contrast, SDD-AGE and size exclusion chromatography are more labor intensive and provide more sensitive information on aggregate assembly such as relative oligomer size. Finally, while size exclusion chromatography does not distinguish SDS-soluble from SDS-insoluble aggregates, it is the most sensitive method for separating aggregates based on oligomer size and allows for identification of protein:protein interactions that are lost during solubilization in SDS. A comprehensive approach (using methods from both categories) is ideal when trying to understand how cellular factors such as molecular chaperones influence amyloid assembly pathways and proteotoxicity.
6. Final Conclusions
The connection between amyloid fibril formation and neurodegeneration is a significant matter of debate. Despite enormous effort over several decades, our basic understanding of the amyloid assembly pathway is still lacking and the complex interplay between protein quality control networks and protein aggregation remains unclear. Budding yeast has emerged as a powerful model system to investigate mechanistic details underlying how assembly of amyloid-like aggregates contributes to cell death. A wide variety of cellular assays are available to dissect this problem utilizing the power of yeast genetics as well as biochemical manipulation of yeast extracts. While yeast cells cannot recapitulate every feature specific to neurons studies in S. cerevisiae can provide a sound foundation of mechanistic understanding to test specific hypothesizes in more complex model organisms.
Acknowledgments
We thank members of the Cyr lab for thoughtful discussions in preparation of this manuscript. This work was supported by funds from the NIH including a predoctoral fellowship to D.W.S. (5F31AG032790) and a grant to D.M.C. (5R01GM067785).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Carrell RW, Lomas DA. Lancet. 1997;350:134–8. doi: 10.1016/S0140-6736(97)02073-4. [DOI] [PubMed] [Google Scholar]
- 2.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Nature. 2005;435:773–8. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiti F, Dobson CM. Annu Rev Biochem. 2006;75:333–66. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 4.Haass C, Selkoe DJ. Nat Rev Mol Cell Biol. 2007;8:101–12. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 5.Treusch S, Cyr DM, Lindquist S. Cell Cycle. 2009;8:1668–74. doi: 10.4161/cc.8.11.8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robakis NK. Neurodegener Dis. 2010;7:32–7. [Google Scholar]
- 7.Khurana V, Lindquist S. Nat Rev Neurosci. 2010;11:436–49. doi: 10.1038/nrn2809. [DOI] [PubMed] [Google Scholar]
- 8.Shorter J, Lindquist S. Nat Rev Genet. 2005;6:435–50. doi: 10.1038/nrg1616. [DOI] [PubMed] [Google Scholar]
- 9.Jones GW, Tuite MF. Bioessays. 2005;27:823–32. doi: 10.1002/bies.20267. [DOI] [PubMed] [Google Scholar]
- 10.Sondheimer N, Lindquist S. Mol Cell. 2000;5:163–72. doi: 10.1016/s1097-2765(00)80412-8. [DOI] [PubMed] [Google Scholar]
- 11.Derkatch IL, Bradley ME, Hong JY, Liebman SW. Cell. 2001;106:171–82. doi: 10.1016/s0092-8674(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 12.Wickner RB, Dyda F, Tycko R. Proc Natl Acad Sci USA. 2008;105:2403–8. doi: 10.1073/pnas.0712032105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel BK, Liebman SW. J Mol Biol. 2007;365:773–82. doi: 10.1016/j.jmb.2006.10.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vitrenko YA, Pavon ME, Stone SI, Liebman SW. Curr Genet. 2007;51:309–19. doi: 10.1007/s00294-007-0127-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Resende CG, Outeiro TF, Sands L, Lindquist S, Tuite MF. Mol Microbiol. 2003;49:1005–17. doi: 10.1046/j.1365-2958.2003.03608.x. [DOI] [PubMed] [Google Scholar]
- 16.Halfmann R, Alberti S, Lindquist S. Trends Cell Biol. 2010 doi: 10.1016/j.tcb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meriin AB, Zhang X, He X, Newnam GP, Chernoff YO, Sherman MY. J Cell Biol. 2002;157:997–1004. doi: 10.1083/jcb.200112104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Douglas PM, Treusch S, Ren HY, Halfmann R, Duennwald ML, Lindquist S, Cyr DM. Proc Natl Acad Sci U S A. 2008 doi: 10.1073/pnas.0802593105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guthrie C, Fink GR. Guide to yeast genetics and molecular and cell biology. Part B. Academic Press; San Diego, Calif: 2002. [Google Scholar]
- 20.Steinkraus KA, Kaeberlein M, Kennedy BK. Annu Rev Cell Dev Biol. 2008;24:29–54. doi: 10.1146/annurev.cellbio.23.090506.123509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.St John TP, Scherer S, McDonell MW, Davis RW. J Mol Biol. 1981;152:317–34. doi: 10.1016/0022-2836(81)90245-x. [DOI] [PubMed] [Google Scholar]
- 22.Douglas PM, Treusch S, Ren HY, Halfmann R, Duennwald ML, Lindquist S, Cyr DM. Proc Natl Acad Sci U S A. 2008;105:7206–11. doi: 10.1073/pnas.0802593105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jung G, Masison DC. Curr Microbiol. 2001;43:7–10. doi: 10.1007/s002840010251. [DOI] [PubMed] [Google Scholar]
- 24.Ferreira PC, Ness F, Edwards SR, Cox BS, Tuite MF. Mol Microbiol. 2001;40:1357–69. doi: 10.1046/j.1365-2958.2001.02478.x. [DOI] [PubMed] [Google Scholar]
- 25.Cox BS, Byrne LJ, Tuite MF. Prion. 2007;1:170–8. doi: 10.4161/pri.1.3.4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaganovich D, Kopito R, Frydman J. Nature. 2008;454:1088–95. doi: 10.1038/nature07195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sipe JD, Cohen AS. J Struct Biol. 2000;130:88–98. doi: 10.1006/jsbi.2000.4221. [DOI] [PubMed] [Google Scholar]
- 28.Kawai-Noma S, Pack CG, Kojidani T, Asakawa H, Hiraoka Y, Kinjo M, Haraguchi T, Taguchi H, Hirata A. J Cell Biol. 2010;190:223–31. doi: 10.1083/jcb.201002149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimura Y, Koitabashi S, Fujita T. Cell Struct Funct. 2003;28:187–93. doi: 10.1247/csf.28.187. [DOI] [PubMed] [Google Scholar]
- 30.Kryndushkin DS, Alexandrov IM, Ter-Avanesyan MD, Kushnirov VV. J Biol Chem. 2003;278:49636–43. doi: 10.1074/jbc.M307996200. [DOI] [PubMed] [Google Scholar]
- 31.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Cell. 1997;90:549–58. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 32.Douglas PM, Summers DW, Ren HY, Cyr DM. Mol Biol Cell. 2009;20:4162–73. doi: 10.1091/mbc.E09-02-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Summers DW, Douglas PM, Ren HY, Cyr DM. J Biol Chem. 2009;284:3628–39. doi: 10.1074/jbc.M807369200. [DOI] [PMC free article] [PubMed] [Google Scholar]