

Abstract
We report in full detail our studies on the catalytic, asymmetric α-fluorination of acid chlorides, a practical method that produces an array of α-fluorocarboxylic acid derivatives in which improved yield and virtually complete enantioselectivity are controlled through electrophilic fluorination of a ketene enolate intermediate. We discovered, for the first time, that a third catalyst, a Lewis acidic lithium salt, could be introduced into a dually-activated system to amplify yields of aliphatic products, primarily through activation of the fluorinating agent. Through our mechanistic studies (based on kinetic data, isotopic labeling, spectroscopic measurements, and theoretical calculations) we were able to utilize our understanding of this “trifunctional” reaction to optimize the conditions and obtain new products in good yield and excellent enantioselectivity.
Introduction
In recent years, the use of fluorinated molecules has greatly increased in medicinal and pharmaceutical chemistry alike; we believe that chemists now rely on fluorination as a prime weapon in their search for efficacy.1,2 All too often though, they depend upon synthetic chemistry that has barely kept pace, especially in the area of asymmetric fluorination.3 The number of drug candidates that contain fluorine has increased dramatically over the past few years, and the ability to install fluorine (in many cases alpha to a carbonyl group)4 with stereocontrol has been increasing in importance.1 Because of the unique properties of the fluorine atom, including its high electronegativity, small atomic radius, and high C–F bond strength, strategic fluorination can drastically alter the metabolism of a drug, its bioavailability, activity, or a host of other relevant properties. For example, one fluorine atom, placed tactically and enantioselectively within the candidate molecule, can lower the basicity of a nearby amino group and enhance N-H acidity; it may increase the acidity of a nearby carboxylic acid; it can block racemization at a chiral center or inhibit cytochrome P450 promoted oxidation of the fluorine center and nearby C-H bonds; and it may enhance binding within a receptor protein.5 Add to that the fact that nowadays new chiral drugs must be optically pure or else have an extraordinary reason not to be, and methods for the asymmetric installation of fluorine become more important.
Great strides in enantioselective α-fluorination have been made in the past few years in which β-keto esters, imides, and aldehydes serve as substrates to produce products in high enantioselectivity and yield.6 On the other hand, an appealing addition to the synthetic repertoire is a practical method for the enantioselective α-fluorination of ketene enolates (occurring at the complementary carboxylate oxidation state) that directly produces simple, optically enriched α-fluorinated carboxylic acid derivatives (Figure 1).7 Such a process would provide an entré to the synthesis of optically enriched fluorinated drugs, natural products, and other molecules of biochemical and synthetic interest.
Figure 1. Catalytic, Enantioselective, α-Fluorination of Acid Chlorides.
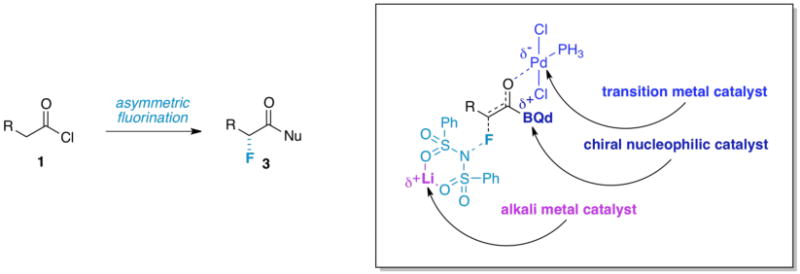
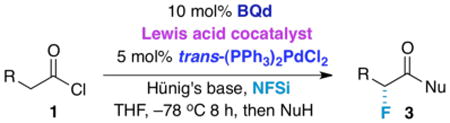
In this paper, we present an asymmetric fluorination method that employs three catalysts working cooperatively (a chiral nucleophile, a transition metal catalyst, and an alkali metal Lewis acid, Figure 1) to afford virtually optically pure products with improved yields. In the search for higher yield and broader scope, we describe how the fluorination method has evolved from an unsatisfactory monofunctional system to a bifunctional procedure, and finally – with the inclusion of an alkali metal Lewis acid cocatalyst – a trifunctional process8. Although mechanistically complex, the method is operationally simple and practical. Our rationale for a trifunctional catalytic system arose from an initial mechanistic study that is supported by theoretical calculations, and verified by experimental findings.
Earlier Work
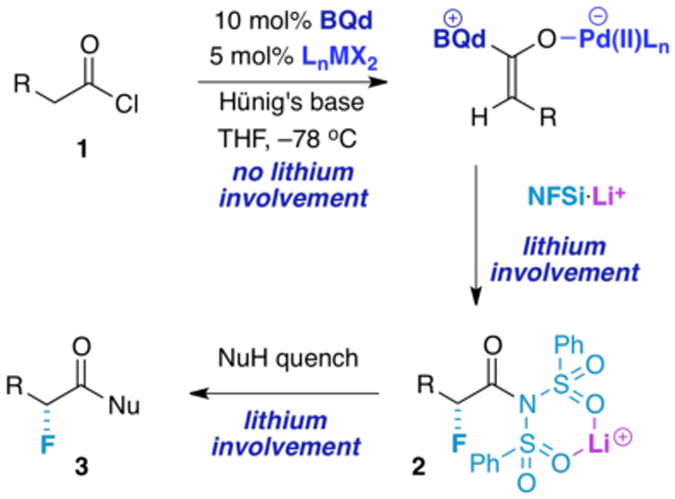
Some time ago, we began our search for a catalytic, enantioselective fluorination method with a monofunctional catalytic system (that employed benzoylquinine or benzoylquinidine as a chiral catalyst, Figure 2)9 to generate α-fluorinated products from acid chlorides 1. This unsatisfactory experiment gave only small amounts of product, an outcome we rationalized by the capricious and unselective activity of the intermediate zwitterionic (ketene) enolate intermediate. This result motivated us to develop a bifunctional approach with the addition of an enolate coordinating transition metal complex10 [typically (dppp)NiCl2, trans-(PPh3)2PdCl2, or (PPh3)2PtCl2)] as cocatalyst.11 Chiral ketene enolate intermediates 4 (from acid chlorides), further activated by a transition metal-based cocatalyst, were efficiently fluorinated with commercially available N-fluorodibenzenesulfonimide (NFSi) to afford the presumed acyl imide 2. This intermediate was then quenched in situ by a variety of nucleophiles (NuH) to produce configurationally stable α-fluorinated carboxylic acid derivatives 3. Depending on the work-up conditions, a variety of highly optically active α-fluorinated carboxylic acid derivatives are made available by choice of quenching nucleophile (Figure 2).12
Figure 2. Preliminary Synthetic Outline.
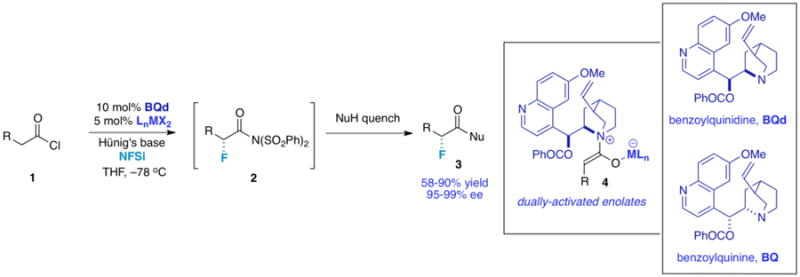
A standard reaction includes NFSi, Hünig's base, and acid chloride 1 (1 equiv. each), 0.1 equiv. cinchona alkaloid catalyst, and 0.05 equiv. metal complex cocatalyst [(dppp)NiCl2, trans-(PPh3)2PdCl2, or (PPh3)2PtCl2)] in THF at –78 °C for 6-12 h, followed by a quench with the desired nucleophile, standard work-up, and chromatography. With this method we could consistently produce products in superb ee (enantiomeric excess) and good to excellent yield only where R is an aromatic ring or otherwise conjugated system (Figure 3). Along with the aromatic substrates, much of the scope results from the quenching nucleophile; α-fluorinated carboxylic acids, amides, esters, and even peptides are all accessible depending on the nucleophile employed to quench the reaction. For example, a water quench affords α-fluoro carboxylic acids, compounds that potentially are of broad utility as derivatizing agents. Weinreb amides can also be readily obtained, thus allowing access to α-fluoro ketones.13,14 As per usual for cinchona alkaloid-promoted ketene enolate reactions, both antipodes of the product are available in similarly high ee by selection of either BQd (benzoylquinidine) or its “pseudoenantiomer” BQ (benzoylquinine).15 On the other hand, most aliphatic substrates (R = alkyl) worked poorly, initially producing desired fluorinated products in low yield. As our major remaining goal was to provide a more broadly applicable method of α-fluorination, the introduction of a third catalyst to the system was investigated. We now report in detail a trifunctional (or “tricomponent”) method that increases product yield and scope while maintaining excellent enantioselectivity, and recount our rationale behind this approach as well.
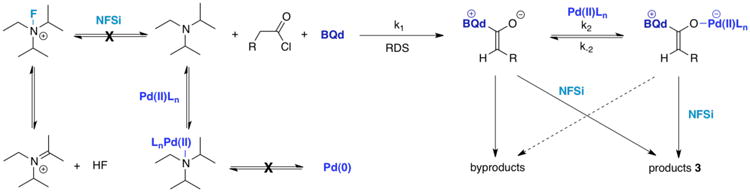
Figure 3. Initial Mechanistic Outline.
Results and Discussion
Initial Kinetic Studies on the Bifunctional System
Intensive kinetic study on catalytic, asymmetric reactions has proven to be an excellent starting point to optimize reaction conditions, as well as to postulate a mechanism.16 Our investigations began by altering the concentration of a single reagent and measuring the rate of product formation as compared to the unmodified reaction. For example, to five dry flasks were added trans-(PPh3)2PdCl2 (0.05 equiv.) and benzoylquinidine (BQd, 0.1 equiv.). Under a nitrogen atmosphere, 1 mL THF was added to each flask and the solutions were cooled to −78 °C. Hünig's base (1.1 equiv.) was added neat to each mixture, followed by a solution of N-fluorodibenzenesulfonimide (NFSi, 1 equiv.) in 0.33 mL THF. After 5 min, a solution of phenylacetyl chloride (1 equiv.) in 0.66 mL THF was added to each reaction mixture. At time intervals of 30, 40, 50, 60, and 70 s, MeOH (0.5 mL) was injected to quench each reaction separately, and all were then allowed to warm to 25 °C overnight. The measured rates were then extrapolated to 0% conversion.
A surprise emerged while discerning the order of each reagent – the rate of reaction is inversely proportional to the concentration of Hünig's base in the regime of 0.5-1.1 equiv. whereas it is proportional at low concentrations (0.01-0.05 equiv.). A number of rationales are conceivable: namely, Hünig's base may be responsible for ketene dimerization; it may react with NFSi in a nonproductive way; or it may bind to the metal cocatalyst, or act to convert the Pd(II) catalyst to Pd(0) at high concentrations (Figure 3). Each of these possibilities should reveal a quantifiable trace. For example, ketene dimers can be readily isolated, but no dimer products were identified in any reaction mixtures. Likewise, when NFSi was mixed with Hünig's base at −78 °C, no interaction between the two species was detected by 19F NMR (although they do react readily at 25 °C). Outside of the unusual rate dependence on Hünig's base, BQd and the acid chloride each displayed a proportional dependence on the rate of acid chloride consumption, while there was no observed dependence on this rate with NFSi or the Pd catalyst. The results from the bifunctional kinetic experiments are summarized in Table 1.
Table 1. The Effect of Reagents on the Initial Rate of Reaction.
| entry | reagent | rate dependence |
|---|---|---|
| 1 | BQd | [BQd]a.b |
| 2 | acid chloride | [acid chloride]a,b |
| 3 | NFSi | 0a |
| 4 | trans-(PPh3)2PdCl2 | 0a |
| 5 | Hünig's base (0.5-1.1 equiv.) | 1/[Hünig's base]b |
| 6 | Hünig's base (0.01-0.05 equiv.) | [Hünig's base]b |
When the concentration of the specified reagent is altered, the initial rate of acid chloride consumption changes by the factor indicated.
When the concentration of the specified reagent is altered, the initial of product formation changes by the factor indicated.
KIE Studies
Kinetic isotope effect (KIE) studies can also shed light on the reaction mechanism.17 For example, observation of a primary KIE upon introduction of deuterium labels in the α-position of the acid chloride would indicate rate-determining dehydrohalogenation, whereas an inverse secondary isotope effect could point instead towards rate limiting fluorination (scenario a, Figure 4). The rate of product formation for α,α-dideuteriophenylacetylchloride compared with phenylacetyl chloride gave an observed KIE of 3.12, which indicates that dehydrohalogenation may be rate limiting.
Figure 4. Isotope Labeling Studies.
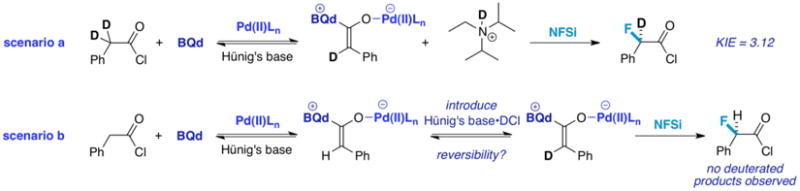
There also exists another possibility – the observed isotope value could be indicative of an equilibrium isotope effect. We hypothesized that reversible enolate formation (and the effect of the cocatalyst thereupon) could be documented by introduction of the DCl salt of Hünig's base, and quantification of the label in the product (scenario b, Figure 4). In the experiment, no incorporation of deuterium was seen in THF as the solvent. As methylene chloride also works well with this reaction system and possesses the ability to solubilize Hünig's base salts, it was tested to determine whether the lack of incorporation occurred due to insolubility. Still, no incorporation was seen, suggesting that reversible enolate formation does not occur, thus negating the possibility of an equilibrium isotope effect. Finally, through analysis of the rate of acid chloride consumption, it was evident that the rate of reaction is proportional to the concentration of the acid chloride and base, confirming dehydrohalogenation as the rate determining step.
All in all, these kinetic data suggest that a slow addition of Hünig's base might be beneficial to take advantage of its peculiar concentration dependence on the initial rate of product formation. In a standard fluorination of octanoyl chloride, an increased yield of 60% (from 40%) was obtained upon slow addition of Hünig's base over 12 h (quenched with aniline, >99% ee). For hydrocinnamoyl chloride, the yield increases from 4% to 22% to 35% when the slow addition is increased from zero time to 12 h to 24 h, respectively (aniline quenched, 99% ee). This result prompted a series of experiments to determine the optimum slow addition length of Hünig's base. When increasing the slow addition time to 24 h, the yield did not increase past 60% for any aliphatic acid chloride tested with the notable exception of the quirky substrate phthalimidopropionyl chloride (which contains a metal binding site),12 and longer slow additions over 48 h showed no signs of improvement. Most aliphatic acid chlorides treated with NFSi and a slow addition of Hünig's base showed similar improvements in yield. While the slow addition of Hünig's base was an important discovery, we envisioned that a Lewis acid could selectively enhance the electrophilicity of NFSi and thereby increase the yield of the desired product. Nevertheless, the kinetic measurements were less illuminating than desired as the first (and least interesting) step proved to be rate determining.
A Trifunctional System
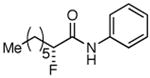
The possibility of trifunctional catalysis came to mind when investigating ways to improve the yield of aliphatic acid chlorides. We speculated that a second Lewis acid could specifically coordinate with the NFSi, thereby increasing its electrophilicity. Among various metals tested for this purpose (Table 2),18 earlier work on Lewis acid catalysis by Nelson19 inspired us to try both LiClO4 and LiPF6, which turned out to be the only two metal salts that gave unambiguously increased yields (no difference in yield was seen by changing the counterion).20 Additionally, when 10 mol% LiClO421 was added to a standard reaction with isovaleryl chloride (no slow addition), an increase in yield of the fluorinated product from 15% to 50% was observed (entries 14, 15). This demonstrates the potential power of the trifunctional catalytic system, at least for reactions that suffer from low yield. When lithium addition is combined with slow addition of Hünig's base over 12 h, the yield increases further to 73% (Table 2, entry 15). Table 3 shows the products of a variety of aliphatic acid chlorides that were tested under the optimized conditions (trifunctional catalysis with slow addition of base) that gave good yield and excellent enantioselectivity. It is important to highlight that octanoyl chloride gave 83% yield (Table 3, entry 1) when 10 mol% LiClO4 was used in addition to the slow addition of Hünig's base, up from 60% yield with solely the slow addition. In every instance, the combination of the slow addition and the catalyst trio resulted in a remarkable increase in yield from what was often originally a trace amount of product. Slow addition by itself typically produces yields about 25%-50% less than lithium addition.
Table 2. Trifunctional Catalysis Screening.
| ||||
|---|---|---|---|---|
|
| ||||
| entrya | third catalyst, (equiv.) | R | NuH | % yield |
| 1 | none | CH2Ph | MeOH | <4 |
| 2 | LiPF6-THF, (0.1) | CH2Ph | MeOH | 21 |
| 3 | LiCI04,(1) | CH2Ph | MeOH | trace |
| 4 | LiCI04, (0.1) | CH2Ph | MeOH | 21 |
| 5 | La(OTf)3,(0.1) | CH2Ph | MeOH | 8 |
| 6 | Sm(OTf)3, (0.1) | CH2Ph | MeOH | 5 |
| 7 | Yb(OTf)3, (0.1) | CH2Ph | MeOH | 3 |
| 8 | LiCI04, (0.1)b | CH2Ph | MeOH | trace |
| 9 | (PPh3)2Pd(CI04)2, (0.05)b | Ph | MeOH | trace |
| 10 | none | CH2Ph | PhNH2 | 4 |
| 11 | none | CH2Ph | PhNH2 | 22c |
| 12 | LiCI04, (0.1) | CH2Ph | PhNH2 | 39c |
| 13 | LiCI04, (0.1) | CH2Ph | PhNH2 | 44d |
| 14 | none | i-Pr | PhNH2 | 15 |
| 15 | LiCI04, (0.1) | i -Pr | PhNH2 | 50 |
| 16 | LiCI04, (0.1) | i -Pr | PhNH2 | 73c |
Reaction conditions: 1 equiv. NFSi, 1 equiv. acid chloride, 0.1 equiv. BQd, 0.05 equiv. frans-(PPh3)2PdCI2, 1.1 equiv. Hünig's base, THF, −78 °C, followed by nucleophilic quench at −78 °C after 8 h.
No trans-(PPh3)2PdCI2 catalyst.
Slow addition of Hünig's base over 12 h.
Slow addition of Hünig's base over 24.
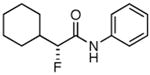
Table 3. Products from α-Aliphatic Acid Chloride Substrates under Optimized Conditions.
| entrya | product | yield | ee (de) |
|---|---|---|---|
| 1 |
|
83% | >99% |
| 2 |
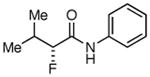
|
73% | >99% |
| 3 |
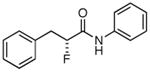
|
44% | >99% |
| 4 |
|
53% | >99% |
| 5b |
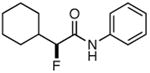
|
55% | >99% |
| 6 |
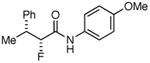
|
40% | (>99%) |
| 7 |
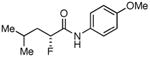
|
46% | >99% |
| 8b |
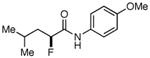
|
56% | >99% |
| 9 |
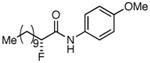
|
66% | >99% |
Reaction conditions: 1 equiv. NFSi, 1 equiv. acid chloride, 0.1 equiv. LiClO4, 0.1 eq. BQd, 0.05 equiv. trans-(PPh3)2PdCl2, 1.1 equiv. Hünig's base (slow addition over 12 h), THF, −78 °C, followed by nucleophilic quench after slow addition at −78 °C.
BQ was used instead of BQd.
“Knockout” Kinetic Experiments: Subdivision of the Reaction into Steps
As the rate determining step occurs so early in the reaction sequence, it leaves the most interesting steps (which crucially affect the chemoselectivity of the reaction) “downstream.” Given the complexity of the reaction, it makes sense to subdivide the overall reaction into four discrete processes: dehydrohalogenation of the acid chloride 1 (which is rate determining); fluorination to form a chiral acylammonium-sulfonimide 5 (enantioselectivity-determining); acylation of the sulfonimide anion to form 2, and finally, transacylation by an adventitious nucleophile to provide 3. The downside of such a subdivision is that each of the pieces studied separately departs from a holistic ideal, and could change its character when separated from the others.
The nature of the first step is clear enough from our overall kinetic study. What about the second, enantioselectivity-determining step? In prior work, we have been able to gain additional insight into complex reactions involving ketenes by preformation of the ketene22 to study reaction rates that happen after the rate determining step. A simple approach was thus taken to understand the complexities of the fluorination step. In the event, we employed high concentrations of BQ and the preformed ketene (pseudo first order conditions). In this scenario, we don't have to worry about catalyst turnover due to slow acylation of sulfonimide; in essence, we obviate this step, as a reaction quench where an adventitious alcohol leads to the observed product. In order to “knock out” the RDS (rate determining step) and preform the ketene, five dry flasks were supplied with trans-(PPh3)2PdCl2 (0.05 equiv., 0.5 mL THF) and cooled with liquid N2. Excess methylketene was generated23 from a solution of THF (1.5 mL) and distilled into each flask, then warmed to −78 °C. A solution of N-fluorodibenzenesulfonimide (NFSi, 1 equiv.) in 0.33 mL THF was added to the reaction mixture, followed by benzoylquinine (BQ) (1.1 equiv.). At time intervals of 30, 60, 90, 120, and 150 s a solution of benzyl alcohol and HCl (1.1 equiv. benzyl alcohol) was injected to quench each reaction, and all were then allowed to warm to 25 °C overnight. The data are shown in Table 4.
Table 4. Investigations on the Rate of Fluorination with Preformed Methylketene.
| entry | reagent changea | rate factor increaseb |
|---|---|---|
| 1 | [NFSi] → 2× [NFSi] | 2 |
| 2 | [trans-(PPh3)2PdCl2] → 2× [trans-(PPh3)2PdCl2] | 2 |
| 3 | no LiClO4 → 0.1 equiv LiClO4 | 1.2 |
Reaction conditions: 1 equiv. NFSi, excess methylketene, 1.1 equiv. BQd, 0.05 equiv. trans-(PPh3)2PdCl2, THF, −78 °C, followed by nucleophilic quench at −78 °C.
Rate of product formation.
By starting with a preformed ketene, the rate-limiting step was observed to be the reaction of the ketene enolate with NFSi. The rate of reaction was also found to be dependent on the concentration of PdII. However, a strict proportionality was observed; for example a doubling of the Pd(II) concentration resulted in a doubling in rate. On the other hand, the addition of lithium to the “knock out” reaction increases the rate by 20% while increasing the yield by 30%. These data indicate that lithium acts not only to increase the rate of formation of the desired product, but may act as well to suppress possible side reactions (dimerization and polymerization).
The Role of Li+
These findings prompted us to undertake a spectroscopic inquiry into the possible role of LiClO4. Binding between LiClO4 and NFSi (each 0.2 mmol) in THF (1 mL) was observed by 19F NMR by a shift from 37.89 ppm to 40.25 ppm at 25 °C, suggesting (along with the aforementioned rate data) Lewis acid activation of the fluorinating agent. Another possible effect of the lithium salt is that an ion methathesis replaces Cl- with ClO4- on the Pd(II) catalyst. However, when the Pd cocatalyst was replaced with (PPh3)2Pd(ClO4)2 or (PPh3)2Pd(ClO4)Cl, the reaction gave only trace amounts of product (Table 2, entry 9), suggesting that the lithium salt does not perturb the transition metal catalyst and that both metals need to act independently of each other in order to administer their effect.
Interestingly, UV measurements show that the addition of LiClO4 (1 equiv., 0.0045 mmol) to phenylethylketene (1 equiv., 0.0045 mmol) in the presence of BQd (1 equiv., 0.0045 mmol) in THF (3 mL) diminishes the concentration of the ketene enolate at −78 °C by 8.5% (Figure 6, EQ-B). By contrast, Pd(II) acts to promote ketene enolate concentration.24 When 1 equiv. of the lithium salt is added to a fluorination reaction of hydrocinnamoyl chloride with slow addition of Hünig's base, only trace amounts of product were obtained (Table 1, entry 3). This was a surprising result because a large increase in lithium was expected to demonstrate better binding with NFSi, resulting in improved yield. One possibility is that LiClO4 binds to BQd, quenching both catalysts (Figure 5, EQ-C), but when 1 equiv. BQd and LiClO4 were mixed together in THF, no binding was observed by 1H NMR or IR spectroscopy. Another option is that lithium preferentially binds to the ketene (as opposed to the enolate). This would explain the increase in ketene concentration (and decrease in ketene enolate concentration) that was observed by UV. Since these isolated situations don't always account for our experimental observations, another scenario was considered. As NFSi binds to lithium, this must allow enough of the ketene to operate independently of lithium when all the reagents are added together. When excess lithium was used, it may severely reduce the concentration of ketene enolate so much so that the reaction barely proceeds. A balance must be achieved with the third catalyst to achieve a superior benefit in yield, where 10 mol% was found to be the optimum concentration for substrates.
Figure 6. Quenching of LiClO4 by TMEDA.
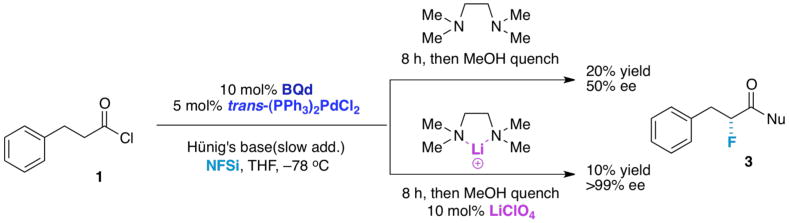
Figure 5. Possible Roles of LiClO4.
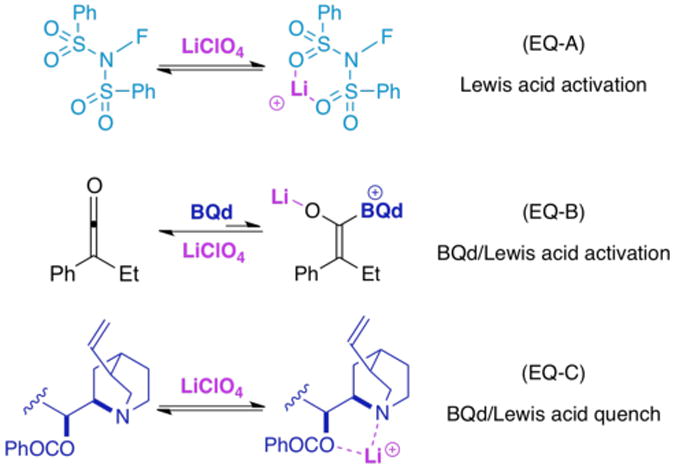
When 1 equiv. of TMEDA is added to a fluorination reaction (1 equiv. hydrocinnamoyl chloride, NFSi, and Hünig's base, plus 0.1 equiv. BQd at −78 °C in THF), the yield increases to 20% from trace amounts, but the ee erodes to 50%. When 1 equiv. of lithium is added under the same reaction conditions, the yield drops to 10% (from 20%), but the ee is restored to >99%. The added TMEDA mimics the catalytic role of BQd, but lacks the ability to induce optical activity to the product. Addition of lithium sequesters the TMEDA, preventing the achiral base from participating in the reaction, and restoring the ee (Figure 6).25 Remarkably, lithium does not hinder BQ or BQd in a similar way, but acts only to promote the reaction.
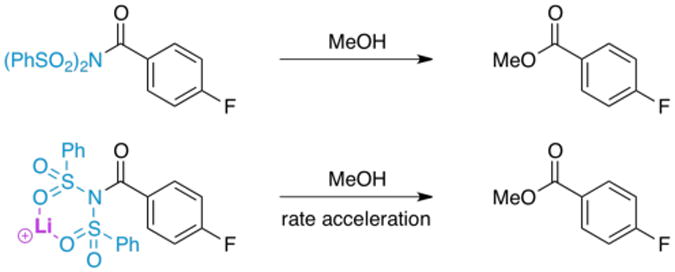
The rate enhancement imparted by lithium in our kinetic study of the ketene enolate fluorination step (ca. 20%), while significant, may not explain completely the increase in yield. One other putative role of lithium may be as a catalyst for the acylation of the quenching nucleophile. To shed light on this possibility, the methanolysis of an acylsulfonimide control was also studied (Figure 7). If lithium binds to NFSi and increases its electrophilicity, it should also bind to acylsulfonimides in a similar fashion and increase their susceptibility to nucleophilic attack. In the event, the acylsulfonimide was allowed to react with MeOH in THF at 25 °C, in one instance with 10 mol% lithium perchlorate present and in the other instance with no lithium source. The extent of methanolysis was monitored by 19F NMR; as expected, the inclusion of lithium ion in the reaction increases the rate of methyl ester formation, giving 4.9% conversion without lithium and 8.4% conversion with lithium after 15 min of stirring, indicating an approximate two-fold rate increase when extrapolated to 0% conversion. This suggests that lithium may act to increase rates and yields by acylsulfonimide activation in situ as well, and that its role in the overall reaction is quite complex. However, the fact that lithium ion exerts only marginal increases in yields for arylketene-type substrates suggests that this effect should be moderate at most.
Figure 7. Methanolysis of an Acylsulfonimide.
Transition Metal Roles
(PPh3)2PdCl2 was also tested for possible binding to BQd. Not surprisingly, when investigated by 1H, 13C, and 31P NMR and IR spectroscopy, no apparent binding was observed in THF at 25 °C. Next, (PPh3)2PdCl2 was examined for any potential interaction with NFSi. Monitoring by 19F NMR and IR shows no change in the chemical shift of NFSi in THF at 25 °C or its absorbance profile, which suggests that no binding to NFSi occurs.
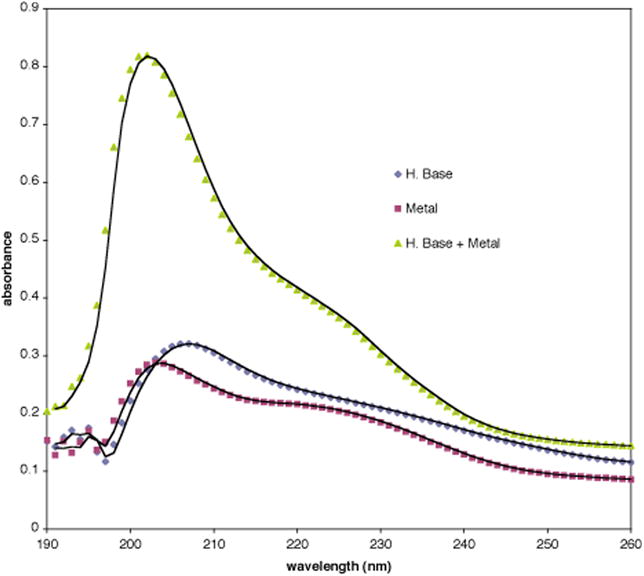
When Hünig's base is mixed with an equimolar amount of [(tris(4-butylphenyl)phosphine)2]PtCl2, (used as a highly soluble surrogate for trans-(PPh3)2PdCl2 that performs equally well in fluorination reactions), and monitored by UV spectroscopy, the resulting absorbance at 202 nm is larger than the individual absorbances at 202 nm of each reagent added together, an observation that points toward metal complexation (Figure 8).26 The λmax shifts slightly upon mixing to a single maximum at 202 nm at 25 °C, (the λmax of Hunig's base is at 207 nm and the λmax of the metal is at 203 nm). Another question concerns whether the metal catalyzes the oxidation of Hünig's base; acetone can usually be found (clearly identifiable by 13C NMR) in crude reaction mixtures in which Hünig's base is oxidized,27 yet none was observed by NMR in the mixture. The role of Hünig's base is as the terminal HCl acceptor in the shuttle base system where it regenerates the kinetic base, BQd; however, if its concentration builds beyond that which is necessary for its primary function, it may bind to the metal catalyst, thereby reducing its efficacy.28
Figure 8. UV-VIS Spectrum of Metal–Hünig's Base Mixtures.
Lastly, the rate of starting material consumption on the entire reaction was again studied, this time with the addition of the new alkali metal (Figure 9). The kinetic experiments show that the rate of acid chloride consumption has no dependence on the concentration of lithium perchlorate, confirming that it plays no identif part in the rate limiting dehydrohalogenation step.
Figure 9. Rate Experiments with Lithium Cocatalyst.
 |
(1) |
Transacylation Experiments
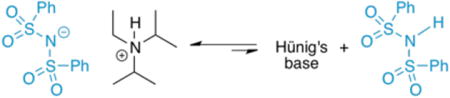
One outstanding ambiguity is how transacylation of the intermediate acyl ammonium salt (Figure 10, 5) occurs.29 Transfer of fluorine to the ketene enolate is expected to form a tight ion pair that can undergo transacylation. However, in THF solvent, Hünig's·HCl salt could protonate the sulfonimide ion rapidly, and one would expect such proton transfers to be fast. A control experiment showed that when (PhSO2)2NH is treated with 1 equiv. Hünig's base in THF at 25 °C, deprotonation of the sulfonimide occurs almost quantitatively by 1H NMR (eq 1).
Figure 10. Transacylation Crossover Experiments.
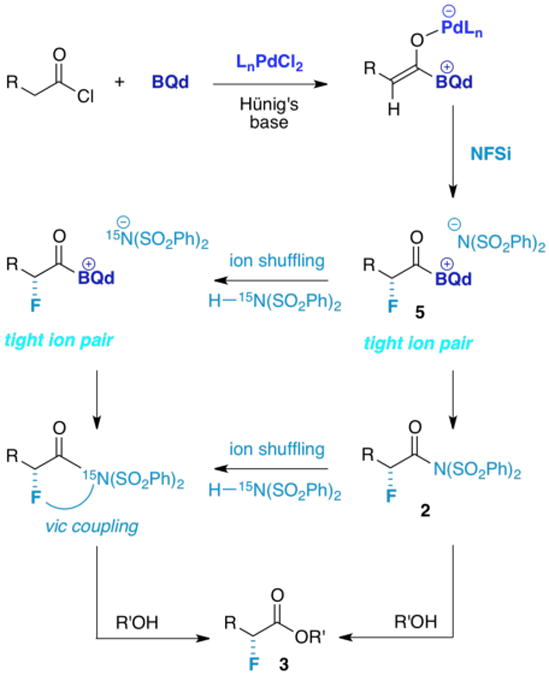
One way to shed light on catalyst turnover is a crossover experiment, employing 15N labeled sulfonimide30 (Figure 10). “Shuffling” of sulfonimide ions through solvent separation of ion pairs should result in incorporation of the label, which can be detected in the intermediate acylsulfonimide by a 19F–15N or 1H– 15N vicinal coupling during an in situ NMR experiment.31 Indeed, when 0.15 equiv. of 15N labeled dibenzenesulfonimide is added to a standard fluorination reaction with 1 equiv. phenylacetyl chloride, incorporation is seen in the splitting of the 19F NMR signals of the crude, unquenched reaction at 25 °C. However, when the reaction is monitored at −78 °C, no incorporation is seen. This is consistent with a tight ion pair at low temperature that does not solvent separate; instead, it turns over the chiral catalyst through transacylation and does not “shuffle.” Not surprisingly, species 2 was found to be stable enough to exist at 25 °C in solution, but it could not be isolated efficiently. For example, quenching with methanol at −78 °C after the reaction was allowed to reach 25 °C, still resulted in formation of 3 in identical yield.
Computational Studies
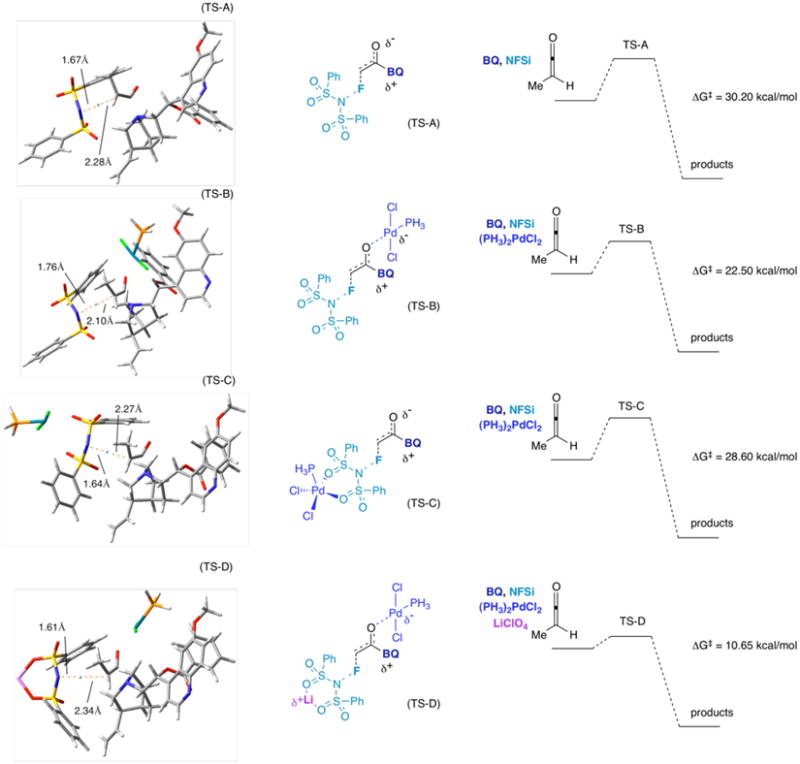
Computational chemistry is often used to account for stereochemical and energetic preferences,32 and we took advantage of modeling programs to gain insight into the observed enantioselectivity and reaction energetics. Several calculations were performed on various possible transition states for fluorination at the Kohn Sham hybrid-DFT B3LYP level (Figure 11).33,34 In two of the transition state (TS) metal-coordinated cases (TS-B, and TS-D), the Pd(II) is bound to the ketene enolate oxygen. These transition states revealed that TS-B is lower in free energy than the uncoordinated case (TS-A) by 7.7 kcal/mol. When compared to starting fragments, TS-D is lower than TS-B by 11.8 kcal/mol, and 19.5 kcal/mol lower than TS-A (we know that the Pd(II) binds to the ketene enolate, enhancing its chemoselectivity and increasing its concentration). TS-C is only slightly lower in energy than TS-A (1.6 kcal/mol), suggesting that TS-C is unlikely in light of the other possible transition states that are much lower in energy. TS-D is the lowest energy transition state (and thus the most likely pathway), lengthening the partial C-F bond in an evidently “earlier,” more exothermic, transition state. The lower energy of TS-D suggests that the addition of both Li(I) and Pd(II) could result in a lower transition state energy that favors the desired product formation, a fact that was also demonstrated experimentally. Replacing BQ with BQd gives similar results, and TS-A, TS-B, TS-C, and TS-D then give Δ G‡ = 30.1, 22.3, 28.4, 10.5 kcal/mol respectively. Transition state calculations using BQ predict the stereochemistry at the fluorinated carbon as S while BQd gives R; this corresponds to what is observed experimentally.
Figure 11. Transition State Calculations.
A Unified Mechanistic Scenario
The complete mechanistic outline is proposed in Figure 12. First the acid chloride undergoes a rate-determining dehydrohalogenation to form the basic ketene enolate involving the chiral nucleophilic catalyst. Next, the ketene enolate can either react with NFSi, NFSi bound to lithium, or bind to the palladium cocatalyst, which can then go on to react with NFSi in the same fashion. Note that the kinetic data on the “knock out” reaction reflect these conclusions; although the rates of reaction increase with each introduction of a cocatalyst, the reaction also proceeds to a measurable extent in the absence of the cocatalysts, especially in the case of Li+. Thus, the “monofunctional” and “bifunctional” reaction terms must be taken into account in the overall equation.
Figure 12. A Unified Mechanistic Proposal.
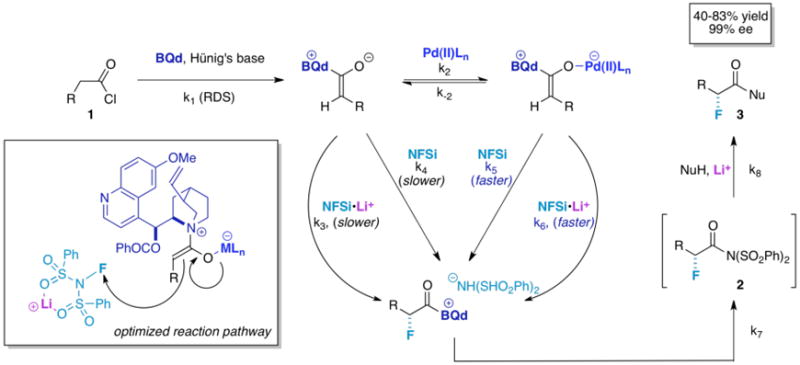
In Figure 12, side reactions that happen at the ketene enolate stage producing unwanted side products are not shown. The four kinetic pathways represented by (k3, k4, k5, k6) after the RDS each have a different rate. Addition of Pd to the reaction increases the rate of product formation, so k5[Pd] is larger than k4. The rate dependence on Pd (II) shows the Pd coordinated reaction pathways are dominant compared to the transition metal free pathways. Since addition of lithium and Pd increases the rate of product formation beyond the rate represented by k5, pathways involving k6 or k3 must be faster than k5.
Additionally, when lithium is added without Pd, the reaction affords product very slowly, which indicates that k3 represents a slow path. Of course, the pathway involving k4 is a minor contributor as well. The kinetic data also agree that the rate of product formation is much greater if not dominant for addition of lithium with Pd vs. the rate of product formation with only lithium. This leaves the trifunctional pathway, characterized by k6, as potentially the fastest. The resulting rate equation for the trifunctional system, focusing on the isolated fluorination step after “knock out,” reflects contributions from competing bifunctional and monofunctional pathways, is shown in eq 2, with the dominant pathways highlighted in blue. In the final reaction step, the chiral
| (2) |
catalyst is released through transacylation of the bis(sulfonimide). The resulting acylsulfonimide intermediate is then quenched by a nucleophile to yield product, a process that is catalyzed by lithium ion as well (3).
Conclusion
We have chronicled the evolution of a practical procedure for the catalytic, asymmetric α-fluorination of acid chlorides. Through the knowledge that we gained from reaction development in tandem with mechanistic studies, we were able to develop a new trifunctional catalytic system that affords, in wide scope, optically active, fluorinated amides, esters, and other carboxylic acid derivatives in fair to high yield and excellent enantiomeric excess directly, from a variety of acid chlorides. In so doing, we are able to propose a mechanistic scenario that is accounts for the very diverse aspects of this polyfunctional catalytic system, and to provide a basis for the further study of polycomponent catalysis.
Experimental
Optimized α-Fluorination Procedure
To a dry 10 mL round bottom flask equipped with a stir bar was added trans-Pd(PPh3)2Cl2 (5.8 mg, 0.0083 mmol), benzoylquinidine (BQd, 7.1 mg, 0.0166 mmol), and LiClO4 (1.7 mg, 0.0166 mmol). Under a nitrogen atmosphere, 0.3 mL THF was added and the solution was cooled to −78 °C. A solution of N-fluorodibenzenesulfonimide (NFSi, 52.3 mg, 0.166 mmol) in 0.4 mL THF was added, followed by a solution of isovaleryl chloride (20.0 mg, 0.166 mmol) in 0.6 mL THF. A solution of Hünig's base (0.03 mL, 0.18 mmol) in 0.7 mL THF was added via syringe pump over 20 h and the reaction was maintained at −78 °C for 2 additional hours. Aniline (0.018 mL, 0.2 mmol) was added at −78 °C and the reaction was allowed to warm to 25 °C overnight. The solvents were removed and the crude mixture purified by column chromatography, eluting with a mixture of EtOAc and hexanes to give 23.9 mg of (R)-2-fluoro-3-methyl-N-phenylbutanamide (73% yield, >99% ee).
Supplementary Material
Acknowledgments
T.L. thanks the NIH (Grant GM064559) and the John Simon Guggenheim Memorial Foundation for support. D.H.P. thanks Johns Hopkins University for a Zeltmann Graduate Fellowship.
Footnotes
Supporting Information Available: General experimental procedures compound characterization, and complete Reference 34a. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Liu P, Sharon A, Chu CK. J Fluorine Chem. 2008;129:743–766. doi: 10.1016/j.jfluchem.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Park BK, Kitteringham NR. Drug Metab Rev. 1994;26:605–643. doi: 10.3109/03602539408998319. [DOI] [PubMed] [Google Scholar]; (c) Smart BE. J Fluorine Chem. 2001;109:3–11. [Google Scholar]; (d) Ojima I. Fluorine in Medicinal Chemistry and Chemical Biology. Wiley-Blackwell; Chichester, U.K.: 2009. [Google Scholar]; (e) Chambers RD. Fluorine in Organic Chemistry. Wiley; New York: 1973. [Google Scholar]
- 2.For recent reviews of the pharmacology of fluorinated derivatives, see: Thomas CJ. Curr Top Med Chem. 2006;6:1529–1543. doi: 10.2174/156802606777951109.Ismail F. J Fluorine Chem. 2002;118:27–33.
- 3.Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 4.Cahard D, Xu X, Couve-Bonnaire S, Pannecoucke X. Chem Soc Rev. 2010;39:558–568. doi: 10.1039/b909566g. [DOI] [PubMed] [Google Scholar]
- 5.(a) Kirk KL. J Fluorine Chem. 2006;127:1013–1029. [Google Scholar]; (b) Bégué JP, Bonnet-Delpon D. Biological Impacts of Fluorination: Pharmaceuticals Based on Natural Products. In: Tressand A, Haufe G, editors. Fluorine and Health: Molecular Imaging, Biomedical Materials and Pharmaceuticals. First. Elsevier; Amsterdam, The Netherlands: 2008. pp. 553–622. [Google Scholar]; (c) Smart BE. J Fluorine Chem. 2001;109:3–11. [Google Scholar]; (d) Schlosserm M, Michel D. Tetrahedron. 1996;52:99–108. [Google Scholar]; (e) Resnati C, Soloshonok VA, editors. ACS Monograph. Vol. 187. American Chemical Society; Washington D.C.: 1995. Chemistry of Organic Fluorine Compounds. II. A Critical Review; pp. 1119–1125. [Google Scholar]; f Amii H, Uneyama K. Chem Rev. 2009;109:2119–2183. doi: 10.1021/cr800388c. [DOI] [PubMed] [Google Scholar]; Ismail FMD. J Fluorine Chem. 2002;118:27–33. [Google Scholar]; (h) Park BK, Kitteringham NR, O'Niell PM. Annu Rev Pharmacol Toxicol. 2001;41:443–470. doi: 10.1146/annurev.pharmtox.41.1.443. [DOI] [PubMed] [Google Scholar]
- 6.(a) Ibrahim H, Brunet VA, O'Hagan D. Angew Chem Int Ed. 2008;47:1179–1182. doi: 10.1002/anie.200704700. [DOI] [PubMed] [Google Scholar]; (b) Pihko PM. Angew Chem Int Ed. 2006;45:544–547. doi: 10.1002/anie.200502425. [DOI] [PubMed] [Google Scholar]; (c) Togni A. Chem Commun. 2004:1147–1155. [Google Scholar]; (d) Ma JA, Cahard D. Chem Rev. 2004;104:6119–6146. doi: 10.1021/cr030143e. [DOI] [PubMed] [Google Scholar]; (e) Ishimaru T, Shibata N, Horikawa T, Yasuda N, Toru T. Angew Chem Int Ed. 2008;47:4157–4161. doi: 10.1002/anie.200800717. [DOI] [PubMed] [Google Scholar]; (f) Shibata N, Ishimaru t, Nakamura S, Toru T. J Fluorine Chem. 2007;128:469–483. [Google Scholar]; (g) Shibata N, Kohno J, Takai K, Ishimaru T, Nakamura S, Toru T, Kanemasa S. Angew Chem Int Ed. 2005;117:4204–4207. doi: 10.1002/anie.200501041. [DOI] [PubMed] [Google Scholar]; (h) Suzuki T, Hamashima Y, Sodeoka M. Angew Chem Int Ed. 2007;46:5435–5439. doi: 10.1002/anie.200701071. [DOI] [PubMed] [Google Scholar]; (i) Hamashima Y, Sodeoka M. Synlett. 2006;10:1467–1478. [Google Scholar]; (j) Franzen J, Marigo M, Fielenbach D, Wabnitz TC, Kjærsgaard A, Jørgensen KA. J Am Chem Soc. 2005;127:18296–18304. doi: 10.1021/ja056120u. [DOI] [PubMed] [Google Scholar]; (k) Beeson TD, MacMillan DWC. J Am Chem Soc. 2005;127:8826–8828. doi: 10.1021/ja051805f. [DOI] [PubMed] [Google Scholar]; (l) Hamashima Y, Yagi K, Takano H, Tamas L, Sodeoka M. J Am Chem Soc. 2002;124:14530–14531. doi: 10.1021/ja028464f. [DOI] [PubMed] [Google Scholar]; (m) Hintermann L, Togni A. Angew Chem Int Ed. 2000;39:4359–4362. doi: 10.1002/1521-3773(20001201)39:23<4359::AID-ANIE4359>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 7.Bellezza F, Cipiciani A, Riccib G, Ruzziconib R. Tetrahedron. 2005;61:8005–8012. [Google Scholar]
- 8.(a) Poe SL, Kobašlija M, McQuade DT. J Am Chem Soc. 2007;129:9216–9221. doi: 10.1021/ja071706x. [DOI] [PubMed] [Google Scholar]; (b) Poe SL, Kobašlija M, McQuade DT. J Am Chem Soc. 2006;128:15586–15587. doi: 10.1021/ja066476l. [DOI] [PubMed] [Google Scholar]; (c) Broadwater SJ, Roth SL, Price KE, Kobašlija M, McQuade DT. Org Biomol Chem. 2005;3:2899–2906. doi: 10.1039/b506621m. [DOI] [PubMed] [Google Scholar]
- 9.(a) Paull DH, Abraham CJ, Scerba MT, Alden-Danforth E, Lectka T. Acc Chem Res. 2008;41:655–663. doi: 10.1021/ar700261a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Paull DH, Alden-Danforth E, Wolfer J, Dogo-Isonagie C, Abraham CJ, Lectka T. J Org Chem. 2007;72:5380–5382. doi: 10.1021/jo070472x. [DOI] [PubMed] [Google Scholar]; (c) France S, Wack H, Taggi AE, Hafez AM, Wagerle TR, Shah MH, Dusich CL, Lectka T. J Am Chem Soc. 2004;126:4245–4255. doi: 10.1021/ja039046t. [DOI] [PubMed] [Google Scholar]; (d) Peng J, Cui HL, Chen YC. Sci China Chem. 2011;54:81–86. [Google Scholar]; (e) Li XJ, Peng FZ, Li X, Wu WT, Sun ZW, Li YM, Zhang SX, Shao ZH. Chem Asian J. 2011;6:220–225. doi: 10.1002/asia.201000561. [DOI] [PubMed] [Google Scholar]; (f) Blaszczyk R, Gaida A, Zawadzki S, Czubacka E, Gaida T. Tetrahedron. 2010;66:9840–9848. [Google Scholar]; (g) Han X, Zhong F, Lu Y. Adv Synth Catal. 2010;352:2778–2782. [Google Scholar]; (h) Shae PL, Chen XY, Ye S. Angew Chem Int Ed. 2010;49:8412–8416. doi: 10.1002/anie.201003532. [DOI] [PubMed] [Google Scholar]; (i) Kuang YY, Niu JZ, Chen FE. Helv Chim Acta. 2010;93:2094–2099. [Google Scholar]
- 10.(a) Sodeoka M, Hamashima Y. Chem Commun. 2009;39:2778–2782. doi: 10.1039/b911015a. [DOI] [PubMed] [Google Scholar]; (b) Hamashima Y, Sasamoto N, Umebayashi N, Sodeoka M. Chem Asian J. 2008;3:1443–1455. doi: 10.1002/asia.200800120. [DOI] [PubMed] [Google Scholar]; (c) Dubs C, Hamashima Y, Sasamoto N, Seidel TM, Suzuki S, Hashizume D, Sodeoka M. J Org Chem. 2008;73:5859–5871. doi: 10.1021/jo800800y. [DOI] [PubMed] [Google Scholar]; (d) Sodeoka M, Hamashima Y. Pure Appl Chem. 2008;80:763–776. [Google Scholar]; (e) Monguchi D, Beemelmanns C, Hashizume D, Hamashima Y, Sodeoka M. J Organomet Chem. 2008;693:867–873. [Google Scholar]; (f) Suzuki T, Goto T, Hamashima Y, Sodeoka M. J Org Chem. 2007;72:246–250. doi: 10.1021/jo062048m. [DOI] [PubMed] [Google Scholar]; (h) Sasamoto N, Dubs C, Hamashima Y, Sodeoka M. J Am Chem Soc. 2006;128:14010–14011. doi: 10.1021/ja065646r. [DOI] [PubMed] [Google Scholar]
- 11.Ferraris D, Young B, Dudding T, Lectka T. J Am Chem Soc. 1998;120:4548–4549. [Google Scholar]
- 12.Paull DH, Scerba MT, Alden-Danforth E, Widger LR, Lectka T. J Am Chem Soc. 2008;130:17260–17261. doi: 10.1021/ja807792c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis FA, Kasu PVN. Tetrahedron Lett. 1998;39:6135–6138. [Google Scholar]
- 14.(R)-N-(benzyloxy)-2-fluoro-2-(4-methoxyphenyl)-N-methylacetamide was synthesized in 45% yield, >99% ee. See Supporting Information for details.
- 15.(a) Paull DH, Weatherwax A, Lectka T. Tetrahedron. 2009;65:6771–6803. doi: 10.1016/j.tet.2009.05.079. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) France S, Weatherwax A, Lectka T. Eur J Org Chem. 2005:475–479. [Google Scholar]
- 16.(a) Taggi AE, Hafez AH, Wack H, Young B, Ferraris D, Lectka T. J Am Chem Soc. 2002;124:6626–6635. doi: 10.1021/ja0258226. [DOI] [PubMed] [Google Scholar]; (b) Rosner T, Le Bars J, Pfaltz A, Blackmond DG. J Am Chem Soc. 2001;123:1848–1855. doi: 10.1021/ja003191e. [DOI] [PubMed] [Google Scholar]; (c) Kitamura M, Tsukamoto M, Bessho Y, Yoshimura M, Kobs U, Widhalm M, Noyori R. J Am Chem Soc. 2002;124:6649–6667. doi: 10.1021/ja010982n. [DOI] [PubMed] [Google Scholar]; (d) Nielsen LPC, Stevenson CP, Blackmond DG, Jacobsen EN. J Am Chem Soc. 2004;126:1360–1362. doi: 10.1021/ja038590z. [DOI] [PubMed] [Google Scholar]; (e) Jacobsen EN, Marko I, France MB, Svendsen JS, Sharpless KB. J Am Chem Soc. 1989;111 [Google Scholar]; (f) Blackmond DG. J Am Chem Soc. 2001;123:545–553. doi: 10.1021/ja002562o. [DOI] [PubMed] [Google Scholar]; (g) Blackmond DG, Hodnett NS, Lloyd-Jones GC. J Am Chem Soc. 2006;128:7450–7451. doi: 10.1021/ja062173f. [DOI] [PubMed] [Google Scholar]; (h) Buono FG, Blackmond DG. J Am Chem Soc. 2003;125:8978–8979. doi: 10.1021/ja034705n. [DOI] [PubMed] [Google Scholar]; (i) Mueller JA, Sigman MS. J Am Chem Soc. 2003;125:7005–7013. doi: 10.1021/ja034262n. [DOI] [PubMed] [Google Scholar]; (h) Parsons AT, Smith AG, Neel AJ, Johnson JS. J Am Chem Soc. 2010;132:9688–9692. doi: 10.1021/ja1032277. [DOI] [PubMed] [Google Scholar]; (k) Shintani R, Tsuji T, Park S, Hayashi T. J Am Chem Soc. 2010;132:7508–7513. doi: 10.1021/ja1023223. [DOI] [PubMed] [Google Scholar]; (l) Novstrup KA, Travia NE, Medvedev GA, Stanciu C, Switzer JM, Thomson KT, Delgass WN, Abu-Omarm MM, Caruthers JM. J Am Chem Soc. 2010;132:558–566. doi: 10.1021/ja906332r. [DOI] [PubMed] [Google Scholar]; (m) Yin CX, Finke RG. J Am Chem Soc. 2005;127:13988–13996. doi: 10.1021/ja052998+. [DOI] [PubMed] [Google Scholar]
- 17.(a) Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. University Science Books; Sausalito, CA: 2006. [Google Scholar]; (b) Giagou T, Meyer MP. Chem Eur J. 2010;16:10616–10628. doi: 10.1002/chem.201001018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qu R, Sun C, Wang C, Ji C, Sun Y, Guan L, Yu M, Cheng G. Europ Poly J. 2005;41:1525–1530. [Google Scholar]
- 19.(a) Xu X, Wang K, Nelson SG. J Am Chem Soc. 2007;129:11690–11691. doi: 10.1021/ja074845n. [DOI] [PubMed] [Google Scholar]; (b) Zhu C, Shen X, Nelson SG. J Am Chem Soc. 2004;126:5352–5353. doi: 10.1021/ja0492900. [DOI] [PubMed] [Google Scholar]
- 20.(a) Capo M, Saa JM. J Am Chem Soc. 2004;126:16738–16739. doi: 10.1021/ja045203s. [DOI] [PubMed] [Google Scholar]; (b) Leonova E, Makarov M, Klemenkova Z, Odinets I. Helv Chim Acta. 2010;93:1990–1999. [Google Scholar]; (c) Chen C, Jordan RF. Organometallics. 2010;29:3679–3680. [Google Scholar]; (d) Wang G, Zhao J, Zhou Y, Wang B, Qu J. J Org Chem. 2010;75:5326–5329. doi: 10.1021/jo100674f. [DOI] [PubMed] [Google Scholar]; (e) Willot M, Chen JC, Zhu J. Synlett. 2009:577–580. [Google Scholar]; (f) Suresh AS, Sandhu JS. Synthetic Commun. 2008;38:3655–3661. [Google Scholar]; (g) Hatano M, Ikeno T, Matsumura T, Torii S, Ishihara K. Adv Synth Catal. 2008;350:1776–1780. [Google Scholar]; (h) Messer R, Fuhrer CA, Haener R. Nucleos, Nucleot Nucl. 2007;26:701–704. doi: 10.1080/15257770701490704. [DOI] [PubMed] [Google Scholar]; (i) Moussaoui Y, Ben Salem R. Phys Chem News. 2005;22:75–79. [Google Scholar]; (j) Yousefi Rozbeh, Azizi N, Saidi MR. J Organomet Chem. 2005;690:76–78. [Google Scholar]; (k) Kinart WJ, Sniec E, Tylak I, Kinart CM. Phys Chem Liq. 2000;38:193–201. [Google Scholar]; (l) Desimoni G, Faita G, Righetti PP, Tacconi G. Tetrahedron. 1991;47:8399–8406. [Google Scholar]
- 21.(a) Henry KJ, Grieco PA. J Chem Soc, Chem Commun. 1993;6:510–512. [Google Scholar]; (b) Grieco PA, Beck JP. Tetrahedron Lett. 1993;34:7367–7370. [Google Scholar]; (c) Grieco PA, Collins JL, Handy ST. Synlett. 1995:1155–1157. [Google Scholar]; (d) Grieco PA, Kaufman MD, Daeuble JF, Saito N. J Am Chem Soc. 1996;118:2095–2096. [Google Scholar]; (e) Grieco PA, May SA, Kaufman MD. Tetrahedron Lett. 1998;39:7047–7050. [Google Scholar]; (f) May SA, Grieco PA, Lee HH. Synlett. 1997:493–494. [Google Scholar]; (g) Hunt KW, Grieco PA. Org Lett. 2001;3:481–484. doi: 10.1021/ol0003836. [DOI] [PubMed] [Google Scholar]
- 22.Hafez AM, Taggi AE, Wack H, Esterbrook J, Lectka T. Org Lett. 2001;3:2049–2051. doi: 10.1021/ol0160147. [DOI] [PubMed] [Google Scholar]
- 23.Calter MA, Guo X. J Org Chem. 1998;63:5308–5309. [Google Scholar]
- 24.Abraham CJ, Paull DH, Bekele T, Scerba MT, Dudding T, Lectka T. J Am Chem Soc. 2008;130:17085–17094. doi: 10.1021/ja806818a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Ma Y, Hoepker AC, Gupta L, Faggin MF, Collum DB. J Am Chem Soc. 2010;132:15610–15623. doi: 10.1021/ja105855v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ramirez A, Sun X, Collum DB. J Am Chem Soc. 2006;128:10326–10336. doi: 10.1021/ja062147h. [DOI] [PubMed] [Google Scholar]; (c) Liou LR, McNeil AJ, Ramirez A, Toombes GES, Gruver JM, Collum DB. J Am Chem Soc. 2008;130:4859–4868. doi: 10.1021/ja7100642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.UV data were collected using acetonitrile as the solvent in order to observe the region of interest. No displacement of catalyst ligands by the solvent was observed by 31P NMR.
- 27.Schreiber SL. U. S. Patent. 1982;4:322–537.
- 28.Taggi AE, Wack H, Hafez AM, France S, Lectka T. Org Lett. 2002;4:627–629. doi: 10.1021/ol0172525. [DOI] [PubMed] [Google Scholar]
- 29.(a) Fu S, Lian X, Ma T, Chen W, Zheng M, Zeng W. Tetrahedron Lett. 2010;51:5834–5837. [Google Scholar]; (b) El-Ayache NC, Li SH, Warnock M, Lawrence DA, Emal CD. Bioorgan Med Chem Lett. 2010;20:966–970. doi: 10.1016/j.bmcl.2009.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Thomas BH, Shafer G, Ma JJ, Tu MH, DesMarteau DD. J Fluorine Chem. 2004;125:1231–1240. [Google Scholar]; (d) DesMarteau DD, Zhu S, Pennington WT, Gotoh Y, Witz M, Zuberi S. J Fluorine Chem. 1989;45:24. [Google Scholar]; (e) Foropoulos JF, DesMarteau DD. Inorg Chem. 1984;23:3720–3723. [Google Scholar]; (f) Koppel IA, Taft RW, Anvia F, Zhu SZ, Hui LQ, Sung KS, DesMarteau DD, Yagupolskii YL, Yagupolskii LM. J Am Chem Soc. 1994;116:3047–3057. [Google Scholar]
- 30.(a) Bose AK, Kugajevsky I. Tetrahedron. 1967;23:1489–1497. [Google Scholar]; (b) Meij R, Stufkens DJ, Vrieze K, Van Gerresheim W, Stam CH. J Organomet Chem. 1979;164:353–370. [Google Scholar]
- 31.Del Bene JE, Perera SA, Bartlett RJ, Yanez M, Mo O, Elguero J, Alkorta I. J Phys Chem A. 2003;107:3126–3131. [Google Scholar]
- 32.For a recent review, see: Balcells D, Maseras F. New J Chem. 2007;31:333–343.
- 33.Calculations were performed using the Gaussian '09 and GaussView v5.08 programs at the Kohn Sham hybrid-DFT B3LYP level of theory and the GenECP method was employed using a 6-31G(d) basis set for all atoms (i.e. C, N, O, F, S, P, Cl) expect palladium which was computed using the well-known Los Alamos LAN2DZ basis set.
- 34.(a) Frisch MJ. Gaussian 09, Revision C B01. Gaussian, Inc.; Wallingford CT: 2004. [Google Scholar]; (b) Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]; (c) Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; (d) Hratchian HP, Schlegel HB. In: Theory and Applications of Computational Chemistry: The First 40 Years. Dykstra CE, Frenking G, Kim KS, Scuseria G, editors. Elsevier; Amsterdam: 2005. pp. 195–249. [Google Scholar]; (e) Hratchian HP, Schlegel HB. J Chem Theory and Comput. 2005;1:61–69. doi: 10.1021/ct0499783. [DOI] [PubMed] [Google Scholar]; (f) Hratchian HP, Schlegel HB. J Chem Phys. 2004;120:9918–24. doi: 10.1063/1.1724823. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.