

Abstract
Sequence, which Nature uses to spectacular advantage, has not been fully exploited in synthetic copolymers. To investigate the effect of sequence and stereosequence on the physical properties of copolymers a family of complex isotactic, syndiotactic and atactic repeating sequence poly(lactic-co-glycolic acid) copolymers (RSC PLGAs) were prepared and their NMR and thermal behavior was studied. The unique suitability of polymers prepared from the bioassimilable lactic and glycolic acid monomers for biomedical applications makes them ideal candidates for this type of sequence engineering. Polymers with repeating units of LG, GLG and LLG (L = lactic, G = glycolic) with controlled and varied tacticities were synthesized by assembly of sequence specific, stereopure dimeric, trimeric and hexameric segmer units. Specifically labeled deuterated lactic and glycolic acid segmers were likewise prepared and polymerized. Molecular weights for the copolymers ranged from Mn = 12-40 kDa by size exclusion chromatography in THF. Although the effects of sequence-influenced solution conformation were visible in all resonances of the 1H and 13C NMR spectra, the diastereotopic methylene resonances in the 1H NMR (CDCl3) for the glycolic units of the copolymers proved most sensitive. An octad level of resolution, which corresponds to an astounding 31-atom distance between the most separated stereocenters, was observed in some mixed sequence polymers. Importantly, the level of sensitivity of a particular NMR resonance to small differences in sequence was found to depend on the sequence itself. Thermal properties were also correlated with sequence.
Introduction
The creation of polymers with a high degree of sequence- and/or stereo-control represents an exciting frontier in materials science due to both the paucity of examples of polymers with high values in these two categories and to the potential benefits to be realized from the creation of new polymers from readily available and well-understood monomers.1-11 A valuable lesson on the key role of sequence/stereochemical control can be learned from Nature, which exploits stereospecific sequences of amino acid monomers to form proteins with a myriad of properties and functions. This lesson is further echoed in the recent reports describing the unique behavior of sequenced foldamers12 and in the benefits already derived from the relatively modest degrees of sequence control present in conventional polymer architectures e.g. block copolymers and stereocontrol e.g. isotactic polypropylenes.13-16 Finally, there are clear advantages to be realized from the rearrangement of “old” monomers to create new polymers: economic, in that the infrastructure that already exists for the large scale synthesis of these old monomers translates into low cost and high availability and pragmatic, in that the knowledge base that already exists regarding the suitability of these monomers for particular applications translates into an efficient path to application.
The extensive investigation of poly(lactic-co-glycolic acid)s (PLGAs) for in vivo applications requiring slow polymer degradation renders them ideal candidates for the investigation of the role of sequence in polymer properties. The popularity of these polymers for applications such as stem cell scaffolding and drug delivery vehicles stems from the fact that bulk structures made from PLGAs hydrolyze at a moderate rate in the body and the products, lactic and glycolic acid, are bioassimilable.17-25 Although other polymers are employed for these applications, studies on PLGAs and PLAs represent a significant proportion of all work in the area. The ubiquity of the materials and the special suitability of the monomers to bioengineering applications make the idea of creating repeating sequence copolymers (RSCs) from these monomers particularly attractive.
Our approach to the investigation of the role of sequence on the properties of PLGAs is to create and then analyze a family of RSCs. Our methodology, which involves the condensation of preformed segmers, allows for the synthesis of polymers with structural- and stereo-sequences more complex than those previously prepared. Selected examples are presented in Figure 1. Previous efforts to prepare sequenced PLAs and PLGAs have relied on the ring-opening polymerization (ROP) of the cyclic lactide, glycolide, and methyl glycolide dimers.26-40 Despite the laudable success achieved though the sophisticated design of selective catalysts and the exploitation of chain-end control the number and types of sequences that can be prepared is limited by the dimeric form of the ROP monomer and by challenges inherent in programming catalysts to deliver a pattern more complex than alternation. While the strategy we employ in this paper of pre-assembling a sequenced segmer by a condensation mechanism is arguably less efficient than ROP, it is convergent and molecular weights > 20 kDa are routinely achievable. Most importantly this approach is general—any RSC PLGA envisioned can be prepared.
Figure 1.

Comparison of complex sequences prepared by segmer assembly with simpler PLA and PLGA microstructures prepared by ring-opening polymerization.26-31,40
The first benefit realized from our creation of a family of PLGA RSCs and the primary focus of this paper is the creation of a partial “Rosetta Stone” for the interpretation of NMR data of polymers with complex stereochemical patterns in general and for PLGAs in particular. The most relevant precedents for this work are the extensive investigations of the NMR for PLA of varying tacticities.29-31,33,41-53 It is worth noting that in the PLA system the 13C NMR spectroscopic shifts have been found to be most sensitive to the relationships between distant stereocenters; differences in relative stereochemistries up to 5-6 units away from the nucleus under observation can be detected.41,43,44,47,49,51,54 Although the NMR data for PLGAs has likewise been studied, the added complexity introduced by having two variables, sequence and stereochemistry, as well as the fairly modest control of these variables achievable using the common ROP synthetic approach, has inhibited progress. These studies have primarily been limited to the partial assignment of local sequence within otherwise random copolymers.34,36,39,40,45,55-58 Our approach to preparing PLGAs, which allows for nearly perfect sequence and stereocontrol, greatly expands the database and offers, thereby, a significant advance in the understanding of PLGA NMR data as well as some interesting new conclusions of a more general nature pertaining to sequenced copolymers.
Results
Naming conventions
Segmers are named by listing the monomers in sequence order from the C-side to the O-side using the abbreviations in Table 1. BnLLracG is, therefore, a trimer of stereopure l-lactic acid, rac-lactic acid and glycolic acid that bears a benzyl protecting group on the carboxylic acid (C-side) terminus. Additionally, the deuterium labeled lactic and glycolic acids are labeled Ld,rac and Gd2, respectively. Polymers are named from the exact segmer used. Thus, the polymer prepared from LLG is named poly LLG rather than poly GLL despite the homology of the two sequences after polymerization i.e. …LLGLLGLLGLLGLLGLLG…
Table 1. Naming conventions for segmers and polymers.
| Symbol | Definition |
|---|---|
| L | L-Lactic unit (S configuration) |
| LR | D-Lactic unit (R configuration) |
| Lrac | rac-Lactic unit |
| Ld,rac | α-d-rac-Lactic unit |
| G | Glycolic unit |
| Gd2 | α-d,d-Glycolic unit |
| Bn | Benzyl protecting group |
| SiR3 | Silyl protecting group |
Synthesis
A series of PLGA RSCs based on dimeric, trimeric, tetrameric and hexameric repeating units have been prepared by condensation of the pre-formed segmers. Segmers were assembled in a convergent fashion by reacting partly protected subunits to form completely protected products which, after subsequent deprotection of both termini, yielded the desired segmers as α-hydroxy carboxylic acids. The synthesis of the LLG and LRLG segmers are presented as representative examples.
Starting from the previously reported dimer, Bn-LG-SiR3,59 catalytic hydrogenation was used to remove the benzyl group and generate the partially deprotected LG-SiR3 unit in a 95% yield (Scheme 1). Benzyl (S)-lactate (Bn-L) or benzyl (R)-lactate (Bn-LR) were coupled to LG-SiR3 to generate di-protected trimers Bn-LLG-SiR3 and Bn-LRLG-SiR3 in 84 and 72% yields, respectively. Removal of both protecting groups gave segmers LLG and LRLG in 83% and 67% yields. Using the same approach the segmers LG, LracG, GLG, GLracG, LracLracG, LracLG, LLracG, and LLRG were prepared. Hexameric units such as LRLGLLG were synthesized by coupling partially deprotected trimeric segmers Bn-LRLG and LLG-SiR3 followed by subsequent deprotections in 75% yield overall. LLGLLRG and GLGLR were prepared similarly from the appropriate trimeric and dimeric precursors. It should be noted that in the preparation of LracLracG from the racemic precursors, there is a slight preference, 60:40, for the formation of LLG units with different stereocenters i.e. LLRG and LRLG are favored over LLG and LRLRG. This preference can be clearly seen in the copolymer spectrum as well. The synthesis of the oligomer LracLG also results in a slight bias towards LRLG over LLG but it is difficult to detect by NMR once polymerized. Complete synthetic schemes for all segmers and deuterium labeled compounds can be found in the supporting information.
Scheme 1.

Polymerization conditions, utilizing mild esterification reagents 1,3 diispropylcarbodiimide (DIC) and 4-(dimethylamino)pyridinium p-toluenesulfonate (DPTS), were adapted from Stupp and Akutsu (Scheme 2).60,61 In this way, for example, poly LLG was prepared by the DIC/DPTS-mediated condensation of the fully deprotected LLG segmer. All polymers were isolated as colorless solids by precipitation into methanol and purified by re-precipitation from methylene chloride into methanol to give yields ranging from 50 to 99% (Table 2). It should be noted that we did not observe significant sequence preferences when assembling segmers with racemic units into polymers. We have found that the inherent but slight preferences for alternation of stereochemical centers that were observed in selected segmer preparations can be minimized in the polymer preparation by coupling segmers bearing L groups on one terminus and G's on the other.
Scheme 2.

Table 2. PLGA repeating sequence copolymer characterization data.
| Polymer | Repeating Patterna | Yield (%)b | THF | CHCl3 | CHCl3 | CHCl3 | ||
|---|---|---|---|---|---|---|---|---|
| Mn (kDa)c | PDIc | Mn (kDa)c | PDIc | Absolute Mnd | DPe,f | |||
| LG |

|
63 | 27.4 | 1.3 | 33.3 | 1.3 | 13.4 | 512 (206) |
| LracG |
|
52 | 28.8 | 1.3 | 34.3 | 1.4 | --- | 527 |
| GLG |
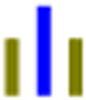
|
78 | 26.2 | 1.2 | 36.2 | 1.4 | 19.4 | 577 (309) |
| GLracG |

|
60 | 21.4 | 1.3 | 27.5 | 1.4 | --- | 439 |
| LLG |
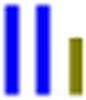
|
70 | 41.2 | 1.2 | 41.8 | 1.3 | 23.1 | 620 (343) |
| LLRG |
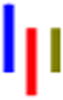
|
71 | 29.0 | 1.4 | 42.3 | 1.3 | --- | 628 |
| LRLG |
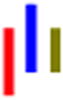
|
59 | 30.6 | 1.4 | 39.8 | 1.4 | --- | 591 |
| LracLracG |
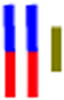
|
65 | 30.5 | 1.4 | 35.2 | 1.3 | --- | 522 |
| LLracG |
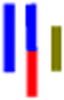
|
50 | 17.8 | 1.4 | 19.3 | 1.6 | --- | 286 |
| LracLG |
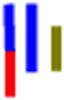
|
83 | 27.4 | 1.4 | 40.5 | 1.4 | 25.9 | 601 (384) |
| Ld,racLG |

|
99 | 32.8 | 1.3 | 31.7 | 1.5 | --- | 468 |
| LLd,racG |

|
62 | 29.6 | 1.4 | 33.7 | 1.4 | --- | 498 |
| GLGd2 |

|
52 | 15.2 | 1.4 | 25.3 | 1.5 | --- | 400 |
| GLGLR |
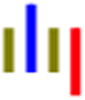
|
65 | 12.3 | 1.5 | 21.1 | 1.4 | --- | 324 |
| LLGLLRG |
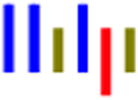
|
70 | 30.0 | 1.4 | 32.0 | 1.5 | --- | 475 |
| LRLGLLG |
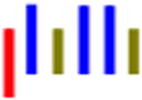
|
63 | 30.1 | 1.4 | 39.8 | 1.3 | --- | 591 |
 (S)-lactic unit,
(S)-lactic unit,
 (R)-lactic unit,
(R)-lactic unit,
 rac-lactic unit,
rac-lactic unit,
 glycolic unit
glycolic unit
 deuterated unit;
deuterated unit;
Isolated after 2× precipitation in MeOH;
Determined by SEC relative to PS standards;
Determined by SEC-MALLS;
DP from SEC data based on number of lactic and glycolic monomers;
(DP) from SEC-MALLS data based on number of lactic and glycolic monomers.
Molecular weights for the polymers were determined by both size exclusion chromatography (SEC) and multi-angle laser light scattering (MALLS). Relative molecular weights were obtained by SEC in both THF and CHCl3 vs. polystyrene standards with the number average molecular weights (Mn) ranging from 12.0 – 41.2 kDa in THF and 19.3 – 42.3 kDa in CHCl3. Absolute Mns were determined for poly LG, GLG, LLG and LracLG, using SEC-MALLS. The dramatic lack of correlation between the SEC molecular weights in the two solvents and the differences between these molecular weights and the absolute Mns determined by MALLS, makes it clear that the Rg of the polymers is extremely sequence and solvent dependent. The SEC molecular weights must, therefore, be regarded with special care. By interpolation of the SEC and SEC-MALLS data obtained, however, we can say with a high degree of confidence that the majority of the polymers prepared have an absolute molecular weight > 15 kDa which corresponds to a DP > 200 (based on the count of glycolic and lactic units). This conclusion is further substantiated by the lack of endgroups observed in the NMRs of these polymers. Previous studies by others on related polymers have shown microstructure dependent SEC behavior.62,63 A more detailed study of Rg vs. sequence is underway but is not the focus of this paper.
The molecular weights of the RSCs, while lower than those routinely achieved by ring-opening of lactides and glycolides, are respectable for a condensation polymerization on the 1-2 g scale that we are currently employing. Moreover, it worth noting again that polymers with the sequence complexity of those reported herein cannot be produced by any known ROP catalytic system.
Microstructural Analysis of RSCs Prepared from Dimeric Segmers
Although this article focuses on the polymers with trimer-based sequences, poly LLG and poly GLG, we will begin with a brief discussion of the simpler, dimeric RSCs based on poly LG. While a portion of this work was reported in a previous article,59 we have since prepared a key member of the series, poly GLGLR, that was not available at the time of that publication.
Our major discovery in the study of the alternating poly LxG series polymers for poly LG, LracG and GLGLR was the surprisingly high sensitivity of the glycolic methylene protons for the relative stereochemistry of the neighboring lactic units (Figure 2). The difference in the methylene signals of isotactic poly LG (δ 4.86 and 4.63, Δ = 0.23 ppm) and syndiotactic poly GLGLR (δ 4.81 and 4.69, Δ = 0.12 ppm) are particularly illustrative. While each spectrum contains a pair of doublets, as would be predicted for the clearly diastereotopic methylene protons, the chemical shifts are dramatically different.
Figure 2.
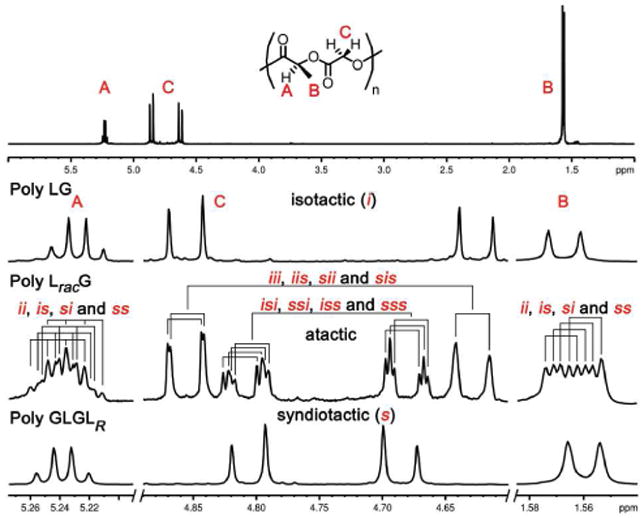
(Top) Full spectrum of poly LG; (Bottom) expansions of selected regions for poly LG, LracG and GLGLR. 1H NMR spectra at 600 MHz in CDCl3.
Each of these alternating copolymers possesses a unique stereosequence that can be encoded using the widely accepted convention of assigning relative stereochemistries in polymers as either i for neighboring units with the same absolute stereochemistry or s for the opposite. Using this coding system, and focusing on a tetrad level of resolution, the stereochemistry of poly LG can, for example, be expressed as iii and poly GLGLR as sss (Figure 3 and Table 3).
Figure 3.

Example of tetrad stereosequence encoding for poly LG (all isotactic) and GLGLR (all syndiotactic).
Table 3. Tetrad assignments for stereosequences of polymers from the poly LG series.
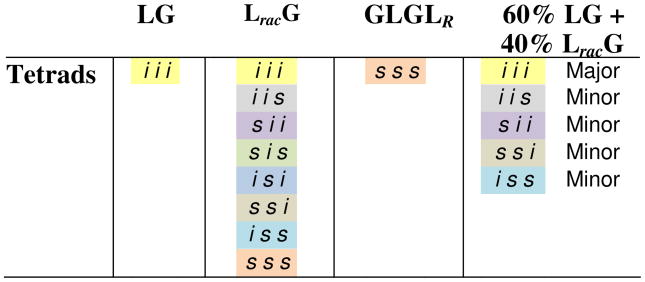
|
We focus on the tetrad level because we can clearly see this level of resolution for some sequences present in the more complex spectrum of atactic poly LracG (Figure 2). Using the data from the stereopure polymers and from a sequence-weighted copolymer prepared by mixing LG and LracG in a 60:40 ratio (see ref 59 for details on this copolymer, see Table 3 for the expected/observed tetrads), we were able to make a nearly complete assignment of the signals. We see that the methylene resonances divide into two regions based on the relative stereochemistry of the closest neighboring lactic units: i-centered (outer signals) and s-centered (inner signals). The resonances for each of the s-centered tetrads, sss, ssi, iss and isi, can be clearly differentiated while those for the i-centered tetrads, iii, iis, sii and sis, overlap such that the effective resolution is expressed at a lesser diad or triad level.
The other resonances in the spectra of the dimeric polymers also show stereosequence-dependent chemical shifts, though none exhibit as high a level of sensitivity/interpretability as the methylene protons. A clear chemical shift difference was noted, for example, between the methine and methyl groups from syndiotactic poly GLGLR and isotactic poly LG. The smaller chemical shift range, however, limits the resolution of the stereosequences present in poly LracG to the triad level. In the 13C NMR spectra (see supporting information) the L-carbonyl resonances were clearly resolved to the triad level while the G-carbonyl, L-methyl and L-methine resonances were nearly resolved triads. The s-carbonyl resonances for both L and G appeared upfield relative to the i-stereoisomer. In contrast, the s-methine and s-methyl resonances were downfield of the i-versions. Interestingly, the methylene resonance, which was so sensitive in the 1H NMR data, was only a broad singlet in the 13C NMR spectrum.
Microstructural Analysis of RSCs Prepared from Trimeric GLxG Segmers
In moving from a dimer-based LG copolymer to a trimer-based copolymer, additional complexity was introduced both in the architecture and in the resulting NMR spectra. The third monomer presents the possibility of two different structural sequences, GLG or LLG, and multiplies the number of possible stereoisomers. It should be noted again that the choice to refer to a particular sequence as GLG rather than GGL was based on the unit actually used in the synthesis. Once polymerized, of course, such differences are irrelevant since poly GLG would necessarily be the same in all respects as poly GGL except in the identity of the endgroups; both would have a repeating sequence of –GGLGGLGGLGGLGGLGGL- in the backbone.
Analysis of the NMR spectra of poly GLG in both the stereopure and racemic forms required that we first assign the resonances for the two chemically distinct G monomers. The G units are inequivalent due to the intrinsic lack of symmetry of the polyester chain, which has a distinct C- and O-terminus for each polymer and for each sequence within the polymer. We can differentially label the G's then as either being connected to the C-terminus of the lactic residue (GC) or the O-terminus (GO). This inequivalence was expressed both in the 1H NMR spectrum (two pairs of doublets, Figure 4) and in the 13C NMR spectrum (C=O, δ 166.5 and 166.4; δ 60.9 and 60.7, see supporting information for 13C NMR spectra). The assignment of the resonances of poly GLG was facilitated by comparison with the spectrum of the polymer that was selectively deuterated at GO, poly GLGd2. The inner pair of doublets were absent from the 1H NMR spectrum and the downfield methylene resonance was not present in 13C NMR spectrum. Further confirmation of the assignment came from the 2D Heteronuclear Multiple Bond Coherence (HMBC) NMR spectrum of poly GLG in which a 3-bond correlation of the L-carbonyl with the outer pair of doublets from the methylene protons of GC was observed along with the correlation between the GO carbonyl and the L-methine (Figure 5).41,46,64,65
Figure 4.
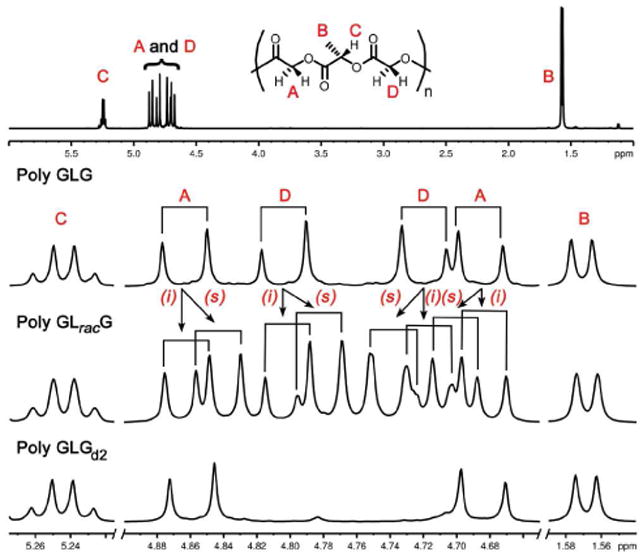
(Top) Full spectrum of poly GLG; (Bottom) expansions of selected regions for poly GLG, GLracG and GLGd2. 1H NMR spectra at 600 MHz in CDCl3.
Figure 5.
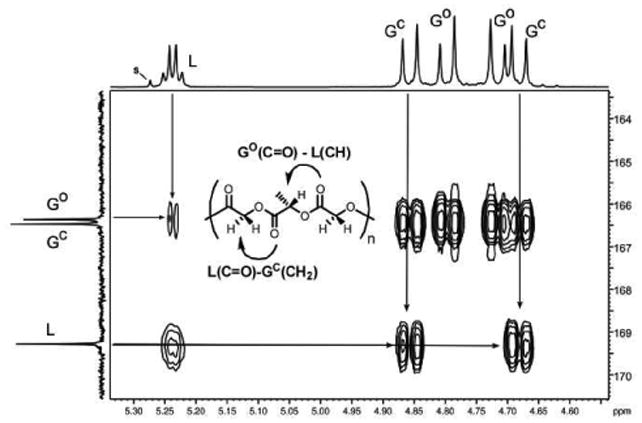
2D HMBC 1H-13C correlation NMR spectrum for poly GLG in CDCl3 (700 MHz, 1H; 175 MHz 13C). The detailed cross peaks correspond to 3-bond correlations between the L-carbonyl with the GC methylenes and the GO carbonyl with the L-methine.
Spectra of poly GLracG established that the chemical shifts of the G resonances were only modestly sensitive to the tacticity of the polymer. More sensitive than the methyl and methine resonances, which exhibited only a slight broadening, the methylene region exhibited 8 pairs of doublets, which given the inequivalence of the two G units, is consistent with a dyad level of resolution. In other words the chemical shifts of the G resonances depend only upon the relative stereochemistries of the two closest L units. The peaks labeled as i-centered resonances corresponded well with the isotactic poly GLG standard. The slightly shifted pattern of two pairs of doublets was assigned to the s dyad. Partially resolved dyads were likewise visible in the 13C NMR spectrum where the lactic carbonyl appeared to broaden while 3 resonances, two at ca. δ 166.5 and a broadened resonance about δ 166.4 were observed for the glycolic carbonyls.
Microstructural Analysis of RSCs Prepared from Trimeric LxLxG Segmers
In moving from the poly GLG series to the other trimeric series, poly LLG, we must again address the nomenclature and consider the stereochemical implications of the second L unit in the series. The two L units are necessarily inequivalent and are designated as LO or LC in relation to the G unit. LO is connected to the G through the O-terminus and the LC is connected through the C-terminus. Since the monomer used to prepare these polymers is LLG rather than LGL, however, the LO unit will be listed first in the series i.e. LOLCG. With these designations in mind, the stereochemical variants of poly LxLxG can be considered. In Figure 6, our approach to assigning tacticity patterns is illustrated. In the case of poly LRLGLLG, for example, the sequence yields two types of G-centered octads, one of which has an issiiss pattern. A subset tetrad pattern of sii is recognized from the larger pattern if the relationships from only the nearest four neighbor L's are considered. Interestingly, the same pattern can be reasonably used as a reference for either LO or LC despite the shift in the “center” of the sequence. The lack of symmetry in the number of relationships considered relevant on each side of the L center is countered by the fact that the relationships chosen for the polyad maximize the number of adjacent vs. distant interactions, on the assumption (substantiated by the data), that the adjacent relationships will be more influential than the distant ones.
Figure 6.

Example of octad-level stereosequence encoding for poly LRLSGLSLSG. The central tetrads for each type of unit are defined as including one distant and two adjacent relationships.
In considering the stereochemical possibilities for the poly LxLxG series we have found it useful to divide the polymers into three families: LO-variable (poly LxLG); LC-variable (poly LLxG); and (LO+LC)-variable (poly LxLxG). Table 4 shows the stereochemical possibilities for the first two families at the tetrad, hexad and octad levels. It is important to note that poly LLG, LRLG, and LRLGLLG exhibit all of the stereochemical variations possible in poly LracLG at the tetrad level. Likewise poly LLG, LLRG, and LLGLLRG exhibit the possible tetrad level variations in poly LLracG. Stereosequence-specific shading in Table 4 illustrates the hierarchical relationship of the hexads and octads to their parent tetrads.
Table 4.
Listing of select polyads for specific poly LxLxG repeating sequence copolymers. Tetrads subsets are identified by color within the hexad and octad tacticity patterns.

|
The 1H NMR spectra of the glycolic methylene regions of these trimeric series are spectacularly informative—octad-level resolution of individual stereosequences is observed in some cases. By comparison with the spectra of poly LLG, LRLG, and LRLGLLG, and LracLG for the LO-variable family (Figure 7) and poly LLG, LLRG, and LLGLLRG and LLracG for the LC-variable family (Figure 8), it was determined that the chemical shifts of these resonances were inherently hierarchical and facile to interpret.
Figure 7.
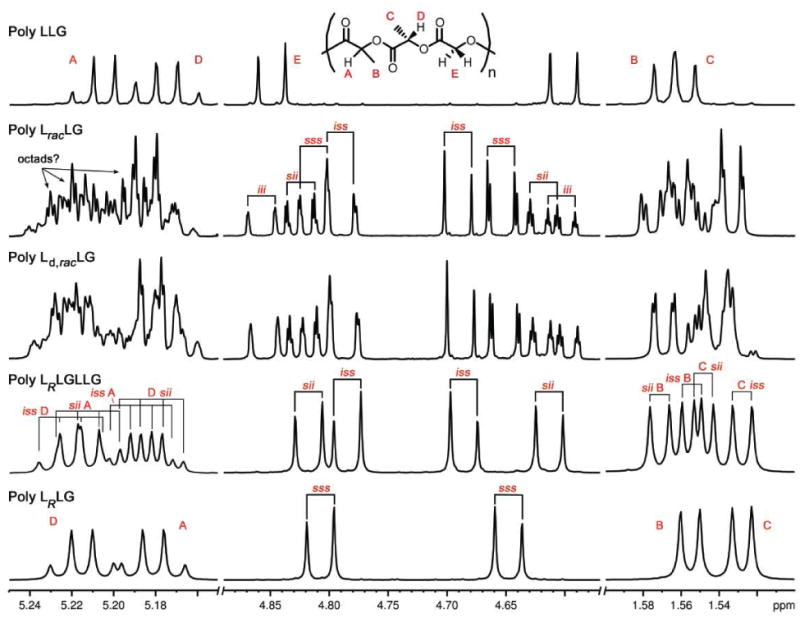
1H NMR spectra of LO-variable LLG polymers at 700 MHz in CDCl3. Comparisons of the expansions of selected regions for poly LLG, LracLG, Ld,racLG, LRLGLLG, and LRLG.
Figure 8.
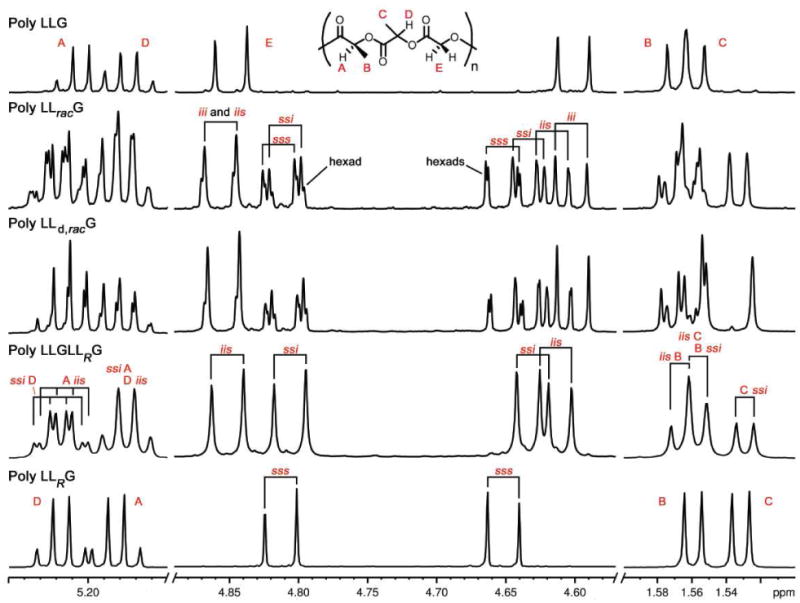
1H NMR spectra of LC-variable LLG polymers at 700 MHz in CDCl3. Comparisons of the expansions of selected regions for poly LLG, LLracG, LLd,racG, LLGLLRG, and LLRG.
Beginning by focusing on the LO-variable family (Figure 7), we can assign the resonances from the stereopure standards. The spectrum of the simplest sequence, isotactic poly LLG, exhibits a pair of well-separated doublets for the diastereotopic G methylene protons. Poly LRLG, the fully syndiotactic sequence also exhibits a single pair of doublets while poly LRLGLLG exhibits two pairs of doublets which is consistent with expectations for this more complex sequence (see Table 4). Although these are stereopure standards with an “infinite” pattern, the resonances are labeled in the figures at the tetrad level for reasons that become clear in the analysis of the racemic variants (vide infra).
The analysis of spectrum of poly LracLG reveals the useful hierarchical nature of the shift pattern in the methylene region: the gross shifts are determined by the central tetrad relationships while the fine shifts are determined by more distant ones. The methylene region of this polymer manifests as well-separated sets of pairs of doublets, one pair for each of the resolved stereosequences. By comparison of the stereosequence standards and the spectrum of poly LracLG, it is clear that the resonances are grouped by the central tetrad relationships, iii, sss, iss, and sii into four well-separated regions. Within these tetrad-controlled shift regions, the “fine” chemical shifts are then determined by the relative stereochemistries of L units beyond the central four.
The degree of resolution of longer range relationships in the methylene region is clearly sequence dependent. Focusing on the upfield proton of the G methylene of poly LracLG several levels of resolution can be identified (Figure 9). In the case of the iii set, for example, there are four nearly resolved doublets, the number that would be expected if there was a doublet present for each of the four possible octad level sequences. Although we do not have standards with sufficient complexity to label the individual sequences, the extremely high level of resolution is striking. The other three sets of doublets in the same region of this spectrum exhibit varying sensitivities ranging from the tetrad to hexad to octad level.
Figure 9.
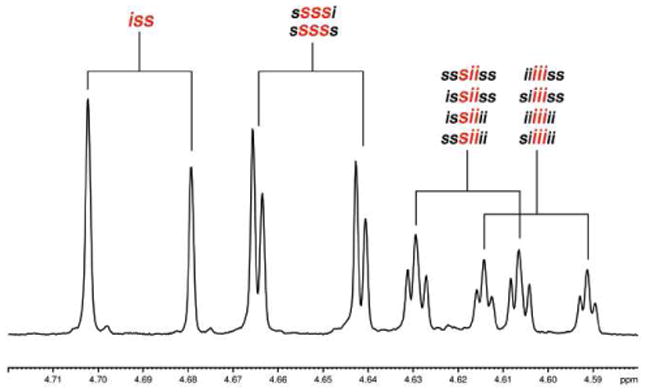
1H NMR expansion for the upfield diastereotopic proton of the glycolic methylenes of poly LracLG (full spectrum in Figure 7). The level of sensitivity for sequence ranges from tetrad (sii, iii) to hexad (sss) to octad (iss) depending on the core tetrad sequence.
A similar hierarchical pattern is observed in the methylene region of poly LLracG (Figure 8). Sets of pairs of doublets corresponding to the expected tetrads, iii, sss, ssi, and iis, can be easily assigned. Within these sets, further resolution of certain sequences is observed. It should be noted that the spectra of poly LRLG and LLRG (Figures 7 & 8) are identical as would be predicted from their enantiomeric relationship.
Changing the NMR solvent from CDCl3 to d6-DMSO dramatically decreases the resolution of the spectra (see supporting information for figure depicting the difference). The chemical shift range of the methylene (and other regions) is decreased and sequence resolution is reduced.
We were also able to make significant progress in assigning the methyl and methine regions of the trimeric series of polymers. These regions are inherently more difficult to interpret because of the presence of the two inequivalent lactic units, the relatively small chemical shift range, and the lack of an hierarchical shift pattern. Primary differentiation of LO vs. LC in the fully sequenced polymers poly LLG, LRLG and LRLGLLG was accomplished using a combination of 2D NMR techniques. Methine resonances were assigned by 2D HMBC NMR spectra where a correlation between the G carbonyl and the LC methine was observed. Frustrating facile assignment, the relative chemical shifts of the LO and LC methines were not consistent throughout the stereochemical series. As Figure 10 illustrates, in isotactic poly LLG the LC resonance is upfield of the LO resonance but the order is reversed in syndiotactic poly LRLG. In polymers that contain mixed stereochemical relationships, poly LRLGLLG, and multiple mixed relationships, poly LracLG and LracLracG, the 2D correlations showed that the chemical shifts of the LO and LC methines were extremely sequence dependent and likely to invert chemical shift order from sequence to sequence. By coupling the assignments of the methine region with 2D COSY NMR spectroscopy, however, the methyl region could be assigned. Unlike the methine resonances, the LO methyl resonances were always downfield of the corresponding LC resonances.
Figure 10.
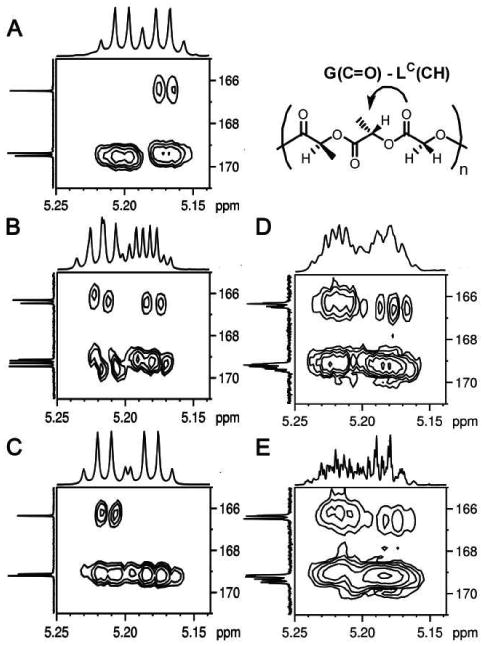
2D HMBC 1H-13C correlation NMR spectra at 700 MHz in CDCl3 for poly LLG (A), LRLGLLG (B), LRLG (C), LracLracG (D) and LracLG (E).
Although complete interpretation of the methine and methyl regions for poly LracLG and LLracG could not be accomplished, it was clear that the sequence resolution of both regions was greater than a tetrad level. It should be noted that neither homonuclear decoupling nor 2D NMR was of much use in interpreting these spectra further. The chemical shift envelope for each type of signal was large enough to render the complete homonuclear decoupling of the regions impractical while the small chemical shift range and sequence-dependent shift inversions (vide supra) hindered the interpretation of 2D spectra. The relatively modest simplification of the spectra for the selectively deuterated polymers poly Ld,racLG and LLd,racG, however, establishes the sensitivity of these signals for the stereochemical relationships of lactic units beyond the tetrad level in the chain. Moreover, by simple counting of the resonances and the comparison of the spectra with the stereosequence standards, it is clear that some sequences are actually resolved to a hexad or even octad level.
Chemical shifts in the 13C NMR spectra of poly LracLG and LLracG showed primarily a tetrad level of resolution. Partial assignment was accomplished by comparison with the stereopure and deuterated RSCs (Figure 11 and 12) and by 2D HMBC NMR as described earlier. Analysis of the stereopure RSCs established that the LO carbonyl resonance was generally downfield of the LC resonance and the L carbonyl resonances were more sensitive to stereochemistry than the 13C NMR resonances for other groups. The presence of >8 lactic carbonyl signals in the spectra of both poly LracLG and LLracG was consistent with a higher than tetrad (mixed hexad/octad) level of resolution, although it was not possible to make individual assignments due to overlap. It is also of note that, in both the L and G carbonyl regions, the i-centered resonances were downfield of the s-centered resonances. Methine LO and LC resonances were assigned by comparison to the deuterium labeled polymers as the deuterium-substituted carbons exhibited much lower intensities due to slow relaxation. These signals clearly resolved to a slightly more than tetrad level for RSCs incorporating racemic units. The methyl region also showed a greater than tetrad level of resolution but the small chemical shift range made individual assignments in this region impractical.
Figure 11.
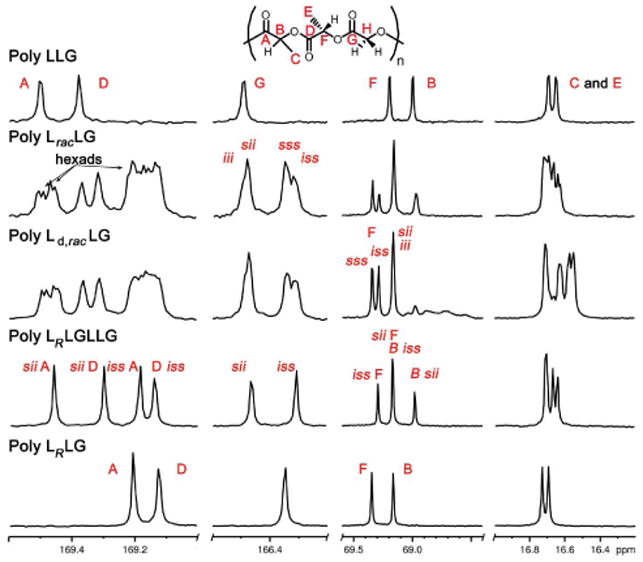
13C NMR spectra of LO-variable LLG polymers at 175 MHz in CDCl3. Comparisons of the expansions of selected regions for poly LLG, LracLG, Ld,racLG, LRLGLLG, and LRLG.
Figure 12.
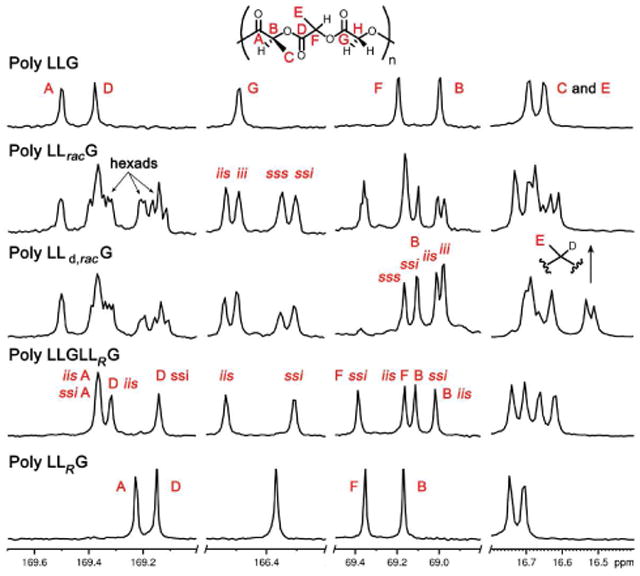
13C NMR spectra of LC-variable LLG polymers at 175 MHz in CDCl3. Comparisons of the expansions of selected regions for poly LLG, LLracG, LLd,racG, LLGLLRG, and LLRG.
The chemical shifts for stereopure polymers are compiled in Tables S1 and S2, which are located in the Supporting Information. It should be emphasized that these are stereopure samples and exact matches to unknowns will not be expected unless the sequences of the spectra compared are homologous at or beyond the level of resolution observed. For convenience, however, the polyads with more complex sequences have been labeled at the tetrad level.
Using these spectral tables as a guide and focusing on the methylene region, it is possible to interpret the spectra of samples that contain more than one structural sequence and of samples that contain multiple stereosequences. In a sample comprising a mixture of different structural sequences, in this case a mixture of poly LG, GLG and LLG, resonances for each sequence can easily be distinguished (Figure 13).
Figure 13.
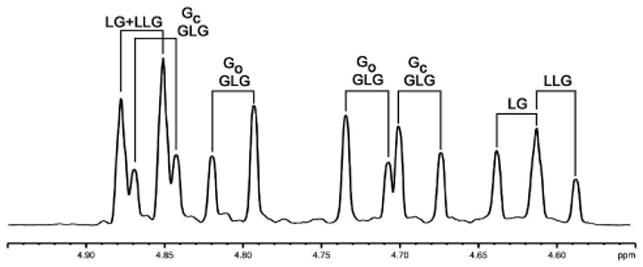
Glycolic methylene region of a mixed 1H NMR spectrum for mixed sample (1:1:1) of poly LG, GLG and LLG at 600 MHz in CDCl3.
A more stringent test is the assignment of the methylene region of poly LracLracG. This spectrum is complicated due to the number of possible polyads (8 at the tetrad level) and the presence of multiple levels of resolution for each polyad. Nevertheless, it is possible to make a fairly complete assignment of the sequences present at the tetrad level (Figure 14).
Figure 14.
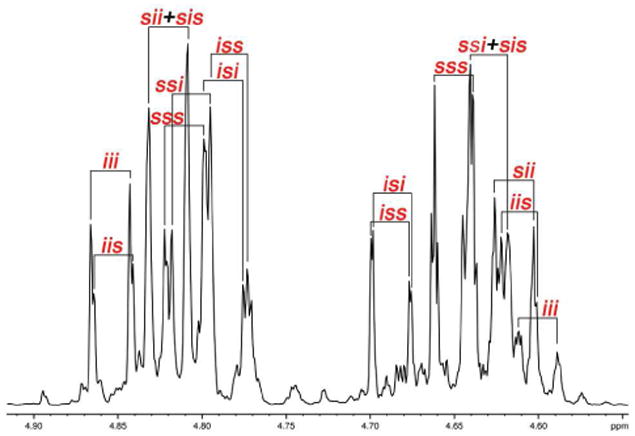
Glycolic methylene region of poly LracLracG at 700 MHz in CDCl3.
MALDI-TOF Mass Spectroscopy
MALDI TOF analysis confirms the sequence fidelity of the PLGA RSCs. Using conditions optimized for PLGAs,66,67 the analysis of selected copolymers was carried out. The masses of the chains present in poly LG, for example, differ in molecular weight by increments of exactly 58 amu, corresponding with the glycolic unit, and 72 amu, corresponding to the lactic unit (Figure 15). Moreover, chains differing by exactly one segmer weight (L + G = 130 amu) dominate, which is consistent with synthesis by assembly of segmers. Analogously, the envelope of masses present in the spectrum of poly GLG differ in molecular weight by a pattern of G, L and G molecular weights with a predomination of chains differing by exactly a segmer weight. Unfortunately, an analysis of the absolute molecular weights did not correspond to chemically reasonable endgroups. It seems likely that there is some degradation of the samples in the experiment. The full spectra for all samples can be found in the supporting information.
Figure 15.
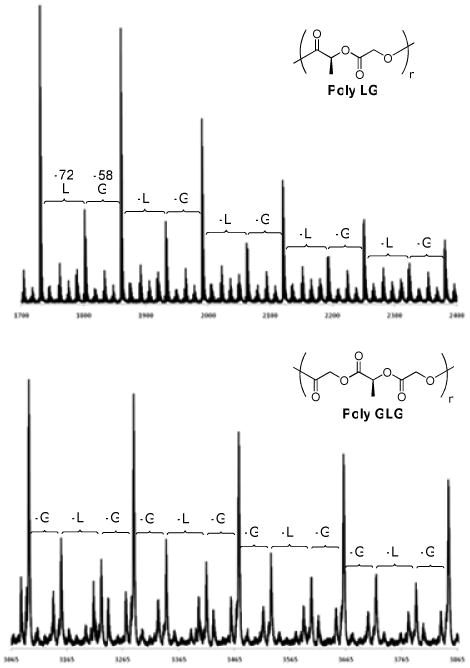
MALDI TOF patterns for poly LG (top) and poly GLG (bottom).
Thermal Properties
Differential scanning calorimetry (DSC) was performed on polymer samples and annealed films. The glass transition temperature (Tg) varied slightly due to changes in the sequence composition (Table 5). When the L content increased from 50% in poly LG to 67% in poly LLG the Tg remained constant around 57 °C. However, increasing the G content to 67% in poly GLG the Tg decreased slightly to 50 °C. Within the LG and LLG series the Tg was dependent on stereochemistry. Isotactic polymers possessed the highest Tg (57 °C), the atactic polymers possessed intermediate Tgs (55-51 °C) and the syndiotactic polymers possessed the lowest Tgs (48-50 °C). For the higher G content GLG series, changes in stereochemistry did not change Tgs; poly GLG and GLracG both exhibit a Tg of 50 °C. Interestingly, melting transitions were only found for the syndiotactic LLG polymers, poly LRLG and LLRG, at 154 °C and 158 °C, respectively.
Table 5. Thermal properties for RSC PLGAs.
| Precipitate | Annealeda | |||
|---|---|---|---|---|
|
|
|
|||
| Polymer | Tg (°C) | Tm (°C) | Tg (°C) | Tm (°C) |
| LG | 57b | ND | 49c | ND |
| LracG | 55b | ND | 48c | ND |
| GLGLR | 50b | ND | --- | --- |
| GLG | 50b | ND | 43c | ND |
| GLracG | 50b | ND | --- | --- |
| LLG | 57b | ND | 50c | 114c |
| LRLG | 50b | 154c | --- | --- |
| LLRG | 48b | 158c | 48c | 155c |
| LLGLL G | 52b | ND | 48c | ND |
| LRLGLLG | 52b | ND | 48c | ND |
| LLG | 51b | ND | 47c | ND |
| LracLG | 51b | ND | 48c | ND |
| LLracG | 53b | ND | 48c | ND |
Polymer films were drop-cast into DSC pans, dried under vacuum and annealed at 85°C for 3 h;
Transitions were measured in second heating cycle;
Transitions were measured in the first cycle.
Polymer films were prepared by drop-casting from methylene chloride into DSC pans, drying under vacuum and annealing at 85 °C for 3 h. Annealing had little effect on the transitions for poly LLRG and LRLG but the Tgs for all of the remaining annealed samples dropped and became less sequence sensitive. A new melting transition appeared for poly LLG at 114 °C, which is much lower than that observed for poly LLRG and LRLG.
Although direct homologs of these RSCs have not been previously reported, the behavior of these sequences can be examined relative to the known PLA sequences. Coates and coworkers have reported the Tm for syndiotactic PLA as 153 °C, which is close to those of poly LLRG and LRLG. The Tm's for isotactic and stereocomplexed PLA are much higher at 175 °C and 230 °C, respectively.30,31
Given the regularity of the remaining stereopure polymers, the lack of crystallization is somewhat surprising. Although annealing did promote crystallization in the case of poly LLG, other sequences did not exhibit melting points despite repeated efforts. We do not rule out the potential for crystallinity, however, as we suspect that we may not have discovered the proper thermal conditions to promote the longer range organization of the chains. Notably, Coates and coworkers did not observe crystal formation for the predominantly heterotactic sequenced PLA.31
Discussion
We are able routinely to prepare RSC PLGAs with DPs > 200 on a per monomer basis with a > 95% sequence fidelity. Previously, we were successful in preparing RSC PLGAs using DCC/DMAP but the DPs were significantly lower.59 The newly adopted DIC/DPTS method, first developed by Stupp and Moore, utilizes DPTS instead of DMAP in order to suppress chain terminating N-acylurea byproducts, neutralize the reaction pH and minimize depolymerization.60,68 Similar methods were used by Hawker and coworkers in the preparation of well-defined oligomers of lactic acid and caprolactone.69,70 Examination of the NMR spectra establishes that under the specified reaction conditions, neither sequence scrambling nor epimerization are significant problems although in select samples mistakes (< 5%) can be observed. The spectrum of poly LG (Figure 2, methylene region), for example, shows contamination by syndiotactic sequences.
Sequence was found to have a dramatic effect on the thermal properties of PLGA. In random PLGAs the material properties are primarily dependent on the length of the lactic blocks which generate preferential packing domains.39 Others have reported that as glycolic content increases the Tg drops to as low as 36 °C at a 50% glycolic unit incorporation.39,56,71 RSC PLGAs, on the other hand, maintained Tgs at or above 50 °C even when the glycolic unit content exceeded 60%. As long as the uniformity of the structural sequence remained, altering the stereosequence only slightly lowered the Tg. Most of the polymers remained amorphous even after annealing. Poly LLG, LRLG and LLRG were the only polymers that displayed a melting transition and poly LLG only crystallized after annealing. Although sequence- and stereoregularity should promote crystallinity, it is clear that it is challenging for these polymers to exhibit long range order based on these relatively short sequences.
Sequence and NMR
Our synthetic approach, which allowed for the creation of an unprecedented array of polymer microstructures, facilitates the understanding of the NMR assignments for specific PLGA sequences. The polymers prepared serve as a partial Rosetta Stone for the assignment of PLGA NMR data. Prior assignments of the NMR resonances for PLGA have been based on random copolymers (and a very small set of easily prepared well-defined homo- and copolymers). While this approach has led to significant understanding for a variety of polymers, it is worth noting that controversies over assignments of stereochemistry in the closely related but obviously simpler PLA system have taken years and the input of several groups to resolve.41,43,46,72 The fact that PLGA has the potential for both structural and stereosequence variability complicates the issue significantly. However, we have been able, using our family of RSCs, not only to make assignments but also to develop a deeper understanding of the issues involved in the interpretation of NMRs of RSCs.
While the complexity of PLGAs make the assignment of NMR resonances more challenging than in PLAs, our studies have also shown that the copolymer structure offers compensatory advantages that facilitate interpretation. The most important advantage offered by the copolymer is the exquisite sensitivity of the methylenic protons of the glycolic units to sequence and stereochemistry. The chemical shift range is relatively large and the shifts appear to depend on stereosequence in a hierarchical fashion: the gross chemical shift is determined by the central polyad while the fine chemical shift depends on longer range relationships. The sensitivity of this signal to stereosequence is much greater than any other 1H NMR resonance or the classically more informative 13C NMR resonances. The second compensatory advantage of PLGAs over PLAs (and other well-studied vinyl polymers) is a function of the fact that the resonances for the two monomers are necessarily distributed over a larger chemical shift range than the resonances for any single monomer and the fact that the polymers are unsymmetric— each monomer and each polymer has distinct C and O termini. Using 2D NMR, these differences were exploited to allow for the definitive assignment of resonances.
The most intriguing single result of the NMR studies of these RSC PLGAs, and one that is applicable beyond the narrow scope of these polymers, is the unambiguous demonstration that chemical shift resolution in a polymer bearing multiple sequences depends on the exact sequence. Although investigators who have previously analyzed polymers with complex stereosequences have proposed shift assignments based on different levels of resolution in some cases,42,47 the range of sequence specific polymers available in our system gives a particularly dramatic validation of the hypothesis. The clearest illustration in our system was found in the methylene region of poly LracLG. Examining the upfield proton, four groups of doublets are visible (Figure 9). The leftmost group that corresponds to the iss tetrad is a simple doublet with no fine structure; this signal is resolved only to the tetrad level. The next group is a barely resolved pair of doublets that corresponds to the gross shift of the sss tetrad but which is resolved to a hexad level. The final groups, which exhibit gross chemical shifts consistent with iii and sii tetrads, exhibit four nearly resolved pairs of doublets which correspond to the much higher octad level of resolution. An octad level of resolution maps to an impressive distance along the backbone of 31 atoms between the most separated stereocenters. Such sequence-dependent resolution occurs throughout these spectra although individual signals exhibit varying levels of sensitivity.
Another important lesson learned from the study of RSC PLGAs is that there are some inherent limitations in our ability to make accurate assignments--limitations that are general to the use of exact sequence RSCs as a key to the interpretation of the NMR spectra of samples with mixed sequence chains, no matter what polymer is involved. The first order approach to assigning spectra, and one that works in some cases, is to assign the resonances for specific sequences by comparison of the chemical shifts with those of a stereopure standard. Our analysis of the extensive database of shifts for the polymers described herein, however, highlights the potential for misinterpretation in systems where the sensitivity of a resonance to stereochemistry is high and not yet understood. The difficulties are illustrated in the simulated spectrum depicted in Figure 16. If a stereosequence with only “i” relationships, for example, is compared to a sample that contains several stereosequences, no peak with a 1:1 shift correspondence to the standard will be found despite the fact that the sample contains a significant (25% in this case) proportion of the “i” sequence in the form of “iiiii” at a hexad level of resolution. A similar problem would occur if the mixed sample contained only iiiis and siiis sequences. Despite the fact that both share the same iii central tetrad, the shift of neither peak would correspond with that of the “all i” standard.
Figure 16.
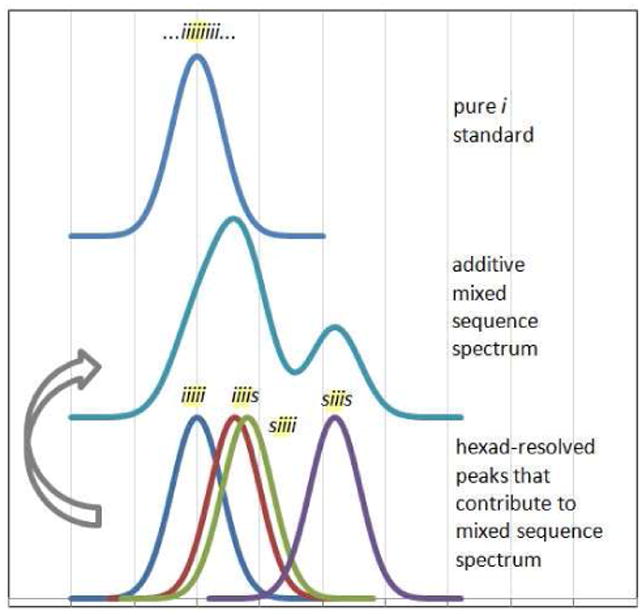
Simulated NMR spectra highlighting the challenges inherent in comparing a standard with perfect stereosequence control to a sample with multiple resolved sequences. Although all sequences share the same “
 ” central tetrad and exhibit similar chemical shifts, the shifts of the nearly hexad-resolved sequences create a pattern that does not show an easily interpretable correspondence with the standard.
” central tetrad and exhibit similar chemical shifts, the shifts of the nearly hexad-resolved sequences create a pattern that does not show an easily interpretable correspondence with the standard.
The lack of correlation between stereopure standards and the resonances for individual sequences can thus be attributed to a combination of two phenomena: 1) a full set of standards is not available i.e. sequences longer than those prepared as standards can be distinguished in mixtures and 2) the ultimate sensitivity of the chemical shift for a particular sequence in a mixture e.g. tetrad, hexad, etc. is determined by the inherent resolution of the NMR spectrum. We see the effects of both of these phenomena in our spectra in that the shifts of our standards do not exactly match those observed in our mixed sequence polymers. To achieve a 100% correlation it would have been necessary in the case of poly LracLracG, for example, to prepare all 32 possible octad sequences. As the synthesis of all of these sequenced polymers is impractical, it was necessary to extrapolate from the observed trends from the available standards in assigning certain sequences. The second phenomenon that results in chemical shift miscorrelation, the fact that the certain chemical shift differences are on the same order of magnitude as the peak widths, is a universally recognized spectroscopic challenge and could be overcome by a combination of expanding the database of sequences and “fitting” the mixed spectra. Although the lack of long-sequence standards prevented an analysis of this depth, these phenomena were taken into account when assignments were made.
The broad manifestation of these chemical shift correlation phenomena in our data can readily be observed in the stacked plots of 1H NMR spectra for the PLGA RSCs (Figures 8 & 9). The fact that peaks labeled as arising from the same tetrad do not always “line up” is due to the peak matching problems just described, not to poor chemical shift calibration. It should be noted that these issues disproportionally complicate the interpretation of the methine and methyl regions because those regions span a smaller chemical shift range and are not as hierarchical as the methylene region.
The analysis of the NMR data for these polymers has also given us some insight into the specific interpretation of the spectra of PLGAs. In particular, we note that the shift range observed for differing stereosequences within the same structural sequence overlaps exactly with the chemical shift range observed for differing structural sequences. Given the similarity of the monomers involved it is, perhaps, not surprising that structural sequence does not introduce a larger perturbation. The bottom line is that, for PLGAs, stereosequence is extremely important in determining the NMR spectral pattern.
Sequence and Conformation
One inescapable conclusion to be drawn from the solution phase behavior of these PLGA RSCs, both NMR and SEC, is that the conformations of these polyesters are sequence dependent. Conformational differences must, for example, be responsible for the differences in chemical environment exhibited by the diastereotopic methylene protons imbedded in different stereosequences. Conformation is also likely to be responsible for the sequence dependence of the SEC—the Rg is determined by the shape assumed by the polymer in solution.
Although the homology of the monomers in these polymers with those in amino acids, specifically alanine and glycine, renders the sequence-dependence of the conformations unsurprising, the fact that the effect is so strong, given the lack of amide-mediated hydrogen bonding, is perhaps less expected. Indeed, it is common practice to substitute the ester analog of an amino acid into a peptide to determine the importance of a particular amino acid to the tertiary structure of a protein.73,74
Although we cannot yet correlate our NMR spectra with specific conformational preferences the data suggests that there is much to be learned. The fact that we observe a sequence-specific sensitivity to stereochemistry is, for example, intriguing. Such behavior could arise either because specific sequences create conformations that place the diastereotopic methylene protons in better positions to “observe” distant stereochemical relationships or it could arise because certain sequences simply have stronger conformational preferences (or both). Also relevant to the understanding of the rules governing the preferred conformations in this system is the observation that the chemical shift difference between the two methylenic protons on a particular carbon was consistently smaller for s-centered polyads than i-centered polyads. An analogous trend has been well-documented in vinyl polymers such as polypropylene.75,76
Conclusions
The solution phase conformations for RSC PLGAs were found to be extremely sequence and stereosequence dependent, analogous to peptides. To access these RSC PLGAs, we have developed a methodology that allows the preparation of complex sequenced copolymers of lactic and glycolic acids from simple mono-protected building blocks. The method is general, convergent, and allows for the synthesis of sequences unavailable by other methods such as ROP. Although RSC synthesis can be used to make stereopure NMR standards, the inherent resolution of specific sequences must be taken into account as the level of resolution for a particular nucleus may be sequence dependent. Improved thermal properties were also a direct result of the uniformity of polymer sequence. We plan to extend this work by undertaking conformational modeling and bulk characterization studies.
Supplementary Material
Acknowledgments
This research was supported by NSF (0809289), Research Corp (RA0356), and the University of Pittsburgh. The authors would like to thank Drs. Robbert Duchateau and Saskia Huijser, for MALDI-TOF analysis, Dr. Krishnan Damodaran for NMR consultation, Dr. Joel Gillespie for materials characterization, Jian Li for synthetic contributions, and Drs. David J. Earl, Megan M. Spence, W. Seth Horne, and Mr. Benjamin N. Norris for helpful discussions. The authors also acknowledge NIH grant 1S10RR017977-01 for support of TOF MS.
Footnotes
Supporting Information Available: Detailed procedures and full characterization of all synthetic intermediates and polymers are provided, along with figures for the 13C spectra of poly GLG and LG series, the 1H spectra for selected polymers in DMSO, and full MADLI-TOF spectra for poly LG and GLG. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Badi N, Lutz JF. Chem Soc Rev. 2009;38:3383–3390. doi: 10.1039/b806413j. [DOI] [PubMed] [Google Scholar]
- 2.Thomas CM. Chem Soc Rev. 2010;39:165–173. doi: 10.1039/b810065a. [DOI] [PubMed] [Google Scholar]
- 3.Ueda M. Prog Polym Sci. 1999;24:699–730. [Google Scholar]
- 4.Datta B, Schuster GB. J Am Chem Soc. 2008;130:2965–2973. doi: 10.1021/ja0726106. [DOI] [PubMed] [Google Scholar]
- 5.Pfeifer S, Lutz JF. J Am Chem Soc. 2007;129:9542–9543. doi: 10.1021/ja0717616. [DOI] [PubMed] [Google Scholar]
- 6.Pfeifer S, Zarafshani Z, Badi N, Lutz JF. J Am Chem Soc. 2009;131:9195–9197. doi: 10.1021/ja903635y. [DOI] [PubMed] [Google Scholar]
- 7.van Hest JCM, Tirrell DA. Chem Commun. 2001:1897–1904. doi: 10.1039/b105185g. [DOI] [PubMed] [Google Scholar]
- 8.Baughman TW, Sworen JC, Wagener KB. Macromolecules. 2006;39:5028–5036. [Google Scholar]
- 9.Ward RE, Meyer TY. Macromolecules. 2003;36:4368–4373. [Google Scholar]
- 10.Copenhafer JE, Walters RW, Meyer TY. Macromolecules. 2008;41:31–35. [Google Scholar]
- 11.Kramer JW, Treitler DS, Dunn EW, Castro PM, Roisnel T, Thomas CM, Coates GW. J Am Chem Soc. 2009;131:16042–16044. doi: 10.1021/ja9075327. [DOI] [PubMed] [Google Scholar]
- 12.Hill DJ, Mio MJ, Prince RB, Hughes TS, Moore JS. Chem Rev. 2001;101:3893–4012. doi: 10.1021/cr990120t. [DOI] [PubMed] [Google Scholar]
- 13.Coates GW. Chem Rev. 2000;100:1223–1252. doi: 10.1021/cr990286u. [DOI] [PubMed] [Google Scholar]
- 14.Berthet MA, Zarafshani Z, Pfeifer S, Lutz JF. Macromolecules. 2009;43:44–50. [Google Scholar]
- 15.Hadjichristidis N. Block Copolymers: Synthetic Strategies, Physical Properties, and Applications. 2002 [Google Scholar]
- 16.Rogers ME, Long TE. Synthetic Methods in Step-Growth Polymers. 2003 [Google Scholar]
- 17.Langer R. Acc Chem Res. 2000;33:94–101. doi: 10.1021/ar9800993. [DOI] [PubMed] [Google Scholar]
- 18.Anderson JM, Shive MS. Adv Drug Deliv Rev. 1997;28:5–24. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 19.Athanasiou KA, Niederauer GG, Agrawal CM. Biomaterials. 1996;17:93–102. doi: 10.1016/0142-9612(96)85754-1. [DOI] [PubMed] [Google Scholar]
- 20.Hutmacher DW. Biomaterials. 2000;21:2529–2543. doi: 10.1016/s0142-9612(00)00121-6. [DOI] [PubMed] [Google Scholar]
- 21.Lunt J. Polym Degrad Stab. 1998;59:145–152. [Google Scholar]
- 22.Shoichet MS. Macromolecules. 2010;43:581–591. [Google Scholar]
- 23.Williams DF. Biomaterials. 2009;30:5897–5909. doi: 10.1016/j.biomaterials.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 24.Matthew HWT. Polym Biomater. (2nd) 2002:167–186. [Google Scholar]
- 25.Seal BL, Otero TC, Panitch A. Materials Science & Engineering, R Reports. 2001;R34:147–230. [Google Scholar]
- 26.Dechy-Cabaret O, Martin-Vaca B, Bourissou D. Chem Rev. 2004;104:6147–6176. doi: 10.1021/cr040002s. [DOI] [PubMed] [Google Scholar]
- 27.Dove AP. Chem Commun. 2008:6446–6470. doi: 10.1039/b813059k. [DOI] [PubMed] [Google Scholar]
- 28.Stanford MJ, Dove AP. Chem Soc Rev. 2010;39:486–494. doi: 10.1039/b815104k. [DOI] [PubMed] [Google Scholar]
- 29.Ovitt TM, Coates GW. J Polym Sci Part A: Polym Chem. 2000;38:4686–4692. [Google Scholar]
- 30.Ovitt TM, Coates GW. J Am Chem Soc. 1999;121:4072–4073. [Google Scholar]
- 31.Ovitt TM, Coates GW. J Am Chem Soc. 2002;124:1316–1326. doi: 10.1021/ja012052+. [DOI] [PubMed] [Google Scholar]
- 32.Chisholm MH, Delbridge EE, Gallucci JC. New J Chem. 2004;28:145–152. [Google Scholar]
- 33.Kasperczyk J, Bero M. Polymer. 2000;41:391–395. [Google Scholar]
- 34.Bero M, Dobrzynski P, Kasperczyk J. Polymer Bulletin (Berlin) 1999;42:131–139. [Google Scholar]
- 35.Choi SH, Park TG. J Biomat Sci Polym E. 2002;13:1163–1173. doi: 10.1163/156856202320813864. [DOI] [PubMed] [Google Scholar]
- 36.Dobrzynski P, Kasperczyk J, Janeczek H, Bero M. Polymer. 2002;43:2595–2601. [Google Scholar]
- 37.Dong CM, Qiu KY, Gu ZW, Feng XD. J Polym Sci Part A: Polym Chem. 2000;38:4179–4184. [Google Scholar]
- 38.Dong CM, Qiu KY, Gu ZW, Feng XD. J Polym Sci Part A: Polym Chem. 2001;39:357–367. [Google Scholar]
- 39.Grijpma DW, Nijenhuis AJ, Pennings AJ. Polymer. 1990;31:2201–6. [Google Scholar]
- 40.Kricheldorf HR, Kreiser I. Makromolekul Chem. 1987;188:1861–73. [Google Scholar]
- 41.Zell MT, Padden BE, Paterick AJ, Thakur KAM, Kean RT, Hillmyer MA, Munson EJ. Macromolecules. 2002;35:7700–7707. [Google Scholar]
- 42.Thakur KAM, Kean RT, Hall ES, Kolstad JJ, Munson EJ. Macromolecules. 1998;31:1487–1494. [Google Scholar]
- 43.Thakur KAM, Kean RT, Hall ES, Kolstad JJ, Lindgren TA, Doscotch MA, Siepmann JI, Munson EJ. Macromolecules. 1997;30:2422–2428. [Google Scholar]
- 44.Thakur KAM, Kean RT, Hall ES, Doscotch MA, Munson EJ. Anal Chem. 1997;69:4303–4309. doi: 10.1021/ac970792o. [DOI] [PubMed] [Google Scholar]
- 45.Kasperczyk J. Macromol Symp. 2001;175:19–31. [Google Scholar]
- 46.Kasperczyk JE. Polymer. 1999;40:5455–5458. [Google Scholar]
- 47.Bero M, Kasperczyk J, Jedlinski ZJ. Makromolekul Chem. 1990;191:2287–96. [Google Scholar]
- 48.Kricheldorf HR, Boettcher C, Tonnes KU. Polymer. 1992;33:2817–2824. [Google Scholar]
- 49.Chabot F, Vert M, Chapelle S, Granger P. Polymer. 1983;24:53–9. [Google Scholar]
- 50.Zhang L, Nederberg F, Messman JM, Pratt RC, Hedrick JL, Wade CG. J Am Chem Soc. 2007;129:12610–12611. doi: 10.1021/ja074131c. [DOI] [PubMed] [Google Scholar]
- 51.Spassky N, Wisniewski M, Pluta C, LeBorgne A. Macromol Chem Phys. 1996;197:2627–2637. [Google Scholar]
- 52.Lillie E, Schulz RC. Makromolekul Chem. 1975;176:1901–6. [Google Scholar]
- 53.Espartero JL, Rashkov I, Li SM, Manolova N, Vert M. Macromolecules. 1996;29:3535–3539. [Google Scholar]
- 54.Kasperczyk J, Bero M. Makromolekul Chem. 1993;194:913–25. [Google Scholar]
- 55.Dobrzynski P, Kasperczyk J, Janeczek H, Bero M. Macromolecules. 2001;34:5090–5098. [Google Scholar]
- 56.Gao QW, Lan P, Shao HL, Hu XC. Polym J. 2002;34:786–793. [Google Scholar]
- 57.Kasperczyk J. Polymer. 1996;37:201–203. [Google Scholar]
- 58.Kister G, Cassanas G, Vert M. Polymer. 1998;39:3335–3340. [Google Scholar]
- 59.Stayshich RM, Meyer TY. J Polym Sci Part A: Polym Chem. 2008;46:4704–4711. [Google Scholar]
- 60.Moore JS, Stupp SI. Macromolecules. 1990;23:65–70. [Google Scholar]
- 61.Akutsu F, Inoki M, Uei H, Sueyoshi M, Kasashima Y, Naruchi K, Yamaguchi Y, Sunahara M. Polym J (Tokyo) 1998;30:421–423. [Google Scholar]
- 62.Kang S, Zhang G, Aou K, Hsu SL, Stidham HD, Yang X. J Chem Phys. 2003;118:3430–3436. [Google Scholar]
- 63.Penco M, Donetti R, Mendichi R, Ferruti P. Macromol Chem Phys. 1998;199:1737–1745. [Google Scholar]
- 64.Bovey FA, Mirau PA. Acc Chem Res. 1988;21:37–43. [Google Scholar]
- 65.Agarwal S, Naumann N, Xie X. Macromolecules. 2002;35:7713–7717. [Google Scholar]
- 66.Huijser S, Staal BBP, Huang J, Duchateau R, Koning CE. Angew Chem Int Ed. 2006;45:4104–4108. doi: 10.1002/anie.200600594. [DOI] [PubMed] [Google Scholar]
- 67.Huijser S, Staal BBP, Huang J, Duchateau R, Koning CE. Biomacromolecules. 2006;7:2465–2469. doi: 10.1021/bm060466v. [DOI] [PubMed] [Google Scholar]
- 68.Nederberg F, Connor EF, Glausser T, Hedrick JL. Chem Commun. 2001:2066–2067. doi: 10.1039/b106125a. [DOI] [PubMed] [Google Scholar]
- 69.Takizawa K, Nulwala H, Hu J, Yoshinaga K, Hawker CJ. J Polym Sci Part A: Polym Chem. 2008;46:5977–5990. [Google Scholar]
- 70.Takizawa K, Tang CB, Hawker CJ. J Am Chem Soc. 2008;130:1718–1726. doi: 10.1021/ja077149w. [DOI] [PubMed] [Google Scholar]
- 71.Park PIP, Jonnalagadda S. J Appl Polym Sci. 2006;100:1983–1987. [Google Scholar]
- 72.Chisholm MH, Iyer SS, Matison ME, McCollum DG, Pagel M. Chem Commun. 1997:1999–2000. [Google Scholar]
- 73.Deechongkit S, Dawson PE, Kelly JW. J Am Chem Soc. 2004;126:16762–16771. doi: 10.1021/ja045934s. [DOI] [PubMed] [Google Scholar]
- 74.Gallo EA, Gellman SH. J Am Chem Soc. 1993;115:9774–9788. [Google Scholar]
- 75.Bovey FA. Acc Chem Res. 1968;1:175–85. [Google Scholar]
- 76.Flory PJ, Fujiwara Y. Macromolecules. 1969;2:327–35. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.