

Abstract
Objective. This study was to evaluate the effect of riluzole on methylmercury- (MeHg-) induced oxidative stress, through promotion of glutathione (GSH) synthesis by activating of glutamate transporters (GluTs) in rat cerebral cortex. Methods. Eighty rats were randomly assigned to four groups, control group, riluzole alone group, MeHg alone group, and riluzole + MeHg group. The neurotoxicity of MeHg was observed by measuring mercury (Hg) absorption, pathological changes, and cell apoptosis of cortex. Oxidative stress was evaluated via determining reactive oxygen species (ROS), 8-hydroxy-2-deoxyguanosine (8-OHdG), malondialdehyde (MDAs), carbonyl, sulfydryl, and GSH in cortex. Glutamate (Glu) transport was studied by measuring Glu, glutamine (Gln), mRNA, and protein of glutamate/aspartate transporter (GLAST) and glutamate transporter-1 (GLT-1). Result. (1) MeHg induced Hg accumulation, pathological injury, and apoptosis of cortex; (2) MeHg increased ROS, 8-OHdG, MDA, and carbonyl, and inhibited sulfydryl and GSH; (3) MeHg elevated Glu, decreased Gln, and downregulated GLAST and GLT-1 mRNA expression and protein levels; (4) riluzole antagonized MeHg-induced downregulation of GLAST and GLT-1 function and expression, GSH depletion, oxidative stress, pathological injury, and apoptosis obviously. Conclusion. Data indicate that MeHg administration induced oxidative stress in cortex and that riluzole could antagonize this situation through elevation of GSH synthesis by activating of GluTs.
1. Introduction
Methylmercury (MeHg) is a well-recognized environmental contaminant with established health risk to human beings by fish and marine mammal consumption [1]. MeHg can easily cross the blood-brain barriers and cause central nervous system (CNS) damage. The mechanism underlying MeHg neurotoxicity is not fully understood. The situation of imbalance between the generation and the elimination of reactive oxygen species (ROS) named oxidative stress, which is characterized by oxidizing biological macromolecules including lipoid, cellular protein, and nucleic acid [2]. There is growing evidence indicated that oxidative stress plays a critical role in the pathogenesis of MeHg neurotoxicity both in vivo and in vitro [3, 4]. For example, cultured microglial cells, astrocytes, and neurons exposed to MeHg and striatal synaptosomes prepared from rats injected with MeHg, demonstrate oxidative stress [5].
Glutathione (GSH) is the major non-enzymatic antioxidant that scavenges ROS and inhibits oxidative stress. GSH also has a vital role for modulating MeHg toxicity by conjugation and efflux of MeHg. In brain, neurons can not directly acquire GSH from extracellular space. Consumed GSH is resynthesized with L-glutamate, L-cysteine, and glycine. The glutamate moiety as precursors for neuronal GSH synthesis is provided by astrocyte glutamate transporters (GluTs) and system Xc− (cystine-glutamate exchanger) [6]. De Bundel et al. [7] reported that mice lacking the specific xCT subunit of system Xc− (xCT−/−) do not have a lower hippocampus GSH content and not induce oxidative stress in vivo. Therefore, we hypothesized that GluTs might act protective effects on antioxidant defense to MeHg neurotoxicity.
Classically, glutamate/aspartate transporter (GLAST) and glutamate transporter-1 (GLT-1), high-affinity astrocyte glutamate transporters (GluTs), are responsible for clearing extracellular glutamate [8]. Recently, it is shown that the functions of these transporters maintain neuronal, astrocyte, microglia, macrophage, and nonneural cells antioxidant defenses [9–13]. Meanwhile, it demonstrated that L-trans-pyrrolidine-2, 4-dicarboxylate (PCD), a substrate inhibitor of the transporters, triggers astrocytes demise in vivo and in vitro. This PDC-induced astrocyte death is independent of excitotoxicity but involves GSH depletion and oxidative stress [14].
Riluzole is a neuroprotective agent approved by food and drug administration (FDA) for use in slowing the progression of amyotrophic lateral sclerosis (ALS) [15]. Lately, we have confirmed that riluzole promoted the function of GLAST and GLT-1 after Mn poisoning [16]. And it was detailed by Frizzo et al. [17] and Fumagalli et al. [18] that riluzole could enhance the uptake of glutamate (Glu). Interestingly, it was also found that riluzole reduced different oxidative stress events following spinal cord injury (SCI) in rats [19]. Therefore, we here use riluzole as the activator of GluTs to clarify the role of Glu transport playing in the occurrence of MeHg-induced oxidative stress.
We hypothesize that riluzole trigger GSH synthesis via promoting Glu transport to antioxidant defense after MeHg exposure. Therefore, the present study was carried out to evaluate the effects of MeHg administration on the parameters of Hg concentration, morphological features, apoptosis, ROS, malondialdehyde (MDA), protein sulfydryl, carbonyl, 8-hydroxy-2-deoxyguanosine (8-OHdG), GSH, Glu, glutamine (Gln) levels, GLAST and GLT-1 mRNA expression and protein levels, and the influence of riluzole on the effects elicited by MeHg.
2. Materials and Methods
2.1. Chemicals
Methylmercury chloride was purchased from Laboratory of Dr. Ehrenstorfer-Schafers (Augsburg, Germany). Analysis kits of sulfydryl, carbonyl, Glu, and Gln were provided by Jiancheng Bioengineering Institute (Nanjing, China). Annexin V-FITC/PI double-stained apoptosis detection kit was purchased from Keygen Biotech Co. (Nanjing, China). Trizol reagent, Real-Time PCR kit, DNA marker and primers were provided by TaKaRa Biotechnology Co., Ltd. (Dalian, China). Goat polyclonal antibodies against GLAST and GLT-1, mouse monoclonal antibody against β-actin, rabbit monoclonal antibody against 8-OHdG were provided by Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Strept avidin-biotin complex (SABC) immunohistochemistry kit was provided by Boster Biochemical Reagent Co., Ltd (Wuhan, China). Riluzole, Fura-2/AM, 2, 7-dichlorofluorescin-diacetate (DCFH-DA), and Folin phenol reagent were purchased from Sigma Chemical Co. (Saint Louis, MO, USA), while other chemicals were provided by local chemical suppliers. All chemicals were of analytical grade, HPLC grade, or the best pharmaceutical grade.
2.2. Animals
The study was performed on wistar rats (170–190 g body weight at the beginning of experiment; N = 80; equal numbers of male and female), which were supplied by Experimental Animal Center, China Medical University, Shenyang, China. All the animals were housed in an air-conditioned room with controlled temperature (25 ± 2°C) and maintained on a 12 h/12 h light cycle (07:00 on–19:00 off). They were allowed free access to food and water. All experimental procedures were conducted in conformity with the institutional guidelines for the care and use of laboratory animals in China Medical University, Shenyang, China, and conformed to the National Institutes of Health Guide for Care and Use of Laboratory Animals (publication no. 85-23, revised 1985). All efforts were made to minimize the number of animals used and their suffering.
2.3. Treatment
Eighty rats were randomly assigned to four groups by weight, including control group, riluzole alone group, MeHg alone group, and riluzole + MeHg group. Rats in control and MeHg alone groups were administrated with 0.9% NaCl subcutaneously (s.c.). Rats of riluzole alone group and riluzole + MeHg group were s.c. administrated with 21.35 μmol/kg riluzole. Two hours later, rats in control group and riluzole alone group were intraperitoneally (i.p.) injected with 0.9% NaCl, rats in MeHg alone and riluzole + MeHg groups were i.p. injected with 12 μmol/kg CH3ClHg respectively. The volume of administration was 5 mL/kg. The pretreatment was given every two days, while the injection was given everyday, lasting for 4 weeks.
2.4. Tissue Collection
At 24 h after the last injection, all the rats in each group were sacrificed by decapitation after anesthetized. Firstly, six of the rats in each group were the brain capsule on ice bath removed and the cortex separated, 100 mg of the cortex was digested by 2 mL nitric acid for 12 h in order to determine the Hg levels, the other cortex was prepared for the 5% or 10% homogenate in order to determine the Glu, GSH, MDA, sulfydryl, and carbonyl levels. Four of the other rats in each group, the cortex were prepared for single-cell suspension in order to investigate the apoptosis and ROS. Four of the rest rats in each group were extracted the total RNA and protein for the determination of GLAST and GLT-1 mRNA and protein. The cortex of last six rats in each group were cut 5 mm thickness tissue pieces and fixed in 4% paraformaldehyde in order to observe the 8-OHdG levels and the pathological changes. All the substance contents and enzyme activities were normalized to the protein which was measured by the method of Lowry [20], using bovine serum albumin (BSA) as standard.
2.5. Measurement of Mercury Concentration
The Hg concentration was detected with cold vapor atomic fluorescence spectrometry (CVAFS). Cortex tissue (100 mg) was digested with 2.0 mL nitric acid for 12 h. Then 2.0 mL of 50% H2SO4 and 3.0 mL of saturated KMnO4 were added. Samples were kept in boiling water bath for 1 h, and the volume was raised up to 10 mL by distilled water. 1.0 mL of sample was mixed with 1.0 mL of dehydrated alcohol and 2.0 mL of 20% stannous chloride and then analyzed by F732 Mercury Analyzer (Huaguang Factory, Shanghai, China). The levels of Hg in cortex were expressed as μg·g−1 tissue.
2.6. Pathological Analysis
The cortex tissue was removed after the rats were perfused into left ventricles with buffered 4% paraformaldehyde. Part of cortex in each rat was cut into 4 mm thickness pieces and fixed in 4% paraformaldehyde. Regular haematoxylin and eosin (HE) staining was performed for morphological observation with AX-70 microscope (Olympus, Japan).
2.7. Apoptosis Assay
The tissue of cortex (100 mg per sample) was made into single-cell suspensions according to method of Villalba et al. [21]. Cells apoptosis was determined by flow cytometer (FCM) assay using Annexin V/PI apoptosis detection kit. The single-cell suspension (1 × 106 cells/mL) was suspended in 200 μL ice-cold binding buffer and then 10 μL horseradish peroxidase FITC labeled Annexin V and 5 μL propidium iodide (PI) were added. The cell suspension was incubated in darkness at room temperature for 15 min. Apoptosis rate was determined by flow cytometer (BD Co., USA). In this study, both FITC and PI negative cells were considered as normal cells. FITC-single positive and PI negative cells were defined as early apoptotic cells, while both FITC and PI positive cells were considered as late apoptotic or necrosis cells.
2.8. Determination of ROS Formation
Intracellular ROS generation was measured by a flow cytometer with an oxidation-sensitive DCFH-DA fluorescent probe, after single-cell suspensions were made. DCFH-DA is a nonfluorescent compound that is freely taken up into cells. DCFH is oxidized to fluorescent dichlorofluorescein (DCF) by the action of cellular oxidants. The suspension was loaded by DCFH-DA solution with a final concentration of 50 μM and was incubated for 30 min at 37°C. Then samples were centrifuged at 1000 rpm for 5 min (4°C), and cells were resuspended with phosphate buffered saline (PBS, pH 7.2–7.4). The fluorescence was detected by flow cytometer (with excitation 488 nm and emission 525 nm). For each treatment, 1 × 105 cells were counted, and the experiment was performed in triplicate.
2.9. MDA Assay
The homogenate of cortex was added into the reaction mixture containing 0.1 mol/L phosphate buffer and 0.1 mol/L FeCl3 in a total volume of 1.0 mL (pH = 7.4). The reaction was stopped by the addition of 1.0 mL 10% TCA, following which of 1.0 mL 0.67% TBA was added and all the tubes were placed in a boiling water bath for 20 min. At the end, the tubes were shifted to an ice bath and centrifuged at 2500 g for 10 min. The amount of MDA formed in each of the samples was assessed by measuring the optical density of the supernatant at 535 nm using tetraethoxypropane (TEP) as standard. MDA content was expressed as nmol·mg−1 protein.
2.10. Sulfydryl and Carbonyl Assay
The sulfydryl and carbonyl contents were measured using assay kits and operated in accordance with the instruction strictly. The sulfydryl content was expressed as μmol·g−1 protein while the carbonyl was nmol·mg−1 protein.
2.11. Immunohistochemical Analysis of 8-OHdG
The 8-OHdG levels were measured using the method of immunohistochemistry. The cortex tissue was fixed by 4% paraformaldehyde for 24 h. Then it was dehydrated with gradient alcohol which was followed by xylene. After that the tissue was embedded by low-melting-point paraffin and cut by Leitz1512 microtome (Leitz Co., Germany) for 5 μm thickness each. For the immunohistochemistry reaction, the slice was rinsed with phosphate buffer for three times, 5 min for each, and then incubated in 3% (v/v) H2O2 at room temperature for 10 min. After that we repaired the antigen and sealed the slice with 10% (v/v) nonimmune goat serum. Twenty minutes later, we removed the serum and added in 100 μL 1 : 100 dilution of 8-OHdG monoclonal antibody, 37°C for 30 min. Then rinsed the slice and biotinylated goat anti-rabbit IgG was added for 10 min at 37°C. SABC reagent was added for 10 min at 37°C. Then staining with 3,3-diaminobenzidine (DAB) was performed. After that the slice was counterstained with hematoxylin, dehydrated after hydrochloric alcohol differentiation, and sealed by neutral gum finally. Took photos by autoexposure microscopy (Olympus, Japan) and analyzed the image by the MetamorphPDP10PBX51 graphic analysis system. For two rats in each group, three occasions for each rat, was calculated the threshold area (%) and the integral optical density (IOD) of the 8-OHdG-immune response-positive-cells.
2.12. Glutathione Assay
GSH in cortex homogenate was determined by reaction with 1, 2-dithio-bis nitro benzoic acid (DTNB). Briefly, 0.9 mL of 10% cortex homogenate was added in 0.1 mL of 50% trichloroacetic acid, and samples were centrifuged at 3000 rpm for 15 min. After that, 0.1 mL of supernatant was added in 4.4 mL of 0.1 M PBS and 0.5 mL of 0.04% DTNB to a total volume of 5.0 mL (pH 7.4). The absorbance of the solution was measured spectrophotometrically at 412 nm. The content of GSH was expressed as μmol·g−1 protein.
2.13. Real-Time PCR for GLAST and GLT-1 mRNA Expression
Total mRNA was extracted from the cortex of rats in each group. Isolation was performed using the Trizol method. It was finally redissolved in RNase-free water. OD260/OD280 of total RNA was between 1.6 and 1.8. Semiquantitative reverse transcription was performed by PrimeScript RT reagent Kit with DNAeraser. Reverse transcription was performed using Applied Biosystems 2720 Thermal Cycler (Applied Bios stems, Singapore). Reverse transcription of 5 μg of total RNA was performed in a final volume 20 μL, using 1 μL TransScript RT, 25 units ribonuclease inhibitor, 0.1 μg random primer (N9) as a primer. 1 μL 10 mM dNTP, 4 μL 5xRT buffer, and RNase-free ddH2O to 20 μL in one cycle: 25°C for 10 min, 42°C for 50 min, and 70°C for 15 min with subsequent cooling to 4°C. The reaction was incubated at 37°C for 15 min, then 85°C for 5 sec. The Gene-specific primers were designed by TaKaRa Co. The primers for GLAST and GLT-1 were designed by invitrogen corporation based on the previously reported sequences for GLAST sense: 5′-GGGTTTTCATTGGAGGGTTGC-3′ and antisense: 5′-CCACGGGTTTCTCTGGTTCAT-3′, for GLT-1 sense: 5′-GGGTCATCCTGGATGGAGGT-3′ and antisense: 5′-CGTGTCGTCATAAACGGACTG-3′. β-actin was used as an internal control. The primer sequence used for β-actin-sense: 5′-GGAGATTACTGCCCTGGCTCCTA-3′, antisense: 5′-GACTCATCGTACTCCTGCTGCTG-3′. The 50 μL PCR reaction mixture contained 25 μL of 2x PCR buffer, 2 μL of PCR Forward primer (10 μM), 2 μL of PCR Reverse primer (10 μM), 1 μL of ROX Reference Dye II (50x), 4 μL of template DNA, and 16 μL dH2O. The initial denaturation was carried out at 95°C for 30 sec, which was followed by amplification in 40 cycles, 95°C for 3 sec, and 60°C for 34 sec using the 7500 Real-Time PCR system (BD Co., USA). The relative expression analysis was carried out using the method of 2−ΔΔCT. For which ΔCT (test) = CT (target, test) − CT (reference, test) , ΔCT (calibrator) = CT (target, calibrator) − CT (reference, calibrator) , and ΔΔCT = ΔCT (Test) − ΔCT (calibrator) . The relative expression was calculated by 2−ΔΔCT.
2.14. Western Blotting for GLAST and GLT-1 Protein
The total protein was extracted from cortex using RIPA buffer (10 mM Na2HPO4, pH 7.2, 150 mM NaCl, 1% sodium deoxicolate, 1% Nonidet P-40, 0.1% SDS) containing protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 0.2 mM 1,10-phenanthroline, 10 μg/mL pepstatin A, 10 μg/mL leupeptin, 10 μg/mL aprotinin, and 10 mM benzamidine). Protein concentrations were determined with BCA reagent. Equal amounts of protein (40 μg) were separated by 8% polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Ternicula, CA). PVDF membranes blocked overnight at 4°C in TBST containing 5% bovine serum albumin fraction V. Following this, the membranes were rinsed briefly in TBST (Tris-buffered saline with 0.1% Tween 20, 150 mM NaCl, 20 mM Tris) for 1 h, and incubated with GLAST (1 : 500), GLT-1 (1 : 300), and β-actin (1 : 500) primary antibody in TBST for 2 h at room temperature. The secondary anti-goat or anti-mouse HRP-conjugated antibodies were applied at dilution of 1 : 5000. Bands were visualized using the ECL kit. To quantify the staining, densitometric analysis were performed using Flurchen V2.0 Stand Alone software. The relative intensity of each sample protein levels were normalized by that of β-actin.
2.15. Glu and Gln Analysis
The levels of Glu and Gln were determined using assay kits in accordance with the instructions strictly. The content of Glu was expressed as μmol·g−1 protein. The levels of Gln was expressed as mmol·g−1 protein.
2.16. Statistical Analysis
Measurement of the parameters was conducted in triplicate experiment, and the means from three experiments were used for statistical analysis. Results were presented as means ± standard deviation. All statistical analysis was performed using the SPSS software, version 11.5. Data were analyzed using one-way analysis of variance followed by the post hoc Duncan multiple range test when F was significant. Statistical significance was set at P < 0.05.
3. Results
3.1. Effects of Riluzole on Hg Concentrations after MeHg Exposure
To confirm MeHg exposure and estimate the riluzole pretreatment on alteration of Hg absorption, CVAFS was employed to analyze the Hg concentration of rat cerebral cortex. Comparing with the control group, there was no significant difference of Hg concentration absorbed in cerebral cortex in the riluzole alone group; elevation of Hg levels in cerebral cortex was found in both MeHg alone and riluzole + MeHg groups; when compared with MeHg alone group, there was no obvious difference of Hg concentration in riluzole + MeHg group (Table 1).
Table 1.
Effects of MeHg exposure and riluzole pretreatment on Hg, MDA, sulfydryl, and carbonyl levels.
| Groups | Hg (μg·g−1) | MDA (nmol·mg−1·pro) | Sulfydryl (μmol·g−1·pro) | carbonyl (nmol·mg−1·pro) |
|---|---|---|---|---|
| Control | 1.15 ± 0.24 | 2.30 ± 0.16 | 46.19 ± 6.86 | 2.57 ± 0.46 |
| Riluzole | 1.07 ± 0.26 | 2.46 ± 0.56 | 48.17 ± 6.99 | 2.80 ± 0.75 |
| MeHg | 15.05 ± 2.73∗∗ | 3.85 ± 0.55∗∗ | 33.21 ± 4.12∗∗ | 6.15 ± 1.35∗∗ |
| Riluzole + MeHg | 13.75 ± 1.95∗∗ | 2.31 ± 0.17## | 38.93 ± 6.21∗# | 5.13 ± 1.09∗∗# |
Data are mean ± SD, n = 6 each.
*P < 0.05, ∗∗ P < 0.01 denote statistical significance compared with control group.
# P < 0.05, ## P < 0.01 denote statistical significance compared with MeHg alone group.
3.2. Effects of Riluzole on Pathological Injury and Apoptosis after MeHg Exposure
To confirm MeHg exposure and to clarify the preventive effect of riluzole on morphological changes of cerebral cortex, HE staining was used. In both control group (Figure 1(a)) and riluzole alone group (Figure 1(b)), it was found that cells were intact and regular, accompanied with abundant cytoplasm. There was one or two round and big nuclei. In MeHg alone group (Figure 1(c)), degeneration and necrosis were observed. And the cells of cerebral cortex were swelling and vacuolation, which appeared nuclei condensation even missing. Cells were sparse and the outline was fuzzy. In the riluzole + MeHg group (Figure 1(d)), the injury was significantly diminished. There were less neuronal loss and fewer dead cells. To observe apoptosis caused by MeHg exposure and riluzole preventive effects, single-cell suspensions of cerebral cortex were made and FCM analysis was used to quantify the rates of cell apoptosis through double staining of Annexin V-FITC and PI. It shows that almost all cells were viable (no staining) in both control (Figure 2(a)) and riluzole alone groups (Figure 2(b)). Many apoptotic cells (Annexin V+ and PI−) were found in MeHg alone group (Figure 2(c)). It showed MeHg increased apoptosis rates 12.8-fold as compared to the normal control. Less apoptosis cells were found in riluzole + MeHg group (Figure 2(d)). It showed that riluzole reduced apoptosis rates by 46% as compared to MeHg exposure.
Figure 1.
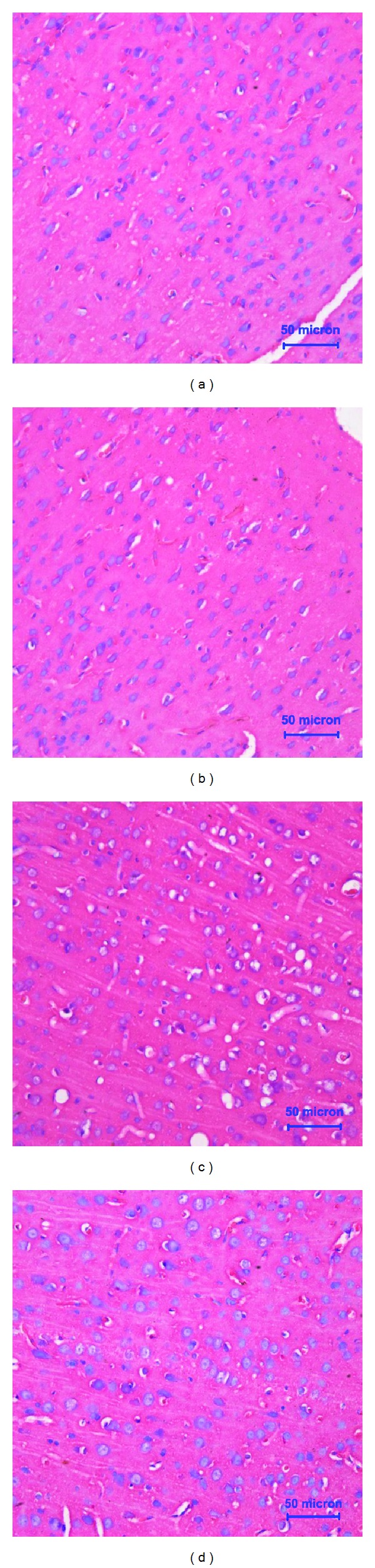
Light microphotographs showed that the morphological changes of cerebral cortex after MeHg exposure and/or riluzole pretreatment. Photographs of control (a), riluzole alone (b), MeHg alone (c), and riluzole + MeHg (d) were in this figure. The sections were stained with HE staining. Magnification × 40.
Figure 2.
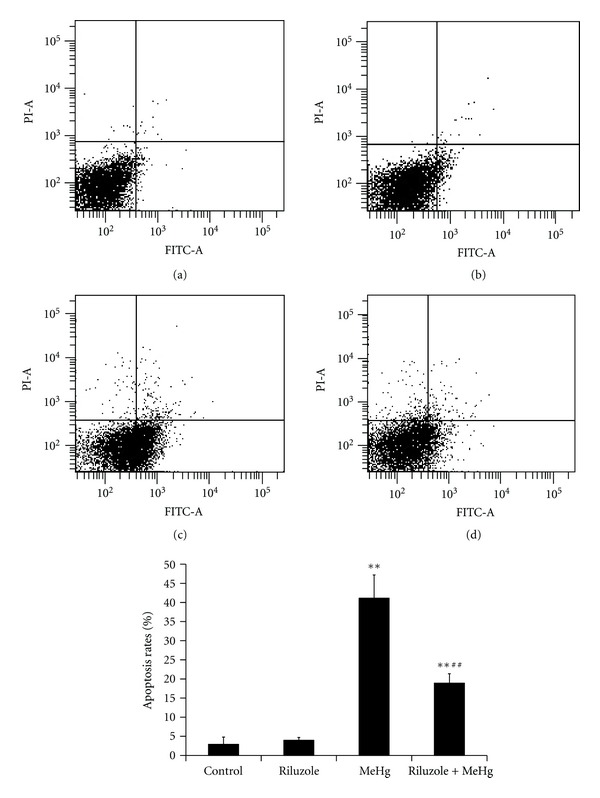
Alterations of hepatic apoptosis after MeHg exposure and/or riluzole pretreatment were shown in this figure. Scatter plots for control (a), riluzole alone (b), MeHg alone (c), and riluzole + MeHg (d) were in this figure. Data are mean ± SD for four animals in each group. **P < 0.01 denotes statistical significance compared with control group; ## P < 0.01 denotes statistical significance compared with MeHg alone group.
3.3. Effects of Riluzole on Oxidative Stress after MeHg Exposure
To observe MeHg-caused intracellular ROS changes and the preventive effects of riluzole, FCM analysis was used to measure intracellular ROS by staining of DCFH-DA. It was found that there was no significant difference of ROS between control and riluzole alone groups. MeHg increased intracellular ROS levels 3.12-fold as compared to the control. Riluzole reduced the production of intracellular ROS by 56.06% as compared to MeHg exposure (Figure 3). It was found that MeHg increased levels of MDA and carbonyl to 1.67-fold and 2.39-fold of control, while it decreased sulfydryl levels to 72% as compared to the control. It was also found that riluzole reduced MDA and carbonyl to 60% and 83.41% comparing with MeHg alone group, and sulfydryl elevated to 1.17-fold (Table 1). The formation of 8-OHdG, an indicator of oxidative damage to DNA, was examined in rat cerebral cortex. There was no observed immunoactivity in cerebral cortex in both control (Figure 4(a)) and riluzole alone groups (Figure 4(b)). Generation of 8-OHdG in DNA in a large mount of cells was observed in the MeHg alone group (Figure 4(c)). Although there were a few 8-OHdG positive cells in riluzole + MeHg group (Figure 4(d)), the number was decreased obviously according to MeHg alone group. The 8-OHdG immunoactivity were quantitatively evaluated by MetamorphPDP10PBX51 graphic analysis system of the threshold area (%) (Figure 4(e)) and the average integrated optical density (IOD) (Figure 4(f)). There was no significant difference of the threshold area and the average integrated optical density between control group and riluzole alone group. The threshold area and the average IOD of 8-OHdG in the MeHg alone group were significantly higher than the control. The threshold area and the average IOD in riluzole + MeHg group were significantly lower than the MeHg alone group.
Figure 3.
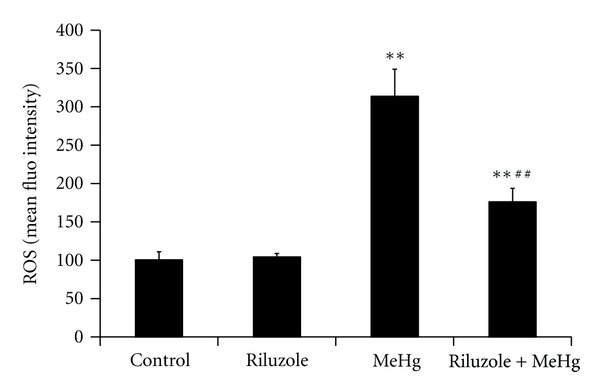
The changes of intracellular ROS levels in cerebral cortex after MeHg exposure and/or riluzole pretreatment were shown in this figure. Data are mean ± SD for four animals in each group. **P < 0.01 denotes statistical significance compared with control group; ## P < 0.01 denotes statistical significance compared with MeHg alone group.
Figure 4.
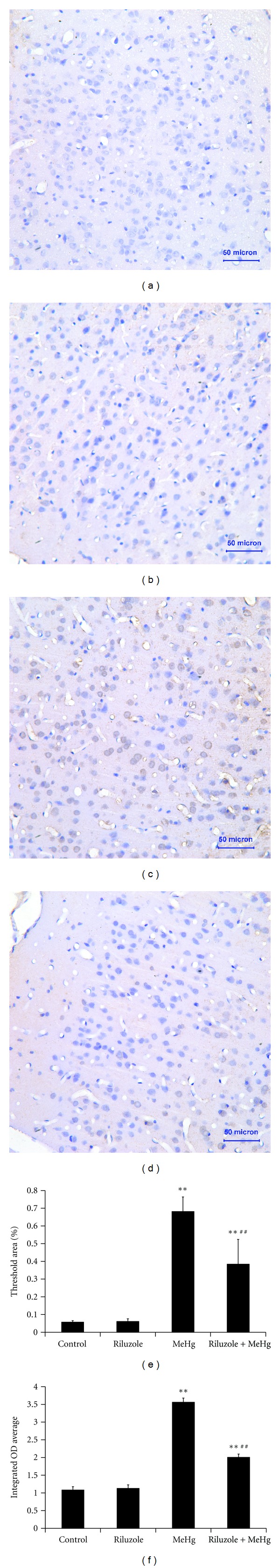
The figure showed the immunohistochemistry assay of 8-OHdG in cerebral cortex after MeHg exposure and riluzole pretreatment. Light microphotographs were for control (a), riluzole alone (b), MeHg alone (c), and riluzole + MeHg (d) were observed respectively, the sections were stained with SABC, and the magnification was set at ×40. Data are mean ± SD for six animals in each group. The effects of MeHg exposure and riluzole pretreatment on 8-OHdG threshold area (e) and integral optical density (f) were also shown in this figure. **P < 0.01 denotes statistical significance compared with control group; ## P < 0.01 denotes statistical significance compared with MeHg alone group.
3.4. Effects of Riluzole on GSH, Glu, and Gln Levels after MeHg Exposure
Comparing with the control group, and GSH and Gln levels increased 1.19-fold and 1.38-fold significantly in the riluzole alone group, GSH and Gln levels decreased to 66.71% and 65.38% obviously in the MeHg alone group. When compared with the MeHg alone group, GSH and Gln concentration elevated 1.30-fold and 1.47-fold significantly in riluzole + MeHg group (Table 2). Comparing with the control group, Glu levels decreased to 80% significantly in riluzole alone group, Glu levels increased 1.26-fold obviously in the MeHg alone group. When compared with the MeHg alone group, Glu concentration decreased to 75.8% significantly in riluzole + MeHg group (Table 2).
Table 2.
Effects of MeHg exposure and riluzole pretreatment on GSH, Glu, and Gln levels.
| Groups | GSH (μmol·g−1pro) | Glu (μmol·g−1 pro) | Gln (mmol·g−1 pro) |
|---|---|---|---|
| Control | 32.77 ± 4.01 | 56.76 ± 10.23 | 0.26 ± 0.02 |
| Riluzole | 39.16 ± 7.46∗ | 45.41 ± 6.90∗ | 0.36 ± 0.05∗∗ |
| MeHg | 21.86 ± 2.70∗∗ | 71.49 ± 11.98∗∗ | 0.17 ± 0.02∗∗ |
| Riluzole + MeHg | 28.34 ± 3.43# | 54.19 ± 3.08## | 0.25 ± 0.04## |
Data are mean ± SD, n = 6 each.
*P < 0.05, ∗∗ P < 0.01 denote statistical significance compared with control group;
# P < 0.05, ## P < 0.01 denote statistical significance compared with MeHg alone group.
3.5. Effects of Riluzole on GLAST and GLT-1 mRNA and Protein after MeHg Exposure
GLAST and GLT-1 mRNA expression were semiquantified by real-time RT-PCR. In riluzole alone group, GLAST and GLT-1 mRNA expressions elevated to 1.2-fold and 1.34-fold of control (Figure 5). MeHg exposure decreased the mRNA expression of GLAST and GLT-1 to 75.84% and 75.71% of control (Figure 5). Comparing with MeHg alone group, GLAST and GLT-1 mRNA increased 1.15-fold and 1.22-fold in riluzole + MeHg group (Figure 5).
Figure 5.
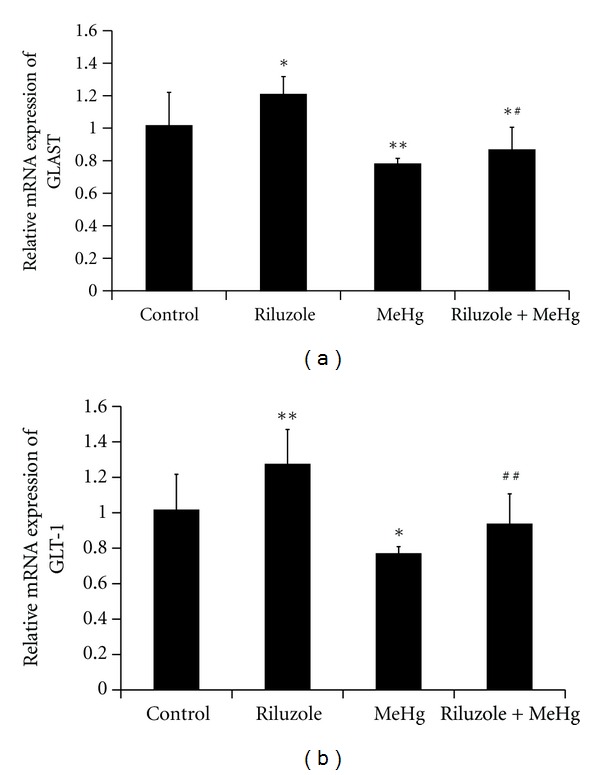
The alteration of GLAST mRNA (a) and GLT-1 mRNA (b) in cerebral cortex after MeHg exposure and riluzole pretreatment was shown in this figure. Data are mean ± S.D. for four animals in every group and each RNA preparation was run three times by real time-PCR, *P < 0.05, **P < 0.01 denote statistical significance compared with control group; # P < 0.05, ## P < 0.01 denote statistical significance compared with MeHg alone group.
GLAST and GLT-1 protein were determined by western blotting analysis. In riluzole alone group, GLAST and GLT-1 protein levels increased to 1.27-fold and 1.17-fold of control (Figure 6). MeHg exposure decreased GLAST and GLT-1 protein levels to 67.66% and 67.79% of control (Figure 6). Comparing with MeHg alone group, GLAST and GLT-1 protein increased 1.16-fold and 1.14-fold in riluzole + MeHg group (Figure 6).
Figure 6.
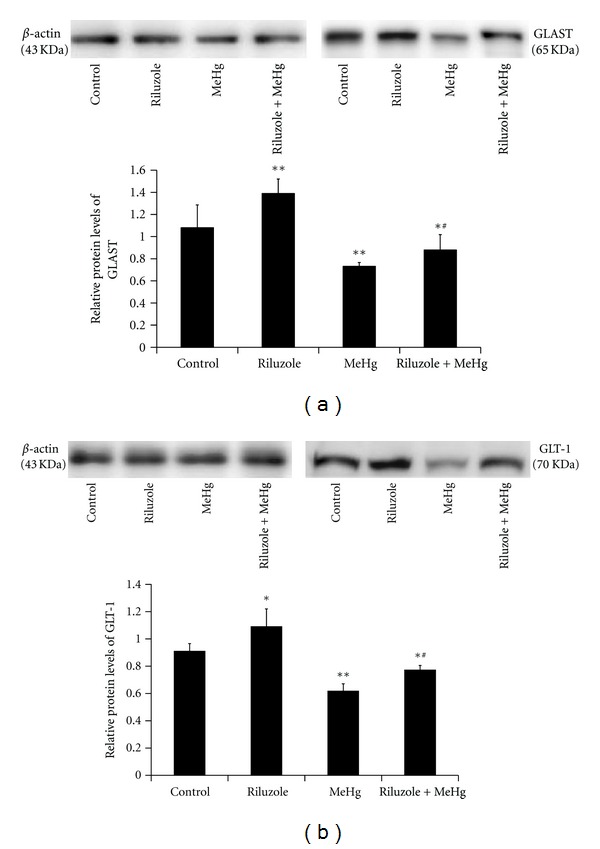
The alteration of GLAST protein (a) and GLT-1 protein (b) in cerebral cortex after MeHg exposure and riluzole pretreatment was shown in this figure. Data are mean ± S.D. for four animals in every group and each RNA or protein preparation was run three times by real time-PCR or western blotting, *P < 0.05, **P < 0.01 denote statistical significance compared with control group; # P < 0.05, ## P < 0.01 denote statistical significance compared with MeHg alone group.
4. Discussion
MeHg has been thought to induce oxidative stress in central nervous system (CNS) with the depletion of GSH [22]. Recently, some studies demonstrated that GluTs acted antioxidant defense through elevating intracellular GSH by their ability to transport GSH precursor (Glu) [14]. Riluzole, the neuroprotective drug currently used for ALS therapy, had been confirmed to serve as the functional properties on increasing GluTs levels, elevating affinity of Glu to GluTs, and enhancing Glu uptake [17, 18, 23]. And according to our earlier studies about manganese-caused excitotoxicity, we interestingly found that riluzole was an effective GluTs activator that can up-regulate GLAST and GLT-1 expression [16]. The present study was designed to pretreat with riluzole into rats before MeHg exposure, in order to find the mechanism of MeHg-induced oxidative stress and possible effect of GluTs on activating anti-oxidative defense against MeHg neurotoxicity.
In this study, it was found that Hg accumulated in cerebral cortex after MeHg exposure. Total Hg levels in MeHg alone group were 13 times higher than those in control group. The results were similar with the conclusion drawn from mice model [24]. It might be as a result of its lipophilic nature that MeHg goes across blood-brain barriers and absorbs in cerebral cortex of brain. However, there were no obvious changes on the total Hg levels between MeHg alone group and the riluzole + MeHg group. It suggested that riluzole acted no effects on Hg transport and excretion.
It was found that MeHg aggravated morphological damage and apoptosis rates significantly in our study. This result on morphological features was consistent with the study of Stoltenburg-Didinger and Markwort [25]. They reported that pathological features of somatosensory cortex appeared in the prenatally MeHg-treated rats. Our result of apoptosis was similar with the reports that 30 ppm MeHg in drinking water for 8 weeks administrating to mice caused neuropathological changes, such as neurons loss, astrocytes and microglia/macrophages proliferation, and necrosis and apoptosis in the cerebral cortex, particularly the deep layer of primary motor cortex and prelimbic cortex [26]. Our result of riluzole-decreased neuropathological damage supported the reports that it increased the neuronal density in excitotoxic prefrontal cortex [27]. And our results of riluzole-inhibited apoptosis were similar with the reports basing on morphine exposure rats [28] and HIV-1 infected patients [29].
To investigate the mechanisms of MeHg-induced pathological damage and apoptosis in cerebral cortex, we observed ROS production, MDA contents, carbonyl and sulfydryl levels, and 8-OHdG expression. It was found that MeHg increased ROS, MDA, carbonyl, and 8-OHdG and decreased sulfydryl significantly. The results suggested that MeHg-caused excessive ROS production was the killer to the normal histological structures and function of cerebral cortex by promoting lipid peroxidation, protein modification, and DNA strand breaks. These findings are in agreement with results of previous study in vivo [30]. Additionally, riluzole showed the obvious inhibition of MeHg oxidative stress. These results were similar with the conclusion basing on inhibitory of riluzole on oxidative stress processes in a spinal cord injury model [19], and in the ALS model [31].
Many researches of MeHg-induced oxidative stress mechanisms focused on the mitochondrial dysfunction, calcium overloading, and alteration of Nrf2 protein [32, 33]. Here, the study interested in endogenous glutathione (GSH) that is one of most abundant and essential thiol tripeptides present in mammalian cells for scavenging ROS. It is also the electron donor for the reduction of peroxides by glutathione peroxidase [34]. We evaluated and found an obvious decrease of GSH levels in cerebral cortex after MeHg exposure. It is for the ability of MeHg to interact with GSH and to lead to the formation of GS-HgCH complex. This interaction decreases the levels of GSH and the oxidized glutathione ration (GSH/GSSG), which contributes to the occurrence of oxidative stress and cerebral cortex injury [35]. Other studies revealed that the GSH content is higher in cerebral cells than other different regions cells in the CNS [36, 37]. This may be the reason for the excessive accumulation of MeHg in cerebral cortex. The similar involvement of GSH in the neurotoxicity of MeHg was also determined previously, showing that the elevated oxidative stress is related with the depleted intracellular GSH levels [38, 39]. Furthermore, N-acetylcysteine (NAC), GSH precursors, also was found to protect against MeHg neurotoxicity [40]. In this study, riluzole pretreatment brought obvious elevation of GSH levels. It may be because of riluzole increasing intracellular Glu, a substrate of the precursors, to synthesize GSH [6]. This gives us enlightenment and evidences to further reveal the mechanism of MeHg-induced GSH depletion and oxidative stress.
Within the family of excitatory amino acid transporters (EAATs), the glutamate transporters, GLAST and GLT-1, are the primary players in mediating glutamate uptake into astrocytes in the mammalian CNS [41]. Therefore, it was hypothesized that GLAST and GLT-1 acted an important role in maintaining GSH in the CNS. In the present study, it was found that both mRNA expression and protein levels of these two key GluTs were decreased obviously in MeHg alone group. Up to now, there were only a few studies in vivo focusing on this. The similar findings of GLT-1 protein were shown by Mutkus et al. [42]. They found that GLT-1 protein levels decreased obviously following 5 μM MeHg exposure in CHO-K1 cells. However, their results about mRNA expression of GLAST and GLT-1, and GLAST protein were different with our data. We also determined and found that total Glu levels in cerebral cortex increased significantly after MeHg exposure. Our studies were consistent with the conclusion drawn from the dialysis fluid and the hippocampus [43, 44]. Our findings also supported the earlier reports that suggested Hg inhibited the ability of astrocytes to uptake [3H]-L-Glu [45].
The preset study that determined and found mRNA expression and protein levels of GLAST and GLT-1 in riluzole + MeHg group were higher than in MeHg alone group. The active effects of riluzole may be under a variety of mechanisms including promoting mRNA expression, elevating protein and/or decreasing GluTs protein turnover. Riluzole has already been reported to increase GluTs function and expression in astrocytes or other cellular systems [46]. Our results supported the conclusion that riluzole enhanced Glu uptake and GluTs activity [17, 18]. Following the activation of GluTs, we also found that riluzole inhibited Glu production and promoted Gln generation during MeHg exposure. This may be caused by actions of riluzole on decreasing Glu release correlating with its ability to inhibit sodium currents [47].
In 1996, Dringen and Hamprecht [48] found the function of GluTs in inhibiting GSH synthesis in astrocytes. Our results suggested that downregulation of GLAST and GLT-1 played important roles in MeHg-caused oxidative stress through limitation of GSH generation. This was supported by the pervious finding that L-trans-pyrrolidine-2,4-dicarboxylate (PDC), a substrate inhibitor of GluTs, induced death of differentiated astrocytes by oxidative stress linking to GSH depletion [14]. However, in this previous study, it was also found that GLAST involved more preferentially than GLT-1 in mediating PDC-induced toxicity in the cultured astrocytes. It may be because that the levels of GLT-1 relative to GLAST may not be sufficient to significantly contribute to transport activity or that GLT-1 may not be active in the astrocytes culture model [49]. But contribution of GLAST and GLT-1 under conditions of altering transport in adult animal brain cannot be excluded in these cultured models. Therefore, our findings would provide more evidence to study on the effect of GluTs on GSH synthesis in vivo.
In conclusion, MeHg exposure increased total Hg, pathological damage, apoptosis rates, ROS production, lipid peroxidation, protein modification, DNA strand breaks, and GSH depletion, inhibited GLAST and GLT-1 mRNA expression and protein levels, and GSH synthesis in cerebral cortex. Pretreatment with riluzole, an activator of GluTs, antagonized MeHg-induced downregulation of GLAST and GLT-1, GSH depletion, oxidative stress, and pathological injury significantly. We drew a conclusion that riluzole acted protective effects on triggering GSH synthesis to antagonize MeHg oxidative stress via activation of GluTs. The study presents some evidence to clarify the mechanisms of MeHg-induced oxidative stress and further to prevent MeHg neurotoxicity.
Acknowledgment
The Grants from the National Natural Science Foundation of China (no. 81172631) supported this work financially.
References
- 1.Turner MD, Marsh DO, Smith JC, et al. Methylmercury in populations eating large quantities of marine fish. Archives of Environmental Health. 1980;35(6):367–378. doi: 10.1080/00039896.1980.10667521. [DOI] [PubMed] [Google Scholar]
- 2.Farina M, Aschner M, Rocha JBT. Oxidative stress in MeHg-induced neurotoxicity. Toxicology and Applied Pharmacology. 2011;256(3):405–417. doi: 10.1016/j.taap.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aschner M, Syversen T, Souza DO, Rocha JBT, Farina M. Involvement of glutamate and reactive oxygen species in methylmercury neurotoxicity. Brazilian Journal of Medical and Biological Research. 2007;40(3):285–291. doi: 10.1590/s0100-879x2007000300001. [DOI] [PubMed] [Google Scholar]
- 4.Shanker G, Aschner M. Methylmercury-induced reactive oxygen species formation in neonatal cerebral astrocytic cultures is attenuated by antioxidants. Molecular Brain Research. 2003;110(1):85–91. doi: 10.1016/s0169-328x(02)00642-3. [DOI] [PubMed] [Google Scholar]
- 5.Yee S, Choi BH. Oxidative stress in neurotoxic effects of methylmercury poisoning. NeuroToxicology. 1996;17(1):17–26. [PubMed] [Google Scholar]
- 6.Had-Aissouni L. Toward a new role for plasma membrane sodium-dependent glutamate transporters of astrocytes: maintenance of antioxidant defenses beyond extracellular glutamate clearance. Amino Acids. 2012;42(1):181–197. doi: 10.1007/s00726-011-0863-9. [DOI] [PubMed] [Google Scholar]
- 7.de Bundel D, Schallier AS, Loyens E, et al. Loss of system xc − does not induce oxidative stress but decreases extracellular glutamate in hippocampus and influences spatial working memory and limbic seizure susceptibility. The Journal of Neuroscience. 2011;31(15):5792–5803. doi: 10.1523/JNEUROSCI.5465-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang J, Takeuchi H, Doi Y, et al. Excitatory amino acid transporter expression by astrocytes is neuroprotective against microglial excitotoxicity. Brain Research. 2008;1210:11–19. doi: 10.1016/j.brainres.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 9.Aoyama K, Watabe M, Nakaki T. Modulation of neuronal glutathione synthesis by EAAC1 and its interacting protein GTRAP3-18. Amino Acids. 2012;42(1):163–169. doi: 10.1007/s00726-011-0861-y. [DOI] [PubMed] [Google Scholar]
- 10.Lewerenz J, Maher P, Methner A. Regulation of xCT expression and system xc − function in neuronal cells. Amino Acids. 2012;42(1):171–179. doi: 10.1007/s00726-011-0862-x. [DOI] [PubMed] [Google Scholar]
- 11.Persson M, Rönnbäck L. Microglial self-defence mediated through GLT-1 and glutathione. Amino Acids. 2012;42(1):207–219. doi: 10.1007/s00726-011-0865-7. [DOI] [PubMed] [Google Scholar]
- 12.Gras G, Samah B, Hubert A, Léone C, Porcheray F, Rimaniol AC. EAAT expression by macrophages and microglia: still more questions than answers. Amino Acids. 2012;42(1):221–229. doi: 10.1007/s00726-011-0866-6. [DOI] [PubMed] [Google Scholar]
- 13.Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system xc − : cystine supplier and beyond. Amino Acids. 2012;42(1):231–246. doi: 10.1007/s00726-011-0867-5. [DOI] [PubMed] [Google Scholar]
- 14.Ré DB, Boucraut J, Samuel D, Birman S, Kerkerian-Le Goff L, Had-Aissouni L. Glutamate transport alteration triggers differentiation-state selective oxidative death of cultured astrocytes: a mechanism different from excitotoxicity depending on intracellular GSH contents. Journal of Neurochemistry. 2003;85(5):1159–1170. doi: 10.1046/j.1471-4159.2003.01752.x. [DOI] [PubMed] [Google Scholar]
- 15.Cheah BC, Vucic S, Krishnan AV, Kiernan MC. Riluzole, neuroprotection and amyotrophic lateral sclerosis. Current Medicinal Chemistry. 2010;17(18):1942–1959. doi: 10.2174/092986710791163939. [DOI] [PubMed] [Google Scholar]
- 16.Deng Y, Xu Z, Xu B, et al. The protective effect of riluzole on manganese caused disruption of glutamate-glutamine cycle in rats. Brain Research. 2009;1289:106–117. doi: 10.1016/j.brainres.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 17.Frizzo ME, Dall’Onder LP, Dalcin KB, Souza DO. Riluzole enhances glutamate uptake in rat astrocyte cultures. Cellular and Molecular Neurobiology. 2004;24(1):123–128. doi: 10.1023/B:CEMN.0000012717.37839.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fumagalli E, Funicello M, Rauen T, Gobbi M, Mennini T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. European Journal of Pharmacology. 2008;578(2-3):171–176. doi: 10.1016/j.ejphar.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 19.Mu X, Azbill RD, Springer JE. Riluzole improves measures of oxidative stress following traumatic spinal cord injury. Brain Research. 2000;870(1-2):66–72. doi: 10.1016/s0006-8993(00)02402-1. [DOI] [PubMed] [Google Scholar]
- 20.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. The Journal of Biological Chemistry. 1951;193(1):265–275. [PubMed] [Google Scholar]
- 21.Villalba M, Martinez-Serrano A, Borner C, Blanco P, Satrustegui J. NMDA-induced increase in [Ca2+](i) and 45Ca2+ uptake in acutely dissociated brain cells derived from adult rats. Brain Research. 1992;570(1-2):347–353. doi: 10.1016/0006-8993(92)90600-e. [DOI] [PubMed] [Google Scholar]
- 22.Stringari J, Nunes AKC, Franco JL, et al. Prenatal methylmercury exposure hampers glutathione antioxidant system ontogenesis and causes long-lasting oxidative stress in the mouse brain. Toxicology and Applied Pharmacology. 2008;227(1):147–154. doi: 10.1016/j.taap.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carbone M, Duty S, Rattray M. Riluzole elevates GLT-1 activity and levels in striatal astrocytes. Neurochemistry International. 2012;60(1):31–38. doi: 10.1016/j.neuint.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roos DH, Puntel RL, Lugokenski TH, et al. Complex methylmercury-cysteine alters mercury accumulation in different tissues of mice. Basic & Clinical Pharmacology & Toxicology. 2010;107(4):789–792. doi: 10.1111/j.1742-7843.2010.00577.x. [DOI] [PubMed] [Google Scholar]
- 25.Stoltenburg-Didinger G, Markwort S. Prenatal methylmercury exposure results in dendritic spine dysgenesis in rats. Neurotoxicology and Teratology. 1990;12(6):573–576. doi: 10.1016/0892-0362(90)90064-j. [DOI] [PubMed] [Google Scholar]
- 26.Fujimura M, Usuki F, Sawada M, Takashima A. Methylmercury induces neuropathological changes with tau hyperphosphorylation mainly through the activation of the c-jun-N-terminal kinase pathway in the cerebral cortex, but not in the hippocampus of the mouse brain. NeuroToxicology. 2009;30(6):1000–1007. doi: 10.1016/j.neuro.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 27.Risterucci C, Coccurello R, Banasr M, Stutzmann JM, Amalric M, Nieoullon A. The metabotropic glutamate receptor subtype 5 antagonist MPEP and the Na+ channel blocker riluzole show different neuroprotective profiles in reversing behavioral deficits induced by excitotoxic prefrontal cortex lesions. Neuroscience. 2006;137(1):211–220. doi: 10.1016/j.neuroscience.2005.08.054. [DOI] [PubMed] [Google Scholar]
- 28.Ji X, Wang W, Cheng J, et al. Free radicals and antioxidant status in rat liver after dietary exposure of environmental mercury. Environmental Toxicology and Pharmacology. 2006;22(3):309–314. doi: 10.1016/j.etap.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 29.Achour A, M’Bika JP, Biquard JM. Enhanced endogenous type I interferon cell-driven survival and inhibition of spontaneous apoptosis by Riluzole. Virology. 2009;386(1):160–167. doi: 10.1016/j.virol.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 30.Carvalho MC, Franco JL, Ghizoni H, et al. Effects of 2,3-dimercapto-1-propanesulfonic acid (DMPS) on methylmercury-induced locomotor deficits and cerebellar toxicity in mice. Toxicology. 2007;239(3):195–203. doi: 10.1016/j.tox.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 31.Jackson M, Lladó J, Rothstein JD. Therapeutic developments in the treatment of amyotrophic lateral sclerosis. Expert Opinion on Investigational Drugs. 2002;11(10):1343–1364. doi: 10.1517/13543784.11.10.1343. [DOI] [PubMed] [Google Scholar]
- 32.Ni M, Li X, Yin Z, et al. Methylmercury induces acute oxidative stress, altering Nrf2 protein level in primary microglial cells. Toxicological Sciences. 2010;116(2):590–603. doi: 10.1093/toxsci/kfq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dreiem A, Seegal RF. Methylmercury-induced changes in mitochondrial function in striatal synaptosomes are calcium-dependent and ROS-independent. NeuroToxicology. 2007;28(4):720–726. doi: 10.1016/j.neuro.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dringen R. Metabolism and functions of glutathione in brain. Progress in Neurobiology. 2000;62(6):649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- 35.Ballatori N, Clarkson TW. Developmental changes in the biliary excretion of methylmercury and glutathione. Science. 1982;216(4541):61–63. doi: 10.1126/science.7063871. [DOI] [PubMed] [Google Scholar]
- 36.Kaur P, Aschner M, Syversen T. Role of glutathione in determining the differential sensitivity between the cortical and cerebellar regions towards mercury-induced oxidative stress. Toxicology. 2007;230(2-3):164–177. doi: 10.1016/j.tox.2006.11.058. [DOI] [PubMed] [Google Scholar]
- 37.Adachi T, Kunimoto M. Acute cytotoxic effects of mercuric compounds in cultured astrocytes prepared from cerebral hemisphere and cerebellum of newborn rats. Biological and Pharmaceutical Bulletin. 2005;28(12):2308–2311. doi: 10.1248/bpb.28.2308. [DOI] [PubMed] [Google Scholar]
- 38.Lee YW, Ha MS, Kim YK. Role of reactive oxygen species and glutathione in inorganic mercury-induced injury in human glioma cells. Neurochemical Research. 2001;26(11):1187–1193. doi: 10.1023/a:1013955020515. [DOI] [PubMed] [Google Scholar]
- 39.Shanker G, Syversen T, Aschner JL, Aschner M. Modulatory effect of glutathione status and antioxidants on methylmercury-induced free radical formation in primary cultures of cerebral astrocytes. Molecular Brain Research. 2005;137(1-2):11–22. doi: 10.1016/j.molbrainres.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 40.Kaur P, Aschner M, Syversen T. Glutathione modulation influences methyl mercury induced neurotoxicity in primary cell cultures of neurons and astrocytes. NeuroToxicology. 2006;27(4):492–500. doi: 10.1016/j.neuro.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 41.Kanner BI. Structure and function of sodium-coupled GABA and glutamate transporters. Journal of Membrane Biology. 2006;213(2):89–100. doi: 10.1007/s00232-006-0877-5. [DOI] [PubMed] [Google Scholar]
- 42.Mutkus L, Aschner JL, Syversen T, Aschner M. Methylmercury alters the in vitro uptake of glutamate in GLAST- and GLT-1-transfected mutant CHO-K1 cells. Biological Trace Element Research. 2005;107(3):231–245. doi: 10.1385/BTER:107:3:231. [DOI] [PubMed] [Google Scholar]
- 43.Juárez BI, Martínez ML, Montante M, Dufour L, García E, Jiménez-Capdeville ME. Methylmercury increases glutamate extracellular levels in frontal cortex of awake rats. Neurotoxicology and Teratology. 2002;24(6):767–771. doi: 10.1016/s0892-0362(02)00270-2. [DOI] [PubMed] [Google Scholar]
- 44.Zanoli P, Cannazza G, Baraldi M. Prenatal exposure to methyl mercury in rats: focus on changes in kynurenine pathway. Brain Research Bulletin. 2001;55(2):235–238. doi: 10.1016/s0361-9230(01)00460-9. [DOI] [PubMed] [Google Scholar]
- 45.Brookes N, Kristt DA. Inhibition of amino acid transport and protein synthesis by HgCl2 and methylmercury in astrocytes: selectivity and reversibility. Journal of Neurochemistry. 1989;53(4):1228–1237. doi: 10.1111/j.1471-4159.1989.tb07419.x. [DOI] [PubMed] [Google Scholar]
- 46.Azbill RD, Mu X, Springer JE. Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Research. 2000;871(2):175–180. doi: 10.1016/s0006-8993(00)02430-6. [DOI] [PubMed] [Google Scholar]
- 47.Bellingham MC. A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade? CNS Neuroscience & Therapeutics. 2011;17(1):4–31. doi: 10.1111/j.1755-5949.2009.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dringen R, Hamprecht B. Glutathione content as an indicator for the presence of metabolic pathways of amino acids in astroglial cultures. Journal of Neurochemistry. 1996;67(4):1375–1382. doi: 10.1046/j.1471-4159.1996.67041375.x. [DOI] [PubMed] [Google Scholar]
- 49.Schlag BD, Vondrasek JR, Munir M, et al. Regulation of the glial Na+-dependent glutamate transporters by cyclic AMP analogs and neurons. Molecular Pharmacology. 1998;53(3):355–369. doi: 10.1124/mol.53.3.355. [DOI] [PubMed] [Google Scholar]