

Abstract
Diabetic nephropathy is the leading cause of end-stage kidney disease worldwide but current treatments remain suboptimal. This review examines the evidence for inflammation in the development and progression of diabetic nephropathy in both experimental and human diabetes, and provides an update on recent novel experimental approaches targeting inflammation and the lessons we have learned from these approaches. We highlight the important role of inflammatory cells in the kidney, particularly infiltrating macrophages, T-lymphocytes and the subpopulation of regulatory T cells. The possible link between immune deposition and diabetic nephropathy is explored, along with the recently described immune complexes of anti-oxidized low-density lipoproteins. We also briefly discuss some of the major inflammatory cytokines involved in the pathogenesis of diabetic nephropathy, including the role of adipokines. Lastly, we present the latest data on the pathogenic role of the stress-activated protein kinases in diabetic nephropathy, from studies on the p38 mitogen activated protein kinase and the c-Jun amino terminal kinase cell signalling pathways. The genetic and pharmacological approaches which reduce inflammation in diabetic nephropathy have not only enhanced our understanding of the pathophysiology of the disease but shown promise as potential therapeutic strategies.
1. Introduction
Diabetic nephropathy (DN) has not been traditionally considered an inflammatory disease. However, recent studies have shown that kidney inflammation is crucial in promoting the development and progression of DN. Inflammation may be a key factor which is activated by the metabolic, biochemical, and haemodynamic derangements known to exist in the diabetic kidney. In this paper, we discuss the evidence for inflammation in DN and the lessons we have learned from novel experimental anti-inflammatory therapies. The main areas covered include the role of immune and inflammatory cells, inflammatory cytokines, and stress-activated protein kinases. We also briefly review the controversy around the role of immune complexes and immune deposition in DN.
2. Inflammatory Cells
In human DN, macrophages and T cells accumulate in the glomeruli and interstitium, even in the early stages of the disease. Recruitment of leukocytes involves three steps: (a) selectin-dependent leukocyte rolling on the endothelium, (b) chemokine-dependent integrin activation and leukocyte adhesion, and (c) transmigration of leukocytes across the endothelium [1]. Proinflammatory cytokines produced by leukocytes such as interleukin-1 (IL-1), tumour necrosis factor-α (TNF-α), and interferon-γ (INF-γ) can induce resident renal cells to produce a spectrum of chemokines. Elements of the diabetic milieu such as high glucose and advanced glycation end products (AGEs) are also potent stimulators of chemokine production. These chemokines include interleukin-8 (CXCL8), monocyte-chemoattractant protein-1 (MCP-1), INF-γ inducible protein (CXCL10), macrophage inflammatory protein-1α (MIP-1α/CCL3), and RANTES (CCL5). The elaborated chemokines then further direct the migration of additional leukocytes into the kidney and set up an inflammatory cycle.
2.1. Macrophages
Macrophages are key inflammatory cells mediating kidney inflammation in experimental and human diabetes. Activated macrophages elaborate a host of proinflammatory, profibrotic, and antiangiogenic factors. These macrophage-derived products include but are not limited to TNF-α, IL-1, IL-6, reactive oxygen species (ROS), plasminogen activator inhibitor-1 (PAI-1), matrix metalloproteinases, transforming growth factor-β (TGF-β), platelet-derived growth factor (PDGF), angiotensin II, and endothelin [1]. In experimental diabetic mice, macrophage accumulation and activation are associated with prolonged hyperglycaemia, glomerular immune complex deposition, increased chemokine production, and progressive fibrosis [2, 3]. In human type 2 diabetes, kidney macrophage accumulation is associated with the degree of glomerular sclerosis [4]. In another human study, interstitial macrophage accumulation correlated strongly with serum creatinine, proteinuria, and interstitial fibrosis at the time of biopsy, and inversely with the renal function decline (slope of 1/serum creatinine) over the following 5 years [5]. These human data support animal studies in suggesting a pathological role for macrophages in DN.
Strategies impairing kidney leukocyte recruitment have added evidence that macrophages mediate diabetic kidney injury. Increased kidney expression of intercellular adhesion molecule-1 (ICAM-1) has been noted in models of type 1 and type 2 diabetes. ICAM-1 serves as a ligand for LFA-1 on monocytes, facilitating leukocyte adhesion and transmigration. Diabetic ICAM-1 knockout mice showed significant reduction in albuminuria, glomerular, and tubulointerstitial injury, in association with a reduction in macrophage accumulation in the kidney [6]. However, ICAM-1 deficiency affects macrophages and lymphocytes, so these studies do not distinguish the roles of macrophages and lymphocytes.
Like ICAM-1, MCP-1 is also significantly increased in DN and expression levels correlate with the number of infiltrating interstitial macrophages. Studies suggest that renal MCP-1 is involved in the direction of macrophage migration into the diabetic kidney, while proteinuria itself may contribute to this upregulation of MCP-1 [7]. Blockade of the MCP-1 receptor (CCR-2) with a selective antagonist ameliorated diabetic glomerular sclerosis [8]. Similarly, diabetic db/db mice and streptozotocin-(STZ-) induced diabetic mice which are deficient in the gene encoding MCP-1 (Ccl2) are protected from renal injury [9, 10]. To further test the pathogenic role of macrophages, we used c-fms blockade to selectively target macrophages during the progression of experimental DN [11]. c-fms is the receptor for colony stimulating factor-1 (CSF-1), the major cytokine promoting macrophage accumulation, activation, and survival. We administered a neutralizing anti-c-fms monoclonal antibody to diabetic db/db mice with established albuminuria. This inhibition suppressed inflammation in the diabetic kidney, as evidenced by the reduction in macrophage accumulation, activation, and proliferation. Along with the effects on macrophages, there was a reduction in MCP-1, TNF-α, and c-Jun amino terminal kinase (JNK) activation. This resulted in a reduction in glomerular hyperfiltration, tubulointerstitial injury, and fibrosis.
The evidence so far indicates that infiltrating macrophages are associated with chronic, low-grade inflammation. Macrophages can interact with resident renal cells to generate a proinflammatory microenvironment that amplifies tissue injury and promotes scarring. As we have shown, macrophage-mediated injury is amenable to novel secondary prevention strategies.
2.2. Lymphocytes
A T cell infiltrate into diabetic kidneys has long been appreciated but not well studied. The role of T cells in kidney disease is better characterized in crescentic glomerulonephritis, such as antiglomerular basement membrane (GBM) disease [12]. Kidney infiltration by CD4+ and CD8+ T cells has been noted in diabetic db/db mice and diabetic NOD mice [2, 13]. In the latter study, glomerular B-cells were also found to be increased. A kidney T cell influx is common among young patients with early type 1 diabetes, especially those with a shorter duration of diabetes, and correlates with renal function and albuminuria [14]. This T cell accumulation has been noted in the juxtaglomerular apparatus of these patients but the functional role of T cells in this compartment is unclear.
It has been reported that the homing of Th1 cells in glomeruli is P-selectin and ICAM-1 dependent and associated with increased levels of IFN-γ and macrophage migration inhibitory factor (MIF) in crescentic Th1-mediated glomerulonephritis [15]. Although the mechanisms of Th1 cell migration in models of DN have not been reported yet, levels of ICAM-1 and P-selectin are increased within the diabetic kidney. As T cells constitutively express LFA-1, and ICAM-1 expression is found on renal endothelial, epithelial, and mesangial cells, it is likely that this interaction plays a significant role in T cell migration into the kidney. This is supported by a study showing that glomerular accumulation of CD4+ T cells was decreased in ICAM-1-deficient db/db mice [6].
Human and rat T cells also express receptors for AGEs. The activation of CD4+ and CD8+ T cells by AGEs can initiate INF-γ secretion by T cells, which will induce further inflammation and oxidative stress within renal tissues [16]. T cells also have the capacity to recruit and activate macrophages through Th1-driven INF-γ production. However, the functional role of T cells varies depending on the model studied. For example, α3 (IV) collagen/RAG1 double-knockout mice were not protected from the development of glomerulonephritis but were protected from interstitial fibrosis [17]. On the other hand, inhibition of regulatory T cells (Tregs) with anti-CD25 monoclonal antibody worsened renal injury in models of ischaemia-reperfusion [18].
To further explore the role of lymphocytes, we studied the kidney outcomes in recombination activating gene-1- (RAG1-) deficient mice made diabetic with STZ injections [19]. RAG1-deficient mice lack mature T and B cells and thus lymphocytes are not recruited into the diabetic kidney. RAG1-deficient mice were not protected from histological injury, renal fibrosis, or reduced creatinine clearance. However, RAG1−/− diabetic mice showed significant attenuation of albuminuria, associated with preservation of podocytes, and a reduction in glomerular macrophage activation. This would suggest that lymphocytes had a lesser role in inflammation but were involved in the pathogenesis of albuminuria. The glomeruli of RAG1−/− diabetic mice were also devoid of immunoglobulin staining, in contrast to their RAG1 intact counterparts. This led us to consider the possible role of B cells and immunoglobulin deposition as a potential contributor to glomerular inflammation and albuminuria (see next section).
One subset of T cells which has raised intense interest is the Forkhead box P3 (CD4+CD25+Foxp3+) regulatory T cell (or Treg). These cells may participate in dampening the inflammation in the diabetic kidney, given the worsening of renal injury when Tregs are inhibited in ischaemia-reperfusion models. STZ-diabetic mice have increased peripheral Treg in the circulation, spleen, and lymph nodes. However, these cells appear to be dysfunctional [20]. Tregs are also increased in the kidneys of STZ-diabetic mice [19]. When diabetic db/db mice were depleted of Tregs, they demonstrated enhanced kidney inflammation, leading to worsening albuminuria and glomerular hyperfiltration. Furthermore, adoptive transfer of Tregs significantly improved insulin sensitivity and reduced nephropathy in the db/db mice [21]. These findings suggest that the manipulation of Treg number or function may be useful in attenuating inflammation in DN.
2.3. Immune Deposition
A number of modified proteins which develop in diabetes are potentially immunogenic. This includes human immune responses to oxidized low-density lipoproteins (LDL), which may subsequently result in the formation of antioxidized LDL immune complexes [22, 23]. In patients with type 1 diabetes, these immune complexes are associated with macroalbuminuria [24]. Another study of type 1 diabetes demonstrated significant positive correlations between IgG antibody concentration isolated from circulating immune complexes and the serum creatinine and albumin excretion [25]. These antibodies were mostly of the proinflammatory IgG1 and IgG3 isotypes. IgG antibodies were proportionally greater than IgM by a ratio of 8 : 1. These immune complexes have been shown in vitro to stimulate production of MCP-1 and CSF-1 [26], and promote glomerular fibrosis by stimulating collagen production by mesangial cells [27]. Oxidized LDL immune complexes are also capable of activating the classical pathway of complement and inducing proinflammatory cytokine production by human macrophages, including IL-1, IL-6, and TNF-α [28]. These responses occur through the ligation of Fcγ receptors on mesangial cells and macrophages and may involve the activation of the p38 mitogen-activated protein kinase (p38 MAPK), JNK, and protein kinase C (PKC) pathways [27].
Circulating immune complexes and glomerular IgG deposition have long been recognized in diabetic rodent models [13, 29]. Similarly, immune deposits have also been described in histological studies of DN [30]. In an analysis of 567 kidney biopsies from patients with type 1 and 2 diabetes, approximately 30% of glomerular disease present was immune complex and secondary focal glomerulosclerosis [31]. This does raise a valid question as to the role of these immune complexes in the pathogenesis of DN. However, these immune deposition and immune complexes have usually been dismissed as concurrent or unrelated diseases. This is reflected by the stance of the Animal Models of Diabetic Complications Consortium (AMDCC) on excluding immune deposition in models of DN [32]. It may require a major paradigm shift before researchers are able to finally lay this issue to rest. Our study of RAG1-deficient mice suggests the possible involvement of immune deposition in promoting albuminuria. We are currently undertaking further studies in this area.
3. Inflammatory Cytokines
As our knowledge of DN expands, a number of inflammatory cytokines have emerged as being closely involved in the pathogenesis of DN. Some of the major inflammatory cytokines which are believed to play an important role in DN are discussed here (summarised in Table 1). The role of cytokines and growth factors in diabetic kidney disease has also been specifically reviewed elsewhere [33, 34].
Table 1.
Cytokines involved in Diabetic Kidney Inflammation.
| Cytokine | Role in Diabetic Kidney Inflammation |
|---|---|
| ICAM-1 | Adhesion molecule facilitating leukocyte-endothelial adhesion and infiltration into diabetic kidneys |
| VCAM-1 | Adhesion molecule facilitating leukocyte-endothelial adhesion and infiltration into diabetic kidneys |
| MCP-1 | Chemoattractant which promotes macrophage recruitment into diabetic kidneys |
| TNF-α | Promotes production of reactive oxygen species, induces cell injury, and increases endothelial permeability |
| IL-1 | Stimulates expression of cell adhesion molecules and profibrotic growth factors and increases endothelial permeability |
| IL-6 | Promotes mesangial proliferation, glomerular hypertrophy, fibronectin production and increases endothelial permeability |
| IL-18 | Increases production of other cytokines (ICAM-1, IL-1, TNF-α) and induces apoptosis of endothelial cells |
| Adiponectin | Reduces oxidative stress, production of TNF-α, and leukocyte-endothelial adhesion |
| Leptin | Induces oxidative stress, inflammation, hypertrophy, and proliferation of vascular smooth muscle cells, and impairs endothelial function |
| Resistin | Promotes expression of MCP-1, VCAM-1, endothelin-1, and proliferation of vascular smooth muscle cells |
3.1. TNF-α
TNF-α is mainly produced by monocytes, macrophages, and T cells. However, resident renal cells are also able to produce TNF-α, including mesangial, glomerular, endothelial, dendritic, and renal tubular cells [35–38]. TNF-α expression is increased in the kidneys of experimental diabetic rats [39]. The effects of TNF-α include promotion of local reactive oxygen species (ROS) generation [40–42], increasing albumin permeability [42], and the induction of cytotoxicity, apoptosis, and necrosis [43, 44]. TNF-α is implicated in the recruitment of monocyte-macrophages, reducing glomerular filtration rate (GFR) by haemodynamic changes [45–48], as well as altering endothelial permeability [49]. In line with experimental data, patients with type 2 diabetes have 3-4 times greater serum levels of TNF-α compared to nondiabetic patients, and these levels are higher in diabetic patients with microalbuminuria compared with those that have normoalbuminuria [50, 51]. Similarly, urinary TNF-α excretion correlates well with the clinical markers of DN and progression of disease [52].
3.2. MCP-1
MCP-1 promotes monocyte and macrophage migration and activation, upregulates expression of adhesion molecules, and promotes the expression of other proinflammatory cytokines [53, 54]. MCP-1 increases progressively in diabetic kidneys in animals models [2, 55]. It is produced by various cells in the kidney, including monocyte-macrophages, mesangial cells, podocytes, and tubular cells [10, 55, 56]. Recent studies have convincingly demonstrated the causative role of MCP-1 in experimental DN. In models of type 1 and 2 diabetes, renal injury was attenuated in MCP-1-deficient animals [9, 10]. Patients with type 2 diabetes and nephropathy excrete high levels of MCP-1 in the urine, which correlates with albuminuria and N-acetyl-β-D-glucosaminidase (NAG) excretion as a marker of tubular injury [7]. Interestingly, inhibition of ACE or the mineralocorticoid receptor also suppresses renal MCP-1 production [57, 58]. However, it remains to be determined if direct inhibition of MCP-1 will be more effective.
3.3. ICAM-1
ICAM-1 is a cell surface glycoprotein involved in leukocyte attachment to the endothelium as the ligand for LFA-1(integrin) on leukocytes. ICAM-1 is also present on the membranes of macrophages and lymphocytes. ICAM-1 expression is increased in models of type 1 and 2 diabetes [59, 60]. It can be induced by hyperglycaemia, AGEs, oxidative stress, hyperlipidaemia, hyperinsulinaemia, and proinflammatory cytokines [1]. Recent studies have demonstrated that mice deficient in ICAM-1 are protected against macrophage accumulation and nephropathy in models of type 1 and 2 diabetes [6, 61]. A soluble form of ICAM-1 has been described as increased in patients with type 2 diabetes and DN [62]. However, there is a lack of data from human studies on the role of ICAM-1 in DN.
3.4. VCAM-1
Vascular cell adhesion molecule-1 (VCAM-1) is another molecule involved in leukocyte-endothelial adhesion, which facilitates leukocyte recruitment into the kidney during inflammation. VCAM-1 expression is increased in the kidneys of patients with DN [63] and in diabetic rodents [64, 65]. During diabetes, VCAM-1 expression is detected on vascular endothelium and infiltrating cells in the kidney [64]. Increasing plasma levels of soluble VCAM-1 are associated with the progression of albuminuria in patients with type 1 and type 2 diabetes [66, 67], suggesting that sVCAM-1 may be a useful biomarker of diabetic renal injury. However, it is unclear whether circulating sVCAM-1 levels correlate with inflammation in diabetic kidneys.
3.5. Interleukin-1
Increased expression of IL-1 is found in experimental DN [68, 69]. IL-1 is able to enhance ICAM-1, (VCAM-1), and E-cadherin expression [70, 71]. Furthermore, IL-1 is able to induce endothelial cell permeability, alter glomerular haemodynamics by affecting prostaglandin synthesis, stimulate mesangial and fibroblast proliferation, and induce TGF-β1 production [72–74]. Polymorphisms of the IL-1 β and IL-1 receptor genes were found to be associated with an increased risk of end-stage kidney disease in Korean patients with type 2 diabetes [75]. However, polymorphisms in the IL-1 gene cluster were not found to contribute to the genetic susceptibility of DN in Caucasian patients with type 1 diabetes [76]. Further investigation is required to determine the importance of IL-1 in human DN.
3.6. Interleukin-6
IL-6 is produced by endothelial cells, leukocytes, adipocytes, and mesangial cells. Experimental studies have shown IL-6 overexpression in diabetic kidneys, which correlate with kidney hypertrophy and albumin excretion [68, 77]. IL-6 has been suggested to mediate endothelial permeability, mesangial proliferation, and increased fibronectin expression [78–81]. IL-6 is increased in patients with type 1 and 2 diabetes with DN, and IL-6 levels are higher in patients with overt proteinuria compared to microalbuminuria or normoalbuminuria [82, 83]. Increased IL-6 is also associated with GBM thickening in type 2 diabetes patients and mesangial expansion in kidney biopsies of diabetic patients [79, 84].
3.7. Interleukin-18
IL-18 is a potent inflammatory cytokine that induces IFN-γ [85] and the production of other proinflammatory cytokines (IL-1 and TNF-α), upregulation of ICAM-1, as well as apoptosis of endothelial cells [86–88]. Tubular epithelial cells are the major source of IL-18 but recent studies have also demonstrated IL-18 production from infiltrating monocyte-macrophages and T cells [89, 90]. Serum and urinary IL-18 are increased in type 2 diabetes patients and correlate with urinary albumin excretion [50, 91].
3.8. Adipokines
Adiponectin, leptin, and resistin are cytokines produced by adipose tissue. Adiponectin regulates insulin sensitivity, and also has anti-inflammatory and antioxidant properties. Adiponectin suppresses TNF-α-induced upregulation of endothelial cell adhesion molecules and interferes with leukocyte rolling and adhesion [92]. Adiponectin also suppresses leukocyte colony formation and reduces TNF-α secretion by macrophages [93, 94]. Adiponectin may also interfere with receptor activation for platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), and epidermal growth factor (EGF) [95]. In diabetic db/db mice, ezetimibe treatment normalized adiponectin levels and enhanced kidney expression of adiponectin receptor 1 [96]. With this, there was a 50% reduction in albuminuria and improved glomerular hypertrophy. STZ-induced diabetic rats overexpressing adiponectin showed improvements in markers of endothelial function, including lower levels of endothelin-1, plasminogen activator inhibitor-1, and inducible nitric oxide synthase [97]. This was associated with preservation of nephrin, lower TGF-β levels and a reduction in proteinuria. In human type 1 and 2 diabetes, serum adiponectin levels are already elevated and positively correlated with both albuminuria and serum creatinine [98–100]. Thus, it is unclear if further manipulation of adiponectin levels would be beneficial in humans.
In contrast to adiponectin, leptin exerts proinflammatory effects. Experimentally, these effects include stimulation of inflammatory signalling pathways and oxidative stress, impairment of endothelial function and platelet aggregation, and hypertrophy and proliferation of vascular smooth muscle cells [101]. The data in humans is less clear. In type 2 diabetes, both low and high serum leptin levels were risk factors for declining renal function. Furthermore, lower serum leptin levels were associated with progression of albuminuria [102]. Another study noted micro- and macroalbuminuria patients with type 2 diabetes had higher leptin levels than normoalbuminuric patients [103]. However, leptin levels may also be elevated in end-stage kidney disease due to reduced degradation.
Resistin promotes expression of endothelin-1, VCAM-1, and MCP-1. Resistin also promotes proliferation of vascular smooth muscle cells via extracellular regulated kinase (ERK) and Akt signalling pathways and inhibits insulin signalling and endothelial nitric oxide synthase (eNOS) activation [101]. The role of resistin in human DN is unclear. In type 2 diabetes patients, resistin was elevated in patients receiving loop diuretics but not thiazides. Serum resistin levels also increase in advanced CKD [104].
4. Stress-Activated Protein Kinases (SAPKs)
p38 MAPK and JNK are stress-activated protein kinases (SAPKs). Receptor activation by various stress stimuli on the cell surface trigger intracellular signalling involving a cascade of phosphorylation by MAP kinase kinase kinase (MAP3K), a MAP kinase kinase (MAP2K), and finally the MAPK/SAPK [105]. There is overlap and redundancy in the ability of different MAP3K's to activate p38 MAPK or JNK. However, direct activation of p38 MAPK and JNK can only occur through MKK3/6 and MKK4/7, respectively.
4.1. p38 MAPK
p38 MAPK has four isoforms (p38α, β, γ, and δ) which are all expressed by kidney cells. However, activation of p38α is most strongly associated with renal inflammation and injury. Recent clinical studies have demonstrated that kidney p38 MAPK activity is increased and associated with the development of DN [106, 107]. Renal biopsies from patients with established type 2 diabetes display prominent glomerular and tubulointerstitial p38 MAPK signalling despite treatment with angiotensin inhibitors [106]. In diabetic animal models, p38 MAPK activity rapidly increases in glomeruli and tubules after the induction of hyperglycaemia, and is also found in the accumulating kidney interstitial cells associated with advanced nephropathy. Studies of nondiabetic kidney disease have shown that pharmacological inhibition of p38 MAPK suppressed inflammation and fibrosis [108, 109].
In vitro studies have identified specific kidney cells and mechanisms of renal injury that may be affected by p38 MAPK signalling during diabetes. Exposure to high glucose activates p38 MAPK in human mesangial cells [110], mouse podocytes [111], and rat proximal tubular cells [112]. Similarly, glycated albumin can stimulate p38 MAPK phosphorylation in cultured fibroblasts [113]. Activation of p38 MAPK has been shown to promote apoptosis of rat mesangial cells exposed to methylglyoxal [114] and apoptosis of mouse podocytes following stimulation with TGF-β [115] and AGEs [116, 117]. In addition, p38 MAPK signalling can contribute to proinflammatory and profibrotic responses. p38 MAPK activation enhances production of MCP-1 by vascular endothelial cells [118], induces local angiotensinogen production in rat tubular cells [112], stimulates TGF-β-induced fibronectin accumulation in renal interstitial fibroblasts [119] and collagen production in mouse mesangial cells [120], increases TGF-β expression in renal tubular cells [121], and promotes synthesis of vascular endothelial growth factor (VEGF) induced by angiotensin II [122, 123]. Studies have also shown that p38 MAPK signalling mediates high-glucose-induced tubular hypertrophy [121] and transactivation of the epidermal growth factor receptor required for dedifferentiation of proximal tubular epithelial cells following oxidant injury [124].
Functional blocking studies are required to determine the pathogenic role of p38 MAPK signalling in diabetes and its complications. Inhibitors of p38α and p38β have been used in a number of nondiabetic models to reduce proteinuria and inflammation [105]. In rats with STZ-induced diabetes, a p38 MAPK inhibitor (FR167653) ameliorated the increased glomerular fibronectin mRNA and protein and reduced mesangial cell apoptosis. Similar results were obtained in high-glucose stimulated cultured rat mesangial cells [125]. However, the investigators did not study the effects of p38 MAPK inhibition on kidney inflammatory cells, albumin excretion, tubulointerstitial injury, or renal function.
We utilized a genetic approach to studying the p38 MAPK pathway in DN. As genetic deletion of p38α is lethal, our strategy was to target the immediate upstream kinases that regulate p38 MAPK signalling. The MKK3 and MKK6 kinases provide a parallel and independent mechanism of phosphorylating p38 MAPK but their relative contribution to the increased p38 MAPK activity associated with DN was unknown. MKK3 appeared to be the most attractive target because MKK3-p38 MAPK signalling has been shown to be nonredundant in some pathological processes in vitro [126, 127]. We studied the effects of MKK3-p38 MAPK inhibition on kidney outcomes in Mkk3-gene-deficient diabetic db/db mice [128]. In the absence of MKK3 signalling, we noted an attenuation of Ccl2 expression (hence MCP-1 levels) and interstitial macrophage accumulation. The result was a reduction in albuminuria with concomitant podocyte preservation and reduced mesangial cell activation. Glomerular sclerosis and tubulointerstitial injury were also attenuated. This study demonstrated a pathogenic role of the MKK3-p38 signalling pathway in the progression of DN and may be a viable target for intervention. There are also nonredundant functions of the upstream kinases which confirm previous in vitro findings.
4.2. JNK
There are three main JNK isoforms. JNK1 and JNK2 are expressed in the kidney but JNK3 is limited to the nervous system [129]. Phosphorylated JNK translocates into the nucleus and activates transcription factors and cellular responses such as inflammation or apoptosis. For example, in vitro studies have demonstrated that inhibition of JNK ameliorates the induction of apoptosis by oxidative stress in tubular epithelial cells [130]. A unique JNK target is the phosphorylation of Ser63 and Ser73 in the NH2-terminal domain of c-Jun, which can be used as a surrogate marker of JNK activity [131]. JNK-dependent signalling is important in normal development because genetic deficiency of both JNK1 and JNK2 are foetal lethal. JNK can be activated by various elements of the diabetic milieu, including hyperglycaemia, AGEs, angiotensin II, ROS, and proinflammatory cytokines (IL-1, TNF-α) [132].
JNK may be important in CSF-1 signalling through c-fms, thus promoting monocyte-macrophage differentiation, development, survival, and function [133, 134]. In animal models of anti-GBM disease and unilateral ureteric obstruction, treatment with the JNK inhibitor CC-401 reduced renal injury through modulation of macrophage activation [135, 136]. In biopsy samples of human DN, JNK activation correlated with interstitial macrophage accumulation, kidney injury molecule-1 (KIM-1) expression, interstitial fibrosis, and loss of renal function [137, 138].
We examined the effects of JNK blockade in a STZ model of diabetes in spontaneously hypertensive rats by administering a JNK inhibitor (CC-930) at the onset of detectable kidney JNK activation (phosphorylated-JNK) and albuminuria [139]. JNK inhibition resulted in a reduction in macrophage accumulation and Ccl2 mRNA (encoding MCP-1). However, JNK inhibition exacerbated albuminuria in association with accelerated loss of glomerular nephrin and podocin. Similar negative outcomes were reported in db/db mice treated with a TAT-JNK inhibitor peptide, which exacerbated albuminuria and nephrin loss despite improvements in insulin sensitivity [140]. Together, these studies demonstrate that blockade of JNK signalling causes significant injury to podocytes while also suppressing kidney inflammation in animal models of early DN. Whether the benefits of JNK inhibition outweigh its effects on podocyte damage in more advanced stages of diabetic renal injury remains to be determined.
4.3. Kidney Inflammation in Type 1 versus Type 2 Diabetic Nephropathy
Analysis of renal biopsies from type 1 and type 2 diabetic patients who develop DN indicates that the inflammatory infiltrate is similar in both groups [5], which is consistent with studies in diabetic animal models [2, 3]. In both type 1 and type 2 models, kidney inflammation in diabetic rodents correlates strongly with the development of hyperglycaemia and glycated haemoglobin, and is driven by an increased kidney production of chemokines and proinflammatory cytokines [2, 3], and induction of kidney SAPK signalling [106]. In addition, the coexistence of hypertension or hyperlipidaemia exacerbates kidney inflammation in both type 1 and type 2 diabetes [137, 141–144].
5. Conclusion
Inflammation plays an essential role in the progression of DN. Recent evidence indicates that innate immunity, rather than adaptive immunity, is the major driving factor in the inflammatory response in diabetic kidneys. The main components of this immune response (infiltrating cell types, cytokines, signalling pathways) are described in this paper (summarized in Figure 1). Our current knowledge indicates that elements of the diabetic milieu (hyperglycaemia, AGEs, immune complexes) can activate kidney cells via induction of SAPK signalling, resulting in the release of chemokines and upregulation of cell adhesion molecules. These events facilitate the kidney infiltration of monocytes and lymphocytes, which become activated in the diabetic kidney and secrete injurious molecules, such as proinflammatory cytokines and reactive oxygen species. This leukocyte activity amplifies the inflammatory response and promotes cell injury and the development of fibrosis. Better understanding of the inflammatory response in diabetic kidneys is expected to identify novel anti-inflammatory strategies for the potential treatment of human DN.
Figure 1.
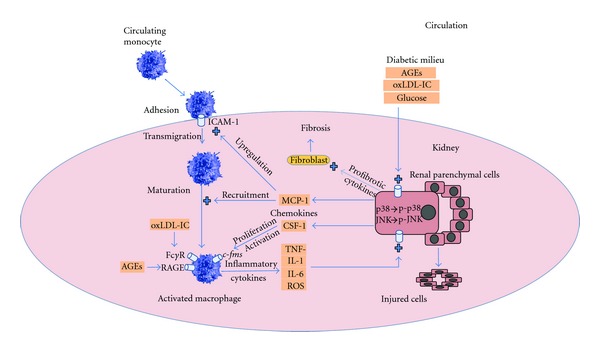
The inflammatory amplification loop in the diabetic kidney. Circulating immune cells such as monocytes are recruited into the diabetic kidney due to upregulation of adhesion molecules such as ICAM-1. Chemokines such as MCP-1 act as chemoattractants which promote accumulation of the immune cells in the kidney. These immune cells are activated by numerous signals such as the ligation of c-fms by CSF-1, receptor for AGE by AGEs, and the Fcγ receptors by antioxidized LDL immune complexes. CSF-1 also promotes the maturation, proliferation, and survival of monocyte-macrophages. Activated immune cells act as inflammatory cells and elaborate proinflammatory cytokines and reactive oxygen species (ROS), which trigger a cell signalling cascade mediated by the stress-activated protein kinases, p38 MAPK, and JNK. These kidney cells then respond by the production of chemokines such as MCP-1 and CSF-1, and profibrotic factors such as TGF-β which increase extracellular matrix production by mesangial cells and interstitial fibroblasts. Ultimately, there is cellular injury and progressive fibrosis within the diabetic kidney.
Abbreviations
- AGEs:
Advanced glycation end products
- CSF-1:
Colony stimulating factor-1
- DN:
Diabetic nephropathy
- ERK:
Extracellular cell regulated kinase
- GBM:
Glomerular basement membrane
- ICAM-1:
Intercellular cell adhesion molecule-1
- IL:
Interleukin
- INF-α:
Interferon-α
- JNK:
c-Jun amino-terminal kinase
- MAPK:
Mitogen-activated protein kinase
- MCP-1:
Monocyte chemoattractant protein-1
- SAPK:
Stress-activated protein kinase
- STZ:
Streptozotocin
- TGF-β:
Transforming growth factor-β
- TNF-α:
Tumour necrosis factor-α
- VCAM-1:
Vascular cell adhesion molecule-1.
References
- 1.Galkina E, Ley K. Leukocyte recruitment and vascular injury in diabetic nephropathy. Journal of the American Society of Nephrology. 2006;17(2):368–377. doi: 10.1681/ASN.2005080859. [DOI] [PubMed] [Google Scholar]
- 2.Chow F, Ozols E, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney International. 2004;65(1):116–128. doi: 10.1111/j.1523-1755.2004.00367.x. [DOI] [PubMed] [Google Scholar]
- 3.Chow FY, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in streptozotocin-induced diabetic nephropathy: potential role in renal fibrosis. Nephrology Dialysis Transplantation. 2004;19(12):2987–2996. doi: 10.1093/ndt/gfh441. [DOI] [PubMed] [Google Scholar]
- 4.Furuta T, Saito T, Ootaka T, et al. The role of macrophages in diabetic glomerulosclerosis. American Journal of Kidney Diseases. 1993;21(5):480–485. doi: 10.1016/s0272-6386(12)80393-3. [DOI] [PubMed] [Google Scholar]
- 5.Nguyen D, Ping F, Mu W, Hill P, Atkins RC, Chadban SJ. Macrophage accumulation in human progressive diabetic nephropathy. Nephrology. 2006;11(3):226–231. doi: 10.1111/j.1440-1797.2006.00576.x. [DOI] [PubMed] [Google Scholar]
- 6.Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Tesch GH. Intercellular adhesion molecule-1 deficiency is protective against nephropathy in type 2 diabetic db/db mice. Journal of the American Society of Nephrology. 2005;16(6):1711–1722. doi: 10.1681/ASN.2004070612. [DOI] [PubMed] [Google Scholar]
- 7.Morii T, Fujita H, Narita T, et al. Association of monocyte chemoattractant protein-1 with renal tubular damage in diabetic nephropathy. Journal of Diabetes and its Complications. 2003;17(1):11–15. doi: 10.1016/s1056-8727(02)00176-9. [DOI] [PubMed] [Google Scholar]
- 8.Kanamori H, Matsubara T, Mima A, et al. Inhibition of MCP-1/CCR2 pathway ameliorates the development of diabetic nephropathy. Biochemical and Biophysical Research Communications. 2007;360(4):772–777. doi: 10.1016/j.bbrc.2007.06.148. [DOI] [PubMed] [Google Scholar]
- 9.Chow FY, Nikolic-Paterson DJ, Ma FY, Ozols E, Rollins BJ, Tesch GH. Monocyte chemoattractant protein-1-induced tissue inflammation is critical for the development of renal injury but not type 2 diabetes in obese db/db mice. Diabetologia. 2007;50(2):471–480. doi: 10.1007/s00125-006-0497-8. [DOI] [PubMed] [Google Scholar]
- 10.Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Rollin BJ, Tesch GH. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney International. 2006;69(1):73–80. doi: 10.1038/sj.ki.5000014. [DOI] [PubMed] [Google Scholar]
- 11.Lim AKH, Ma FY, Nikolic-Paterson DJ, Thomas MC, Hurst LA, Tesch GH. Antibody blockade of c-fms suppresses the progression of inflammation and injury in early diabetic nephropathy in obese db/db mice. Diabetologia. 2009;52(8):1669–1679. doi: 10.1007/s00125-009-1399-3. [DOI] [PubMed] [Google Scholar]
- 12.Tipping PG, Holdsworth SR. T cells in crescentic glomerulonephritis. Journal of the American Society of Nephrology. 2006;17(5):1253–1263. doi: 10.1681/ASN.2005091013. [DOI] [PubMed] [Google Scholar]
- 13.Xiao X, Ma B, Dong B, et al. Cellular and humoral immune responses in the early stages of diabetic nephropathy in NOD mice. Journal of Autoimmunity. 2009;32(2):85–93. doi: 10.1016/j.jaut.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 14.Moriya R, Manivel JC, Mauer M. Juxtaglomerular apparatus T-cell infiltration affects glomerular structure in Type 1 diabetic patients. Diabetologia. 2004;47(1):82–88. doi: 10.1007/s00125-003-1253-y. [DOI] [PubMed] [Google Scholar]
- 15.Odobasic D, Kitching AR, Semple TJ, Timoshanko JR, Tipping PG, Holdsworth SR. Glomerular expression of CD80 and CD86 is required for leukocyte accumulation and injury in crescentic glomerulonephritis. Journal of the American Society of Nephrology. 2005;16(7):2012–2022. doi: 10.1681/ASN.2004060437. [DOI] [PubMed] [Google Scholar]
- 16.Imani F, Horii Y, Suthanthiran M, et al. Advanced glycosylation endproduct-specific receptors on human and rat T- lymphocytes mediate synthesis of interferon γ: role in tissue remodeling. Journal of Experimental Medicine. 1993;178(6):2165–2172. doi: 10.1084/jem.178.6.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.LeBleu VS, Sugimoto H, Miller CA, Gattone VH, Kalluri R. Lymphocytes are dispensable for glomerulonephritis but required for renal interstitial fibrosis in matrix defect-induced Alport renal disease. Laboratory Investigation. 2008;88(3):284–292. doi: 10.1038/labinvest.3700715. [DOI] [PubMed] [Google Scholar]
- 18.Kinsey GR, Sharma R, Huang L, et al. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. Journal of the American Society of Nephrology. 2009;20(8):1744–1753. doi: 10.1681/ASN.2008111160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim AKH, Ma FY, Nikolic-Paterson DJ, Kitching AR, Thomas MC, Tesch GH. Lymphocytes promote albuminuria, but not renal dysfunction or histological damage in a mouse model of diabetic renal injury. Diabetologia. 2010;53(8):1772–1782. doi: 10.1007/s00125-010-1757-1. [DOI] [PubMed] [Google Scholar]
- 20.Zhen Y, Sun L, Liu H, et al. Alterations of peripheral CD4 + CD25 + Foxp3 + T regulatory cells in mice with STZ-induced diabetes. Cellular and Molecular Immunology. 2012;9(1):75–85. doi: 10.1038/cmi.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eller K, Kirsch A, Wolf AM, et al. Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes. 2011;60(11):2954–2962. doi: 10.2337/db11-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mironova M, Virella G, Lopes-Virella MF. Isolation and characterization of human antioxidized LDL autoantibodies. Arteriosclerosis, Thrombosis, and Vascular Biology. 1996;16(2):222–229. doi: 10.1161/01.atv.16.2.222. [DOI] [PubMed] [Google Scholar]
- 23.Lopes-Virella MF, Virella G. The role of immune and inflammatory processes in the development of macrovascular disease in diabetes. Frontiers in Bioscience. 2003;8:s750–s768. doi: 10.2741/1141. [DOI] [PubMed] [Google Scholar]
- 24.Atchley D, Lopes-Virella M, Zheng D, Kenny D, Virella G. Oxidized LDL-anti-oxidized LDL immune complexes and diabetic nephropathy. Diabetologia. 2002;45(11):1562–1571. doi: 10.1007/s00125-002-0962-y. [DOI] [PubMed] [Google Scholar]
- 25.Virella G, Carter RE, Saad A, Crosswell EG, Game BA, Lopes-Virella MF. Distribution of IgM and IgG antibodies to oxidized LDL in immune complexes isolated from patients with type 1 diabetes and its relationship with nephropathy. Clinical Immunology. 2008;127(3):394–400. doi: 10.1016/j.clim.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hora K, Satriano JA, Santiago A, et al. Receptors for IgG complexes activate synthesis of monocyte chemoattractant peptide 1 and colony-stimulating factor 1. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(5):1745–1749. doi: 10.1073/pnas.89.5.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdelsamie SA, Li Y, Huang Y, et al. Oxidized LDL immune complexes stimulate collagen IV production in mesangial cells via Fc gamma receptors I and III. Clinical Immunology. 2011;139(3):258–266. doi: 10.1016/j.clim.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saad AF, Virella G, Chassereau C, Boackle RJ, Lopes-Virella MF. OxLDL immune complexes activate complement and induce cytokine production by MonoMac 6 cells and human macrophages. Journal of Lipid Research. 2006;47(9):1975–1983. doi: 10.1194/jlr.M600064-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Abrass CK. Evaluation of the presence of circulating immune complexes and their relationship to glomerular IgG deposits in streptozotocin-induced diabetic rats. Clinical and Experimental Immunology. 1984;57(1):17–24. [PMC free article] [PubMed] [Google Scholar]
- 30.Ainsworth SK, Hirsch HZ, Brackett NC. Diabetic glomerulonephropathy: histopathologic, immunofluorescent, and ultrastructural studies of 16 cases. Human Pathology. 1982;13(5):470–478. doi: 10.1016/s0046-8177(82)80030-0. [DOI] [PubMed] [Google Scholar]
- 31.Haider DG, Peric S, Friedl A, et al. Kidney biopsy in patients with diabetes mellitus. Clinical Nephrology. 2011;76(3):180–185. doi: 10.5414/cn106955. [DOI] [PubMed] [Google Scholar]
- 32.Brosius FC, Alpers CE, Bottinger EP, et al. Mouse models of diabetic nephropathy. Journal of the American Society of Nephrology. 2009;20(12):2503–2512. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu C-C, Sytwu H-K, Lin Y-F. Cytokines in diabetic nephropathy. Advances in Clinical Chemistry. 2012;56:55–74. doi: 10.1016/b978-0-12-394317-0.00014-5. [DOI] [PubMed] [Google Scholar]
- 34.Chiarelli F, Gaspari S, Marcovecchio ML. Role of growth factors in diabetic kidney disease. Hormone and Metabolic Research. 2009;41(8):585–593. doi: 10.1055/s-0029-1220752. [DOI] [PubMed] [Google Scholar]
- 35.Jevnikar AM, Brennan DC, Singer GG, et al. Stimulated kidney tubular epithelial cells express membrane associated and secreted TNFα . Kidney International. 1991;40(2):203–211. doi: 10.1038/ki.1991.201. [DOI] [PubMed] [Google Scholar]
- 36.Nakamura T, Fukui M, Ebihara I, et al. mRNA expression of growth factors in glomeruli from diabetic rats. Diabetes. 1993;42(3):450–456. doi: 10.2337/diab.42.3.450. [DOI] [PubMed] [Google Scholar]
- 37.Sugimoto H, Shikata K, Wada J, Horiuchi S, Makino H. Advanced glycation end products-cytokine-nitric oxide sequence pathway in the development of diabetic nephropathy: aminoguanidine ameliorates the overexpression of tumour necrosis factor-α and inducible nitric oxide synthase in diabetic rat glomeruli. Diabetologia. 1999;42(7):878–886. doi: 10.1007/s001250051241. [DOI] [PubMed] [Google Scholar]
- 38.Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney International. 2007;71(7):619–628. doi: 10.1038/sj.ki.5002132. [DOI] [PubMed] [Google Scholar]
- 39.Mensah-Brown EPK, Obineche EN, Galadari S, et al. Streptozotocin-induced diabetic nephropathy in rats: the role of inflammatory cytokines. Cytokine. 2005;31(3):180–190. doi: 10.1016/j.cyto.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 40.Radeke HH, Meier B, Topley N, Floge J, Habermehl GG, Resch K. Interleukin 1-α and tumor necrosis factor-α induce oxygen radical production in mesangial cells. Kidney International. 1990;37(2):767–775. doi: 10.1038/ki.1990.44. [DOI] [PubMed] [Google Scholar]
- 41.Koike N, Takamura T, Kaneko S. Induction of reactive oxygen species from isolated rat glomeruli by protein kinase C activation and TNF-α stimulation, and effects of a phosphodiesterase inhibitor. Life Sciences. 2007;80(18):1721–1728. doi: 10.1016/j.lfs.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 42.Mccarthy ET, Sharma R, Sharma M, et al. TNF-α increases albumin permeability of isolated rat glomeruli through the generation of superoxide. Journal of the American Society of Nephrology. 1998;9(3):433–438. doi: 10.1681/ASN.V93433. [DOI] [PubMed] [Google Scholar]
- 43.Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. Journal of Immunology. 1988;141(8):2629–2634. [PubMed] [Google Scholar]
- 44.Boyle JJ, Weissberg PL, Bennett MR. Tumor necrosis factor-α promotes macrophage-induced vascular smooth muscle cell apoptosis by direct and autocrine mechanisms. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(9):1553–1558. doi: 10.1161/01.ATV.0000086961.44581.B7. [DOI] [PubMed] [Google Scholar]
- 45.Baud L, Ardaillou R. Tumor necrosis factor in renal injury. Mineral and Electrolyte Metabolism. 1995;21(4-5):336–341. [PubMed] [Google Scholar]
- 46.Gomez-Chiarri M, Ortiz A, Lerma JL, et al. Involvement of tumor necrosis factor and platelet-activating factor in the pathogenesis of experimental nephrosis in rats. Laboratory Investigation. 1994;70(4):449–459. [PubMed] [Google Scholar]
- 47.Marsden PA, Brenner BM. Transcriptional regulation of the endothelin-1 gene by TNF-α . American Journal of Physiology, Cell Physiology. 1992;262(4):C854–C861. doi: 10.1152/ajpcell.1992.262.4.C854. [DOI] [PubMed] [Google Scholar]
- 48.Marsden PA, Ballermann BJ. Tumor necrosis factor α activates soluble guanylate cyclase in bovine glomerular mesangial cells via an L-arginine-dependent mechanism. Journal of Experimental Medicine. 1990;172(6):1843–1852. doi: 10.1084/jem.172.6.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wójciak-Stothard B, Entwistle A, Garg R, Ridley AJ. Regulation of TNF-α-induced reorganization of the actin cytoskeleton and cell-cell junctions by Rho, Rac, and Cdc42 in human endothelial cells. Journal of Cellular Physiology. 1998;176(1):150–165. doi: 10.1002/(SICI)1097-4652(199807)176:1<150::AID-JCP17>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 50.Moriwaki Y, Yamamoto T, Shibutani Y, et al. Elevated levels of interleukin-18 and tumor necrosis factor-α in serum of patients with type 2 diabetes mellitus: relationship with diabetic nephropathy. Metabolism: Clinical and Experimental. 2003;52(5):605–608. doi: 10.1053/meta.2003.50096. [DOI] [PubMed] [Google Scholar]
- 51.Navarro JF, Mora C, Macía M, García J. Inflammatory parameters are independently associated with urinary albumin in type 2 diabetes mellitus. American Journal of Kidney Diseases. 2003;42(1):53–61. doi: 10.1016/s0272-6386(03)00408-6. [DOI] [PubMed] [Google Scholar]
- 52.Navarro JF, Mora C, Muros M, García J. Urinary tumour necrosis factor-α excretion independently correlates with clinical markers of glomerular and tubulointerstitial injury in type 2 diabetic patients. Nephrology Dialysis Transplantation. 2006;21(12):3428–3434. doi: 10.1093/ndt/gfl469. [DOI] [PubMed] [Google Scholar]
- 53.Gu L, Tseng SC, Rollins BJ. Monocyte chemoattractant protein-1. Chemical Immunology. 1999;72:7–29. doi: 10.1159/000058723. [DOI] [PubMed] [Google Scholar]
- 54.Viedt C, Orth SR. Monocyte chemoattractant protein-1 (MCP-1) in the kidney: does it more than simply attract monocytes? Nephrology Dialysis Transplantation. 2002;17(12):2043–2047. doi: 10.1093/ndt/17.12.2043. [DOI] [PubMed] [Google Scholar]
- 55.Kato S, Luyckx VA, Ots M, et al. Renin-angiotensin blockade lowers MCP-1 expression in diabetic rats. Kidney International. 1999;56(3):1037–1048. doi: 10.1046/j.1523-1755.1999.00643.x. [DOI] [PubMed] [Google Scholar]
- 56.Gruden G, Setti G, Hayward A, et al. Mechanical stretch induces monocyte chemoattractant activity via an NF-κB-dependent monocyte chemoattractant protein-1-mediated pathway in human mesangial cells: inhibition by rosiglitazone. Journal of the American Society of Nephrology. 2005;16(3):688–696. doi: 10.1681/ASN.2004030251. [DOI] [PubMed] [Google Scholar]
- 57.Amann B, Tinzmann R, Angelkort B. ACE inhibitors improve diabetic nephropathy through suppression of renal MCP-1. Diabetes Care. 2003;26(8):2421–2425. doi: 10.2337/diacare.26.8.2421. [DOI] [PubMed] [Google Scholar]
- 58.Han SY, Kim CH, Kim HS, et al. Spironolactone prevents diabetic nephropathy through an anti-inflammatory mechanism in type 2 diabetic rats. Journal of the American Society of Nephrology. 2006;17(5):1362–1372. doi: 10.1681/ASN.2005111196. [DOI] [PubMed] [Google Scholar]
- 59.Sugimoto H, Shikata K, Hirata K, et al. Increased expression of intercellular adhesion molecule-1 (ICAM-1) in diabetic rat glomeruli: glomerular hyperfiltration is a potential mechanism of ICAM-1 upregulation. Diabetes. 1997;46(12):2075–2081. doi: 10.2337/diab.46.12.2075. [DOI] [PubMed] [Google Scholar]
- 60.Matsui H, Suzuki M, Tsukuda R, Iida K, Miyasaka M, Ikeda H. Expression of ICAM-1 on glomeruli is associated with progression of diabetic nephropathy in a genetically obese diabetic rat, Wistar fatty. Diabetes Research and Clinical Practice. 1996;32(1-2):1–9. doi: 10.1016/0168-8227(96)01209-0. [DOI] [PubMed] [Google Scholar]
- 61.Okada S, Shikata K, Matsuda M, et al. Intercellular adhesion molecule-1-deficient mice are resistant against renal injury after induction of diabetes. Diabetes. 2003;52(10):2586–2593. doi: 10.2337/diabetes.52.10.2586. [DOI] [PubMed] [Google Scholar]
- 62.Güler S, Cakir B, Demirbas B, et al. Plasma soluble intercellular adhesion molecule 1 levels are increased in type 2 diabetic patients with nephropathy. Hormone Research. 2002;58(2):67–70. doi: 10.1159/000064664. [DOI] [PubMed] [Google Scholar]
- 63.Seron D, Cameron JS, Haskard DO. Expression of VCAM-1 in the normal and diseased kidney. Nephrology Dialysis Transplantation. 1991;6(12):917–922. doi: 10.1093/ndt/6.12.917. [DOI] [PubMed] [Google Scholar]
- 64.Ina K, Kitamura H, Okeda T, et al. Vascular cell adhesion molecule-1 expression in the renal interstitium of diabetic KKAy mice. Diabetes Research and Clinical Practice. 1999;44(1):1–8. doi: 10.1016/s0168-8227(99)00011-x. [DOI] [PubMed] [Google Scholar]
- 65.Wang F, Li M, Cheng L, et al. Intervention with cilostazol attenuates renal inflammation in streptozotocin-induced diabetic rats. Life Sciences. 2008;83(25-26):828–835. doi: 10.1016/j.lfs.2008.09.027. [DOI] [PubMed] [Google Scholar]
- 66.Clausen P, Jacobsen P, Rossing K, Jensen JS, Parving HH, Feldt-Rasmussen B. Plasma concentrations of VCAM-1 and ICAM-1 are elevated in patients with Type 1 diabetes mellitus with microalbuminuria and overt nephropathy. Diabetic Medicine. 2000;17(9):644–649. doi: 10.1046/j.1464-5491.2000.00347.x. [DOI] [PubMed] [Google Scholar]
- 67.Rubio-Guerra AF, Vargas-Robles H, Lozano Nuevo JJ, Escalante-Acosta BA. Correlation between circulating adhesion molecule levels and albuminuria in type-2 diabetic hypertensive patients. Kidney and Blood Pressure Research. 2009;32(2):106–109. doi: 10.1159/000210554. [DOI] [PubMed] [Google Scholar]
- 68.Navarro JF, Milena FJ, Mora C, León C, García J. Renal pro-inflammatory cytokine gene expression in diabetic nephropathy: effect of angiotensin-converting enzyme inhibition and pentoxifylline administration. American Journal of Nephrology. 2007;26(6):562–570. doi: 10.1159/000098004. [DOI] [PubMed] [Google Scholar]
- 69.Sassy-Prigent C, Heudes D, Mandet C, et al. Early glomerular macrophage recruitment in streptozotocin-induced diabetic rats. Diabetes. 2000;49(3):466–475. doi: 10.2337/diabetes.49.3.466. [DOI] [PubMed] [Google Scholar]
- 70.Brady HR. Leukocyte adhesion molecules and kidney diseases. Kidney International. 1994;45(5):1285–1300. doi: 10.1038/ki.1994.169. [DOI] [PubMed] [Google Scholar]
- 71.Park CW, Kim JH, Lee JW, et al. High glucose-induced intercellular adhesion molecule-1 (ICAM-1) expression through an osmotic effect in rat mesangial cells is PKC-NF-κB-dependent. Diabetologia. 2000;43(12):1544–1553. doi: 10.1007/s001250051567. [DOI] [PubMed] [Google Scholar]
- 72.Pfeilschifter J, Pignat W, Vosbeck K, Marki F. Interleukin 1 and tumor necrosis factor synergistically stimulate prostaglandin synthesis and phospholipase A2 release from rat renal mesangial cells. Biochemical and Biophysical Research Communications. 1989;159(2):385–394. doi: 10.1016/0006-291x(89)90003-x. [DOI] [PubMed] [Google Scholar]
- 73.Royall JA, Berkow RL, Beckman JS, Cunningham MK, Matalon S, Freeman BA. Tumor necrosis factor and interleukin 1α increase vascular endothelial permeability. American Journal of Physiology, Lung Cellular and Molecular Physiology. 1989;257(6):L399–L410. doi: 10.1152/ajplung.1989.257.6.L399. [DOI] [PubMed] [Google Scholar]
- 74.Vesey DA, Cheung C, Cuttle L, Endre Z, Gobe G, Johnson DW. Interleukin-1 β stimulates human renal fibroblast proliferation and matrix protein production by means of a transforming growth factor-β-dependent mechanism. Journal of Laboratory and Clinical Medicine. 2002;140(5):342–350. doi: 10.1067/mlc.2002.128468. [DOI] [PubMed] [Google Scholar]
- 75.Lee SH, Ihm CG, Sohn SD, et al. Polymorphisms in interleukin-1β and interleukin-1 receptor antagonist genes are associated with kidney failure in Korean patients with type 2 diabetes mellitus. American Journal of Nephrology. 2004;24(4):410–414. doi: 10.1159/000080044. [DOI] [PubMed] [Google Scholar]
- 76.Tarnow L, Pociot F, Hansen PM, et al. Polymorphisms in the interleukin-1 gene cluster do not contribute to the genetic susceptibility of diabetic nephropathy in Caucasian patients with IDDM. Diabetes. 1997;46(6):1075–1076. doi: 10.2337/diab.46.6.1075. [DOI] [PubMed] [Google Scholar]
- 77.Thomson SC, Deng A, Bao D, Satriano J, Blantz RC, Vallon V. Ornithine decarboxylase, kidney size, and the tubular hypothesis of glomerular hyperfiltration in experimental diabetes. Journal of Clinical Investigation. 2001;107(2):217–224. doi: 10.1172/JCI10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Coleman DL, Ruef C. Interleukin-6: an autocrine regulator of mesangial cell growth. Kidney International. 1992;41(3):604–606. doi: 10.1038/ki.1992.91. [DOI] [PubMed] [Google Scholar]
- 79.Suzuki D, Miyazaki M, Naka R, et al. In situ hybridization of interleukin 6 in diabetic nephropathy. Diabetes. 1995;44(10):1233–1238. doi: 10.2337/diab.44.10.1233. [DOI] [PubMed] [Google Scholar]
- 80.Hirano T, Akira S, Taga T, Kishimoto T. Biological and clinical aspects of interleukin 6. Immunology Today. 1990;11(12):443–449. doi: 10.1016/0167-5699(90)90173-7. [DOI] [PubMed] [Google Scholar]
- 81.Ruef C, Budde K, Lacy J, et al. Interleukin 6 is an autocrine growth factor for mesangial cells. Kidney International. 1990;38(2):249–257. doi: 10.1038/ki.1990.193. [DOI] [PubMed] [Google Scholar]
- 82.Aso Y, Yoshida N, Okumura KI, et al. Coagulation and inflammation in overt diabetic nephropathy: association with hyperhomocysteinemia. Clinica Chimica Acta. 2004;348(1-2):139–145. doi: 10.1016/j.cccn.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 83.Saraheimo M, Teppo AM, Forsblom C, Fagerudd J, Groop PH. Diabetic nephropathy is associated with low-grade inflammation in Type 1 diabetic patients. Diabetologia. 2003;46(10):1402–1407. doi: 10.1007/s00125-003-1194-5. [DOI] [PubMed] [Google Scholar]
- 84.Dalla Vestra M, Mussap M, Gallina P, et al. Acute-phase markers of inflammation and glomerular structure in patients with type 2 diabetes. Journal of the American Society of Nephrology. 2005;16(3) supplement 1:S78–S82. doi: 10.1681/asn.2004110961. [DOI] [PubMed] [Google Scholar]
- 85.Okamura H, Tsutsui H, Komatsu T, et al. Cloning of a new cytokine that induces IFN-γ production by T cells. Nature. 1995;378(6552):88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- 86.Dai SM, Matsuno H, Nakamura H, Nishioka K, Yudoh K. Interleukin-18 enhances monocyte tumor necrosis factor α and interleukin-1β production induced by direct contact with T lymphocytes: implications in rheumatoid arthritis. Arthritis and Rheumatism. 2004;50(2):432–443. doi: 10.1002/art.20064. [DOI] [PubMed] [Google Scholar]
- 87.Mariño E, Cardier JE. Differential effect of IL-18 on endothelial cell apoptosis mediated by TNF-α and Fas (CD95) Cytokine. 2003;22(5):142–148. doi: 10.1016/s1043-4666(03)00150-9. [DOI] [PubMed] [Google Scholar]
- 88.Stuyt RJL, Netea MG, Geijtenbeek TBH, Kullberg BJ, Dinarello CA, Van Der Meer JWM. Selective regulation of intercellular adhesion molecule-1 expression by interleukin-18 and interleukin-12 on human monocytes. Immunology. 2003;110(3):329–334. doi: 10.1046/j.1365-2567.2003.01747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Melnikov VY, Ecder T, Fantuzzi G, et al. Impaired IL-18 processing protects caspase-1-deficient mice from ischemic acute renal failure. Journal of Clinical Investigation. 2001;107(9):1145–1152. doi: 10.1172/JCI12089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Melnikov VY, Faubel S, Siegmund B, Scott Lucia M, Ljubanovic D, Edelstein CL. Neutrophil-independent mechanisms of caspase-1- and IL-18-mediated ischemic acute tubular necrosis in mice. Journal of Clinical Investigation. 2002;110(8):1083–1091. doi: 10.1172/JCI15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nakamura A, Shikata K, Hiramatsu M, et al. Serum interleukin-18 levels are associated with nephropathy and atherosclerosis in Japanese patients with type 2 diabetes. Diabetes Care. 2005;28(12):2890–2895. doi: 10.2337/diacare.28.12.2890. [DOI] [PubMed] [Google Scholar]
- 92.Ouedraogo R, Gong Y, Berzins B, et al. Adiponectin deficiency increases leukocyte-endothelium interactions via upregulation of endothelial cell adhesion molecules in vivo. Journal of Clinical Investigation. 2007;117(6):1718–1726. doi: 10.1172/JCI29623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yokota T, Oritani K, Takahashi I, et al. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood. 2000;96(5):1723–1732. [PubMed] [Google Scholar]
- 94.Ouchi N, Kihara S, Arita Y, et al. Adipocyte-derived plasma protein, adiponectin, suppresses lipid accumulation and class A scavenger receptor expression in human monocyte-derived macrophages. Circulation. 2001;103(8):1057–1063. doi: 10.1161/01.cir.103.8.1057. [DOI] [PubMed] [Google Scholar]
- 95.Wang Y, Lam KSL, Xu JY, et al. Adiponectin inhibits cell proliferation by interacting with several growth factors in an oligomerization-dependent manner. Journal of Biological Chemistry. 2005;280(18):18341–18347. doi: 10.1074/jbc.M501149200. [DOI] [PubMed] [Google Scholar]
- 96.Tamura Y, Murayama T, Minami M, Matsubara T, Yokode M, Arai H. Ezetimibe ameliorates early diabetic nephropathy in db/db mice. Journal of Atherosclerosis and Thrombosis. 2012;19(7):608–618. doi: 10.5551/jat.11312. [DOI] [PubMed] [Google Scholar]
- 97.Nakamaki S, Satoh H, Kudoh A, Hayashi Y, Hirai H, Watanabe T. Adiponectin reduces proteinuria in streptozotocin-induced diabetic wistar rats. Experimental Biology and Medicine. 2011;236(5):614–620. doi: 10.1258/ebm.2011.010218. [DOI] [PubMed] [Google Scholar]
- 98.Schalkwijk CG, Chaturvedi N, Schram MT, Fuller JH, Stehouwer CDA. Adiponectin is inversely associated with renal function in type 1 diabetic patients. Journal of Clinical Endocrinology and Metabolism. 2006;91(1):129–135. doi: 10.1210/jc.2005-1117. [DOI] [PubMed] [Google Scholar]
- 99.Saraheimo M, Forsblom C, Fagerudd J, et al. Serum adiponectin is increased in type 1 diabetic patients with nephropathy. Diabetes Care. 2005;28(6):1410–1414. doi: 10.2337/diacare.28.6.1410. [DOI] [PubMed] [Google Scholar]
- 100.Kato K, Osawa H, Ochi M, et al. Serum total and high molecular weight adiponectin levels are correlated with the severity of diabetic retinopathy and nephropathy. Clinical Endocrinology. 2008;68(3):442–449. doi: 10.1111/j.1365-2265.2007.03063.x. [DOI] [PubMed] [Google Scholar]
- 101.Rivero A, Mora C, Muros M, García J, Herrera H, Navarro-González JF. Pathogenic perspectives for the role of inflammation in diabetic nephropathy. Clinical Science. 2009;116(6):479–492. doi: 10.1042/CS20080394. [DOI] [PubMed] [Google Scholar]
- 102.Hanai KO, Babazono T, Mugishima M, et al. Association of serum leptin levels with progression of diabetic kidney disease in patients with type 2 diabetes. Diabetes Care. 2011;34(12):2557–2559. doi: 10.2337/dc11-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fruehwald-Schultes B, Kern W, Beyer J, Forst T, Pfützner A, Peters A. Elevated serum leptin concentrations in type 2 diabetic patients with microalbuminuria and macroalbuminuria. Metabolism: Clinical and Experimental. 1999;48(10):1290–1293. doi: 10.1016/s0026-0495(99)90270-6. [DOI] [PubMed] [Google Scholar]
- 104.Axelsson J, Bergsten A, Qureshi AR, et al. Elevated resistin levels in chronic kidney disease are associated with decreased glomerular filtration rate and inflammation, but not with insulin resistance. Kidney International. 2006;69(3):596–604. doi: 10.1038/sj.ki.5000089. [DOI] [PubMed] [Google Scholar]
- 105.Ma FY, Liu J, Nikolic-Paterson DJ. The role of stress-activated protein kinase signaling in renal pathophysiology. Brazilian Journal of Medical and Biological Research. 2009;42(1):29–37. doi: 10.1590/s0100-879x2008005000049. [DOI] [PubMed] [Google Scholar]
- 106.Adhikary L, Chow F, Nikolic-Paterson DJ, et al. Abnormal p38 mitogen-activated protein kinase signalling in human and experimental diabetic nephropathy. Diabetologia. 2004;47(7):1210–1222. doi: 10.1007/s00125-004-1437-0. [DOI] [PubMed] [Google Scholar]
- 107.Sakai N, Wada T, Furuichi K, et al. Involvement of extracellular signal-regulated kinase and p38 in human diabetic nephropathy. American Journal of Kidney Diseases. 2005;45(1):54–65. doi: 10.1053/j.ajkd.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 108.Stambe C, Atkins RC, Tesch GH, et al. Blockade of p38α MAPK ameliorates acute inflammatory renal injury in rat anti-GBM glomerulonephritis. Journal of the American Society of Nephrology. 2003;14(2):338–351. doi: 10.1097/01.asn.0000048715.12315.fd. [DOI] [PubMed] [Google Scholar]
- 109.Stambe C, Atkins RC, Tesch GH, Masaki T, Schreiner GF, Nikolic-Paterson DJ. The Role of p38α mitogen-activated protein kinase activation in renal fibrosis. Journal of the American Society of Nephrology. 2004;15(2):370–379. doi: 10.1097/01.asn.0000109669.23650.56. [DOI] [PubMed] [Google Scholar]
- 110.Wilmer WA, Dixon CL, Hebert C. Chronic exposure of human mesangial cells to high glucose environments activates the p38 MAPK pathway. Kidney International. 2001;60(3):858–871. doi: 10.1046/j.1523-1755.2001.060003858.x. [DOI] [PubMed] [Google Scholar]
- 111.Susztak K, Raff AC, Schiffer M, Böttinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55(1):225–233. [PubMed] [Google Scholar]
- 112.Zhang SL, Tang SS, Chen X, Filep JG, Ingelfinger JR, Chan JSD. High levels of glucose stimulate angiotensinogen gene expression via the p38 mitogen-activated protein kinase pathway in rat kidney proximal tubular cells. Endocrinology. 2000;141(12):4637–4646. doi: 10.1210/endo.141.12.7844. [DOI] [PubMed] [Google Scholar]
- 113.Daoud S, Schinzel R, Neumann A, et al. Advanced glycation endproducts: activators of cardiac remodeling in primary fibroblasts from adult rat hearts. Molecular Medicine. 2001;7(8):543–551. [PMC free article] [PubMed] [Google Scholar]
- 114.Liu BF, Miyata S, Hirota Y, et al. Methylglyoxal induces apoptosis through activation of p38 mitogen-activated protein kinase in rat mesangial cells. Kidney International. 2003;63(3):947–957. doi: 10.1046/j.1523-1755.2003.00829.x. [DOI] [PubMed] [Google Scholar]
- 115.Kang YS, Park YG, Kim BK, et al. Angiotensin II stimulates the synthesis of vascular endothelial growth factor through the p38 mitogen activated protein kinase pathway in cultured mouse podocytes. Journal of Molecular Endocrinology. 2006;36(2):377–388. doi: 10.1677/jme.1.02033. [DOI] [PubMed] [Google Scholar]
- 116.Chuang PY, Yu Q, Fang W, Uribarri J, He JC. Advanced glycation endproducts induce podocyte apoptosis by activation of the FOXO4 transcription factor. Kidney International. 2007;72(8):965–976. doi: 10.1038/sj.ki.5002456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Porras A, Zuluaga S, Black E, et al. p38α Mitogen-activated Protein Kinase Sensitizes Cells to Apoptosis Induced by Different Stimuli. Molecular Biology of the Cell. 2004;15(2):922–933. doi: 10.1091/mbc.E03-08-0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Takaishi H, Taniguchi T, Takahashi A, Ishikawa Y, Yokoyama M. High glucose accelerates MCP-1 production via p38 MAPK in vascular endothelial cells. Biochemical and Biophysical Research Communications. 2003;305(1):122–128. doi: 10.1016/s0006-291x(03)00712-5. [DOI] [PubMed] [Google Scholar]
- 119.Suzuki H, Uchida K, Nitta K, Nihei H. Role of mitogen-activated protein kinase in the regulation of transforming growth factor-β-induced fibronectin accumulation in cultured renal interstitial fibroblasts. Clinical and Experimental Nephrology. 2004;8(3):188–195. doi: 10.1007/s10157-004-0297-8. [DOI] [PubMed] [Google Scholar]
- 120.Chin BY, Mohsenin A, Li SX, Choi AMK, Choi ME. Stimulation of pro-α1(I) collagen by TGF-β1 in mesangial cells: role of the p38 MAPK pathway. American Journal of Physiology. 2001;280(3):F495–F504. doi: 10.1152/ajprenal.2001.280.3.F495. [DOI] [PubMed] [Google Scholar]
- 121.Fujita H, Omori S, Ishikura K, Hida M, Awazu M. ERK and p38 mediate high-glucose-induced hypertrophy and TGF-β expression in renal tubular cells. American Journal of Physiology. 2004;286(1):F120–F126. doi: 10.1152/ajprenal.00351.2002. [DOI] [PubMed] [Google Scholar]
- 122.Wu DT, Bitzer M, Ju W, Mundel P, Böttinger EP. TGF-β concentration specifies differential signaling profiles of growth arrest/differentiation and apoptosis in podocytes. Journal of the American Society of Nephrology. 2005;16(11):3211–3221. doi: 10.1681/ASN.2004121055. [DOI] [PubMed] [Google Scholar]
- 123.Schiffer M, Bitzer M, Roberts ISD, et al. Apoptosis in podocytes induced by TGF-β and Smad7. Journal of Clinical Investigation. 2001;108(6):807–816. doi: 10.1172/JCI12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhuang S, Yan Y, Han J, Schnellmann RG. p38 kinase-mediated transactivation of the epidermal growth factor receptor is required for dedifferentiation of renal epithelial cells after oxidant injury. Journal of Biological Chemistry. 2005;280(22):21036–21042. doi: 10.1074/jbc.M413300200. [DOI] [PubMed] [Google Scholar]
- 125.Jung DS, Li JJ, Kwak SJ, et al. FR167653 inhibits fibronectin expression and apoptosis in diabetic glomeruli and in high-glucose-stimulated mesangial cells. American Journal of Physiology. 2008;295(2):F595–F604. doi: 10.1152/ajprenal.00624.2007. [DOI] [PubMed] [Google Scholar]
- 126.Wang L, Ma R, Flavell RA, Choi ME. Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for activation of p38α and p38δ MAPK isoforms by TGF-β1 in murine mesangial cells. Journal of Biological Chemistry. 2002;277(49):47257–47262. doi: 10.1074/jbc.M208573200. [DOI] [PubMed] [Google Scholar]
- 127.Wang L, Kwak JH, Kim SI, He Y, Choi ME. Transforming growth factor-β1 stimulates vascular endothelial growth factor 164 via mitogen-activated protein kinase kinase 3-p38α and p38δ mitogen-activated protein kinase-dependent pathway in murine mesangial cells. Journal of Biological Chemistry. 2004;279(32):33213–33219. doi: 10.1074/jbc.M403758200. [DOI] [PubMed] [Google Scholar]
- 128.Lim AKH, Nikolic-Paterson DJ, Ma FY, et al. Role of MKK3-p38 MAPK signalling in the development of type 2 diabetes and renal injury in obese db/db mice. Diabetologia. 2009;52(2):347–358. doi: 10.1007/s00125-008-1215-5. [DOI] [PubMed] [Google Scholar]
- 129.Bogoyevitch MA, Boehm I, Oakley A, Ketterman AJ, Barr RK. Targeting the JNK MAPK cascade for inhibition: basic science and therapeutic potential. Biochimica et Biophysica Acta. 2004;1697(1-2):89–101. doi: 10.1016/j.bbapap.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 130.Wang L, Matsushita K, Araki I, Takeda M. Inhibition of c-Jun N-terminal kinase ameliorates apoptosis induced by hydrogen peroxide in the kidney tubule epithelial cells (NRK-52E) Nephron. 2002;91(1):142–147. doi: 10.1159/000057616. [DOI] [PubMed] [Google Scholar]
- 131.Pulverer BJ, Kyriakis JM, Avruch J, Nikolakaki E, Woodgett JR. Phosphorylation of c-jun mediated by MAP kinases. Nature. 1991;353(6345):670–674. doi: 10.1038/353670a0. [DOI] [PubMed] [Google Scholar]
- 132.Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK)—from inflammation to development. Current Opinion in Cell Biology. 1998;10(2):205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- 133.Himes SR, Sester DP, Ravasi T, Cronau SL, Sasmono T, Hume DA. The JNK are important for development and survival of macrophages. Journal of Immunology. 2006;176(4):2219–2228. doi: 10.4049/jimmunol.176.4.2219. [DOI] [PubMed] [Google Scholar]
- 134.Mao C, Ray-Gallet D, Tavitian A, Moreau-Gachelin F. Differential phosphorylations of Spi-B and Spi-1 transcription factors. Oncogene. 1996;12(4):863–873. [PubMed] [Google Scholar]
- 135.Flanc RS, Ma FY, Tesch GH, et al. A pathogenic role for JNK signaling in experimental anti-GBM glomerulonephritis. Kidney International. 2007;72(6):698–708. doi: 10.1038/sj.ki.5002404. [DOI] [PubMed] [Google Scholar]
- 136.Ma FY, Flanc RS, Tesch GH, et al. A pathogenic role for c-Jun amino-terminal kinase signaling in renal fibrosis and tubular cell apoptosis. Journal of the American Society of Nephrology. 2007;18(2):472–484. doi: 10.1681/ASN.2006060604. [DOI] [PubMed] [Google Scholar]
- 137.De Borst MH, Prakash J, Sandovici M, et al. c-Jun NH2-terminal kinase is crucially involved in renal tubulo-interstitial inflammation. Journal of Pharmacology and Experimental Therapeutics. 2009;331(3):896–905. doi: 10.1124/jpet.109.154179. [DOI] [PubMed] [Google Scholar]
- 138.De Borst MH, Prakash J, Melenhorst WBWH, et al. Glomerular and tubular induction of the transcription factor c-Jun in human renal disease. Journal of Pathology. 2007;213(2):219–228. doi: 10.1002/path.2228. [DOI] [PubMed] [Google Scholar]
- 139.Lim AKH, Ma FY, Nikolic-Paterson DJ, et al. Evaluation of JNK blockade as an early intervention treatment for type 1 diabetic nephropathy in hypertensive rats. American Journal of Nephrology. 2011;34(4):337–346. doi: 10.1159/000331058. [DOI] [PubMed] [Google Scholar]
- 140.Ijaz A, Tejada T, Catanuto P, et al. Inhibition of C-jun N-terminal kinase improves insulin sensitivity but worsens albuminuria in experimental diabetes. Kidney International. 2009;75(4):381–388. doi: 10.1038/ki.2008.559. [DOI] [PubMed] [Google Scholar]
- 141.Mohan S, Reddick RL, Musi N, et al. Diabetic eNOS knockout mice develop distinct macro- and microvascular complications. Laboratory Investigation. 2008;88(5):515–528. doi: 10.1038/labinvest.2008.23. [DOI] [PubMed] [Google Scholar]
- 142.Sato W, Kosugi T, Zhang L, et al. The pivotal role of VEGF on glomerular macrophage infiltration in advanced diabetic nephropathy. Laboratory Investigation. 2008;88(9):949–961. doi: 10.1038/labinvest.2008.60. [DOI] [PubMed] [Google Scholar]
- 143.Lassila M, Seah KK, Allen TJ, et al. Accelerated nephropathy in diabetic apolipoprotein E-knockout mouse: role of advanced glycation end products. Journal of the American Society of Nephrology. 2004;15(8):2125–2138. doi: 10.1097/01.ASN.0000133025.23732.46. [DOI] [PubMed] [Google Scholar]
- 144.Kikuchi Y, Imakiire T, Yamada M, et al. Mizoribine reduces renal injury and macrophage infiltration in non-insulin-dependent diabetic rats. Nephrology Dialysis Transplantation. 2005;20(8):1573–1581. doi: 10.1093/ndt/gfh888. [DOI] [PubMed] [Google Scholar]