

Abstract
Vitamin D deficiency is a major risk factor for central nervous system (CNS) demyelinating diseases including multiple sclerosis (MS) and its animal model, that of experimental autoimmune encephalomyelitis (EAE). Both vitamin D3 and 1, 25-dihydroxyviatmin-D3 (calcitriol) had beneficial effects in EAE/MS. However, the exact cause of vitamin D deficiency in EAE/MS is not clear. Previously, we documented that lovastatin (LOV) provides protection in EAE animals via inhibition of RhoA–ROCK signaling. Herein, we demonstrate that LOV prevents the lowering of circulating 25-hydroxyvitamin-D3 and 1,25-dihydroxyviatmin-D3 levels including 1,25-dihydroxyviatmin-D3 levels in the peripheral lymphoid organs and CNS of treated EAE animals. These effects of LOV were attributed to enhanced expression of vitamin D synthesizing enzyme (1α-hydroxylase) in kidney and the CNS, with corresponding reduction of vitamin D catabolizing enzyme (24-hydorxylase) expression in the CNS of EAE animals via inhibition of RhoA–ROCK signaling. Ex vivo and in vitro studies established that autoreactive Th1/Th17 cells had higher expression of 24-hydroxylase than Th2/T regulatory cells, that was reverted by LOV or ROCK inhibitor. Interestingly, LOV-mediated regulation of vitamin D metabolism had improved vitamin D3 efficacy to confer protection in EAE animals and that was ascribed to the LOV- and calcitriol-induced immunomodulatory synergy. Together, these data provide evidence that interfering with RhoA–ROCK signaling in autoreactive Th1/Th17 cells can improve vitamin D3 efficacy in clinical trials of MS and related neurodegenerative disorders.
Multiple sclerosis (MS) is an immunologically complex neurodegenerative disease marked by trafficking of autoreactive lymphocytes and mononuclear cells into the central nervous system (CNS) with subsequent demyelination due to loss of oligodendrocytes (OLs) and axonal degeneration.1,2 Increasing evidence suggests that pathogenic CD4+ T helper (Th) cells ie, interferon-γ (IFN-γ)–secreting Th1 and interleukin-17 (IL-17)–secreting Th17 cells play a central role in the inflammatory and demyelinating pathology; whereas IL-4–secreting Th2 and regulatory T (Treg) cells keep the autoimmune response under control.2–4 In addition, environmental factors are important in influencing MS risk.5 Therefore, understanding the molecular mechanism(s) induced by environmental factors in immune cells involved in the regulation of inflammatory responses will provide new insights for the management of MS.
Strong inverse relationship between vitamin D metabolite concentrations and MS prevalence has been documented in conjunction with sun exposure.6 Sun exposure is essential to induce the biosynthesis of 25-hydroxyvitamin D3 (25-OH-D3), a substrate of CYP27B1 (1α-hydroxylase), which mainly occurs in the kidney, although numerous cell types/tissues also express CYP27B1 to produce 1,25-dihdroxyvitamin D3 [1,25-(OH)2D3], that provides beneficial effects in MS.7,8 Recently, a positive association has been documented between CYP27B1-gene mutations and MS prevalence,9 supporting the notion that higher in situ 1,25-(OH)2D3 levels are important to limit MS pathogenesis. The transcriptional regulatory functions of 1,25-(OH)2D3 are mediated by the nuclear vitamin D receptor (VDR),10 and genetic epidemiological studies have shown that the VDR b allele correlated well with MS risk in Japan.11,12 1,25-(OH)2D3 is inactivated by mitochondrial enzyme, CYP24A1 (24-hydroxylase) in the kidney, including other cell types/tissues by hydroxylation at 24 carbon position.8 Vitamin D3 and 1,25-(OH)2D3 are documented to inhibit experimental autoimmune encephalomyelitis (EAE; murine model of MS) as well as to reverse established EAE.13–17 Importantly, dietary intake of vitamin D3 and higher circulating levels of 25-OH-D3 are documented to reduce MS prevalence.18,19 In addition, MS clinical trials conducted with higher dose of vitamin D3 for short durations were found to be protective and safe in patients.20–23 However, the underlying mechanism(s) responsible for vitamin D deficiency in MS/EAE is not clear.
Seasonal changes in the circulatory 25-OH-D3 levels were inversely related to the plasma cholesterol and triglycerides levels,24,25 indicating that lowering of plasma lipids can increase the bioavailability of vitamin D metabolites in human patients. Consistent with these findings, the elevated circulatory 25-OH-D3 levels were associated with reduced serum lipid profile in heart disease patients treated with lipid-lowering drugs, statins.26,27 Importantly, statins as montherapy and in combination with presently prescribed MS drugs demonstrated significant reduction of gadolinium lesions in the MS brain.28,29 These effects of statins were ascribed to the activation of autoreactive Th17 cell inhibition and the induction of Th1/Th2 shift in MS patients via lowering of isoprenoids at the cellular level, resulting in inhibition of Rho family small GTPase, RhoA, and its downstream target, Rho kinase (ROCK), as evident from EAE model studies.30–32 RhoA–ROCK signaling controls the variety of cellular processes including cellular signaling, proliferation, and differentiation.33 Considering that statins can increase the circulating levels of 25-OH-D3 in heart disease patients, we proposed to investigate the impact of statin treatment on vitamin D metabolism in EAE animals. To gain more insight into the protective mechanism, we studied the statin-mediated regulation of vitamin D metabolizing enzyme expressions at the cellular level and tested whether statin could improve the efficacy of vitamin D3 in MS clinical trials.
Materials and Methods
Chemicals and Reagents
Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Lovastatin (LOV), Y27643, vitamin D3, and calcitriol were purchased from EMD Chemicals (Philadelphia, PA). Anti-CYP27B1 (recognizes 57 kDa protein band) and anti-CYP24A1 (recognizes 50-kDa protein band) antibodies were purchased from Abcam (Cambridge, MA). Anti–β-actin and, anti-mouse IgG, and anti-rabbit polyclonal IgG secondary antibodies were obtained from Vector Laboratory (Burlingame, CA).
Animals
Female Lewis rats weighing 250 to 300 g were purchased from Charles River Laboratory (Wilmington, MA) and housed in the animal care facility at the Medical University of South Carolina throughout the experiment and provided with food and water ad libitum. During experimentation, rats were fed commercial rat chow containing 0.33 μg/day vitamin D3 and 1% calcium. For experimental studies using vitamin D3–deficient diet, rats were fed synthetic vitamin D3–deficient diet (Charles River Laboratory) 20 days before MBP priming for EAE induction, and rats showed significant reduction of circulating levels of 25-OH-D3 and 1,25-(OH)2D3 on day 20 (data not shown). All animal experimentations were conducted in accord with accepted standards of humane care, as outlined in the ethical guidelines and approved by the Medical University of South Carolina Animal Ethics Committee.
EAE Induction and Evaluation
Procedures used for an induction of EAE are as described previously in our publications, with slight modifications.34,35 In brief, rats received a subcutaneous injection of 30 μg of guinea pig MBP in 0.1 mL of PBS emulsified with equal volume of CFA supplemented with 2 mg/mL of mycobacterium tuberculosis H37Ra (Difco, Detroit, MI) in the hind limb footpads on days 0 and 7. Immediately and again 24 hours later, rats received pertussis toxin (200 ng, intraperitoneally, ip) in 0.1 mL PBS. Pertussis toxin was administered to rats according to standardized protocol in our laboratory for induction of EAE. Similarly, control rats received subcutaneous injection of PBS and CFA emulsion in the hind limb footpads on days 0 and 7. Rats were examined for clinical scores by an experimentally blinded investigator daily. Clinical score assessed on a scale of 0 to 5, as follows: 0, no clinical disease; 1.0, piloerection; 2.0, loss in tail tonicity; 3.0, hind-leg paralysis; 4.0, paraplegia, and 5.0, moribund or dead. Several times during the study, rats were weighed. The clinical data of rats with clinical scores >4.0 were not included for statistical analysis. At the conclusion of the study, the rats were euthanized. A blood sample was obtained, and perfusion was done. The kidneys, lymph nodes, and lumbar spinal cords were removed, snap-frozen with liquid nitrogen, and stored at −70°C before RNA, protein, or 1,25-(OH)2D3 extraction. Alternatively, the lumbar spinal cord of each rat was cut into four small pieces and fixed in 4% paraformaldehyde for histopathology. The blood was centrifuged, and decanted plasma was frozen at −70°C before analysis.
Drug Treatments
LOV (4 mg/mL) was suspended in 0.8% ethanol/0.6 N NaOH/PBS, and pH was adjusted to 7.4 with 1N HCl. Mevalonolactone (5 mg/mL) was suspended in PBS; farnesol (5 mg/mL) was suspended in PBS; farnesyl transferase inhibitor–277 (0.5 mg/mL) was suspended in PBS; geranylgeranyl transferase inhibitor–298 was suspended in ethanol and diluted in PBS (0.5 mg/mL); fasudil was dissolved in 100 μl of DMSO and diluted in PBS (1 mg/mL); and 50 μg/mL of calcitriol was suspended in ethanol and diluted to 1 and 2 μg/mL in PBS. Drugs were administered intraperitoneally (ip) at the specified dose, once daily, using a 1-mL insulin syringe. Treatment was started after the onset of EAE disease (11 to 13 days post-immunization [dpi]) and continued until the lessening of paralytic symptoms (25 dpi). Rat developing EAE with no drug treatment received an injection (ip) of vehicle (0.8% ethanol in PBS), once daily. Control rats received an injection (ip) of vehicle or drug, once daily. For vitamin D3 treatment, vitamin D3 (0.25 mg/kg) suspended in saline was administered to rats by gavage every day. Similarly, for combination treatments, LOV and calcitriol were administered once daily in established EAE rats with clinical disease scores ≥3.0 until the lessening of paralytic symptoms (25 dpi).
25-OH-D3 and 1,25-(OH)2D3 Analysis
Plasma was separated from blood and the concentrations of 25-OH-D3 and 1,25-(OH)2D3 were determined in duplicate by radioimmunoassay (RIA) developed in the laboratory of Bruce Hollis, as documented earlier.36 Laboratory personnel were blinded to the samples. The measurements of 1,25-(OH)2D3 in the tissues of spinal cord were performed by extraction methods published earlier.16 Briefly, tissues were extracted with chloroform-methanol-4% KCl in water (1:2:0.8 v/v) mixture to recover the vitamin D metabolites. The extracts were then vacuum dried, suspended in ethanol, diluted in sample buffer, and analyzed in duplicate for 1,25-(OH)2D3 using rat 1,25-(OH)2D3 ELISA kit (NovaTeinBio, Cambridge, MA). A spike recovery control was performed with each 1,25-(OH)2D3 extraction by adding 100 pg of 1,25-(OH)2D3 to a crushed spinal cord of rat fed a vitamin D3-depleted diet for 20 days.
Stimulation of MBP-Primed Spleen Cells and Characterization of CD4+ T Cells
The spleens of MBP-primed rats were collected after the onset of EAE with clinical score <2.0 or from naive controls and processed to make single-cell suspension followed by the removal of RBCs with 1× pharma lyses (BD Biosciences). Cells were washed twice and suspended in RPMI 1640, and the concentration was adjusted to 5 × 106 cells/mL. Cells were plated in six-well tissue culture plates and incubated with 50 μg/mL of MBP in the presence/absence of LOV (5 μmol/L) at 37°C. After 72 h, nonadherent cells were collected by centrifugation, and CD4+ T cells were positively selected using Dynabeads M-450 CD4 beads (Dynal Biotech, Oslo, Norway). Separation of CD4+ T cells was monitored using flow cytometry analysis, demonstrating >97% purity. Total RNA was purified from collected CD4+ T cells and used for real-time PCR analysis.
In Vitro T Helper Cell Differentiation
Naive CD4+CD25− T cells were purified from the spleen single cell suspensions using magnetic cell sorting (Miltenyi Biotec) with 98% to 99% of purity. Purified CD4+CD25− T cells were stimulated with antibodies to CD3 (5 μg/mL) and CD28 (2 μg/mL) under Th17 cell differentiation conditions (rmIL-6, 20 ng/mL; R&D systems; rhTGF-β1, 3 ng/mL; anti-IL-4, 10 μg/mL; and anti–IFN-γ, 10 μg/mL; BD Biosciences), Th1 cell differentiation conditions (rmIL-12, 10 ng/mL and IL-4, 10 μg/mL; BD Biosciences), Th2 cell differentiation conditions (rmIL-4, 20 ng/mL; rmIL-2, 10 ng/mL, R&D Systems; and anti-IFN-γ, 10 μg/mL), or Treg cell differentiation conditions (rhTGF-β1, 5 ng/mL and rmILs-2, 40 ng/mL; R&D Systems) for 48 hours. This was followed by treatment with LOV (5 μmol/L) for another 24 hours to assess the expression of CYP24A1 using quantitative real-time PCR.
Histopathology
Fixed lumbar spinal cords sections of 5 μm were stained with Luxol fast blue or H&E (Sigma-Aldrich) and examined using light microscopy (Olympus BX-60) and images were acquired with an Olympus digital camera (Optronics; Goleta, CA). The region of spinal cord depicting >10 nuclei was considered as a lesion. The meninges, and the gray and white matter of the sections were scored in a blinded fashion based on the presence or the absence of infiltrating cells in each of the regions of the spinal cord. The histopathology score was recorded as the number of lesions in the spinal cord of each group rats that showed a readily identifiable inflammatory cell infiltrate. Similarly, luxol fast blue–stained spinal cord sections were analyzed for degree of demyelination and scored. The demyelination degree was scored as grade 0, no disease; grade 1, foci of demyelination/axonal loss that was superficial and involved <25% of the lateral columns; grade 2, deep foci that involved >25% of the lateral columns; and grade 3, diffuse and widespread demyelination and axonal loss.
Real-Time PCR and Transcript Analysis
For quantitative assessments of relative mRNA levels, total RNA was prepared using TRIzol LS reagent (Invitrogen, Carlsbad, CA), following the manufacturer's instructions. RNA was reverse transcribed using an MMLV-RT reverse transcription kit with random hexamer primers (Bio-Rad, Hercules CA). Relative levels of transcripts of various genes described in Table 1 were determined by real-time PCR (iCycler IQ, Bio-Rad) using a SYBR green PCR mix (Bio-Rad). Rat-specific primers for each gene were designed using Primer Quest software and oligonucleotides were purchased from Integrated DNA Technologies Inc. (Coralville, IA). Thermal cycling conditions were as follows: activation of iTaq DNA polymerase at 95°C for 10 minutes, followed by 40 cycles of amplification at 95°C for 30 seconds and 60°C for 1 minute. The performance of each real-time PCR run was determined by examining the melting curve. The detection of threshold was set above the mean baseline fluorescence determined by the first 20 cycles. Amplification reaction in which the fluorescence increased above the threshold was defined as positive. Values obtained for each gene of interest were normalized to β-actin or 18S rRNA transcripts as an internal control.
Table 1.
List of Primers Used for Quantitative Real-Time PCR Analysis
| Gene name | Primer sequence |
|---|---|
| β-actin | FP: 5′-AGCTGTGCTATGTTGCCCTAGACT-3′; RP: 5′-ACCGCTCATTGCCGATAGTGATGA-3′ |
| 18S rRNA | FP: 5′-CCAGAGCGAAAGCATTTGCCAAGA-3′; RP: 5′-TCGGCATCGTTTATGGTCGGAACT-3′ |
| CYP24A1 | FP: 5′-TTAGACCCGAACGCTGGCTTGAAA-3′; RP: 5′-AGCAAAGAGCCAAGTGGAGCTGTA-3′ |
| CYP27B1 | FP: 5′-GGATCAGATGTTTGCCTTTGCCCA-3′; RP: 5′-AGGAAGTGGGTTAAGTGATGCCCA-3′ |
| IL-4 | FP: 5′-GGTATCCACGGATGTAACGACAGC-3′; RP: 5′-CCGTGGTGTTCCTTGTTGCCGTAA −3′ |
| IFN-γ | FP: 5′-TTTGAGGTCAACAACCCACAGGTC-3′; RP: 5′-TTTCCGCTTCCTGAGGCTGGATT-3′ |
| IL-6 | FP: 5′-CAGAAGGAGTGGCTAAGGAC-3′; RP: 5′-CACTAGGTTTGCCGAGTAGA-3′ |
| IL-23 | FP: 5′-ATCACCACTGGGAGACTCAACAGA-3′; RP: 5′-TGCGAAGGATCTTGGAACGGAGAA-3′ |
| IL-17A | FP: 5′-TCTGAGCCAGCCAAGAAGAAGTGT-3′; RP: 5′-TTCCAACCCAAACATAGGCAC-3′ |
| FoxP3 | FP: 5′-AGAGTTTCTCAAGCACTGCCAAGC-3′; RP: 5′-TGCATAGCTCCCAGCTTCTCCTTT-3′ |
| RORγt | FP: 5′-AGGCCATTCAGTATGTGGTGGAGT-3′; RP: 5′-TGTGTGGTTGTTGGCATTGTAGGC-3′ |
FP, forward primer; RP, reverse primer.
Western Blotting
Tissues were homogenized in ice-cold lysis buffer (50 mm Tris-HCl, pH 7.4, containing 50 mmol/L NaCl, 1 mmol/L EDTA, 0.5 mmol/L EGTA, 10% glycerol, and protease inhibitor mixture), using a soincator with three to four pulses for 30 seconds each. Samples were centrifuged at 12,000 × g for 10 minutes at 4°C, and protein concentration was determined in the supernatant using Bradford reagent (Bio-Rad). SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting were performed as described previously.34 Autoradiograph of immunoblots was generated using enhanced chemiluminescence detection kits (Amersham Biosciences, Arlington Heights, IL). Densitometeric analysis of autoradiograph was performed using ImageJ software from the NIH (available at http://rsb.info.nih.gov/ij).
Quantification of Cytokines
Slices of the spinal cords (20 to 50 mg) of rats were soincated in 0.5 mL of PBS and centrifuged at 12,000 × g for 10 minutes at 4°C. The protein concentration in each sample supernatant was determined using Bradford reagent (Bio-Rad). To quantify the levels of various cytokines, ELISA kits (eBiosciences Inc., San Diego, CA) were used as per instructions in the product manuals. Recombinant protein of cytokines was used as standard to measure their concentration in test samples. Levels of cytokines in each sample in triplicate were measured using an ELISA reader (UV-VIS spectrophotometer), and data were normalized with protein concentration in each sample.
Statistical Analysis
For multiple comparisons, one-way multiple range analysis of variance followed by Bonferroni post-test was used and P values were determined for each experiment of three to four separate experiments using GraphPad Prism 3.0 software (GraphPad Software, San Diego, CA). Two-way analysis of variance plus Bonferroni post-test was used to compare two groups over multiple time points. Student's t-test was used to compare two groups of matched samples. P values <0.05 were considered significant.
Results
Effect of LOV Treatment on Plasma Levels of 25-OH-D3 and 1,25-(OH)2D3 in EAE Rats in Relation to Clinical Symptoms
The experiments reported here used the acute EAE model to investigate whether LOV-mediated signaling mechanism(s) enhances the systemic levels of vitamin D metabolites to influence MS risk. Earlier, we and others documented that statin-mediated inhibition of Rho family GTPases in immune cells participates in the attenuation of EAE pathogenesis.31,32 Therefore, we first sought to measure the concentrations of vitamin D metabolites in the blood of EAE rats treated with LOV or vehicle on peak clinical exacerbations day (16 dpi) and during the lessening of paralytic symptoms (25 dpi). Rats were fed a commercially available laboratory animal diet. LOV (2 mg/kg) was administered after the onset of neurological impairments and continued until 25 dpi (Figure 1A). As expected, an increase in the neurological impairments in association with histopathological changes due to the infiltration of autoreactive immune cells in the spinal cords of EAE rats 16 dpi were attenuated by treatment with LOV (Table 2). As illustrated in Figure 1B, plasma levels of 25-OH-D3 were reduced significantly in EAE rats treated with vehicle compared with controls 16 dpi and were abrogated by treatment with LOV. This was also the case for 1,25-(OH)2D3 plasma level in EAE rats treated with vehicle compared with controls, which likewise was abrogated by treatment with LOV (Figure 1C). With lessening of clinical symptoms (25 dpi), plasma levels of 25-OH-D3 in EAE rats treated with vehicle were similar to those in controls (Figure 1B), whereas 1,25-(OH)2D3 plasma levels were still lower; albeit not significantly so, in EAE rats treated with vehicle as compared with controls (Figure 1C). On the other hand, 1,25-(OH)2D3 plasma levels were significantly elevated in EAE rats treated with LOV 25 dpi compared with vehicle-treated EAE or control rats (Figure 1C). Moreover, a significant change in the plasma levels of 1,25-(OH)2D3 was also evident in controls treated with LOV rather than vehicle (Figure 1C). These data provide evidence that neurological impairments in association with histopathological changes in CNS due to the infiltration of immune cells are correlated with reduced circulating levels of 25-OH-D3 and 1,25-(OH)2D3 in EAE animals, and were controlled by treatment with LOV.
Figure 1.

Effect of LOV on clinical exacerbations, vitamin D metabolites, and expression of its metabolizing enzymes in EAE rats. EAE was induced in female Lewis rats with guinea pig MBP (30 μg/rat), and LOV (2 mg/kg) or vehicle treatments were initiated as described in Materials and Methods.A: Composite mean ± SEM of the clinical scores of 9 to 12 rats per group evaluated in three separate experiments. Composite mean ± SEM of four to five samples per group of the levels of 25-OH-D3 (B) and 1,25-(OH)2D3 (C) in the plasmas of treated EAE and control rats 16 dpi and 25 dpi. D: Composite mean ± SEM of four to five samples per group analyzed by real-time PCR in duplicate for CYP24A1 and CYP27B1 transcript levels in the kidneys of treated EAE and control rats 16 dpi. E: Representative autoradiograph of four samples per group depicting the levels of CYP27B1, CYP24A1, and β-actin proteins in the kidneys of treated EAE and control rats 16 dpi. F: Histogram depicts the composite mean ± SEM of four samples per group analyzed by Western blotting for CYP24A1 and CYP27B1 proteins normalized with β-actin and then compared with respective controls in the kidneys of treated EAE and control rats 16 dpi. Statistical significance as indicated: *P < 0.05; **P < 0.01; ***P < 0.001; NS, not significant.
Table 2.
Effect of Lovastatin (LOV) and Various Metabolites/Inhibitors of Mevalonate Pathways on the Disease Severity in Experimental Autoimmune Encephalomyelitis (EAE) Animals
| Group | Disease severity | Mortality (%) | Sections with lesions (%) |
|---|---|---|---|
| Vehicle | 3.5 ± 0.5 | 20 | 49 ± 10 |
| LOV | 2.5 ± 1.0⁎⁎ | 0⁎ | 22 ± 10⁎⁎ |
| MEV | 4.0 ± 0.5⁎ | 40⁎⁎ | 70 ± 19⁎⁎ |
| LOV + MEV | 3.2 ± 0.5NS | 20NS | 48 ± 8NS |
| LOV + FOH | 3.5 ± 1.0NS | 30NS | 51 ± 5NS |
| FTI-277 | 3.8 ± 0.5NS | 20NS | 59 ± 11NS |
| GGTI-298 | 2.8 ± 1.0⁎⁎ | 0⁎ | 25 ± 12⁎⁎ |
| Fasudil | 2.5 ± 0.5⁎⁎ | 0⁎ | 21 ± 6⁎⁎ |
| Vitamin D3–deficient diet | |||
| Vehicle | 4.0 ± 1.0 | 30 | 65 ± 5 |
| LOV | 3.2 ± 0.5⁎ | 0⁎⁎ | 55 ± 7⁎ |
| Vitamin D3 | 3.0 ± 1.0⁎⁎ | 0⁎⁎ | 46 ± 6⁎⁎ |
| LOV + vitamin D3 | 2.5 ± 0⁎⁎⁎ | 0⁎⁎ | 20 ± 2⁎⁎ |
Disease severity shown as the composite mean ± SEM (n = 10 to 12) of clinical scores evaluated in three separate experiments. Rats with a clinical score >2 for 2 consecutive days were considered to have EAE. Histological data are of five rats per group, in which three to four sections per rat were examined for the presence of lesions. The region of spinal cord depicting >10 nuclei was considered as lesion. Statistical tests were performed with Student's t-test (disease severity and mortality) and Mann–Whitney rank sum test (histopathological data). Statistical significance:
P < 0.05,
P < 0.01,
P < 0.001 and not significant (NS) versus EAE with vehicle.
Mitochondrial cytochrome P450 enzymes ie, CYP27B1 (1α-hydroxylase) and CYP24A1 (24-hydroxylase) expressed in the proximal tubules of kidney are reported to maintain the systemic concentrations of vitamin D metabolites.8 We reasoned that differences in synthesis or inactivation early in the disease process would have the greatest impact on disease outcome. Therefore, the expressions of the CYP24A1 and CYP27B1 in the kidneys of EAE rats treated/untreated with LOV were examined. Importantly, no change in the level of CYP24A1 transcripts was observed in the kidneys of EAE treated with LOV or vehicle, including control rats treated with LOV or vehicle (Figure 1D), suggesting that EAE pathogenesis has no effect on kidney physiology. However, the level of CYP27B1 transcripts was significantly elevated in the kidneys of EAE rats treated with LOV rather than vehicle (Figure 1D). This LOV-mediated increase in the levels of CYP27B1 transcripts was also evident in the kidneys of control rats treated with LOV rather than vehicle (Figure 1D). Consistent with RNA data, a significant increase in the level of CYP27B1 protein was observed in the kidneys of EAE rats treated with LOV rather than vehicle (Figure 1, E and F). A LOV-mediated increase in the level of CYP27B1 protein was also evident in the kidneys of control rats treated with LOV rather than vehicle (Figure 1, E and F). The observed elevated levels of CYP27B1 protein in the kidneys of EAE rats compared with controls (Figure 1, E and F) is indicative of an induction of compensatory mechanism to maintain 1,25-(OH)2D3 homeostasis in EAE rats. Similar to RNA data, no change in the CYP24A1 protein levels was observed in the kidneys of EAE versus controls (Figure 1, E and F). Moreover, the observed increase in the CYP24A1 protein level in the kidneys of EAE rats treated with LOV (Figure 1, E and F) is reflective of an induction of feedback inhibition loop wherein the 1,25-(OH)2D3 strongly induces the CYP24A1 gene.37 Taken together, these data provide evidence that systemic inflammation does not affect kidney physiology in EAE rats, whereas LOV tends to induce the CYP27B1 gene in the kidneys of treated EAE animals. These data explain the above-described findings relating to how LOV increases the circulatory levels of 1,25-(OH)2D3 in treated EAE rats.
Effect of LOV Treatment on in Situ Levels of 1,25-(OH)2D3 in the Spinal Cords of EAE Rats
The bioavailability of 1,25-(OH)2D3 in the spinal cord is most relevant to disease outcome as EAE pathology is confined to the CNS.1,2 Using methods published earlier,16 we determined in situ levels of 1,25-(OH)2D3 and compared them with the expressions of CYP24A1 and CYP27B1 in the spinal cords of EAE rats treated or not treated with LOV. At 16 dpi, the levels of 1,25-(OH)2D3 in the spinal cords were as follows: 23 ± 3 fmol/g in EAE, 39 ± 7 fmol/g in EAE plus LOV, and 37 ± 2 fmol/g in controls. Consistent with systemic levels of 1,25-(OH)2D3, the spinal cord levels of 1,25-(OH)2D3 were also reduced significantly in EAE rats treated with vehicle than LOV and compared with controls (P < 0.001 and P = 0.002, respectively). Likewise, levels of CYP24A1 transcripts were significantly elevated in the spinal cords of EAE rats treated with vehicle as compared with controls, and that was reversed by treatment with LOV (Figure 2A). Importantly, a significant increase in the level of CYP27B1 transcripts was observed in the spinal cords of EAE rats treated with LOV rather than vehicle (Figure 2A). The CYP27B1 transcripts level was also elevated in the spinal cords of LOV-treated controls rather than those treated with vehicle (Figure 2A). Consistent with RNA data, increased level of CYP24A1 protein in the spinal cords of EAE rats was attenuated by treatment with LOV (Figure 2, B and C). In addition, CYP27B1 protein level was elevated in the spinal cords of LOV-treated control rats rather than those treated with vehicle (Figure 2, B and C). These results confirmed our conclusions that neurological impairments due to an infiltration of autoreactive immune cells were co-related with reduced in situ levels of 1,25-(OH)2D3 in the spinal cords of EAE animals ascribed to its inactivation by 24-hydroxylase (CYP24A1). In contrast, LOV represses the CYP24A1 gene induction with corresponding induction of CYP27B1 gene that may eventually increase in situ levels of 1,25-(OH)2D3 in the spinal cord of EAE animals. These findings suggest that LOV-mediated regulation of the expression of vitamin D metabolizing enzymes in immune cells in the CNS may contribute to limit EAE pathogenesis via induction of the apoptosis of immune cells, as documented in an animal model of arthritis.38
Figure 2.
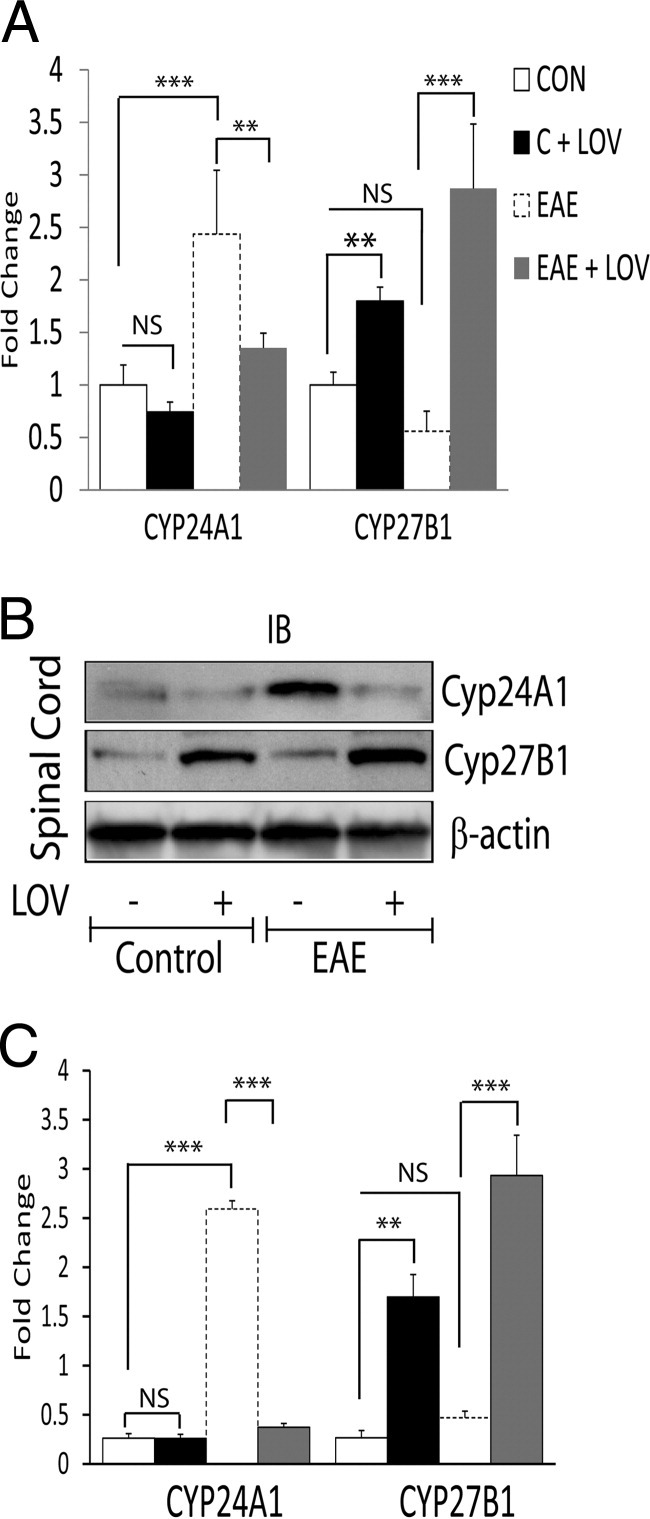
Effect of LOV on CYP24A1 and CYP27B1 expression in the spinal cords of EAE rats. Spinal cords were collected 16 dpi from EAE and control rats treated with LOV or vehicle as detailed in Figure 1 legend. A: Composite mean ± SEM of four to five samples per group of the levels of CYP24A1 and CYP27B1 transcripts analyzed by real-time PCR in the spinal cord tissues. B: Representative autoradiograph depicting the levels of CYP24A1, CYP27B1, and β-actin proteins analyzed by Western blotting in the spinal cord tissues. C: Histogram depicting the composite mean ± SEM of four to five samples per group of the levels of CYP24A1 and CYP27B1 proteins normalized to β-actin and compared with respective controls in the spinal cord tissues. Statistical significance as indicated: **P < 0.01; ***P < 0.001; NS, not significant.
Previous studies have documented that statin-mediated attenuation of CNS inflammation in EAE rats was ascribed to the inhibition of RhoA–ROCK signaling mechanism in immune cells.31,32 We next sought to confirm our hypothesis that LOV-mediated inhibition of RhoA–ROCK signaling participates in the maintenance of vitamin D homeostasis in treated EAE rats. To address this, we used various metabolites or inhibitors of the mevalonate pathway to treat EAE rats (Figure 3A), as described previously.31 LOV-mediated attenuation of the paralytic symptoms in association with decreased CNS inflammation in treated EAE rats was reversed by co-treatment with L-mevalolactone (MEV) or farnesol (FOH) (Table 2). Importantly, LOV-mediated protective effects in EAE rats were mimicked by treatment of EAE rats with geranylgeranyl transferase inhibitor (GGTI-298) or Rho kinase (ROCK) inhibitor (fasudil), but not with inhibitor of farnesyl transferase (FTI-277) (Table 2). Consistent with clinical exacerbations data, LOV-mediated decrease in the level of CYP24A1 transcripts in the spinal cords of treated EAE rats were abrogated by co-treatment with MEV (Figure 3B). Similar was the case for CYP27B1 gene induction, as LOV-mediated increases in the levels of CYP27B1 transcripts in the spinal cords of EAE rats were reversed by co-treatment with MEV (Figure 3B). However, LOV-mediated regulation of the induction of CYP24A1/CYP27B1 genes in the spinal cords of treated EAE rats were mimicked by treatment with GGTI-298 or fasudil, but not with FTI-277 (Figure 3C). Similar to the results of LOV plus MEV experiment, FOH co-treatment also abrogated the LOV-mediated regulation of the induction of CYP24A1/CYP27B1 genes in the spinal cords of EAE rats (Figure 3C). Together, these data confirmed our hypothesis that LOV-mediated increased bioavailability of 1,25-(OH)2D3 via regulation of CYP24A1/CYP27B1 gene inductions in the CNS of EAE animals is ascribed to the inhibition of RhoA–ROCK signaling mechanism.
Figure 3.

Effect of RhoA–ROCK signaling on the expressions of vitamin D–metabolizing enzymes in the spinal cords of EAE rats. EAE was induced in rats as detailed in the Figure 1 legend, and treatment with LOV alone or in combination with various metabolites of mevalonate pathway or specific enzyme inhibitor alone were initiated as described in Materials and Methods.A: Cartoon depicting the mevalonate pathway, and metabolites or enzyme inhibitors used to treat EAE rats are bolded. B: Plot depicts the levels of CYP24A1 and CYP27B1 transcripts analyzed by real-time PCR in duplicate in the spinal cords of EAE rats treated with LOV or metabolites of mevalonate pathway 16 dpi. C: Plot depicts the levels of CYP24A1 and CYP27B1 transcripts analyzed by real-time PCR in duplicate in the spinal cords of EAE and control rats treated with various inhibitors, ie, geranylgeranyl transferase (GGTI-298), farnesyl transferase (FTI-277) and RhoA kinase (fasudil). Results are expressed is the composite mean ± SEM from five rats per group of three separate experiments. Differences were statistical significance as indicated: **P < 0.01; ***P < 0.001; NS, not significant.
Effect of LOV on in Situ Levels of 1,25-(OH)2D3 in the Peripheral Lymphoid Organs of EAE Rats and MBP Sensitized CD4+ T Cells
The development of EAE is associated with infiltration of autoreactive CD4+ T cells to the CNS from peripheral lymphoid organs ie, spleen and draining lymph nodes. The immunosuppressive and apoptotic effects of 1,25-(OH)2D3 in immune cells are measurable by its in situ bioavailability.14 Thus, we next examined the levels of 1,25-(OH)2D3 in the peripheral lymphoid organs of EAE rats. Levels of 1,25-(OH)2D3 were as follows: 28 ± 7 fmol/g in EAE, 57 ± 8 fmol/g in EAE plus LOV, and 55 ± 7 fmol/g in the spleens of rats. Likewise, levels of 1,25-(OH)2D3 were as follows; 18 ± 3 fmol/g in EAE, 40 ± 6 fmol/g in EAE plus LOV, and 35 ± 4 fmol/g in controls in the draining lymph nodes of rats. Consistent with 1,25-(OH)2D3 plasma levels, in situ levels of 1, 25-(OH)2D3 in the peripheral lymphoid organs were significantly reduced in EAE rats compared with controls, and that was significantly rescued by treatment with LOV (P < 0.002 and P < 0.001, respectively). Together these results demonstrate that LOV-mediated increase in in situ levels of 1,25-(OH)2D3 in the peripheral lymphoid organs are contributing to limit the generation of autoreactive CD4+ T cells in EAE rats.
At least three mechanisms might explain how LOV represses the CYP24A1 gene induction in EAE rats more completely than treated with vehicle. LOV might directly/indirectly repress CYP24A1 gene or induces the CYP27B1 gene expression, and/or just the skewing of immune response from Th1-Th2 and Th17-Treg cells. To distinguish these possibilities, we next investigated whether MBP-sensitized CD4+ T cells are responsible for the depletion of 1,25-(OH)2D3 in the peripheral lymphoid organs and the CNS of EAE rats. Spleen cells from MBP-primed EAE 12 dpi and naive rats were cultured ex vivo in the presence or absence of MBP (50 μg/mL) and treated with LOV (5.0 μmol/L) or ROCK inhibitor, Y27643 (2.5 μmol/L), for 72 hours, as described in Materials and Methods. Then, purified CD4+ T cells from the nonadhering cell populations were used to analyze the induction of CYP27B1/CYP24A1 genes with parallel analysis of the induction of various cytokine genes. Interestingly, MBP-sensitized CD4+ T cells from EAE rats demonstrated no increase in the levels of CYP27B1 transcripts as compared with those from naive rats (Figure 4A). On the other hand, both LOV and Y27643 treatment demonstrated significant increase in the levels of CYP27B1 transcripts in MBP-sensitized CD4+ T cells from EAE and naive rats compared with vehicle-treated rats (Figure 4A). MBP-sensitized CD4+ T cells from EAE rats expressed higher levels of CYP24A1 transcripts compared with those from naive rats (Figure 4B). These increased levels of CYP24A1 transcripts were significantly reduced by treatment with LOV or Y27643 in MBP-sensitized CD4+ T cells from EAE rats (Figure 4B). However, no change in the expression of CYP24A1 was observed among MBP-sensitized CD4+ T cells from naive rats (Figure 4B). Because Th1 and Th17 cells can initiate and perpetuate the CNS inflammatory response in EAE, and because their development is under the strict influence of Th2 and Treg cells, respectively,4 we next analyzed the expression of the signatory molecules of these cell types. We found that the observed CYP27B1 and CYP24A1 RNA data were correlated with LOV- or Y27643-mediated skewing of Th1-Th2 and Th17-Treg phenotype responses. It was evident from a significant decrease in the levels of IFN-γ (Figure 4C) and IL-17 (Figure 4E) transcripts with corresponding increase in the levels of IL-4 (Figure 4D) and FoxP3 (Figure 4F) transcripts in LOV or Y27643 treated MBP-sensitized CD4+ T cells from EAE rats rather than naive rats. Because calcitriol is important in inducing the CYP24A1 gene,37 we next found that LOV attenuated the calcitriol-mediated induction of CYP24A1 gene in MBP-sensitized CD4+ T cells (Figure 4G). Furthermore, naive splenic CD4+ T cells that were differentiated under Th1 and Th17 polarizing conditions in vitro demonstrated the increased levels of CYP24A1 transcripts over those differentiated under Th2 and Treg polarization conditions (Figure 4H). Importantly, LOV treatment repressed the CYP24A1 gene induction in these differentiated CD4+ T cells under these polarizing conditions (Figure 4H). Together, these results provide evidence that MBP-sensitized CD4+ Th1 and Th17 cells had higher levels of the CYP24A1 transcripts compared with MBP-sensitized Th2 and Treg cells, which may be contributing to the inactivation of 1,25-(OH)2D3, thereby reducing its in situ levels in the peripheral lymphoid organs and the CNS of EAE animals. In contrast, interference with RhoA–ROCK signaling with LOV or ROCK inhibitor had limited the inactivation of 1,25-(OH)2D3 in autoreactive CD4+ T (Th1/Th17) cells.
Figure 4.

Effect of LOV and ROCK inhibitor on CYP24A1 and CYP27B1 expression in MBP-primed CD4+ T cells. Spleen cells were purified from the spleens of EAE rats by methods detailed under Materials and Methods. Spleen cells were stimulated with MBP (50 μg/mL) in the presence or absence of LOV (5 μmol/L). After 72 hours in culture, nonadhering cells were pelleted from the cultures and CD4+ T cells were purified using specific columns as described in Materials and Methods.A and B: Composite mean ± SEM for three to four samples analyzed in triplicate by real-time PCR for measuring the levels of CYP24A1 and CYP27B1 transcripts in MBP-sensitized CD4+ T cells. C–F: Composite mean ± SEM for IFN-γ, IL-4, IL-17, and FoxP3 transcripts levels in similarly treated MBP-sensitized CD4+ T cells. G: Composite mean ± SEM of three samples analyzed in triplicate by real-time PCR for the measurement of CYP24A1 transcripts in CD4+ T cells treated with calcitriol in the presence/absence of LOV for 24 hours. H: Composite mean of two samples per group analyzed in triplicate by real-time PCR to measure the levels of CYP24A1 transcripts in LOV-treated naive CD4+ T cells differentiated under Th1, Th17, Th2, and Treg polarizing conditions, as detailed in Materials and Methods. Differences were statistical significance as indicated: **P < 0.01 and not significant (NS) versus CON (vehicle); ††P < 0.01 and †††P < 0.001 versus EAE (vehicle); and ***P < 0.001 (G).
Effect of LOV on Immunosuppressive Activity of Vitamin D3 in EAE Rats
To distinguish whether this LOV-mediated increased in situ levels of 1,25-(OH)2D3 contributes to the immunosuppressive activities of vitamin D3 in EAE rats, we fed EAE rats a diet without vitamin D3 before MBP-priming and treatment with LOV or vitamin D3. Earlier, mice fed a diet without vitamin D3 are documented to show severe EAE-related exacerbations.13 Consistently, we also observed severe neurological impairments and inflammation in MBP-primed rats fed a diet deficient in vitamin D3, which were reduced moderately by treatment with LOV (Table 2). Similar was the case for MBP-primed rats treated with vitamin D3 (Table 2). Importantly, a LOV and vitamin D3 combination demonstrated greater protection in EAE rats than did their individual doses (Table 2). Moreover, the mortality rate in EAE rats was also reduced by a LOV plus vitamin D3 combination (Table 2). The skewing of Th1-Th2 phenotype response in the CNS of EAE rats was evident from changes in the transcripts levels of signatory cytokines, ie, IL-4 and IFN-γ, in the spinal cords of EAE rats treated with LOV or vitamin D3 (Figure 5A). This skewing of Th1 to Th2 was further enhanced in EAE rats receiving LOV and vitamin D3 in combination (Figure 5A). Likewise, LOV- or vitamin D3-mediated attenuation of the Th17 phenotypic response in EAE rats was evident from the reduced transcripts levels of Th17 signatory molecules, ie, IL-17A and ROR-γt in the spinal cords of treated EAE rats (Figure 5B). This attenuation of Th17 cells generation was enhanced further in EAE rats receiving LOV and vitamin D3 in combination (Figure 5B). Differentiation of naive T cells to Th17 cells is mediated by the secretion of IL-1β and IL-6 from dendritic cells and that synergizes with IL-23 to secrete IL-17 by CD4+ T cells.3 We found that the elevated levels of IL-23 and IL-6 transcripts in the spinal cords of EAE rats were reduced by treatment with LOV or vitamin D3 (Figure 5C). However, LOV and vitamin D3 in combination demonstrated enhanced reduction of these cytokines in the spinal cords of EAE rats (Figure 5C). Likewise, the generation of Treg cells was evident from the observed changes in the transcript levels of signatory protein, ie, CD25 and transcription factor, FoxP3 in the spinal cords of LOV or vitamin D3 treated EAE rats compared with those that received vehicle (Figure 5D). Combination treatment with LOV and vitamin D3 had enhanced expression of these mediators in the spinal cords of EAE rats (Figure 5D). Together, these data provide evidence that LOV-mediated regulation of vitamin D metabolism has an additive effect to improve the efficacy of vitamin D3 in EAE animals.
Figure 5.

Effect of LOV and vitamin D3 therapy on EAE disease in rats fed a vitamin D3–deficient diet. Adult rats were fed a synthetic diet formulated to provide no vitamin D3 for 20 days before priming to MBP. EAE was induced and treatment with LOV (2 mg/kg, ip) and vitamin D3 (0.25 mg/kg, po) in combination or individually were started after the onset of disease. Composite mean ± SEM of three to four samples per group analyzed in triplicate by real-time PCR for IL-4 and IFN-γ (A), IL-17A and ROR-γt (B), IL-23 and IL-6 (C), and CD25 and FoxP3 (D) transcripts in the spinal cords of EAE and control rats treated with LOV or vitamin D3 16 dpi. Differences were statistical significance as indicated: *P < 0.05; **P < 0.01; ***P < 0.001. NS, not significant.
Effect of LOV and Calcitriol Combination on CNS Inflammation and EAE Development
To further confirm that LOV-mediated increased efficacy of vitamin D3 in EAE animals is ascribable to the increased bioavailability of 1,25-(OH)2D3, we performed the preclinical testing of LOV and 1,25-(OH)2D3 (calcitriol) using their suboptimal doses in combination or alone. We performed our experimentation in rats with established EAE as in clinical settings of MS, therapeutic intervention is often started after the onset of the symptoms (Figure 6A). Administration of LOV and calcitriol in combination during established EAE significantly ameliorated neurological impairments as compared with those in vehicle-treated EAE rats and were better than the effects of their individual treatments (Figure 6B and Table 3). In addition, LOV and calcitriol combination treatment decreased the number of inflammatory lesions in the spinal cords of EAE rats compared with those treated with vehicle, which was better than the effects observed with their respective treatments (Table 3). Likewise, LOV and calcitriol in combination reduced the degree of demyelination in the spinal cords of EAE rats compared with those treated with vehicle, which was, again, greater than the effects seen with their respective treatments (Table 3).
Figure 6.
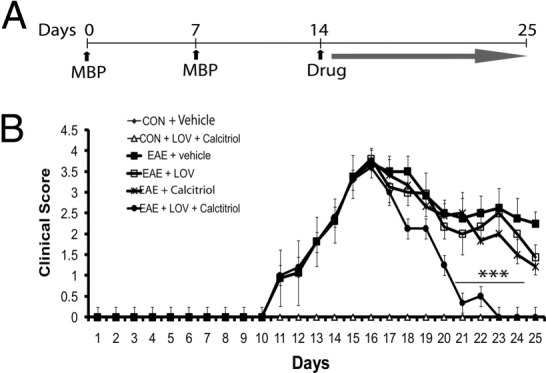
Effect of LOV and calcitriol combination on clinical exacerbation in EAE rats. EAE was induced in adult Lewis female rats as described in Figure 1 legend. Treatment with LOV (1 mg/kg) and calcitriol (1 μg/kg) was initiated individually or in combination in established EAE rats (14 dpi) and continued until the lessening of paralytic symptoms (25 dpi). A: Cartoon depicting the treatment strategy of EAE rats with LOV or calcitriol. B: Composite mean ± SEM of clinical scores of 10 to 12 rats per group evaluated in four separate experiments. LOV and calcitriol combination demonstrated significant lessening of paralytic symptoms 21 to 23 dpi and completely 25 dpi compared with their individual treatments or vehicle. Statistical significance as indicated: ***P < 0.001 versus EAE (vehicle).
Table 3.
Effect of Lovastatin (LOV) and Calcitriol Combination on Disease Severity in Experimental Autoimmune Encephalomyelitis (EAE) Rats
| Group | LOV (mg/kg/wt) | Calcitriol (μg/kg/wt) | Disease recovery, 25 dpi | Degree of demyelination | Sections with lesions (%) |
|---|---|---|---|---|---|
| Control | 0 | 0 | 0 | 0 | 0 |
| Control | 1 | 1 | 0 | 0 | 0 |
| EAE | 0 | 0 | 2.5 ± 1.0 | 2.23 ± 0.7 | 55 ± 15 |
| EAE | 1 | 0 | 2.0 ± 1.1⁎ | 1.7 ± 0.5⁎ | 35 ± 4⁎ |
| EAE | 0 | 1 | 2.1 ± 0.5⁎ | 1.8 ± 0.6⁎ | 42 ± 7⁎ |
| EAE | 1 | 1 | 0.5 + 0.3⁎⁎ | 1.0 ± 0.6⁎⁎⁎ | 6 ± 3⁎⁎ |
Disease severity shown as the composite mean ± SEM (n = 10 to 12) of the clinical cores evaluated in three separate experiments. Histological data are of five rats per group, in which three to four sections per rat were examined for the presence of lesions. The region of spinal cord depicting >10 nuclei was considered as lesion. Demyelination was categorized as score of 0 indicates no disease; grade 1 refers to foci of demyelination/axonal loss that is superficial and involves <25% of the lateral columns; grade 2 denotes deep foci that involve >25% of the lateral columns; and grade 3 denotes diffuse and widespread demyelination/axonal loss. Statistical tests were performed with Student's t-test (disease severity) and Mann–Whitney rank sum test (histopathological data). Statistical significance:
P < 0.05,
P < 0.01, and
P < 0.001 versus EAE plus vehicle.
To validate the observed improvements in neurological impairments, inflammation, and demyelination in EAE rats, we next evaluated the influence of LOV and calcitriol combination on T cell–mediated immunity in treated EAE rats. Administration of LOV plus calcitriol combination attenuated Th17 phenotype responses, as evident from the reduced levels of the IL-1β, TNF-α, IL-17A, and IL-6 proteins in the spinal cords of EAE rats compared with those treated with vehicle only (Table 4). In addition, the administration of LOV plus calcitriol had favored the generation of Treg cells as evident from the increased levels of the TGF-β1 and IL-10 proteins in the spinal cords of EAE rats as compared with those treated with vehicle only (Table 4). Importantly, LOV plus calcitriol combination effects in EAE rats were greater than the effects observed with their respective treatments (Table 4). Together, these data provide evidence that LOV-mediated regulation of vitamin D metabolism improves vitamin D3 efficacy to confer protection in EAE animals ascribable to the immunomodulatory synergy between LOV and 1,25-(OH)2D3 (calcitriol).
Table 4.
Effect of Lovastatin (LOV) and Calcitriol Combination on Cytokines in the Spinal Cords of Experimental Autoimmune Encephalomyelitis (EAE) Rats
| Groups | CON | CON + LOV + calcitriol | EAE | EAE + LOV | EAE + calcitriol | EAE + LOV + calcitriol |
|---|---|---|---|---|---|---|
| IL-1β | 150 ± 41 | 122 ± 35 | 625 ± 26 | 389 ± 38⁎⁎ | 344 ± 75⁎⁎ | 169 ± 39⁎⁎⁎ |
| TNF-α | 6 ± 2.2 | 9.6 ± 2 | 38 ± 4 | 19.3 ± 1.8⁎⁎ | 15.1 ± 3.9⁎⁎ | 10.8 ± 0.2⁎⁎⁎ |
| IL-17A | 30 ± 4.5 | 29 ± 7.5 | 226 ± 15 | 93 ± 0.2⁎⁎ | 116 ± 22.3⁎⁎ | 28 ± 9⁎⁎⁎ |
| IL-6 | 15 ± 0.9 | 22 ± 6 | 129 ± 8 | 46.2 ± 3⁎⁎ | 61 ± 5⁎⁎ | 29 ± 3⁎⁎⁎ |
| TGF-β1 | 10 ± 2 | 13 ± 2.3 | 25 ± 3.6 | 71 ± 11⁎⁎ | 79 ± 7⁎⁎ | 161 ± 12⁎⁎⁎ |
| IL-10 | 7 ± 0.2 | 9 ± 2.1 | 15 ± 2.8 | 45 ± 8⁎⁎ | 53 ± 5.7⁎⁎ | 123 ± 21.4⁎⁎⁎ |
Concentration of cytokines and neurotrophic factors (pg/mg of spinal cord tissue homogenate) shown as composite mean ± SEM (n = 10 to 12) evaluated in three separate experiments. Statistical tests were performed using one-way multiple-range analysis of variance followed by Benferrroni post-test correction. Statistical significance:
P < 0.01 and
P < 0.001 versus EAE plus vehicle.
Discussion
Vitamin D deficiency is a risk factor for MS patients, as dietary intake of vitamin D3 or higher circulatory 1,25-(OH)2D3 levels are reported to reduce MS prevalence.18,19 Our findings provide unprecedented evidence that autoreactive Th1/Th17 CD4+ cells are responsible for vitamin D deficiency in EAE animals, which can be rescued by interfering with the RhoA–ROCK signaling pathway.
Importantly, the reduced circulating levels of 25-OH-D3 and 1,25-(OH)2D3 during the peak clinical day, but without significant change on recovery phase in EAE animals compared with controls (Figure 1, A–C), were consistent with previous published studies in EAE and MS.18,39 This alteration of 25-OH-D3 and 1,25-(OH)2D3 levels in EAE animals were rescued by treatment with LOV (Figure 1, A–C). Likewise, the reduced in situ levels of 1,25-(OH)2D3 and increased CYP24A1 (24-hydroxylase) expressions in the peripheral lymph nodes and spinal cords of EAE animals were consistent with previous study16 and were reversed by LOV with a corresponding increase in CYP27B1 (1α-hydroxylase) expressions in treated EAE animals (Figure 2, A–C). Importantly, EAE disease had no effect on the CYP24A1/CYP27B1 expressions in the kidneys of EAE animals. On the contrary, LOV enhanced CYP27B1 expression in the kidneys of treated EAE and control animals (Figure 1, D–F). This LOV-mediated regulation of CYP24A1/CYP27B1 expression in the CNS of EAE animals (Figure 3, B and C) and in MBP-sensitized CD4+ Th1/Th17 cells in ex vivo and in vitro studies (Figure 4) was ascribed to the inhibition of the RhoA–ROCK signaling pathway. We next found that LOV-mediated regulation of the expression of vitamin D metabolizing enzymes had improved vitamin D3 efficacy to confer protection in EAE animals (Table 2 and Figure 5), which was ascribed to immunomodulatory synergy between LOV- and calcitriol-induced protective activities (Tables 3 and 4 and Figure 6).
Protective effects of 1,25-(OH)2D3 are attributed to its interaction with receptor, VDR, that inhibits the proliferation and differentiation of myelin-reactive CD4+ T cells in EAE40 as well as regulates the expressions of CYP24A1/CYP27B1 in cells.37 The observed reduced in situ levels of 1,25-(OH)2D3 in the peripheral lymphoid organs and the spinal cords of EAE animals were associated with increased number of autoreactive immune cells (Th1/Th17) revealed by their higher signatory transcript levels (Figure 2), expressing higher levels of CYP24A1 that lead to the inactivation of 1,25-(OH)2D3. This was in agreement with a previously documented sexual disparity for vitamin D metabolism in EAE and MS that shows positive correlation with increased expression of CYP24A1 in autoreactive CD4+ T cells and inactivation of 1,25-(OH)2D3 in males over that in females.16,39 Interestingly, LOV-mediated inhibition of CYP24A1 gene expression in Th1/Th17 cells may be allowing 1,25-(OH)2D3 to accumulate in the peripheral lymphoid organs and spinal cords. The exact mechanism by which LOV regulates the expressions of CYP24A1/CYP27B1 in immune cells warrants further investigation. Importantly, our data showed that LOV-mediated regulation of the expression of CYP24A1 and CYP27B1 genes was ascribed to the inhibition of RhoA–ROCK signaling pathway in the spinal cords of EAE animals (Figure 3, B and C) as well as in MBP-sensitized CD4+ T (Th1/Th17) cells (Figure 4, A and B). Importantly, the induction of CYP24A1 and CYP27B1 genes is tightly regulated by 1,25-(OH)2D3-/VDR induced transcriptional activities in cells.37 The dimerization of VDR with retinoid X receptor (RXR) promotes its nuclear localization and subnuclear targeting to the promoter of target gene.41,42 Peroxisome proliferator–activated receptor–γ (PPAR-γ) is known to heterodimerize with RXR as VDR to induce the targeted gene expression in cells.43 We and others reported earlier that statin induces the transcriptional activity of PPAR-γ in cells via inhibition of RhoA–ROCK signaling pathway.44,45 Based on this information, we postulate that statin-induced PPAR-γ transcriptional activity may be participating in the regulation of the expression of vitamin D metabolizing enzyme in treated EAE animals. We reasoned that PPAR-γ activation may compete with VDR for common RXR ligand, ie, 9-cis retinoic acid, which, in turn, represses CYP24A1 gene induction, but switches on CYP27B1 gene induction in cells, as activated VDR is known to repress CYP27B1 gene induction in cells.37 The present findings provide unprecedented evidence that interference with the RhoA–ROCK signaling pathway in immune cells increases 1,25-(OH)2D3, levels both locally and systemically. Therefore, a greater understanding of possible cross-talk between PPAR-γ and VDR in immune cells will suggest new therapeutic interventions for MS management. The availability of conditional knockout mice for cell-specific deletion of RhoA and ROCK will expand our understanding of molecular mechanisms that participate in the maintenance of vitamin D homeostasis.
Both 1,25-(OH)2D3 and statin are documented to attenuate EAE disease development via inhibition of Th17 phenotype response with generation of Treg cells and induction of immunomodulatory Th1 to Th2 shift.13–16,35,40,46 In addition, higher concentrations of 1,25-(OH)2D3 are reported to sensitize the inflammatory cells to apoptotic signals.17,38 A recent study documented that calcitriol-mediated immunomodulatory activities are linked with induction of VDR transcriptional activity in Th1/Th17 cells in EAE animals.15 This signifies the importance of observed skewing of Th1–Th2 and Th17–Treg responses in EAE animals treated with LOV in combination with vitamin D3 or calcitriol (Figures 5 and 6 and Tables 2–4), attributed to their immunomodulatory synergy to confer protection in EAE animals. It is noteworthy that both LOV and 1,25-(OH)2D3 can cross the blood–brain barrier because of their lipophilic nature.47,48 Therefore, the observed protective effects of LOV and 1,25-(OH)2D3 in the CNS of EAE animals are not restricted to immune cells but are found in part in brain cells also, as they are known to express VDR and CYP27B1.49,50 In addition, statins are known to attenuate the activation of brain cells in vitro and in vivo.31,34,51–53 Therefore, further studies are warranted with transgenic/knockout models with cell-type expression of vitamin D metabolizing enzymes to distinguish between the direct effect of these agents on inflammatory immune cells and brain cells that confer protection in EAE model.
Findings from the present study are novel that provide a rationale to test statin or other inhibitors of RhoA–ROCK signaling ie, fasudil or chemical inhibitor of CYP24A1 (presently under investigation in cancer54,55) in combination with reduced dose of vitamin D3 to minimize the potential side effects of its higher dose when prescribed for longer duration in patients with MS. Clinical trials conducted so far with vitamin D3 in small numbers of MS patients have shown efficacy.20,21 Importantly, 1, 25-(OH)2D3 analog in combination with interferon β1 (IFNB-1b) showed superior protection in EAE animals as compared with their respective treatments.23 These findings provide an opportunity to improve current treatment strategies for MS, through combination therapy with an agent that can increase the bioavailability of 1,25-(OH)2D3. Use of statins for MS is still controversial, as results from statin trials are mixed.29,56,57 One study reported that the combination of a statin with IFNB was antagonistic,56 whereas other small studies suggested that there could be benefit.28,29 Likewise, of two recent reports, one showed the adverse effect of statin in combination with IFNB in a phase II clinical trial in MS patients, and the second showed no beneficial effects.58,59 More recently, a multicenter randomized clinical trial with atorvastatin as add-on therapy to IFNB-1b in younger MS patients showed no beneficial effects.60 One possibility of this failure of statin as add-on therapy in combination with IFNB trials could be their antagonizing mechanism of actions in MS.60,61 However, this does not rule out the possibility of testing statin in combination with other MS therapeutics (glatiramer acetate) or disease-modifying agents that demonstrate protection in EAE.62–65 Importantly, the present study suggests that a combination of statin with disease-modifying agent, ie, vitamin D3 as add-on therapy with Food and Drug Administration–approved MS drugs will make them useful for the management of MS.
In summary, the present study demonstrates that reduced circulating levels of 25-OH-D3 and 1,25-(OH)2D3 and in situ levels of 1,25-(OH)2D3 in the peripheral lymphoid organs and CNS of EAE animals can be rescued by interfering with the RhoA–ROCK signaling pathway in autoreactive immune cells. Findings from this study provide evidence that targeting of the RhoA–ROCK signaling pathway in autoreactive CD4+ T cells with statin or other chemical inhibitors as add-on therapy to MS therapeutics will reduce the dose of vitamin D3 in clinical trials of MS and related neurodegenerative disorders, including diabetic neuropathy.
Acknowledgments
We thank Joyce Brian and Carrie Barnes for technical assistance.
Footnotes
Supported by grants from the NIH (NS-22576, NS-37766, 1BX001072, C06-RR015455, and C06-RR018823).
A.S.P. and M.K.P. contributed equally to this work.
References
- 1.Al-Omaishi J., Bashir R., Gendelman H.E. The cellular immunology of multiple sclerosis. J Leukoc Biol. 1999;65:444–452. doi: 10.1002/jlb.65.4.444. [DOI] [PubMed] [Google Scholar]
- 2.Prineas J.W., Barnard R.O., Kwon E.E., Sharer L.R., Cho E.S. Multiple sclerosis: remyelination of nascent lesions. Ann Neurol. 1993;33:137–151. doi: 10.1002/ana.410330203. [DOI] [PubMed] [Google Scholar]
- 3.Bettelli E., Carrier Y., Gao W., Korn T., Strom T.B., Oukka M., Weiner H.L., Kuchroo V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 4.Zhu J., Paul W.E. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev. 2010;238:247–262. doi: 10.1111/j.1600-065X.2010.00951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marrie R.A. Environmental risk factors in multiple sclerosis aetiology. Lancet Neurol. 2004;3:709–718. doi: 10.1016/S1474-4422(04)00933-0. [DOI] [PubMed] [Google Scholar]
- 6.Beretich B.D., Beretich T.M. Explaining multiple sclerosis prevalence by ultraviolet exposure: a geospatial analysis. Mult Scler. 2009;15:891–898. doi: 10.1177/1352458509105579. [DOI] [PubMed] [Google Scholar]
- 7.Hayes C.E. Vitamin D: a natural inhibitor of multiple sclerosis. Proc Nutr Soc. 2000;59:531–535. doi: 10.1017/s0029665100000768. [DOI] [PubMed] [Google Scholar]
- 8.Omdahl J.L., Morris H.A., May B.K. Hydroxylase enzymes of the vitamin D pathway: expression, function, and regulation. Annu Rev Nutr. 2002;22:139–166. doi: 10.1146/annurev.nutr.22.120501.150216. [DOI] [PubMed] [Google Scholar]
- 9.Ramagopalan S.V., Dyment D.A., Cader M.Z., Morrison K.M., Disanto G., Morahan J.M., Berlanga-Taylor A.J., Handel A., De Luca G.C., Sadovnick A.D., Lepage P., Montpetit A., Ebers G.C. Rare variants in the CYP27B1 gene are associated with multiple sclerosis. Ann Neurol. 2011;70:881–886. doi: 10.1002/ana.22678. [DOI] [PubMed] [Google Scholar]
- 10.Haussler M.R., Whitfield G.K., Haussler C.A., Hsieh J.C., Thompson P.D., Selznick S.H., Dominguez C.E., Jurutka P.W. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res. 1998;13:325–349. doi: 10.1359/jbmr.1998.13.3.325. [DOI] [PubMed] [Google Scholar]
- 11.Fukazawa T., Yabe I., Kikuchi S., Sasaki H., Hamada T., Miyasaka K., Tashiro K. Association of vitamin D receptor gene polymorphism with multiple sclerosis in Japanese. J Neurol Sci. 1999;166:47–52. doi: 10.1016/s0022-510x(99)00112-4. [DOI] [PubMed] [Google Scholar]
- 12.Niino M., Fukazawa T., Yabe I., Kikuchi S., Sasaki H., Tashiro K. Vitamin D receptor gene polymorphism in multiple sclerosis and the association with HLA class II alleles. J Neurol Sci. 2000;177:65–71. doi: 10.1016/s0022-510x(00)00336-1. [DOI] [PubMed] [Google Scholar]
- 13.Cantorna M.T., Hayes C.E., DeLuca H.F. 1,25-Dihydroxyvitamin D3 reversibly blocks the progression of relapsing encephalomyelitis, a model of multiple sclerosis. Proc Natl Acad Sci USA. 1996;93:7861–7864. doi: 10.1073/pnas.93.15.7861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spach K.M., Pedersen L.B., Nashold F.E., Kayo T., Yandell B.S., Prolla T.A., Hayes C.E. Gene expression analysis suggests that 1,25-dihydroxyvitamin D3 reverses experimental autoimmune encephalomyelitis by stimulating inflammatory cell apoptosis. Physiol Genomics. 2004;18:141–151. doi: 10.1152/physiolgenomics.00003.2004. [DOI] [PubMed] [Google Scholar]
- 15.Joshi S., Pantalena L.C., Liu X.K., Gaffen S.L., Liu H., Rohowsky-Kochan C., Ichiyama K., Yoshimura A., Steinman L., Christakos S., Youssef S. 1,25-Dihydroxyvitamin D(3) ameliorates Th17 autoimmunity via transcriptional modulation of interleukin-17A. Mol Cell Biol. 2011;31:3653–3669. doi: 10.1128/MCB.05020-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spach K.M., Hayes C.E. Vitamin D3 confers protection from autoimmune encephalomyelitis only in female mice. J Immunol. 2005;175:4119–4126. doi: 10.4049/jimmunol.175.6.4119. [DOI] [PubMed] [Google Scholar]
- 17.Mayne C.G., Spanier J.A., Relland L.M., Williams C.B., Hayes C.E. 1,25-Dihydroxyvitamin D3 acts directly on the T lymphocyte vitamin D receptor to inhibit experimental autoimmune encephalomyelitis. Eur J Immunol. 2011;41:822–832. doi: 10.1002/eji.201040632. [DOI] [PubMed] [Google Scholar]
- 18.Munger K.L., Levin L.I., Hollis B.W., Howard N.S., Ascherio A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA. 2006;296:2832–2838. doi: 10.1001/jama.296.23.2832. [DOI] [PubMed] [Google Scholar]
- 19.Munger K.L., Zhang S.M., O'Reilly E., Hernan M.A., Olek M.J., Willett W.C., Ascherio A. Vitamin D intake and incidence of multiple sclerosis. Neurology. 2004;62:60–65. doi: 10.1212/01.wnl.0000101723.79681.38. [DOI] [PubMed] [Google Scholar]
- 20.Burton J.M., Kimball S., Vieth R., Bar-Or A., Dosch H.M., Cheung R., Gagne D., D'Souza C., Ursell M., O'Connor P. A phase I/II dose-escalation trial of vitamin D3 and calcium in multiple sclerosis. Neurology. 2010;74:1852–1859. doi: 10.1212/WNL.0b013e3181e1cec2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stein M.S., Liu Y., Gray O.M., Baker J.E., Kolbe S.C., Ditchfield M.R., Egan G.F., Mitchell P.J., Harrison L.C., Butzkueven H., Kilpatrick T.J. A randomized trial of high-dose vitamin D2 in relapsing-remitting multiple sclerosis. Neurology. 2011;77:1611–1618. doi: 10.1212/WNL.0b013e3182343274. [DOI] [PubMed] [Google Scholar]
- 22.Kimball S.M., Ursell M.R., O'Connor P., Vieth R. Safety of vitamin D3 in adults with multiple sclerosis. Am J Clin Nutr. 2007;86:645–651. doi: 10.1093/ajcn/86.3.645. [DOI] [PubMed] [Google Scholar]
- 23.Smolders J., Hupperts R., Barkhof F., Grimaldi L.M., Holmoy T., Killestein J., Rieckmann P., Schluep M., Vieth R., Hostalek U., Ghazi-Visser L., Beelke M. Efficacy of vitamin D(3) as add-on therapy in patients with relapsing-remitting multiple sclerosis receiving subcutaneous interferon beta-1a: a phase II, multicenter, double-blind, randomized, placebo-controlled trial. J Neurol Sci. 2011;311:44–49. doi: 10.1016/j.jns.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 24.Kamezaki F., Sonoda S., Tomotsune Y., Yunaka H., Otsuji Y. Seasonal variation in serum lipid levels in Japanese workers. J Atheroscler Thromb. 2010;17:638–643. doi: 10.5551/jat.3566. [DOI] [PubMed] [Google Scholar]
- 25.Woodhouse P.R., Khaw K.T., Plummer M. Seasonal variation of serum lipids in an elderly population. Age Ageing. 1993;22:273–278. doi: 10.1093/ageing/22.4.273. [DOI] [PubMed] [Google Scholar]
- 26.Perez-Castrillon J.L., Vega G., Abad L., Sanz A., Chaves J., Hernandez G., Duenas A. Effects of Atorvastatin on vitamin D levels in patients with acute ischemic heart disease. Am J Cardiol. 2007;99:903–905. doi: 10.1016/j.amjcard.2006.11.036. [DOI] [PubMed] [Google Scholar]
- 27.Ertugrul D.T., Yavuz B., Cil H., Ata N., Akin K.O., Kucukazman M., Yalcin A.A., Dal K., Yavuz B.B., Tutal E. STATIN-D Study: comparison of the influences of rosuvastatin and fluvastatin treatment on the levels of 25 hydroxyvitamin D. Cardiovasc Ther. 2010;2:146–152. doi: 10.1111/j.1755-5922.2010.00141.x. [DOI] [PubMed] [Google Scholar]
- 28.Vollmer T., Key L., Durkalski V., Tyor W., Corboy J., Markovic-Plese S., Preiningerova J., Rizzo M., Singh I. Oral simvastatin treatment in relapsing-remitting multiple sclerosis. Lancet. 2004;363:1607–1608. doi: 10.1016/S0140-6736(04)16205-3. [DOI] [PubMed] [Google Scholar]
- 29.Lanzillo R., Orefice G., Quarantelli M., Rinaldi C., Prinster A., Ventrella G., Spitaleri D., Lus G., Vacca G., Carotenuto B., Salvatore E., Brunetti A., Tedeschi G., Brescia Morra V. Atorvastatin combined to interferon to verify the efficacy (ACTIVE) in relapsing-remitting active multiple sclerosis patients: a longitudinal controlled trial of combination therapy. Mult Scler. 2010;16:450–454. doi: 10.1177/1352458509358909. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X., Jin J., Peng X., Ramgolam V.S., Markovic-Plese S. Simvastatin inhibits IL-17 secretion by targeting multiple IL-17-regulatory cytokines and by inhibiting the expression of IL-17 transcription factor RORC in CD4+ lymphocytes. J Immunol. 2008;180:6988–6996. doi: 10.4049/jimmunol.180.10.6988. [DOI] [PubMed] [Google Scholar]
- 31.Paintlia A.S., Paintlia M.K., Singh A.K., Singh I. Inhibition of rho family functions by lovastatin promotes myelin repair in ameliorating experimental autoimmune encephalomyelitis. Mol Pharmacol. 2008;73:1381–1393. doi: 10.1124/mol.107.044230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dunn S.E., Youssef S., Goldstein M.J., Prod'homme T., Weber M.S., Zamvil S.S., Steinman L. Isoprenoids determine Th1/Th2 fate in pathogenic T cells, providing a mechanism of modulation of autoimmunity by atorvastatin. J Exp Med. 2006;203:401–412. doi: 10.1084/jem.20051129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nobes C.D., Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 34.Paintlia A.S., Paintlia M.K., Khan M., Vollmer T., Singh A.K., Singh I. HMG-CoA reductase inhibitor augments survival and differentiation of oligodendrocyte progenitors in animal model of multiple sclerosis. FASEB J. 2005;19:1407–1421. doi: 10.1096/fj.05-3861com. [DOI] [PubMed] [Google Scholar]
- 35.Paintlia A.S., Paintlia M.K., Singh A.K., Stanislaus R., Gilg A.G., Barbosa E., Singh I. Regulation of gene expression associated with acute experimental autoimmune encephalomyelitis by lovastatin. J Neurosci Res. 2004;77:63–81. doi: 10.1002/jnr.20130. [DOI] [PubMed] [Google Scholar]
- 36.Hollis B.W. Quantitation of 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D by radioimmunoassay using radioiodinated tracers. Methods Enzymol. 1997;282:174–186. doi: 10.1016/s0076-6879(97)82106-4. [DOI] [PubMed] [Google Scholar]
- 37.Kerry D.M., Dwivedi P.P., Hahn C.N., Morris H.A., Omdahl J.L., May B.K. Transcriptional synergism between vitamin D-responsive elements in the rat 25-hydroxyvitamin D3 24-hydroxylase (CYP24) promoter. J Biol Chem. 1996;271:29715–29721. doi: 10.1074/jbc.271.47.29715. [DOI] [PubMed] [Google Scholar]
- 38.Cantorna M.T., Hayes C.E., DeLuca H.F. 1,25-Dihydroxycholecalciferol inhibits the progression of arthritis in murine models of human arthritis. J Nutr. 1998;128:68–72. doi: 10.1093/jn/128.1.68. [DOI] [PubMed] [Google Scholar]
- 39.Correale J., Ysrraelit M.C., Gaitan M.I. Gender differences in 1,25 dihydroxyvitamin D3 immunomodulatory effects in multiple sclerosis patients and healthy subjects. J Immunol. 2010;185:4948–4958. doi: 10.4049/jimmunol.1000588. [DOI] [PubMed] [Google Scholar]
- 40.Mayne C.G., Spanier J.A., Relland L.M., Williams C.B., Hayes C.E. 1,25-Dihydroxyvitamin D3 acts directly on the T lymphocyte vitamin D receptor to inhibit experimental autoimmune encephalomyelitis. Eur J Immunol. 2011;41:822–832. doi: 10.1002/eji.201040632. [DOI] [PubMed] [Google Scholar]
- 41.Bettoun D.J., Burris T.P., Houck K.A., Buck D.W., 2nd, Stayrook K.R., Khalifa B., Lu J., Chin W.W., Nagpal S. Retinoid X receptor is a nonsilent major contributor to vitamin D receptor-mediated transcriptional activation. Mol Endocrinol. 2003;17:2320–2328. doi: 10.1210/me.2003-0148. [DOI] [PubMed] [Google Scholar]
- 42.Prufer K., Racz A., Lin G.C., Barsony J. Dimerization with retinoid X receptors promotes nuclear localization and subnuclear targeting of vitamin D receptors. J Biol Chem. 2000;275:41114–41123. doi: 10.1074/jbc.M003791200. [DOI] [PubMed] [Google Scholar]
- 43.Szeles L., Poliska S., Nagy G., Szatmari I., Szanto A., Pap A., Lindstedt M., Santegoets S.J., Ruhl R., Dezso B., Nagy L. Research resource: transcriptome profiling of genes regulated by RXR and its permissive and nonpermissive partners in differentiating monocyte-derived dendritic cells. Mol Endocrinol. 2010;24:2218–2231. doi: 10.1210/me.2010-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen Y., Wu H., Wang C., Shao H., Huang H., Jing H., Li D. Simvastatin attenuates cardiopulmonary bypass-induced myocardial inflammatory injury in rats by activating peroxisome proliferator-activated receptor gamma. Eur J Pharmacol. 2010;649:255–262. doi: 10.1016/j.ejphar.2010.08.058. [DOI] [PubMed] [Google Scholar]
- 45.Paintlia A.S., Paintlia M.K., Singh A.K., Orak J.K., Singh I. Activation of PPAR-gamma and PTEN cascade participates in lovastatin-mediated accelerated differentiation of oligodendrocyte progenitor cells. Glia. 2010;58:1669–1685. doi: 10.1002/glia.21039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nath N., Giri S., Prasad R., Singh A.K., Singh I. Potential targets of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor for multiple sclerosis therapy. J Immunol. 2004;172:1273–1286. doi: 10.4049/jimmunol.172.2.1273. [DOI] [PubMed] [Google Scholar]
- 47.Botti R.E., Triscari J., Pan H.Y., Zayat J. Concentrations of pravastatin and lovastatin in cerebrospinal fluid in healthy subjects. Clin Neuropharmacol. 1991;14:256–261. doi: 10.1097/00002826-199106000-00010. [DOI] [PubMed] [Google Scholar]
- 48.Kalueff A.V., Minasyan A., Keisala T., Kuuslahti M., Miettinen S., Tuohimaa P. The vitamin D neuroendocrine system as a target for novel neurotropic drugs. CNS Neurol Disord Drug Targets. 2006;5:363–371. doi: 10.2174/187152706784111506. [DOI] [PubMed] [Google Scholar]
- 49.Eyles D.W., Smith S., Kinobe R., Hewison M., McGrath J.J. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat. 2005;29:21–30. doi: 10.1016/j.jchemneu.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 50.Baas D., Prufer K., Ittel M.E., Kuchler-Bopp S., Labourdette G., Sarlieve L.L., Brachet P. Rat oligodendrocytes express the vitamin D(3) receptor and respond to 1,25–dihydroxyvitamin D(3) Glia. 2000;31:59–68. doi: 10.1002/(sici)1098-1136(200007)31:1<59::aid-glia60>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 51.Kawashima S., Yamashita T., Miwa Y., Ozaki M., Namiki M., Hirase T., Inoue N., Hirata K., Yokoyama M. HMG-CoA reductase inhibitor has protective effects against stroke events in stroke-prone spontaneously hypertensive rats. Stroke. 2003;34:157–163. doi: 10.1161/01.str.0000048213.18751.52. [DOI] [PubMed] [Google Scholar]
- 52.Bi X., Baudry M., Liu J., Yao Y., Fu L., Brucher F., Lynch G. Inhibition of geranylgeranylation mediates the effects of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors on microglia. J Biol Chem. 2004;279:48238–48245. doi: 10.1074/jbc.M405442200. [DOI] [PubMed] [Google Scholar]
- 53.Cordle A., Landreth G. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors attenuate beta-amyloid-induced microglial inflammatory responses. J Neurosci. 2005;25:299–307. doi: 10.1523/JNEUROSCI.2544-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iglesias-Gato D., Zheng S., Flanagan J.N., Jiang L., Kittaka A., Sakaki T., Yamamoto K., Itoh T., Lebrasseur N.K., Norstedt G., Chen T.C. Substitution at carbon 2 of 19-nor-1alpha,25-dihydroxyvitamin D(3) with 3-hydroxypropyl group generates an analogue with enhanced chemotherapeutic potency in PC-3 prostate cancer cells. J Steroid Biochem Mol Biol. 2011;127:269–275. doi: 10.1016/j.jsbmb.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 55.Muindi J.R., Yu W.D., Ma Y., Engler K.L., Kong R.X., Trump D.L., Johnson C.S. CYP24A1 inhibition enhances the antitumor activity of calcitriol. Endocrinology. 2010;151:4301–4312. doi: 10.1210/en.2009-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Birnbaum G., Cree B., Altafullah I., Zinser M., Reder A.T. Combining beta interferon and atorvastatin may increase disease activity in multiple sclerosis. Neurology. 2008;71:1390–1395. doi: 10.1212/01.wnl.0000319698.40024.1c. [DOI] [PubMed] [Google Scholar]
- 57.Paul F., Waiczies S., Wuerfel J., Bellmann-Strobl J., Dorr J., Waiczies H., Haertle M., Wernecke K.D., Volk H.D., Aktas O., Zipp F. Oral high-dose atorvastatin treatment in relapsing-remitting multiple sclerosis. PLoS One. 2008;3:e1928. doi: 10.1371/journal.pone.0001928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sorensen P.S., Lycke J., Eralinna J.P., Edland A., Wu X., Frederiksen J.L., Oturai A., Malmestrom C., Stenager E., Sellebjerg F., Sondergaard H.B. Simvastatin as add-on therapy to interferon beta-1a for relapsing-remitting multiple sclerosis (SIMCOMBIN study): a placebo-controlled randomised phase 4 trial. Lancet Neurol. 2011;10:691–701. doi: 10.1016/S1474-4422(11)70144-2. [DOI] [PubMed] [Google Scholar]
- 59.Rudick R.A., Pace A., Rani M.R., Hyde R., Panzara M., Appachi S., Shrock J., Maurer S.L., Calabresi P.A., Confavreux C., Galetta S.L., Lublin F.D., Radue E.W., Ransohoff R.M. Effect of statins on clinical and molecular responses to intramuscular interferon beta-1a. Neurology. 2009;72:1989–1993. doi: 10.1212/WNL.0b013e3181a92b96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kamm C.P., El-Koussy M., Humpert S., Findling O., von Bredow F., Burren Y., Schwegler G., Schott D., Donati F., Muller M., Goebels N., Muller F., Slotboom J., Tettenborn B., Kappos L., Naegelin Y., Mattle H.P. Atorvastatin added to interferon beta for relapsing multiple sclerosis: a randomized controlled trial. J Neurol. 2012 doi: 10.1007/s00415-012-6513-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zamvil S.S., Steinman L. Combining statins with interferon beta in multiple sclerosis: think twice, it might not be all right. Lancet Neurol. 2011;10:672–673. doi: 10.1016/S1474-4422(11)70153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stuve O., Youssef S., Weber M.S., Nessler S., von Budingen H.C., Hemmer B., Prod'homme T., Sobel R.A., Steinman L., Zamvil S.S. Immunomodulatory synergy by combination of atorvastatin and glatiramer acetate in treatment of CNS autoimmunity. J Clin Invest. 2006;116:1037–1044. doi: 10.1172/JCI25805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paintlia A.S., Paintlia M.K., Singh I., Singh A.K. Combined medication of lovastatin with rolipram suppresses severity of experimental autoimmune encephalomyelitis. Exp Neurol. 2008;214:168–180. doi: 10.1016/j.expneurol.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Paintlia A.S., Paintlia M.K., Singh I., Singh A.K. Immunomodulatory effect of combination therapy with lovastatin and 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside alleviates neurodegeneration in experimental autoimmune encephalomyelitis. Am J Pathology. 2006;169:1012–1025. doi: 10.2353/ajpath.2006.051309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luccarini I., Ballerini C., Biagioli T., Biamonte F., Bellucci A., Rosi M.C., Grossi C., Massacesi L., Casamenti F. Combined treatment with atorvastatin and minocycline suppresses severity of EAE. Exp Neurol. 2008;211:214–226. doi: 10.1016/j.expneurol.2008.01.022. [DOI] [PubMed] [Google Scholar]