

Abstract
Fibrinogen (Fg) is significantly up-regulated in the kidney after acute kidney injury (AKI). We evaluated the performance of Fg as a biomarker for early detection of AKI. In rats and mice with kidney tubular damage induced by ischemia/reperfusion (I/R) or cisplatin administration, respectively; kidney tissue and urinary Fg increased significantly and correlated with histopathological injury, urinary kidney injury molecule-1 (KIM-1) and N-acetyl glucosaminidase (NAG) corresponding to the progression and regression of injury temporally. In a longitudinal follow-up of 31 patients who underwent surgical repair of abdominal aortic aneurysm, urinary Fg increased earlier than SCr in patients who developed postoperative AKI (AUC-ROC = 0.72). Furthermore, in a cohort of patients with biopsy-proven AKI (n = 53), Fg immunoreactivity in the tubules and interstitium increased remarkably and was able to distinguish patients with AKI from those without AKI (n = 59). These results suggest that immunoreactivity of Fg in the kidney, as well as urinary excretion of Fg, serves as a sensitive and early diagnostic translational biomarker for detection of AKI.
Acute kidney injury (AKI) is a common disorder that portends adverse outcomes in critically ill and non-critically ill hospitalized patients.1–3 Serum creatinine (SCr) and blood urea nitrogen (BUN) are most commonly used to detect kidney injury in preclinical and clinical studies and in routine clinical care, but have severe limitations in terms of sensitivity and specificity.4,5 Furthermore, BUN and SCr concentrations render a very delayed signal, even after considerable kidney injury, and this delay in the diagnosis of AKI prevents timely patient-management decisions, such as withdrawal or reduction in dose of the toxic agent or administration of therapeutics to mitigate the injury.6,7 Recent international efforts in biomarker identification and evaluation have resulted in the qualification of a subset of second-generation biomarkers for preclinical studies that can outperform or add significant value to existing conventional biomarkers.6,8 Significant effort is continuously being expended not only in evaluating the existing biomarkers in human studies for early detection of AKI, but also in identifying newer, more sensitive biomarkers as a part of the rolling biomarker qualification process.9
Previous research in our laboratory identified urinary fibrinogen (Fg) to be significantly up-regulated after kidney ischemia/reperfusion (I/R)-induced injury in rats, compared with controls, as well as in patients with clinically established multifactorial AKI, compared with healthy volunteers.10 These observations were consistent with published reports showing a positive correlation between urinary fibrin degradation products and proteinuria in patients with glomerulonephritis.11 Urinary fibrin degradation products in combination with N-acetyl-β-d-glucosaminidase (NAG) were also shown to allow a diagnosis of 25 out of 26 acute rejection episodes at least 24 hours before deterioration in renal function. Furthermore, this elevation of fibrin degradation products preceded the rise in NAG in 9 of 11 patients.12 Thus far, however, studies have lacked the ability to systematically and comprehensively evaluate the performance of Fg excretion in the urine and its immunostaining patterns in kidney as translational biomarker for early, sensitive, and specific detection of AKI.
The objective of the present study was to characterize the expression and excretion pattern of Fg relative to BUN, SCr, kidney injury molecule-1 [KIM-1; also known as hepatitis A virus cellular receptor 1 homolog (HAVcr-1)], and N-acetyl-β-d-glucosaminidase (NAG) and to perform histopathological analysis using mechanistically distinct forms of AKI in mice, rats, and humans. Specifically, our aims were to evaluate i) the diagnostic performance of urinary Fg after 30-minute bilateral renal I/R-induced reversible injury in rats and cisplatin (20 mg/kg, i.p.)-induced irreversible acute nephrotoxicity in mice; ii) the specificity of urinary Fg in detecting AKI using a well-established, galactosamine (1.1 g/kg, i.p.)-induced liver injury model13; iii) the predictive potential of urinary Fg as an AKI diagnostic biomarker in humans using a longitudinal follow up of patients who developed AKI after surgery of abdominal aortic aneurysms; and iv) the utility of Fg immunostaining patterns in differentiating patients with or without AKI.
Materials and Methods
Animals
Animals were maintained in a central animal facility over wood chips free of any known chemical contaminants under conditions of 21 ± 1°C and 50% to 80% relative humidity at all times in an alternating 12 hours light-dark cycle. Animals were fed with commercial rodent chow and given water ad libitum; animals were acclimated for 1 week before use. All animal maintenance and treatment protocols were in compliance with the Guide for Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health and were approved by the Harvard Medical School Institutional Animal Care and Use Committee.
Bilateral Renal I/R
Male Wistar rats (280 to 320 g) were subjected to I/R by clamping both renal arteries under anesthesia (30 mg/kg pentobarbital, i.p.) for 30 minutes, as described previously.5 Rats were euthanized by an overdose of phenobarbital (180 mg/kg) and were sacrificed at 3, 6, 12, 18, 24, 72, 120, and 168 hours after reperfusion (n = 4). Sham-surgery rats underwent anesthesia and a laparotomy only and were sacrificed after 24 hours and used as controls.
Cisplatin-Induced Kidney Toxicity
Male BALB/c mice (25 to 29 g) were treated with a single intraperitoneal injection of 20 mg/kg cisplatin (Sigma-Aldrich, St. Louis, MO) in 0.9% saline solution. Mice were euthanized by an overdose of phenobarbital (180 mg/kg) and were sacrificed at 24, 48, or 72 hours after cisplatin administration (n = 5). Control animals were injected with equal volume of 0.9% saline solution and were euthanized on day 1.
Galactosamine-Induced Liver Toxicity
To determine the specificity of Fg as a biomarker for kidney damage, male Sprague-Dawley rats (Harlan Laboratories, Frederick, MD) received a single intraperitoneal injection of either 1.1 g/kg of galactosamine or 0.9% saline solution (n = 6). At 24 hours after galactosamine injection, rats were euthanized by inhalation overdose of isoflurane.
Urine and Blood Collection
At approximately 16 hours (for rats) or 4 hours (for mice) before sacrifice, animals were placed in metabolic cages and urine was collected by adding RNAlater stabilization reagent (1 mL for rats; 40 μL for mice) to the urine collection tube. After collection, urine was centrifuged for 10 minutes at 12,000 × g at 4°C and aliquots were stored at −80°C. Blood was collected in heparinized tubes (for plasma collection) and nonheparinized tubes (for serum collection). After centrifugation at 12,000 × g for 10 minutes at 4°C, plasma was collected and stored in aliquots at −80°C.
Clinical Chemistry, Histopathology, and Histology Scoring
SCr was measured using a Beckman Creatinine Analyzer II and urine creatinine (uCr) was measured using a creatinine assay kit (Cayman Chemical, Ann Arbor, MI), according to the manufacturers' protocols. BUN was measured spectrophotometrically at 340 nm using a commercially available kit (Thermo Fisher Scientific, Rockford, IL), as described previously.14
Sagittal sections of kidney and liver were placed in 10% neutral buffered formalin and embedded into paraffin. For histological evaluation, embedded tissues were cut into 4- to 6-μm sections and stained with H&E. The sections were evaluated under a light microscope by a pathologist (V.B.) in a masked manner, and a severity score grading scale of 0 to 3 was used to grade pathological lesions: 0, no lesions to minimal; 1, mild acute tubular injury manifested by mild tubular distension, a low epithelial lining, and nonspecific degenerative changes of the epithelial cells; 2, moderate acute tubular injury, with frequent single cell necrosis within the epithelial layer; and 3, severe acute tubular injury, demonstrated by the presence of widespread epithelial cell necrosis, low epithelial lining, and accumulation of cellular debris in the tubule lumens.
Urine Analyses
Urinary KIM-1 was measured using established Luminex-based assays.4,5 Levels of KIM-1 in mice urine were determined using a Luminex-based assay recently established by the Bonventre Laboratory (Brigham and Women's Hospital, Boston, MA). Urinary NAG was measured spectrophotometrically using a commercially available kit according to the manufacturer's instructions (Roche Diagnostics, Basel, Switzerland). Fg protein in urine of humans, rats, and mice was measured using commercially available species-specific Luminex-based assay kits from Millipore (Billerica, MA).10 This assay uses two polyclonal antibodies as capture and detecting antibodies, respectively, against Fg protein and is therefore unable to distinguish between the intact Fg and intermediates (such as α/γ, β/γ, or the half-molecule of one each α/β/γ polypeptide chains) or other fibrin degradation products and fibrin-derived peptides.
RNA Extraction and Quantitative Real-Time PCR
At necropsy, tissue was collected, sliced into small fragments, flash-frozen in liquid nitrogen, and stored at −80°C. Kidneys from rats were macroscopically separated into medulla and cortex. Total RNA was isolated from tissue using a TRIzol-chloroform method as described previously.10,14 The concentration of total isolated RNA was measured at 260 nm using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Wilmington, DE), and integrity was determined by 1% agarose gel electrophoresis. One microgram of RNA was reverse-transcribed into cDNA using a QuantiTect reverse transcription kit from Qiagen (Valencia, CA) according to the manufacturer's instructions. The expression profiles of Fgα, Fgβ, Fgγ, and KIM-1 were evaluated with quantitative real-time PCR using a QuantiFast SYBR Green PCR kit (Qiagen) on a CFX96 RT-PCR instrument (Bio-Rad Laboratories, Hercules, CA). Amplification was performed using the following temperature profile: 3 minutes of enzyme activation at 95°C, followed by 40 cycles of 95°C for 10 seconds and 55°C for 30 seconds. GAPDH was used as a housekeeping gene. Primer sequences were as described previously.10
Immunoblotting
Total protein was isolated by homogenization of kidney tissues in radioimmunoprecipitation assay buffer containing complete protease inhibitor cocktail tablets (Roche Applied Science, Indianapolis, IN). Protein concentration was determined using a Pierce BCA protein assay kit (Thermo Fisher Scientific, Rockford, IL), according to manufacturer's instructions.
Equal amounts of protein were loaded onto 12% polyacrylamide gel. The proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane. After blocking in 5% blotting-grade blocker nonfat dry milk (Bio-Rad Laboratories) for 1 hour at room temperature, the membranes were incubated with a polyclonal anti-fibrinogen antibody (1:1000 dilution; Nordic Immunology; Nordic Immunological Laboratories, Eindhoven, Netherlands) at 4°C, overnight. After three washes with Tris-buffered saline/Tween, the blots were incubated in horseradish peroxidase-conjugated secondary antibody (GE Healthcare, Little Chalfont, UK) and developed using an enhanced chemiluminescence kit (GE Healthcare) by autoradiography. A monoclonal anti-β-actin antibody (Cell Signaling Technology, Danvers, MA) was used as a loading control.
Human Studies
Abdominal Aortic Aneurysm
Urine samples were obtained from 31 patients undergoing open surgical repair of abdominal aortic aneurysm at Brigham and Women's Hospital. Urine was collected preoperatively and at multiple time points after aortic cross-clamping: immediately afterwards and at 2 to 6 hours, 6 to 12 hours, 12 to 24 hours, 24 to 28 hours, 48 to 72 hours, and 72 to 96 hours after clamping. Not all participants underwent urine sampling at all time points, because of clinical circumstances. Urine was collected from the urometer of a Foley catheter or from spontaneous voids in the postoperative setting. Urine samples were centrifuged at 5000 × g for 5 minutes and supernatants were aliquoted and frozen at −80°C within 6 hours of collection. The Institutional Review Board approved the protocols for recruitment and sample collection.
Kidney Biopsy Specimens from Patients With and Without AKI
Pathology reports, finalized between January 2008 and March 2011 at Brigham and Women's Hospital and pertinent to patients who underwent the native kidney biopsy procedure for medical kidney disease, were retrieved using the hospital electronic reporting system. Cases with the primary diagnosis of acute tubular necrosis were selected; however, patients with concomitant glomerular pathological findings, significant acute interstitial nephritis, and significant chronic damage (defined as >20% of global glomerulosclerosis or tubulointerstitial scarring) were excluded. The cases that did not have adequate frozen tissue with ≥5 glomeruli in the sample submitted for immunofluorescence studies were also excluded. In all, 53 cases of acute tubular necrosis were available for the present study. For a control group, 59 cases with minimal or mild histological alterations were selected. The control cases all had adequate frozen tissue for immunofluorescence studies and comprised 27 cases of thin glomerular basement membrane disease and 18, 13, and 2 cases with minimal (<10%), 10% to 20%, and 21% to 25% chronic changes (global glomerulosclerosis and/or tubulointerstitial scarring), respectively, and no other significant histological abnormalities.
Kidney Biopsy Specimens from Patients with MCD
Using the same database as for the patients with and without AKI, we identified 14 patients with minimal-change disease (MCD) with known values of urinary protein/creatinine ratio and SCr values at the time of the kidney biopsy. All samples had adequate frozen tissue material for immunofluorescence studies. Of the 14 patients, 7 had a light-microscopic diagnosis of acute tubular necrosis and 7 had no significant findings on light microscopy.
Immunofluorescence Staining and Scoring
At Brigham and Women's Hospital, direct immunofluorescence studies are routinely performed on standard cryostat sections, using fluorescein isothiocyanate-labeled polyclonal antisera specific for human heavy chains of IgG, IgM, and IgA, kappa and lambda light chains (IgK and IgL), C3, albumin, and fibrin-related antigens, on all human kidney biopsies, at the time of biopsy evaluation. Only sections stained for fibrin-related antigens (polyclonal rabbit anti-human antibody; Dako, Carpinteria, CA) were retrieved from the archives and were retrospectively reviewed in the present study by a kidney pathologist (V.B.), using an Olympus BX53 (Tokyo, Japan) vertical illumination fluorescence microscope.
Frozen sections stained for fibrin-related antigens in human kidney biopsy samples were examined under an immunofluorescence microscope and were graded by pattern of staining, without knowledge of the clinical or morphological diagnosis. The luminal and apical staining was graded as 0 (none), 1 (involving rare tubules), or 2 (involving several tubules). The apical staining was graded as 0 (none), 1 (mild), or 2 (marked). The interstitial staining was graded as 1 (mild) or 2 (marked); a few samples that did not demonstrate any interstitial reactivity were graded as 0 (none). The score (mean ± SEM) for each category was plotted using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA).
Statistical Analyses
Results are expressed for individual animals or as means ± SEM. Urinary data were normalized to urinary creatinine. Statistical analyses were performed by Student's t-test using Graph Pad Prism 5 software. Statistical significance was set at P < 0.05. Diagnostic performance was assessed by taking the area underneath the receiver operating characteristic curve (AUC-ROC) using MedCalc version 9.5.0 surface (Mariakerke, Belgium).
Results
Kidney I/R Injury in Rats Results in Significant Up-Regulation and Excretion of Fg Correlating with Histopathological Injury
Thirty minutes of bilateral renal ischemia followed by reperfusion resulted in peak of kidney injury and dysfunction at 24 hours, as measured by increase in SCr, BUN, and histopathological damage (Figure 1, A and B). Kidney parenchyma revealed extensive tubulointerstitial damage at 24 hours, particularly prominent at the corticomedullary junction, with marked injury of the S3 segments of the proximal tubules. The individual tubules showed distension of their lumens and extensive degenerative changes of the epithelial cells, with widespread necrosis and collections of cellular debris within the tubule lumens. At 120 hours and 168 hours after reperfusion, kidneys showed mild tubular distension and prominent reactive changes in the epithelial cell layer. Occasional mitotic figures were detected, and the nuclei were enlarged and revealed prominent nucleoli (Figure 1B).
Figure 1.
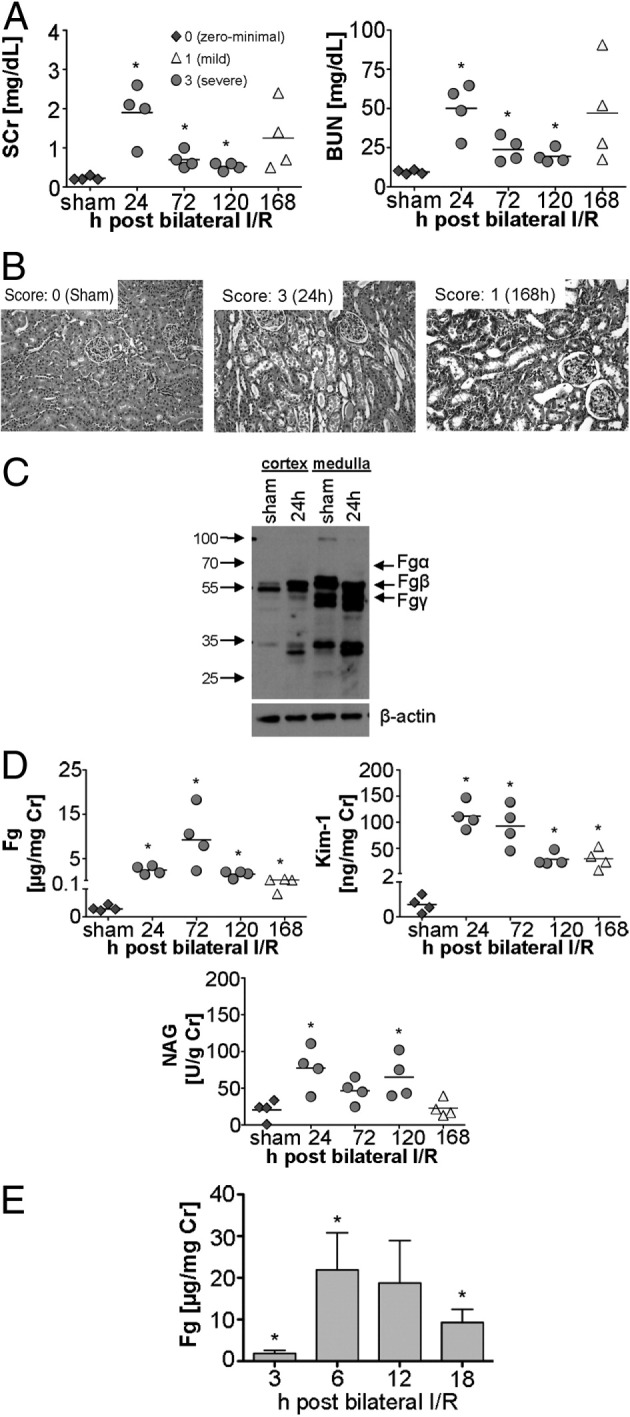
Urinary Fg increases significantly in rats after 30 minutes of bilateral renal I/R injury, correlating with the progression and resolution of AKI. A: Kidney injury parameters SCr and BUN were measured in the plasma of male Wistar rats after 30-minute bilateral renal I/R. B: Representative images of formalin-fixed, H&E-stained kidney histology sections of sham-treated versus injured tissue after 24 and 168 hours of reperfusion (n = 4 per group) show different levels of acute tubular injury. Scoring from 0 (no lesions) to 3 (severe acute tubular injury) is as described under Materials and Methods. C: Western blot analyses for Fg in rat kidney cortex and medulla collected at 24 hours after sham surgery or I/R injury revealed increased immunoreactivity of Fg- and fibrin-derived peptides in cortex and medulla at 24 hours after I/R injury, compared with sham surgery. D: Urinary levels of Fg, KIM-1, and NAG normalized to urinary creatinine over time after I/R injury. Each data point represents an induvidual animal. Colors and symbols indicate histopathology scores of acute kidney injury in the kidney. A black line indicates the mean. E: Urinary Fg levels at early time points (3, 6, 12, and 18 hours) after I/R injury. Data are indicated as mean ± SEM *P < 0.05 Student's t-test. n = 4 per group.
Significantly elevated mRNA expression of all three chains (Fgα, Fgβ, and Fgγ) was observed after I/R in the rat kidney (Table 1). Among the three chains, Fgβ showed the highest expression after 72 hours of reperfusion, with an ∼26-fold increase in cortex and ∼75-fold in medulla, whereas Fgα showed only a ∼5-fold increase in both medulla and cortex (Table 1). The mRNA up-regulation of Fgγ was slightly higher in cortex than in medulla. Immunoreactivity of Fg- and fibrin-derived peptides was considerably increased in cortex and medulla at 24 hours after I/R injury, compared with sham surgery (Figure 1C). Urinary Fg increased significantly after 24 hours (∼80-fold) after reperfusion, peaked at 72 hours (∼315-fold), and by 120 hours had decreased (∼50-fold) (Figure 1D). Urinary excretion of the advanced kidney injury biomarkers KIM-1 and NAG showed the highest levels after 24 hours, compared with sham surgery (Figure 1D).
Table 1.
mRNA Expression of Fgα, Fgβ, and Fgγ Significantly Increases after Ischemia/Reperfusion or Cisplatin-Induced Kidney Tubular Injury
| Treatment | mRNA expression (fold change relative to sham/0 hours) |
|||
|---|---|---|---|---|
| Fgα | Fgβ | Fgγ | Kim-1 | |
| Ischemia/reperfusion, cortex | ||||
| sham | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.2 | 1.0 ± 0.3 |
| 24 hours | 3.0 ± 0.6⁎ | 22.3 ± 7.6⁎ | 17.1 ± 4.0⁎ | 364.7 ± 32.8⁎ |
| 72 hours | 2.9 ± 0.4⁎ | 26.2 ± 5.9⁎ | 8.2 ± 1.1⁎ | 208.9 ± 55.1⁎ |
| 120 hours | 4.4 ± 1.0⁎ | 23.1 ± 7.8⁎ | 13.6 ± 2.2⁎ | 125.8 ± 35.4⁎ |
| 168 hours | 4.6 ± 0.1⁎ | 19.3 ± 4.0 | 8.2 ± 1.3 | 108.5 ± 41.2⁎ |
| Ischemia/reperfusion, medulla | ||||
| sham | 1.0 ± 0.2 | 1.0 ± 0.2 | 1.0 ± 0.2 | 1.0 ± 0.1 |
| 24 hours | 3.8 ± 0.6⁎ | 40.2 ± 8.7⁎ | 4.8 ± 1.6⁎ | 325.9 ± 95.7⁎ |
| 72 hours | 6.1 ± 1.8⁎ | 75.6 ± 24.8⁎ | 9.6 ± 2.4 | 318.2 ± 144.3 |
| 120 hours | 5.0 ± 2.1 | 46.8 ± 23.4 | 9.6 ± 3.9 | 159.1 ± 67.6 |
| 168 hours | 2.9 ± 1.0 | 27.6 ± 11.1⁎ | 2.8 ± 0.9 | 76.7 ± 40.7 |
| Cisplatin, 20 mg/kg | ||||
| 0 hours | 1.0 ± 0.0 | 1.0 ± 0.5 | 1.0 ± 0.2 | 1.0 ± 0.1 |
| 24 hours | 9.6 ± 2.5⁎ | 1.8 ± 0.3 | 25.0 ± 7.8⁎ | 5.0 ± 1.0⁎ |
| 48 hours | 27.8 ± 7.0⁎ | 17.3 ± 6.8⁎ | 183.2 ± 46.6⁎ | 18.1 ± 4.8⁎ |
| 72 hours | 25.3 ± 2.9⁎ | 23.0 ± 7.6⁎ | 242.4 ± 32.3⁎ | 23.9 ± 2.7⁎ |
Data are expressed as mean fold change ± SEM relative to sham-treated rats (I/R) or vehicle-treated mice.
P < 0.05 Student's t-test.
The early diagnostic capability of urinary Fg was assessed using urine samples collected at 3, 6, 12, and 18 hours after 30 minutes of bilateral renal I/R injury (Figure 1E). Urinary Fg excretion was increased as early as 3 hours (∼60-fold), sharply escalating at 6 hours (∼700-fold).
Cisplatin-Induced Nephrotoxicity in Mice Results in Marked Increase in Fg Corresponding to Histological Damage
In an irreversible model of kidney injury in mice induced by a single injection of 20 mg/kg cisplatin, an increase in SCr and BUN indicative of impaired kidney function was observed at 24 to 48 hours and peaked at 72 hours (Figure 2A). Cisplatin-induced tubulointerstitial damage was most prominent in the superficial renal cortex (Figure 2B). The initial injury was manifested by mild tubular distension and a low epithelial lining in most tubules; some tubules revealed vacuolization of cytoplasm (Figure 2B). At 48 hours, injured tubules also revealed single-cell death in some tubules, with karyopyknosis and accumulation of cellular debris in a few tubular lumens. After 72 hours of cisplatin administration, widespread epithelial cell necrosis was observed in the kidneys, with sloughing of the epithelium, denuded tubular basement membranes, and necrotic debris filling the lumens of many tubules (Figure 2B).
Figure 2.
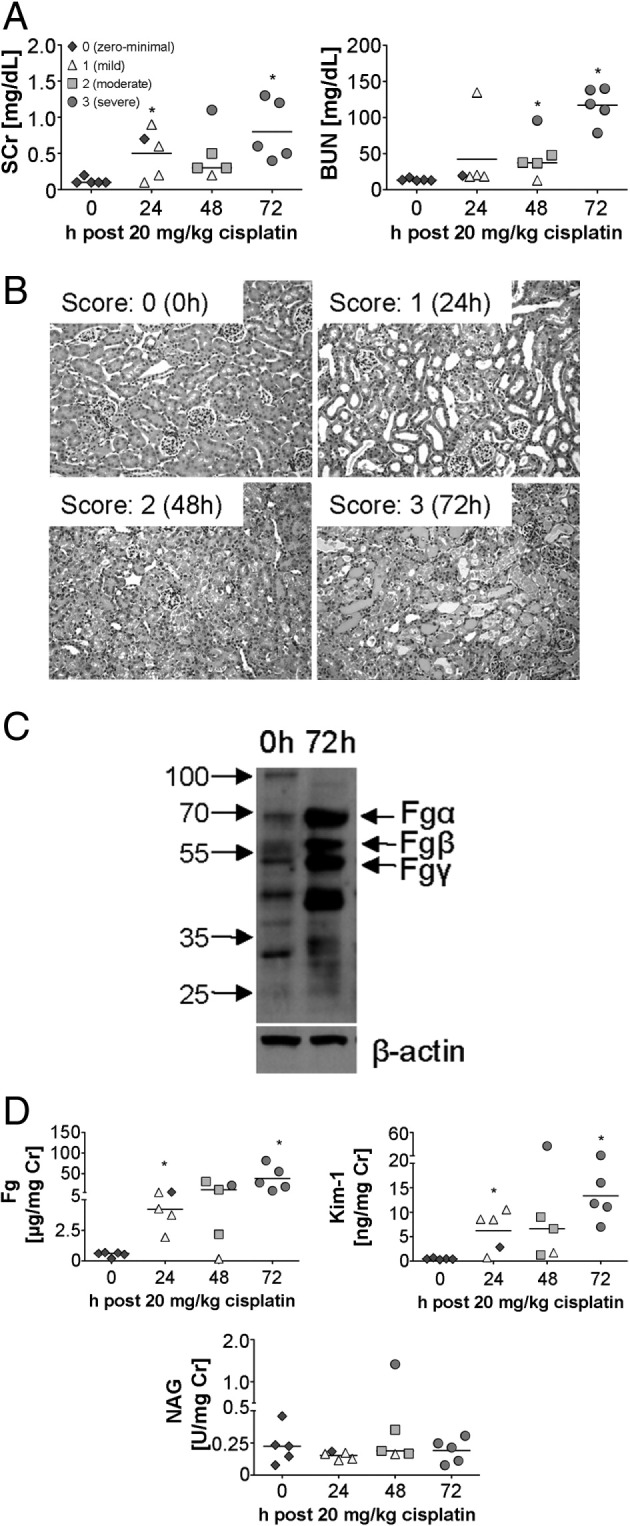
Urinary Fg increases progressively in mice after irreversible kidney toxicity induced by cisplatin. A: Kidney injury parameters SCr and BUN were measured in the plasma of male BALB/c mice administered 20 mg/kg cisplatin. B: Representative formalin-fixed, H&E-stained kidney histology sections of vehicle- versus cisplatin-treated mice after 24, 48, and 72 hours (n = 5 per group) show different levels of acute tubular injury. Scoring from 0 (no lesions) to 3 (severe acute tubular injury) is as described under Materials and Methods. C: Western blot analyses for Fg in mice kidney lysates at 72 hours (peak of injury) after cisplatin administration revealed increase in Fg protein expression. D: Urinary levels of Fg, KIM-1, and NAG normalized to urinary creatinine over time after cisplatin administration. Each data point represents an individual animal. Histopathology score of acute tubular injury in the kidney are as indicated in the legend key. A black line indicates the mean. *P < 0.05 Student's t-test. n = 4 per group.
Increased mRNA expression of Fgα, Fgβ, and Fgγ in kidneys of cisplatin-treated mice was detected as early as 24 hours (Table 1). In contrast to I/R injury, in which Fgβ showed the highest elevation among all three chains in the kidney, administration of cisplatin led to a massive increase in Fgγ (∼250-fold after 72 hours), compared with Fgα (∼25-fold) and Fgβ (∼23-fold). Similar increase in Fg protein expression in the kidney was observed at 72 hours after cisplatin administration (Figure 2C). Approximately 70-fold higher levels of urinary Fg were detected as early as 24 hours after cisplatin administration, corresponding with ∼30-fold increase in urinary KIM-1 concentration, whereas urinary excretion of NAG remained unchanged (Figure 2D).
Specificity of Urinary Fg as Biomarker of Kidney Injury
To evaluate the specificity of Fg as an AKI biomarker, rats were treated with galactosamine (1.1 g/kg, i.p.), a well-established hepatotoxicant. At 24 hours after galactosamine injection, the liver showed extensive hepatocellular damage, with a marked increase in the activity of the liver enzymes alanine aminotransferase (ALT) and aspartate aminotransferase (AST), but with no signs of kidney structural or functional damage (Table 2). Galactosamine treatment did not lead to any increase in mRNA levels of Fgα, Fgβ, and Fgγ in the kidney, nor in urinary excretion of Fg, thereby suggesting increased urinary Fg to be a specific indicator of kidney damage (Table 2).
Table 2.
mRNA Expression of Fgα, Fgβ, and Fgγ and Urinary Excretion of Fibrinogen Does Not Increase after Galactosamine-Induced Liver Toxicity
| Measure | Galactosamine treatment |
|
|---|---|---|
| 0 g/kg | 1.1 g/kg | |
| Histopathological score | ||
| Kidney | 0 | 0 |
| Liver | 0 | 2 |
| Clinical chemistry | ||
| BUN (mg/dL) | 19.9 ± 0.8 | 19.1 ± 1.1 |
| SCr (mg/dL) | 0.5 ± 0.0 | 0.5 ± 0.0 |
| ALT (U/L) | 26.0 ± 1.9 | 422.8 ± 121.9⁎ |
| AST (U/L) | 42.5 ± 3.9 | 246.0 ± 57.5 |
| Gene expression, kidney (fold change relative to control) | ||
| Fgα | 1.0 ± 0.2 | 1.2 ± 0.1 |
| Fgβ | 1.0 ± 0.1 | 1.0 ± 0.0 |
| Fgγ | 1.0 ± 0.1 | 0.9 ± 0.1 |
| Kim-1 | 1.0 ± 0.1 | 1.0 ± 0.1 |
| Urinary excretion | ||
| Fg (ng/mg uCr) | 201.9 ± 38.1 | 26.0 ± 26.0 |
Data are expressed as means ± SEM.
ALT, alanine transaminase; AST, aspartate transaminase; BUN, blood urea nitrogen; SCr, serum creatinine; uCr, urine creatinine.
P < 0.05 Student's t-test.
Increased Urinary Fg in Patients with Postoperative AKI after Abdominal Aortic Aneurysm
Of 31 patients undergoing abdominal aortic aneurysm repair, 7 developed postoperative AKI as defined as ≥50% rise in SCr. Demographic characteristics of patients according to their AKI status are shown in Table 3. Among the seven patients with postoperative AKI, SCr levels tended to rise to ≥50% of baseline values within 48 hours after surgery, whereas urinary Fg, KIM-1, and NAG showed an earlier increase (Figure 3). The diagnostic ability of Fg, KIM-1, and NAG to identify AKI versus no AKI is shown in Table 4. The AUC-ROC exceeded 0.7 for each biomarker at different time points (24 to 48 hours and 48 to 72 hours for Fg, 2 to 6 hours for KIM-1, and 6 to 12 hours for NAG).
Table 3.
Demographic and Clinical Characteristics of 31 Patients Undergoing Abdominal Aortic Aneurysm Repair
| Characteristic | AKI | No AKI |
|---|---|---|
| Sample size | n = 7 | n = 24 |
| Age (years) | 76.7 ± 6.9 | 71.4 ± 9.5 |
| Female (%) | 42.9 | 37.5 |
| Diabetes (%) | 14.2 | 25 |
| Hypertension (%) | 100 | 91.7 |
| Juxtarenal or suprarenal (%) | 28.6 | 33.3 |
| Preoperative SCr (mg/mL) | 1.5 ± 0.7 | 1.1 ± 0.4 |
| Preoperative eGRF (mL/min per 1.73m2) | 58.7 ± 37.5 | 62.2 ± 19.4 |
| Weight (kg) | 90.1 ± 12.8 | 81.5 ± 20.6 |
| Cross-clamp time (min) | 80.9 ± 27.3 | 48.9 ± 19.9 |
Figure 3.
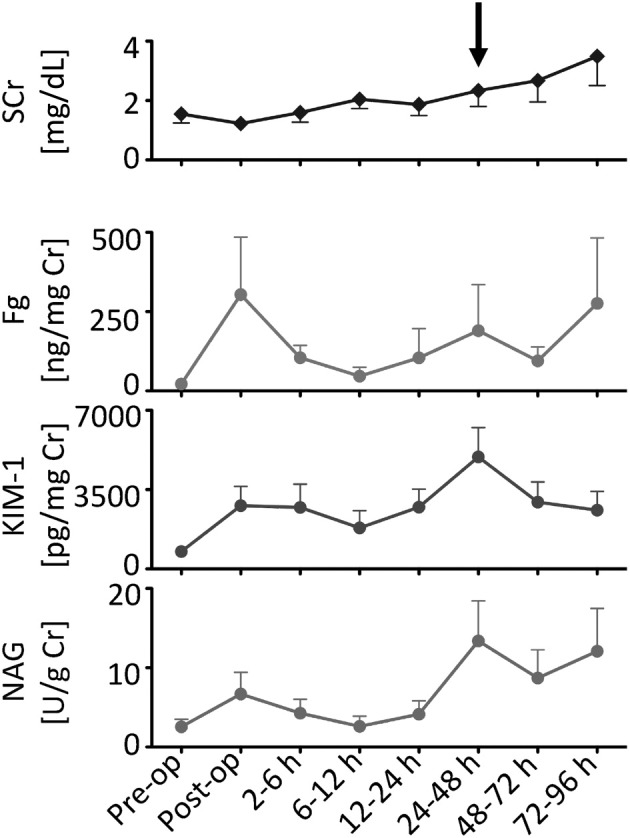
Urinary Fg shows earlier increase among patients with postoperative AKI after abdominal aortic aneurysm, compared with SCr. Shown are SCr levels in comparison with urinary KIM-1, Fg, and NAG normalized to urinary creatinine in seven patients who developed AKI (≥50% rise in SCr over baseline). An arrow indicates the rise in SCr that occurred by 48 hours over baseline. Data are expressed as means ± SEM. n = 7.
Table 4.
Comparative Diagnostic Performance Characteristics of Urinary Biomarkers for the Identification of Established AKI
| Time (hours) | AUC-ROC (95% CI) |
Patients (no.) |
|||
|---|---|---|---|---|---|
| Fg | KIM-1 | NAG | AKI | No AKI | |
| Preoperative | 0.56 (0.36–0.74) | 0.52 (0.33–0.71) | 0.55 (0.36–0.73) | 7 | 22 |
| Postoperative | 0.68 (0.48–0.84) | 0.66 (0.46–0.82) | 0.62 (0.42–0.79) | 6 | 23 |
| 2–6 | 0.63 (0.42–0.81) | 0.75 (0.54–0.90) | 0.51 (0.30–0.71) | 5 | 20 |
| 6–12 | 0.52 (0.26–0.77) | 0.58 (0.32–0.82) | 0.75 (0.48–0.93) | 4 | 12 |
| 12–24 | 0.65 (0.44–0.83) | 0.64 (0.43–0.82) | 0.52 (0.31–0.72) | 6 | 20 |
| 24–48 | 0.72 (0.53–0.87) | 0.55 (0.37–0.73) | 0.61 (0.42–0.78) | 7 | 24 |
| 48–72 | 0.70 (0.51–0.85) | 0.52 (0.34–0.71) | 0.51 (0.32–0.70) | 6 | 24 |
| 72–96 | 0.61 (0.40–0.79) | 0.51 (0.30–0.71) | 0.45 (0.32–0.74) | 5 | 21 |
AUC-ROC, area under the receiver operating characteristic curve.
Immunostaining Patterns of Fg as an Indicator of Kidney Tubular Damage in Patients with Biopsy-Proven Acute Tubular Injury
Morphological diagnosis of acute tubular necrosis (ATN) in human kidney biopsy samples was established based on light microscopic findings. In the absence of kidney injury, the tubules were closely packed together, back to back, and revealed preserved cellular details (Figure 4A), whereas a low epithelial lining and dilatation of the tubules with accumulation of necrotic cells and cellular debris in their lumens, as well as mild interstitial edema, was observed in all cases of acute tubular injury (Figure 4B). Immunoreactivity for Fg/fibrin-related antigens was examined in all compartments of kidney parenchyma including glomeruli, tubules, interstitium, and vasculature. There was no evidence of Fg immunoreactivity in the glomerulus or vasculature; in particular, there were no signs of vascular or glomerular thrombosis or segmental fibrinoid necrosis in any of the cases examined. A distinctly differential immunostaining pattern in the luminal and apical region of the tubules was noted in patients with ATN (n = 53) or without ATN (n = 59) (Figure 4, C to F). Fine granular reactivity in the apical region of the tubular epithelial cell cytoplasm was noted, which became much more pronounced and widespread in the ATN patients and was sometimes, but not necessarily, observed in association with tubular distension and/or luminal staining (Figure 4, F and I). Luminal staining was characterized by the reactivity of accumulated intraluminal Fg, which usually consisted of cellular debris admixed with proteinaceous material. There was a significant increase in luminal immunoreactivity of Fg in ATN patients, compared with those without ATN (Figure 4, C and I). Interstitial staining for Fg was observed in the vast majority of the biopsies, which also showed increased immunoreactivity in the patients with AKI, compared with those without AKI (Figure 4, H and I).
Figure 4.

Comparison of Fg immunostaining patterns in human kidney biopsies in patients with and without AKI. Shown are representative photomicrographs of H&E-stained (A and B) and Fg immunofluorescent-stained (C–H) formalin-fixed, paraffin-embedded kidney tissue sections of human kidney biopsies in patients without AKI (normal) and with AKI. I: Average immunostaining intensity score classified by luminal, apical, and interstitial patterns for 112 patients with and without AKI. Data are expressed as means ± SEM. *P < 0.05 Student's t-test. Original magnification, ×40.
Immunostaining Patterns of Fg Differentiate Patients with MCD Who Develop AKI from Those Who Do Not Develop AKI
The samples from patients with MCD did not reveal significant glomerular pathology under light microscopy (Figure 5, A and C), but all samples demonstrated diffuse effacement of visceral epithelial cell foot processes on electron microscopy (Figure 5, B and D); seven patients also demonstrated signs of acute tubular injury (Figure 5E). Accordingly, all patients presented with nephrotic syndrome and prominent proteinuria (9.86 ± 1.84 g per 24 hours versus 10.14 ± 1.22 g per 24 hours) (Figure 5E), but some were associated with acute renal failure, as indicated by SCr (0.77 ± 0.07 mg/dL versus 4.35 ± 0.71) (Figure 5E). Fg immunoreactivity was significantly increased in the luminal (Figure 5, F, I, and L), apical (Figure 5, G, J, and L), and interstitial regions (Figure 5, H, K, and L) in MCD patients who developed AKI, compared with those who did not develop AKI. Approximately a sixfold increase in luminal staining, a ninefold increase in apical staining, and a 1.5-fold increase in interstitial immunoreactivity of Fg was observed in MCD patients with AKI, compared with those without AKI, which suggests that expression patterns of Fg immunostaining in the kidney can serve as an effective way to diagnose AKI in MCD patients.
Figure 5.

Differences in Fg immunostaining patterns in patients with MCD with or without AKI. Shown are representative photomicrographs of human kidney biopsy sections in patients with MCD but not AKI (A, B, and F–H) or patients with both MCD and AKI (C, D, and I–K). A and C: Kidney sections after PAS staining following formalin fixation. B and D: Electron microscopic images of kidney sections after uranyl acetate and lead citrate treatment. F–K: Representative images of immunofluorescence staining for Fg. E: Total urinary protein and SCr. L: Average immunostaining intensity score classified by luminal, apical, and interstitial patterns for 14 patients with and without AKI. Data are expressed as means ± SEM. *P < 0.05 Student's t-test. Original magnification: ×20 (A and C); ×40 (F–K). Scale bar = 2 μm.
Discussion
The incidence of AKI is on the rise, the mortality rate from AKI remains high, and studies have demonstrated that a single event of AKI increases the risk for developing chronic kidney disease.15–17 One of the major impediments in treating AKI is making an accurate diagnosis that is early enough to alter the course of the disease by allowing effective and timely intervention.4,7 An active international effort is underway to identify sensitive, specific, and early diagnostic biomarkers in blood and urine using rodent models of AKI,5,18,19 as well as clinical studies in humans.20,21 Here we show, first, that urinary Fg serves as an early, sensitive and specific biomarker for AKI in mice, rats and humans irrespective of whether the injury is initiated by I/R or cisplatin administration and, second, that immunoreactivity patterns of Fg effectively differentiate patients with and without AKI.
Fibrinogen is a soluble 340-kDa trinodular dimeric glycoprotein with a hexameric structure composed of three distinct pairs of disulfide-linked polypeptide chains: Fgα, Fgβ, and Fgγ.22,23 Beyond its central role in hemostasis, Fg is also recognized as an acute-phase response protein and is significantly up-regulated on inflammatory conditions in diverse pathological conditions.24–26 We have previously reported Fgβ to be the second highest gene up-regulated among 22,523 genes in rat kidney at 24 hours after 20 minutes of bilateral I/R injury.10 With the present study, we provide further supportive evidence that, after 30-minute bilateral I/R injury in rats, mRNA levels of Fgα, Fgβ, and Fgγ significantly increase at 24 hours, remain elevated till day 5, and then decline by day 7 (Table 1). Interestingly, after cisplatin administration the Fgγ mRNA levels showed much higher increase (∼240 fold), compared with Fgβ (23-fold), at peak of injury (72 hours). It is possible that the differential regulation of Fgβ/Fgγ mRNA is related to the mechanism of injury in both models in which leukocyte-endothelial interactions and endothelial dysfunction play a predominant role in initiating injury in I/R,27,28 whereas cisplatin elicits epithelial cell apoptosis by forming DNA adducts, generating reactive oxygen species, mitochondrial damage, and inflammation.14,29
Plasma levels of Fg increase severalfold on inflammation or tissue damage, and have been implicated as a risk factor for cardiovascular disease,30 stroke,31 Alzheimer's disease,32 and chronic renal failure.33 The question as to whether an increase in urinary Fg can serve as a sensitive, specific and an early diagnostic indicator of a disease has not yet been effectively answered. We have previously shown in a cross-sectional study of individuals with and without kidney damage (n = 25 per group) that urinary Fg performed very well in differentiating between patients with and without AKI or chronic kidney disease (AUC-ROC = 0.98).10 In the present study, urinary levels of Fg increased significantly after kidney damage in rats and mice, followed a temporal profile of incline/decline of injury, and correlated with different grades of histopathology (Figures 1 and 2). An approximately 60-fold increase in urinary Fg levels at 3 hours after 30-minute bilateral I/R injury could indicate early diagnostic capabilities of urinary Fg, but could also be a consequence of plasma leakage due to organ damage. Nonetheless, consistent with these results, in seven patients with postoperative AKI after abdominal aortic aneurysm surgery the SCr levels tended to rise to ≥50% of baseline values within 48 hours after surgery, whereas urinary Fg increased immediately in the postoperative period (AUC-ROC = 0.68), in concordance with tubular injury biomarkers such as urinary KIM-1 (AUC-ROC = 0.66) and NAG (AUC-ROC = 0.62). This suggests that the excretion of Fg in the urine may be an early and sensitive indicator of tubulovascular damage.
We studied patients undergoing abdominal aortic aneurysm surgery, a common setting for hospital-acquired AKI due to ischemia. The relatively small numbers of patients studied precluded definitive conclusions regarding the potential of Fg, KIM-1, or NAG to serve as diagnostic tests. The results presented here should be regarded as preliminary, calling for larger scale validation in larger cohorts. The observation that biomarkers rose early in the postoperative setting suggests that earlier identification of AKI may be possible with novel biomarkers such as Fg, KIM-1, and NAG. Because the distinction between AKI and no AKI is based on SCr, which itself is not a perfect marker of AKI, it should come as no surprise that substantial overlap in biomarker values has been observed in the present study and by others.20,34 Future studies of Fg and other novel biomarkers of AKI should be adequately powered to study moderate to severe AKI, along with other clinical endpoints such as length of stay, mortality, and fluid balance.35,36
Immunoreactive fibrin/Fg staining in vascular and extravascular spaces of tissue occurs in a number of diseases such as atherosclerosis,37 kidney disease,38 tumors,39 and inflammation,40 among others. In a rat model of I/R injury, we have previously found distinct expression patterns of Fgα, Fgβ, and Fgγ chains in the renal tubular epithelial cells, glomeruli, and interstitium at baseline and during the regeneration in the injured kidney.10 Previous studies conducted using human kidney biopsy specimens have demonstrated membranous and interstitial deposition of Fg in glomeruli in subacute and chronic glomerulonephritis.41,42 Here, we report that in patients with a pathological diagnosis of ATN (n = 53) there was no evidence of Fg immunoreactivity in the glomerulus or vasculature, whereas the immunoreactivity in the apical and luminal region of the tubules, as well as in the interstitium, was markedly increased, compared with patients without evidence of ATN (n = 59). Tubular epithelial cells normally do not reveal significant reactivity for fibrin, but occasionally fine granular reactivity in the apical aspect of the tubular epithelial cell cytoplasm can be noted, sometimes but not necessarily in association with tubular distension and/or luminal staining. Immunofluorescence staining for Fg has traditionally been used to look for vascular and glomerular inflammatory or thrombotic lesions in kidney biopsy material,43 but in many kidney pathology laboratories this stain has not been used on a routine basis for diagnosis of AKI. In particular, the use of Fg immunofluorescence staining for evaluation of tubulointerstitial pathological processes in routine kidney biopsies has never been investigated or reported. In our hands, however, Fg immunofluorescence staining has become a very useful tool, not only in identifying microangiopathic vascular and glomerular changes, cellular crescents, or segmental fibrinoid necrosis, but also as an early and sensitive marker of acute tubular injury.
The precise mechanism of development of acute renal failure in the setting of MCD remains unknown, but hemodynamic abnormalities in association with sudden and pronounced hypoalbuminemia, intraparenchymal edema, and pre-existing vascular disease and ischemia have all been noted.44,45 Regardless of the specific mechanism, acute tubular injury with or without epithelial cell necrosis is seen in many patients with both MCD and AKI. The present study showed a striking increase in immunoreactivity of Fg in patients who developed AKI after MCD, compared with patients with MCD who did not develop AKI. Such a finding on immunofluorescence microscopy can be used as an aid in the diagnosis of acute tubular injury in patients with MCD, particularly in cases in which this diagnosis can be limited by the sample size or subtle nature of histopathological changes.
In summary, the present results demonstrate that the increase in kidney tissue expression of Fg, as well as urinary Fg, is conserved across species and thereby serves as a sensitive and early diagnostic translational biomarker for detection of acute kidney injury.
Footnotes
Supported in part by the NIH (Pathway to Independence grant ES016723 and Outstanding New Environmental Scientist award ES017543 to V.S.V.).
CME Disclosure: The authors of this article and the planning committee members and staff have no financial relationships with commercial interest to disclose.
References
- 1.Barrantes F., Feng Y., Ivanov O., Yalamanchili H.B., Patel J., Buenafe X., Cheng V., Dijeh S., Amoateng-Adjepong Y., Manthous C.A. Acute kidney injury predicts outcomes of non-critically ill patients. Mayo Clin Proc. 2009;84:410–416. doi: 10.1016/S0025-6196(11)60559-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chertow G.M., Burdick E., Honour M., Bonventre J.V., Bates D.W. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol. 2005;16:3365–3370. doi: 10.1681/ASN.2004090740. [DOI] [PubMed] [Google Scholar]
- 3.Uchino S., Kellum J.A., Bellomo R., Doig G.S., Morimatsu H., Morgera S., Schetz M., Tan I., Bouman C., Macedo E., Gibney N., Tolwani A., Ronco C., Beginning and Ending Supportive Therapy for the Kidney (BEST Kidney) Investigators Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;294:813–818. doi: 10.1001/jama.294.7.813. [DOI] [PubMed] [Google Scholar]
- 4.Vaidya V.S., Ferguson M.A., Bonventre J.V. Biomarkers of acute kidney injury. Annu Rev Pharmacol Toxicol. 2008;48:463–493. doi: 10.1146/annurev.pharmtox.48.113006.094615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaidya V.S., Ozer J.S., Dieterle F., Collings F.B., Ramirez V., Troth S., Muniappa N., Thudium D., Gerhold D., Holder D.J., Bobadilla N.A., Marrer E., Perentes E., Cordier A., Vonderscher J., Maurer G., Goering P.L., Sistare F.D., Bonventre J.V. Kidney injury molecule-1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat Biotechnol. 2010;28:478–485. doi: 10.1038/nbt.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonventre J.V., Vaidya V.S., Schmouder R., Feig P., Dieterle F. Next-generation biomarkers for detecting kidney toxicity. Nat Biotechnol. 2010;28:436–440. doi: 10.1038/nbt0510-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siew E.D., Ware L.B., Ikizler T.A. Biological markers of acute kidney injury. J Am Soc Nephrol. 2011;22:810–820. doi: 10.1681/ASN.2010080796. [DOI] [PubMed] [Google Scholar]
- 8.Warnock D.G., Peck C.C. A roadmap for biomarker qualification. Nat Biotechnol. 2010;28:444–445. doi: 10.1038/nbt0510-444. [DOI] [PubMed] [Google Scholar]
- 9.Goodsaid F.M., Mendrick D.L. Translational medicine and the value of biomarker qualification. Sci Transl Med. 2010;2 doi: 10.1126/scitranslmed.3001040. 47ps44. [DOI] [PubMed] [Google Scholar]
- 10.Krishnamoorthy A., Ajay A.K., Hoffmann D., Kim T.M., Ramirez V., Campanholle G., Bobadilla N.A., Waikar S.S., Vaidya V.S. Fibrinogen {beta}-derived B{beta}15–42 peptide protects against kidney ischemia/reperfusion injury. Blood. 2011;118:1934–1942. doi: 10.1182/blood-2011-02-338061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naish P., Evans D.J., Peters D.K. Urinary fibrinogen derivative excretion and intraglomerular fibrin deposition in glomerulonephritis. Br Med J. 1974;1:544–546. doi: 10.1136/bmj.1.5907.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia B., Thompson A.E., Tucker S.M., Wing A.J. Urinary excretion of NAG and FDP in acute renal graft rejection. Proc Eur Dial Transplant Assoc. 1975;11:311–319. [PubMed] [Google Scholar]
- 13.Zhou Y., Vaidya V.S., Brown R.P., Zhang J., Rosenzweig B.A., Thompson K.L., Miller T.J., Bonventre J.V., Goering P.L. Comparison of kidney injury molecule-1 and other nephrotoxicity biomarkers in urine and kidney following acute exposure to gentamicin, mercury, and chromium. Toxicol Sci. 2008;101:159–170. doi: 10.1093/toxsci/kfm260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krishnamoorthy A., Clement M.E., O'Leary E., Bonventre J.V., Vaidya V.S. TIM2 gene deletion results in susceptibility to cisplatin-induced kidney toxicity. Toxicol Sci. 2010;118:298–306. doi: 10.1093/toxsci/kfq240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waikar S.S., Liu K.D., Chertow G.M. The incidence and prognostic significance of acute kidney injury. Curr Opin Nephrol Hypertens. 2007;16:227–236. doi: 10.1097/MNH.0b013e3280dd8c35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waikar S.S., Winkelmayer W.C. Chronic on acute renal failure: long-term implications of severe acute kidney injury. JAMA. 2009;302:1227–1229. doi: 10.1001/jama.2009.1364. [DOI] [PubMed] [Google Scholar]
- 17.Winterberg P.D., Lu C.Y. Acute kidney injury: the beginning of the end of the dark ages. Am J Med Sci. 2011 doi: 10.1097/MAJ.0b013e318228aef8. [Epub ahead of press] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dieterle F., Perentes E., Cordier A., Roth D.R., Verdes P., Grenet O., Pantano S., Moulin P., Wahl D., Mahl A., End P., Staedtler F., Legay F., Carl K., Laurie D., Chibout S.D., Vonderscher J., Maurer G. Urinary clusterin, cystatin C, beta2-microglobulin and total protein as markers to detect drug-induced kidney injury. Nat Biotechnol. 2010;28:463–469. doi: 10.1038/nbt.1622. [DOI] [PubMed] [Google Scholar]
- 19.Harpur E., Ennulat D., Hoffman D., Betton G., Gautier J.C., Riefke B., Bounous D., Schuster K., Beushausen S., Guffroy M., Shaw M., Lock E., Pettit S., HESI Committee on Biomarkers of Nephrotoxicity Biological qualification of biomarkers of chemical-induced renal toxicity in two strains of male rat. Toxicol Sci. 2011;122:235–252. doi: 10.1093/toxsci/kfr112. [DOI] [PubMed] [Google Scholar]
- 20.Parikh C.R., Coca S.G., Thiessen-Philbrook H., Shlipak M.G., Koyner J.L., Wang Z., Edelstein C.L., Devarajan P., Patel U.D., Zappitelli M., Krawczeski C.D., Passik C.S., Swaminathan M., Garg A.X., TRIBE-AKI Consortium Postoperative biomarkers predict acute kidney injury and poor outcomes after adult cardiac surgery. J Am Soc Nephrol. 2011;22:1748–1757. doi: 10.1681/ASN.2010121302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shlipak M.G., Coca S.G., Wang Z., Devarajan P., Koyner J.L., Patel U.D., Thiessen-Philbrook H., Garg A.X., Parikh C.R., TRIBE-AKI Consortium Presurgical serum cystatin C and risk of acute kidney injury after cardiac surgery. Am J Kidney Dis. 2011;58:366–373. doi: 10.1053/j.ajkd.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doolittle R.F. Fibrinogen and fibrin. Annu Rev Biochem. 1984;53:195–229. doi: 10.1146/annurev.bi.53.070184.001211. [DOI] [PubMed] [Google Scholar]
- 23.Weisel J.W. Fibrinogen and fibrin. Adv Protein Chem. 2005;70:247–299. doi: 10.1016/S0065-3233(05)70008-5. [DOI] [PubMed] [Google Scholar]
- 24.Adams R.A., Schachtrup C., Davalos D., Tsigelny I., Akassoglou K. Fibrinogen signal transduction as a mediator and therapeutic target in inflammation: lessons from multiple sclerosis. Curr Med Chem. 2007;14:2925–2936. doi: 10.2174/092986707782360015. [DOI] [PubMed] [Google Scholar]
- 25.Schultz D.R., Arnold P.I. Properties of four acute phase proteins: c-reactive protein, serum amyloid A protein, alpha 1-acid glycoprotein, and fibrinogen. Semin Arthritis Rheum. 1990;20:129–147. doi: 10.1016/0049-0172(90)90055-k. [DOI] [PubMed] [Google Scholar]
- 26.Reinhart W.H. Fibrinogen–marker or mediator of vascular disease? Vasc Med. 2003;8:211–216. doi: 10.1191/1358863x03vm494ra. [DOI] [PubMed] [Google Scholar]
- 27.Sabbahy M.E., Vaidya V.S. Ischemic kidney injury and mechanisms of tissue repair. Wiley Interdiscip Rev Syst Biol Med. 2011;3:606–618. doi: 10.1002/wsbm.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonventre J.V. Pathophysiology of acute kidney injury: roles of potential inhibitors of inflammation. Contrib Nephrol. 2007;156:39–46. doi: 10.1159/000102069. [DOI] [PubMed] [Google Scholar]
- 29.Pabla N., Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int. 2008;73:994–1007. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- 30.Danesh J., Collins R., Appleby P., Peto R. Association of fibrinogen, C-reactive protein, albumin, or leukocyte count with coronary heart disease: meta-analyses of prospective studies. JAMA. 1998;279:1477–1482. doi: 10.1001/jama.279.18.1477. [DOI] [PubMed] [Google Scholar]
- 31.Catto A.J., Grant P.J. Risk factors for cerebrovascular disease and the role of coagulation and fibrinolysis. Blood Coagul Fibrinolysis. 1995;6:497–510. doi: 10.1097/00001721-199509000-00001. [DOI] [PubMed] [Google Scholar]
- 32.van Oijen M., Witteman J.C., Hofman A., Koudstaal P.J., Breteler M.M. Fibrinogen is associated with an increased risk of Alzheimer disease and vascular dementia. Stroke. 2005;36:2637–2641. doi: 10.1161/01.STR.0000189721.31432.26. [DOI] [PubMed] [Google Scholar]
- 33.Prinsen B.H., Rabelink T.J., Beutler J.J., Kaysen G.A., De Boer J., Boer W.H., Hagen E.C., Berger R., De Sain-Van Der Velden M.G. Increased albumin and fibrinogen synthesis rate in patients with chronic renal failure. Kidney Int. 2003;64:1495–1504. doi: 10.1046/j.1523-1755.2003.00211.x. [DOI] [PubMed] [Google Scholar]
- 34.Siew E.D., Ware L.B., Gebretsadik T., Shintani A., Moons K.G., Wickersham N., Bossert F., Ikizler T.A. Urine neutrophil gelatinase-associated lipocalin moderately predicts acute kidney injury in critically ill adults. J Am Soc Nephrol. 2009;20:1823–1832. doi: 10.1681/ASN.2008070673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waikar S.S., Betensky R.A., Bonventre J.V. Creatinine as the gold standard for kidney injury biomarker studies? Nephrol Dial Transplant. 2009;24:3263–3265. doi: 10.1093/ndt/gfp428. [DOI] [PubMed] [Google Scholar]
- 36.Waikar S.S., Bonventre J.V. Creatinine kinetics and the definition of acute kidney injury. J Am Soc Nephrol. 2009;20:672–679. doi: 10.1681/ASN.2008070669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bini A., Fenoglio J., Jr, Sobel J., Owen J., Fejgl M., Kaplan K.L. Immunochemical characterization of fibrinogen, fibrin I, and fibrin II in human thrombi and atherosclerotic lesions. Blood. 1987;69:1038–1045. [PubMed] [Google Scholar]
- 38.Hancock W.W., Gee D., De Moerloose P., Rickles F.R., Ewan V.A., Atkins R.C. Immunohistological analysis of serial biopsies taken during human renal allograft rejection: Changing profile of infiltrating cells and activation of the coagulation system. Transplantation. 1985;39:430–438. doi: 10.1097/00007890-198504000-00018. [DOI] [PubMed] [Google Scholar]
- 39.Dvorak H.F., Senger D.R., Dvorak A.M. Fibrin as a component of the tumor stroma: origins and biological significance. Cancer Metastasis Rev. 1983;2:41–73. doi: 10.1007/BF00046905. [DOI] [PubMed] [Google Scholar]
- 40.Kao V.C., Wissler R.W. A study of the immunohistochemical localization of serum lipoproteins and other plasma proteins in human atherosclerotic lesions. Exp Mol Pathol. 1965;4:465–479. doi: 10.1016/0014-4800(65)90011-0. [DOI] [PubMed] [Google Scholar]
- 41.Koffler D., Paronetto F. Immunofluorescent localization of immunoglobulins, complement, and fibrinogen in human diseases: II. Acute, subacute, and chronic glomerulonephritis. J Clin Invest. 1965;44:1665–1671. doi: 10.1172/JCI105273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bini A., Mesa-Tejada R., Fenoglio J.J., Jr, Kudryk B., Kaplan K.L. Immunohistochemical characterization of fibrin(ogen)-related antigens in human tissues using monoclonal antibodies. Lab Invest. 1989;60:814–821. [PubMed] [Google Scholar]
- 43.Jennette J.C., Olson J.L., Schwartz M.M., Silva F.G. ed 6. Lippincott Williams & Wilkins; Philadelphia: 2006. Heptinstall's Pathology of the Kidney; pp. 536–537. ch 12. [Google Scholar]
- 44.Waldman M., Crew R.J., Valeri A., Busch J., Stokes B., Markowitz G., D'Agati V., Appel G. Adult minimal-change disease: clinical characteristics, treatment, and outcomes. Clin J Am Soc Nephrol. 2007;2:445–453. doi: 10.2215/CJN.03531006. [DOI] [PubMed] [Google Scholar]
- 45.Jennette J.C., Falk R.J. Adult minimal change glomerulopathy with acute renal failure. Am J Kidney Dis. 1990;16:432–437. doi: 10.1016/s0272-6386(12)80055-2. [DOI] [PubMed] [Google Scholar]