

Abstract
Proliferative vitreoretinopathy (PVR) is a blinding disease associated with rhegmatogenous retinal detachment, for which there is no satisfactory treatment. Surgery helps in many cases, but, to our knowledge, there are no pharmacological approaches to reduce PVR risk. We report that suppressing expression of p53 was a required event in two assays of PVR (namely, platelet-derived growth factor receptor α–mediated contraction of cells in a collagen gel and retinal detachment in an animal model of PVR). Furthermore, preventing the decline in the level of p53 with agents such as Nutlin-3 protected from retinal detachment, which is the most vision-compromising component of PVR. Finally, Nutlin-3 may be effective in the clinical setting because it prevented human PVR vitreous-induced contraction of cells isolated from a patient PVR membrane. These studies identify Nutlin-3 as a potential PVR prophylaxis.
Proliferative vitreoretinopathy (PVR) is a blinding disease that afflicts 5% to 11% of patients who undergo surgery to correct a rhegmatogenous retinal detachment1; there are between 1700 and 3700 cases of PVR in the United States annually.2,3 The treatment for PVR is repeat surgery,4 which is anatomically successful in only 60% to 80% of cases,5,6 and the procedure carries the risk of recurrence.7–12 Efforts to identify nonsurgical (ie, pharmacological) approaches to treat PVR have not been successful.13–15 Thus, there is a an immediate need for new therapy options for individuals who are afflicted by this blinding disease.
The full-thickness retinal break (eg, tears and holes) that is quintessential to rhegmatogenous retinal detachment results in exposure of cells to vitreous, a rich source of growth factors and cytokines.16 These cells migrate into vitreous, proliferate, and synthesize extracellular matrix proteins.1 This series of events culminates in the formation of a retina-associated membrane, which contracts and thereby causes retinal detachment and vision loss.17
Although cells (retinal pigment epithelial cells, glial cells, and fibroblasts18–20) in PVR membranes express a plethora of cell surface receptors, the platelet-derived growth factor (PDGF) receptor α (PDGFRα) is essential for experimental PVR, and is associated with clinical PVR.21–23 The surprisingly prominent role of PDGFRα in PVR appears to be related to the fact that it can be engaged by a wide spectrum of vitreal agents, which activate it indirectly and thereby trigger a signature set of signaling events that include suppression of p53.24–26
Various forms of cellular stress increase expression and activate p53, a tetrameric transcription factor, and thereby trigger the p53 pathway, which leads to cell cycle arrest, apoptosis, and/or senescence.27 The finding that p53 and/or the p53 pathway is mutated in approximately 50% of solid tumors28 has motivated the development of pharmacological agents that stimulate the p53 pathway. For instance, the small molecule, Nutlin-3, activates the p53 pathway by preventing p53 from interacting with murine double min 2 (Mdm2), also called Hdm2 in humans,29 which reduces the level of p53 by a variety of mechanisms.30–32 Nutlin-3 (RG7112/RO5045337) is in clinical trials for certain tumors in which the Hdm2/p53 pathway is intact.33 Furthermore, ophthalmic formulation of Nutlin-3 has been developed.34
The two goals of this study were as follows: i) to test if the previously noted correlation between the PDGFRα-mediated decline in the level of p53 and the development of PVR was causally related and ii) to test if Nutlin-3–mediated stabilization of p53 is a potential approach to prevent PVR.
Materials and Methods
Major Reagents and Cell Culture
The phospho-Y742 PDGFRα antibody was raised against the phospho-peptide KQADTTQpYVPMLDMK.35 The Ras GTP-activating protein (RasGAP) antibody was crude rabbit antiserum against a GST fusion protein including the SH2-SH3-SH2 region of the human RasGAP.36 Antibodies against PDGFRα, phospho-Akt (S473), Akt, and p53 were obtained from Cell Signaling Technology (Beverly, MA). Secondary antibodies (horseradish peroxidase–conjugated goat anti-rabbit IgG and goat anti-mouse IgG) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Enhanced chemiluminescent substrate for detection of horseradish peroxidase was from Pierce Protein Research Products (Rockford, IL). An ApoAlert annexin V–fluorescein isothiocyanate apoptosis kit and an in situ β-galactosidase assay kit were obtained from Clontech Laboratories, Inc. (Mountain View, CA) and Agilent Technologies (Santa Clara, CA), respectively. Nutlin-3 was obtained from Cayman Chemical (Ann Arbor, MI). Normal rabbit vitreous (RV) was prepared from frozen rabbit eyeballs, as previously described.24 The level of PDGFs in RV is either very low or lower than the level of detection.37,38
RPEM cells are retinal pigment epithelial (RPE) cells derived from a human epiretinal membrane, as previously described.39 Ethics approval was obtained from the Clinical Ethics Research Board (University of British Columbia, Vancouver, BC, Canada) and the Capital Health Authority Research Ethics Board (Halifax, NS) before retaining patient vitreous and/or PVR membranes that would have been otherwise discarded. Primary rabbit conjunctival fibroblasts (RCFs) were obtained and cultured as previously described.40 RCFs that stably expressed the short hairpin RNA (shRNA)–targeting vector specific for green fluorescent protein (GFP), PDGFRα, p53, or PDGFRα/p53 were designated shGFP, shPDGFRα, shp53, and shPDGFRα/p53, respectively. F cells are immortalized mouse embryo fibroblasts derived from PDGFR knockout mice that do not express either of the two PDGFR genes; Fα and Fβ cells are F cells in which PDGFRα or PDGFRβ has been re-expressed.21
Knockdown of PDGFRα and p53
Oligos (5′-GCCAGCTCTTATTACCCTCTA-3′ for PDGFRα, 5′-CGGGCGTAAACGCTTCGAGAT-3′ for p53, and 5′-ACAACAGCCACAACGTCTATA-3′ for GFP) in a hairpin-pLKO.1 retroviral vector, the packaging plasmid (pCMV-dR8.91), the envelope plasmid (VSV-G/pMD2.G), and 293T packaging cells were from Dana-Farber Cancer Institute/Harvard Medical School (Boston, MA). The shRNA lentiviruses were prepared as previously described.26 The viruses were used to infect RCF cells. Successfully infected cells were selected on the basis of their ability to proliferate in media containing puromycin (1 μg/mL). The resulting cells were characterized by using Western blot analysis using antibodies against PDGFRα, p53, and RasGAP (loading control).
Western Blot Analysis
Cells were grown to 90% confluence in serum-containing medium, and then incubated for 24 hours in medium without serum. Cells were stimulated (as detailed for each experiment), washed twice with ice-cold PBS, and lysed in extraction buffer (10 mmol/L Tris-HCl, pH 7.4; 5 mmol/L EDTA; 50 mmol/L NaCl; 50 mmol/L NaF; 1% Triton X-100; 20 μg/mL aprotinin; 2 mmol/L Na3VO4; and 1 mmol/L phenylmethylsulfonyl fluoride). Lysates were clarified by centrifugation at 13,000 × g, 4°C for 15 minutes. Equal amounts of protein were separated by 10% SDS-PAGE, transferred to PVDF membranes, and then subjected to Western blot analysis using indicated antibodies. The signal intensity was determined by densitometry and analyzed with Quantity One software (Bio-Rad, Hercules, CA).
Collagen I Contraction Assay
Cells were trypsinized, washed, and resuspended in 1.5 mg/mL of neutralized collagen I (INAMED, Fremont, CA) (pH 7.2) at a density of 1 × 106 cells/mL for RPEM or 5 × 104 cells/mL for RCFs. The mixture was fractionated into wells of a 24-well plate that had been preincubated overnight with 5 mg/mL bovine serum albumin in PBS. The collagen solution was solidified by incubating at 37°C for 90 minutes, and overlaid with medium containing the desired agents. The media were replaced every day, and the gel diameter was measured on day 3. The gel area was calculated using the formula πr2, where r is the radius of the gel.
Proliferation and Apoptosis Assays
Proliferation and apoptosis were assayed as previously described.24 Briefly, RCFs were seeded into 24-well plates at a density of 50,000 cells per well in Dulbecco's modified Eagle's medium (DMEM) plus 10% fetal bovine serum (FBS). After 6 hours, the cells had attached; the medium was aspirated; the cells were rinsed twice with PBS; and the cells were cultured in serum-free DMEM with or without RV (1:3 dilution). The media were replaced every day. On day 3, the cells were counted in a hemocytometer. At least three independent experiments were performed.
To monitor apoptosis, RCFs were seeded into 6-cm dishes at a density of 1 × 105 cells per dish in DMEM plus 10% FBS. After the cells had attached to the dishes, they were treated as described in the proliferation assay. On day 3, the cells were harvested and stained with fluorescein isothiocyanate–conjugated annexin V and propidium iodide, according to the instructions provided with the apoptosis kit (BD Biosciences, Palo Alto, CA). The cells were analyzed by flow cytometry in Coulter Beckman XL (Caguas, Puerto Rico). At least three independent experiments were performed.
Senescence Assay
RCF cells were plated into a 12-well plate (10,000 cells per well) in DMEM (high glucose) supplemented with 10% FBS. After 6 hours, the medium was replaced with DMEM with or without RV (1:3 dilution) and replenished every 24 hours. On day 3, the β-galactosidase activity was assessed according to the manufacturer's instructions provided with the in situ β-galactosidase assay kit.
Rabbit Model for PVR
PVR was induced in Dutch Belted rabbits, obtained from Covance (Denver, PA), as previously described.25 Briefly, a gas vitrectomy was performed by injecting 0.1 mL of perfluoropropane (C3F8) (Alcon, Fort Worth, TX) into vitreous. One week later, the right eye of rabbits was injected in one of two ways. For the experiment injecting RCFs expressing shRNAs, 0.1 mL of DMEM containing 1 × 105 RCFs that were modified as outlined in Figure 3 were injected, along with 0.1 mL of rabbit platelet-rich plasma. For the Nutlin-3 experiment, all rabbits were injected with 0.1 mL of DMEM containing 1 × 105 unmodified RCFs and 0.1 mL rabbit platelet-rich plasma, and either not injected a third time or injected with vehicle or 0.1 mL of 200 μmol/L Nutlin-3. The vehicle or Nutlin-3 injection was repeated on days 3 and 5. The retinal status was evaluated with an indirect ophthalmoscope fitted with a +30 D fundus lens on days 1, 3, 5, 7, 14, 21, and 28. PVR was graded according to the Fastenberg scale of classification41: stage 0, no disease; stage 1, epiretinal membrane; stage 2, vitreoretinal traction without retinal detachment; stage 3, localized retinal detachment (one to two quadrants); stage 4, extensive retinal detachment (two to four quadrants without complete detachment); and stage 5, complete retinal detachment. On day 28, animals were sacrificed, and eyes were enucleated and frozen at −80°C. All surgical procedures were performed under aseptic conditions and pursuant to the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research. The protocol for the use of animals was approved by the Schepens Animal Care and Use Committee.
Figure 3.

Molecularly suppressing expression of p53 rescues the ability of PDGFRα-deficient cells to contract. A: Western blot analysis of knockdown RCFs. Lentiviruses are used to stably express shRNAs directed against GFP, PDGFRα, or p53 in primary RCFs. The resulting cell lysates are subjected to Western blot analysis using the indicated antibodies. The signal intensity is quantified and expressed as a ratio of the loading control (RasGAP). The data presented are representative of two independent experiments. B: Expression of PDGFRα potentiates RV-mediated suppression of p53. The cells in A are left resting or exposed to vitreous (diluted 1:3 in DMEM) from normal rabbits (RV) for 2 hours and lysed, and total cell lysates are subjected to Western blot analysis with the indicated antibodies. The signal intensity is quantified, and the data presented are representative of three independent experiments. The data are subjected to a paired t-test. The asterisk denotes a statistically significant difference. C: RV induces contraction of RCF-loaded collagen gels, which requires PDGFRα-mediated suppression of p53. The cells in A are subjected to a collagen contraction assay. RV (diluted 1:3 in DMEM) is added on top of the gels on day 0 and replenished every 24 hours; the experiment is terminated on day 3. The panel below the bar graph shows photographs of representative gels; the data in the bar graph are triplicates within a single experiment. The data are subjected to a paired t-test. The asterisk denotes a statistically significant difference. The data presented are representative of three independent experiments. NS, not significant.
Immunohistochemical Data
Rabbit eyeballs were fixed in 10% formalin for 48 hours and embedded in paraffin after dehydration. Subsequently, paraffin sections (4 μm thick) were prepared, dewaxed in xylene, and rehydrated in ethanol, diluted ethanol, and deionized water. Antigen retrieval was performed by boiling the slides for 20 minutes in a citrate-based buffer (Vector Laboratories Inc., Burlingame, CA). The endogenous peroxidase activity was blocked by incubation with 1% H2O2 in methanol for 10 minutes, and the endogenous avidin and biotin binding sites were blocked by incubation with avidin and biotin blocking buffers (Vector Laboratories Inc.). The resulting sections were first incubated in blocking buffer containing 3% goat serum, and then in primary antibody [diluted 1:200 in blocking buffer, anti-p53, from Abcam (Cambridge, MA)] overnight at 4°C. Incubation with secondary antibody (biotinylated goat anti-mouse; Abcam) was for 1 hour at room temperature. Finally, the ABC reagent (Vector Laboratories Inc.) was added for 45 minutes, and the sections were stained with 3,3′-diaminobenzidine (Thermo Scientific, Rockford, IL). The sections were observed and photographed under a microscope.
Statistics
The experimental data were analyzed using an unpaired t-test and one-way analysis of variance and/or post tests. P < 0.05 was considered statistically significant.
Results
Suppressing p53 Is Essential for RV-Induced Contraction and Retinal Detachment
RV contained a variety of non-PDGFs that indirectly activated PDGFRα and thereby chronically stimulated Akt,26 which phosphorylated and activated Mdm2,42 which mediated a decline in the level of p53.43–45 Nutlin-3 antagonized the interaction of Mdm2 and p53, and thereby prevented Mdm2-mediated reduction of p53.29 Because of these properties, we considered whether Nutlin-3 would prevent RV-mediated reduction in the level of p53, prevent contraction of cells in collagen gels, and protect rabbits from developing PVR. We chose primary RCFs for these studies because they robustly contracted collagen gels and induced PVR.
Nutlin-3 effectively blocked the RV-mediated decline in the level of p53 and contraction of collagen gels (Figure 1, A and B). As expected, it had no effect on RV-induced phosphorylation of PDGFRα or activation of Akt (Figure 1A), events that were upstream of the known action of Nutlin-3. The minimum dose to prevent the RV-induced reduction of p53 was 2 mmol (see Supplemental Figure S1A at http://ajp.amjpathol.org), whereas the maximum tolerated dose was 30 mmol (see Supplemental Figure S1B at http://ajp.amjpathol.org). Moreover, multiple intravitreal injections of Nutlin-3 of up to 20 mmol did not produce overt signs of retinal toxicity (see Supplemental Figure S2 at http://ajp.amjpathol.org and data not shown). These results set the stage to test if Nutlin-3 could prevent retinal detachment in an animal model of PVR.
Figure 1.

Nutlin-3 prevents both the RV-induced decline in the level of p53 and contraction. A: The impact of Nutlin-3 on RV-mediated signaling events. RCFs are pretreated with either Nutlin-3 (10 μmol/L) or vehicle for 30 minutes and then exposed to either RV (diluted 1:3 in DMEM) or DMEM for 2 hours. The resulting cell lysates are subjected to Western blot analysis using the indicated antibodies. The numbers are a ratio of p- PDGFRα/PDGFRα, p-Akt/Akt, or p53/RasGAP. The data presented are representative of three independent experiments. B: The impact of Nutlin-3 on RV-mediated contraction. RCFs are subjected to the collagen contraction assay. Nutlin-3 (10 μmol/L) or vehicle was added as indicated. The data are subjected to a paired t-test. The asterisk indicates a statistically significant difference.
As shown in Figure 2, 100% of the animals in both control groups (uninjected, open circles; or injected with vehicle, filled circles) developed complete retinal detachment (stage 5) by day 28. In contrast, 0% of the Nutlin-3–treated animals (Figure 2A) succumbed to even partial retinal detachment (stage 3 or 4), although they formed membranes (stage 1 and 2 in Figure 2A; see also Supplemental Figure S2 at http://ajp.amjpathol.org). More important, the p53 level was higher in epiretinal membranes isolated from Nutlin-3–injected animals compared with vehicle-injected controls (Figure 2B). These observations indicated that Nutlin-3 treatment maintained a high level of p53 expression in cells of the epiretinal membrane and prevented retinal detachment.
Figure 2.

Nutlin-3 prevents retinal detachment and elevates p53 in epiretinal membranes. A: The impact of Nutlin-3 on experimental PVR. RCFs are used to induce PVR, as described in Materials and Methods. The indicated rabbits receive no additional injections (none) or 0.1-mL injections of either vehicle or Nutlin-3 (20 μmol/L) on days 0, 2, and 4. PVR was graded according to the Fastenberg scale of classification 41: stage 0, no disease; stage 1, epiretinal membrane; stage 2, vitreoretinal traction without retinal detachment; stage 3, localized retinal detachment (1 to 2 quadrants); stage 4, extensive retinal detachment (2 to 4 quadrants without complete detachment); stage 5, complete retinal detachment. The data are subjected to an unpaired t-test or a one-way analysis of variance test. The asterisk denotes a statistically significant difference. B: Nutlin-3 enhances expression of p53 in epiretinal membranes. At the end of the experiment (day 28), the eyes are enucleated and paraffin sections of whole eyes are subjected to p53 IHC. Arrows, epiretinal membrane; arrowheads, retina; brown, p53. Left and right panels: A Nutlin-3–injected animal. Middle panel: A rabbit treated with vehicle. Left and middle panels: Stained with the p53 antibody. Right panel: Stained with non–immune IgG. Scale bar = 50 μm.
A molecular approach to assess the importance of reducing p53 for RV-mediated contraction and retinal detachment led to a similar conclusion. The overall strategy of this second approach was to reduce the PVR potential of RCFs by silencing the expression of PDGFRα, and then to test if it could be rescued by silencing the expression of p53. Lentiviral-mediated delivery of shRNAs directed toward either PDGFRα or p53 suppressed expression by at least 80% (Figure 3A). As shown in Figure 3B, RV-mediated suppression of p53 in shPDGFRα cells was substantially reduced, although not completely eliminated. Similarly, RV-induced contraction of cells was largely, although not completely, diminished in shPDGFRα cells (Figure 3C).
To investigate whether the incomplete suppression of RV-mediated contraction was due to the residual expression of PDGFRα, we tested the efficacy of imatinib to interfere with RV-induced contraction. Imatinib completely blocked RV-induced contraction in shGFP cells (see Supplemental Figure S3A at http://ajp.amjpathol.org), which indicated that one of the imatinib targets (abl or c-kit PDGFRα and PDGFRβ46) was essential. Our previously characterized panel of cells that did or did not express PDGFRs21 provided the opportunity to identify the relevant imatinib target. F cells, which were immortalized fibroblasts from mice lacking both PDGFR genes (but harboring all other imatinib targets), contracted weakly to RV, and this response was unaffected by imatinib (see Supplemental Figure S3B at http://ajp.amjpathol.org). Expressing PDGFRβ in these cells did not improve RV-induced contraction or generate sensitivity to imatinib (see Supplemental Figure S3B at http://ajp.amjpathol.org). Finally, expression of PDGFRα improved this RV-stimulated response, which was erased by imatinib (see Supplemental Figure S3B at http://ajp.amjpathol.org). These observations indicated that PDGFRα was the relevant target of imatinib, and suggested that the modest RV-induced contraction seen in shPDGFRα cells (Figure 3C) was because of residual expression of PDGFRα.
As expected from previous studies assessing the importance of PDGFRα for experimental PVR,21,25,47 there was a significantly statistic reduction in the PVR potential of shPDGFRα cells (Figure 4). shPDGFRα cells failed to induce retinal detachment, although they retained their ability to form membranes, which exerted traction of the retina (Figure 4). Molecularly suppressing p53 in shPDGFRα cells fully restored their ability to induce retinal detachment (Figure 4). Thus, two different experimental approaches indicated that reducing the level of p53 was essential for RV-mediated contraction and retinal detachment in an animal model of PVR.
Figure 4.

Molecularly suppressing expression of p53 restores the ability of PDGFRα-deficient cells to induce retinal detachment. The cells described in the legend to Figure 3A are compared for their PVR potential in a rabbit model of this disease. Inset: The type of lenti-shRNA used to modify the cells. PVR is induced as described in Materials and Methods. Each symbol represents the response of an individual rabbit on the indicated day. The data are subjected to an unpaired t-test or a one-way analysis of variance. The asterisk denotes a statistically significant difference.
PDGFRα Does More than Reduce p53 to Promote Contraction
To assess if reducing p53 was the only PDGFRα-mediated event required for contraction in response to RV, we compared this outcome in shGFP and shp53 cells. If it was the only event, then the contraction of shp53 cells would be RV independent. As shown in Supplemental Figure S4 (available at http://ajp.amjpathol.org), this was not the case. Although basal contraction of shp53 cells was enhanced, they responded well to RV. These observations indicated that PDGFRα did more than reduce the level of p53 to mediate RV-dependent contraction.
We asked the same question for three additional cellular responses associated with PVR: proliferation, protection from apoptosis and senescence. Just like contraction, proliferation of shp53 cells was responsive to RV (Figure 5A). In contrast, protection from apoptosis and senescence were fully engaged in unstimulated cells and RV did not further enhance these responses (Figure 5, B and C). These findings indicated that reducing p53 was sufficient to trigger some of the RV-stimulated cellular responses that were associated with PVR (protection from apoptosis and senescence), whereas contraction and proliferation required an event(s) in addition to reducing the level of p53 (Figure 6). The identity of the signaling enzymes responsible for mediating this event(s) is under investigation.
Figure 5.

The importance of PDGFRα and suppression of p53 for RV-dependent cell proliferation and protection from apoptosis and senescence. A: RCFs (shGFP, shPDGFRα, shPDGFRα/shp53, and shp53) are seeded into a 24-well plate at a density of 5 × 104 cells per well in DMEM plus 10% FBS. Six hours after the cells attach, the medium is changed to either 0.5-mL DMEM or rabbit vitreous (diluted 1:3 in DMEM). The media are replaced every day. The cells are counted with a hemocytometer on day 3. The mean ± SD of three independent experiments is shown. *P < 0.05 using a paired t-test. B: The RCFs in A are seeded into 60-mm dishes at a density of 100,000 cells per dish in DMEM plus 10% FBS. Six hours after the cells attach, the medium is changed to either 3-mL DMEM or rabbit vitreous (diluted 1:3 in DMEM). The media are replaced every day. On day 3, the cells are stained with fluorescein isothiocyanate–conjugated annexin V and propidium iodide (PI) in an apoptosis assay kit by following the manufacturer's instructions. Cells that are stained with annexin V and/or PI are detected and quantified by flow cytometry in Coulter Beckman XL. The mean ± SD of three independent experiments is shown. C: The RCFs in A are seeded into a 12-well plate at a density of 10,000 cells per well in DMEM plus 10% FBS. Six hours after the cells attach, the medium is changed to either 1-mL DMEM or rabbit vitreous (diluted 1:3 in DMEM). The media are replaced every day. On day 3, the β-galactosidase activity is measured as outlined in the manufacturer's instructions. Both stained and unstained cells are counted and photographed under an inverted microscope. The mean ± SD of three independent experiments is shown. *P < 0.05 using a paired t-test. NS, not significant. Scale bar = 50 μm.
Figure 6.
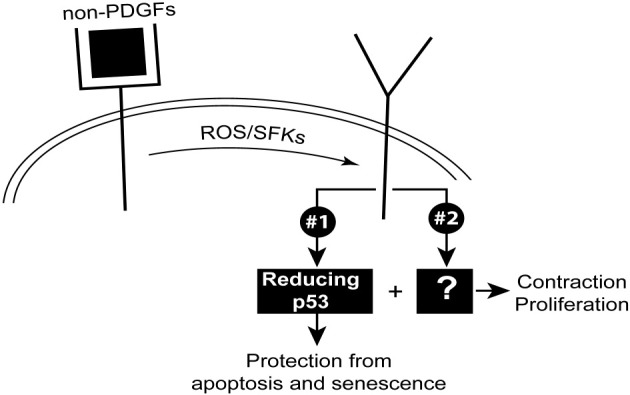
RV-engaged PDGFRα triggers two signaling pathways that drive cellular response intrinsic to PVR: signaling pathway 1, leading to a reduction in the level of p53; this is sufficient for protection from apoptosis and senescence. Contraction and proliferation require an additional set of signaling events that constitute pathway 2. ROS, reactive oxygen species; SFK, src family kinases.
Residual expression of PDGFRα in shPDGFRα cells provided an opportunity to compare cellular responses associated with PVR for their dependence on the level of expression of PDGFRα. In shPDGFR cells, RV was still able to promote contraction (Figure 3C), proliferation (Figure 5A) and survival (Figure 5B), however to a much lesser extent than in control cells. In contrast, RV failed to protect shPDGFR cells from senescence (Figure 5C). We conclude that protection from senescence required a higher level of PDGFR expression than the other three cellular responses.
Relevance to Clinical PVR
To begin to assess the clinical relevance of these findings, we considered the impact of Nutlin-3 on human vitreous (HV)–mediated signaling events and contraction of RPE cells isolated from a human PVR membrane. As shown in Figure 7A, Nutlin-3 prevented the precipitous decline in the level of p53 observed in HV-treated control cells, without affecting upstream signaling events. Furthermore, Nutlin-3 inhibited HV-stimulated contraction of RPE-containing collagen gels (Figure 7B). In addition, p53 was undetectable in epiretinal membranes from patients with PVR (see Supplemental Figure S5 at http://ajp.amjpathol.org). These findings indicated that Nutlin-3 had the potential to protect patients from developing PVR.
Figure 7.

p53 Attenuates HV-driven collagen gel contraction. A: The impact of Nutlin-3 on HV-mediated signaling events. The experiment is the same as in Figure 3A, except RPE cells from a patient epiretinal membrane are used in place of RCF, and HV (a pool of vitreous from five patients with PVR, diluted 1:3 in DMEM) is used in place of RV. The data presented are representative of three independent experiments. B: The impact of Nutlin-3 on HV-mediated contraction. The experiment is the same as in Figure 3B, with the modification noted in Figure 5A. The data are subjected to a paired t-test. The asterisk denotes a statistically significant difference. The data shown are representative of three independent experiments.
Discussion
We report that suppressing the expression of p53 appeared to be a required event in PDGFRα-mediated contraction of cells in a collagen gel and retinal detachment in an animal model of PVR. Furthermore, preventing the decline in the level of p53 with agents such as Nutlin-3 protected from retinal detachment. Finally, Nutlin-3 may be effective in the clinical setting because it prevented human PVR vitreous-induced contraction of cells isolated from a patient PVR membrane.
In the present study, we administered Nutlin-3 in a series of intravitreal injections. Although this approach completely prevented retinal detachment, 50% of the rabbits developed vitreal traction (stage 2) (Figure 2A). Because the last injection of Nutlin-3 was 23 days before the end of the experiment, the level of Nutlin-3 may have decreased to lower than the therapeutic range. The recently developed ocular formulation of Nutlin-3, which can be administered as a subconjunctival injection,34 may be a more effective approach to achieve Nutlin-3–mediated PVR prophylaxis.
Nutlin-3 treatment effectively prevented retinal detachment, and also slowed formation of membranes (stage 1) (Figure 2A), both of which are of clinical benefit. We were intrigued that membranes were able to form in Nutlin-3 animals because Nutlin-3 activates the p53 pathway,29 which counters cellular events that are intrinsic to membrane formation, such as proliferation and survival.48 However, this observation is consistent with reports that elevating p53 does not always cause apoptosis and/or cell cycle arrest.49–51 For instance, p53 is transiently elevated in mitotic cells of normal tissue in healthy animals, and fails to engage the p53 pathway because p53 is held in check by post-translational modifications.52 Additional studies are required to assess if this mechanism explains why membranes form in Nutlin-3–treated animals.
These studies reveal that cellular responses associated with PVR do not have the same requirements. Contraction and proliferation require a decline in the level of p53 and a second PDGFRα-mediated event (s), whereas protection from apoptosis and senescence proceed when only p53 is suppressed (Figure 6).
Epiretinal membranes formed in rabbits injected with cells that were unable to suppress p53 efficiently (shPDGFRα), which was required for optimal RV-mediated proliferation and viability, cellular events that are thought to be essential for membrane formation. Previous investigators have found that proliferation-incompetent cells induce PVR, provided that they are injected at a sufficiently high level.53 Thus, membranes may have formed in rabbits injected with shPDGFRα cells because enough of them were injected.
Both molecular and pharmacological approaches indicate that reducing the level of p53 was permissive for retinal detachment, a process that involves contraction of the retina-associated membrane. A simple explanation for this phenomenon is that p53 suppresses the expression of genes that are required for retinal detachment. For instance, p53 may inhibit production of those extracellular matrix proteins that are required for contraction of the membrane.54 However, such an explanation appears inadequate for the in vitro contraction assays, which contained ample extracellular matrix proteins that are conducive for contraction. Perhaps p53 down-regulates expression of integrins, such as β1,55 whose interaction with extracellular matrix proteins is essential for contraction. Alternatively, there may be a connection to epithelial cell membrane protein and focal adhesion kinase, which are essential for contraction of collagen gels and strongly implicated in PVR.56–60 Identifying the p53-governed genes that are essential for retinal detachment is an ongoing area of investigation.
Finally, just as p53 suppresses cell cycle progression,61 our findings indicate that p53 is the checkpoint of retinal detachment. In contrast to genetic lesions of the p53 pathway that are present in approximately 50% of solid tumors,28 it appears that epigenetic, environmental factors that result in noncanonical activation of PDGFRα drive p53-dependent blinding diseases, such as PVR.
Acknowledgments
We thank Marie Ortega and Jessica Lanzim for their help with the animal studies, Peter Mallen for his assistance in preparing figures, and Bianai Fan for the histological sections.
Footnotes
Supported by a grant from the NIH (EY012509 to A.K.) and the Canadian Institutes of Health Research grants IAO 77736 and MOP 97806 (J.M. and J.C.).
Disclosure: The authors and The Schepens Eye Research Institute (Harvard University) have filed a provisional US patent application related to this work.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at http://dx.doi.org/10.1016/j.ajpath.2012.05.036.
Supplementary data
Minimum effective dose and maximum tolerated dose of Nutlin-3. A: Western blot analysis to test levels of various proteins. RCFs are pretreated with the indicated concentrations of Nutlin-3 for 30 minutes, and then exposed to RV (diluted 1:3 in DMEM) for 2 hours. Cells are lysed, and the resulting lysates are subjected to Western blot analysis using the indicated antibodies. The data shown are representative of three independent experiments. B: Maximum tolerated dose analysis. RCFs are grown to near confluence in DMEM plus 10% FBS, and the medium is replaced with DMEM plus 5% FBS plus the indicated concentration of Nutlin-3. The medium is replaced every 24 hours, and the photographs are taken on day 3. The data shown are representative of two independent experiments. Scale bar = 50 μm.
Epiretinal membranes from rabbits subjected to the PVR protocol. Rabbits that undergo a gas vitrectomy are intravitreally injected with the indicated concentration of Nutlin-3 on days 0, 2, and 4. The rabbits undergo a fundus examination on days 1, 3, 5, 7, and 14. After the last fundus examination, the rabbits are euthanized, and the eyes are enucleated and fixed in 10% formalin. Sections are prepared, stained with H&E, and then photographed. Representative photographs of a Nutlin-3 (A) and a vehicle (B) injected eye are presented. Arrowheads and arrows, retina and epiretinal membrane, respectively. Scale bar = 50 μm. The lack of the retinal pigment epithelial cell layer is an artifact related to processing of the tissue.
Inhibiting PDGFRα kinase activity blocks RV-induced collagen contraction. The indicated RCFs (50,000 cells/mL) (A) or F cells (1 million cells/mL) (B) are subjected to a collagen contraction assay, as described in Materials and Methods. F cells are immortalized mouse embryo fibroblasts derived from PDGFR knockout mice that do not express either of the two PDGFR genes, and Fα and Fβ cells are F cells in which PDGFRα or PDGFRβ is re-expressed.21 After the collagen is solidified (90 minutes), DMEM or RV (diluted 1:3 in DMEM), supplemented with imatinib (10 μmol/L) or its vehicle, is added and replenished every 24 hours; the experiments are terminated on day 3. The panels below the bar graphs show photographs of representative gels; the data presented are representative of three independent experiments. The data are subjected to a paired t-test. The asterisk denotes a statistically significant difference. NS, not significant.
PDGFRα does more than suppress p53 to mediate RV-dependent contraction. A: Western blot analysis of knockdown RCFs. Lentiviruses are used to stably express shRNAs directed against GFP, PDGFRα, or p53 in primary RCFs. The resulting cell lysates are subjected to Western blot analysis using the indicated antibodies. The signal intensity is quantified and expressed as a ratio of the loading control (RasGAP). The data presented are representative of two independent experiments. B: Expression of PDGFRα potentiates RV-mediated suppression of p53. The cells in A are left resting or exposed to vitreous (diluted 1:3 in DMEM) from normal rabbits (RV) for 2 hours and lysed, and total cell lysates are subjected to Western blot analysis with the indicated antibodies. The signal intensity is quantified, and the data presented are representative of three independent experiments. C: RV-induced contraction of RCF-loaded collagen gels, which requires PDGFRα-mediated suppression of p53. The cells (50,000 cells/mL) in A are subjected to a collagen contraction assay. RV (diluted 1:3 in DMEM) is added on top of the gels on day 0 and replenished every 24 hours; the experiments are terminated on day 3. The panel below the bar graph shows photographs of representative gels; the data presented are representative of three independent experiments. The data are subjected to a paired t-test. The asterisk denotes a statistically significant difference. NS, not significant.
p53 is undetectable in human PVR membranes. Epiretinal membranes from patients with PVR who are subjected to p53 IHC. Although the positive control (rat colon cancer) demonstrates that this approach could readily detect p53 protein (B), p53 is undetectable in membranes from seven different patients, three of which are shown (D–F). The green-brown pigment is routinely observed in such samples, which contain many retinal pigment epithelial cells23; they are also present in the section of a PVR membrane that is stained with a nonimmune, isotype-matched control antibody (C). A: A section of rat colon cancer that serves as a negative control for B.
References
- 1.Han D. Proliferative Vitreoretinopathy. In: Albert D.M., Miller J.W., Azar D.T., Blodi B.A., editors. Elsevier Saunders; Philadelphia: 2008. pp. 2315–2324. [Google Scholar]
- 2.Wilkes S.R., Beard C.M., Kurland L.T., Robertson D.M., O'Fallon W.M. The incidence of retinal detachment in Rochester, Minnesota, 1970–1978. Am J Ophthalmol. 1982;94:670–673. doi: 10.1016/0002-9394(82)90013-7. [DOI] [PubMed] [Google Scholar]
- 3.Haimann M.H., Burton T.C., Brown C.K. Epidemiology of retinal detachment. Arch Ophthalmol. 1982;100:289–292. doi: 10.1001/archopht.1982.01030030291012. [DOI] [PubMed] [Google Scholar]
- 4.Charteris D.G. Growth factors in proliferative vitreoretinopathy. Br J Ophthalmol. 1998;82:106. doi: 10.1136/bjo.82.2.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michels R.G., Wilkinson C.P., Rice T.A. Mosby; St Louis: 1990. Retinal Detachment; pp. 669–706. [Google Scholar]
- 6.Mietz H., Heimann K. Onset and recurrence of proliferative vitreoretinopathy in various vitreoretinal diseases. Br J Ophthalmol. 1995;79:874–877. doi: 10.1136/bjo.79.10.874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Girard P., Mimoun G., Karpouzas I., Montefiore G. Clinical risk factors for proliferative vitreoretinopathy after retinal detachment surgery. Retina. 1994;14:417–424. doi: 10.1097/00006982-199414050-00005. [DOI] [PubMed] [Google Scholar]
- 8.Lleó Pérez A., Campos Fernández R., López Santoveña F., Sánchez Lorente G., Hernández Martinez F.J., Navarro Palop C. [Clinical risk factors for proliferative vitreoretinopathy after retinal detachment surgery] Spanish. Arch Soc Esp Oftalmol. 2000;75:741–750. [PubMed] [Google Scholar]
- 9.Nagasaki H., Ideta H., Uemura A., Morita H., Ito K., Yonemoto J. Comparative study of clinical factors that predispose patients to proliferative vitreoretinopathy in aphakia. Retina. 1991;11:204–207. doi: 10.1097/00006982-199111020-00002. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez de la Rua E., Pastor J.C., Aragon J., Mayo-Iscar A., Martinez V., Garcia-Arumi J., Giraldo A., Sanabria-Ruiz Colmenares M.R., Miranda I. Interaction between surgical procedure for repairing retinal detachment and clinical risk factors for proliferative vitreoretinopathy. Curr Eye Res. 2005;30:147–153. doi: 10.1080/02713680490904142. [DOI] [PubMed] [Google Scholar]
- 11.Tseng W., Cortez R.T., Ramirez G., Stinnett S., Jaffe G.J. Prevalence and risk factors for proliferative vitreoretinopathy in eyes with rhegmatogenous retinal detachment but no previous vitreoretinal surgery. Am J Ophthalmol. 2004;137:1105–1115. doi: 10.1016/j.ajo.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 12.Yoshino Y., Ideta H., Nagasaki H., Uemura A. Comparative study of clinical factors predisposing patients to proliferative vitreoretinopathy. Retina. 1989;9:97–100. doi: 10.1097/00006982-198909020-00004. [DOI] [PubMed] [Google Scholar]
- 13.Wiedemann P., Hilgers R.D., Bauer P., Heimann K., Daunomycin Study Group Adjunctive daunorubicin in the treatment of proliferative vitreoretinopathy: results of a multicenter clinical trial. Am J Ophthalmol. 1998;126:550–559. doi: 10.1016/s0002-9394(98)00115-9. [DOI] [PubMed] [Google Scholar]
- 14.Asaria R.H., Kon C.H., Bunce C., Charteris D.G., Wong D., Khaw P.T., Aylward G.W. Adjuvant 5-fluorouracil and heparin prevents proliferative vitreoretinopathy: results from a randomized, double-blind, controlled clinical trial. Ophthalmology. 2001;108:1179–1183. doi: 10.1016/s0161-6420(01)00589-9. [DOI] [PubMed] [Google Scholar]
- 15.Schiff W.M., Hwang J.C., Ober M.D., Olson J.L., Dhrami-Gavazi E., Barile G.R., Chang S., Mandava N. Safety and efficacy assessment of chimeric ribozyme to proliferating cell nuclear antigen to prevent recurrence of proliferative vitreoretinopathy. Arch Ophthalmol. 2007;125:1161–1167. doi: 10.1001/archopht.125.9.1161. [DOI] [PubMed] [Google Scholar]
- 16.Oh K., Hartnett M., Landers I.M. Pathogenic Mechanisms of Retinal Detachment. In: Ryan S., editor. Elsevier Mosby; Philadelphia: 2006. [Google Scholar]
- 17.Campochiaro P. The Pathogenesis of Proliferative Vitreoretinopathy. In: Ryan S., editor. Elsevier Mosby; Philadelphia: 2006. [Google Scholar]
- 18.Campochiaro P.A. Mechanisms in ophthalmic disease: pathogenic mechanisms in proliferative vitreoretinopathy. Arch Ophthalmol. 1997;115:237–241. doi: 10.1001/archopht.1997.01100150239014. [DOI] [PubMed] [Google Scholar]
- 19.Baudouin C., Fredj-Reygrobellet D., Gordon W.C., Baudouin F., Peyman G., Lapalus P., Gastaud P., Bazan N.G. Immunohistologic study of epiretinal membranes in proliferative vitreoretinopathy. Am J Ophthalmol. 1990;110:593–598. doi: 10.1016/s0002-9394(14)77054-0. [DOI] [PubMed] [Google Scholar]
- 20.Vinores S.A., Campochiaro P.A., Conway B.P. Ultrastructural and electron-immunocytochemical characterization of cells in epiretinal membranes. Invest Ophthalmol Vis Sci. 1990;31:14–28. [PubMed] [Google Scholar]
- 21.Andrews A., Balciunaite E., Leong F.L., Tallquist M., Soriano P., Refojo M., Kazlauskas A. Platelet-derived growth factor plays a key role in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 1999;40:2683–2689. [PubMed] [Google Scholar]
- 22.Robbins S.G., Mixon R.N., Wilson D.J., Hart C.E., Robertson J.E., Westra I., Planck S.R., Rosenbaum J.T. Platelet-derived growth factor ligands and receptors immunolocalized in proliferative retinal diseases. Invest Ophthalmol Vis Sci. 1994;35:3649–3663. [PubMed] [Google Scholar]
- 23.Cui J., Lei H., Samad A., Basavanthappa S., Maberley D., Matsubara J., Kazlauskas A. PDGF receptors are activated in human epiretinal membranes. Exp Eye Res. 2009;88:438–444. doi: 10.1016/j.exer.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lei H., Kazlauskas A. Growth factors outside of the platelet-derived growth factor (PDGF) family employ reactive oxygen species/Src family kinases to activate PDGF receptor alpha and thereby promote proliferation and survival of cells. J Biol Chem. 2009;284:6329–6336. doi: 10.1074/jbc.M808426200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lei H., Velez G., Hovland P., Hirose T., Gilbertson D., Kazlauskas A. Growth factors outside the PDGF family drive experimental PVR. Invest Ophthalmol Vis Sci. 2009;50:3394–3403. doi: 10.1167/iovs.08-3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lei H., Velez G., Kazlauskas A. Pathological signaling via platelet-derived growth factor receptor {alpha} involves chronic activation of Akt and suppression of p53. Mol Cell Biol. 2011;31:1788–1799. doi: 10.1128/MCB.01321-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levine A.J., Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hainaut P., Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res. 2000;77:81–137. doi: 10.1016/s0065-230x(08)60785-x. [DOI] [PubMed] [Google Scholar]
- 29.Vassilev L.T., Vu B.T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C., Fotouhi N., Liu E.A. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 30.Prives C. Signaling to p53: breaking the MDM2-p53 circuit. Cell. 1998;95:5–8. doi: 10.1016/s0092-8674(00)81774-2. [DOI] [PubMed] [Google Scholar]
- 31.Ofir-Rosenfeld Y., Boggs K., Michael D., Kastan M.B., Oren M. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol Cell. 2008;32:180–189. doi: 10.1016/j.molcel.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sasaki M., Kawahara K., Nishio M., Mimori K., Kogo R., Hamada K., Itoh B., Wang J., Komatsu Y., Yang Y.R., Hikasa H., Horie Y., Yamashita T., Kamijo T., Zhang Y., Zhu Y., Prives C., Nakano T., Mak T.W., Sasaki T., Maehama T., Mori M., Suzuki A. Regulation of the MDM2-P53 pathway and tumor growth by PICT1 via nucleolar RPL11. Nat Med. 2011;17:944–951. doi: 10.1038/nm.2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Secchiero P., Bosco R., Celeghini C., Zauli G. Recent advances in the therapeutic perspectives of Nutlin-3. Curr Pharm Des. 2011;17:569–577. doi: 10.2174/138161211795222586. [DOI] [PubMed] [Google Scholar]
- 34.Brennan R.C., Federico S., Bradley C., Zhang J., Flores-Otero J., Wilson M., Stewart C., Zhu F., Guy K., Dyer M.A. Targeting the p53 pathway in retinoblastoma with subconjunctival Nutlin-3a. Cancer Res. 2011;71:4205–4213. doi: 10.1158/0008-5472.CAN-11-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lei H., Velez G., Cui J., Samad A., Maberley D., Matsubara J., Kazlauskas A. N-acetylcysteine suppresses retinal detachment in an experimental model of proliferative vitreoretinopathy. Am J Pathol. 2010;177:132–140. doi: 10.2353/ajpath.2010.090604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenkranz S., DeMali K.A., Gelderloos J.A., Bazenet C., Kazlauskas A. Identification of the receptor-associated signaling enzymes that are required for platelet-derived growth factor-AA-dependent chemotaxis and DNA synthesis. J Biol Chem. 1999;274:28335–28343. doi: 10.1074/jbc.274.40.28335. [DOI] [PubMed] [Google Scholar]
- 37.Lei H., Hovland P., Velez G., Haran A., Gilbertson D., Hirose T., Kazlauskas A. A potential role for PDGF-C in experimental and clinical proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci. 2007;48:2335–2342. doi: 10.1167/iovs.06-0965. [DOI] [PubMed] [Google Scholar]
- 38.Pennock S., Rheaume M.A., Mukai S., Kazlauskas A. A novel strategy to develop therapeutic approaches to prevent proliferative vitreoretinopathy. Am J Pathol. 2011;179:2931–2940. doi: 10.1016/j.ajpath.2011.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wong C.A., Potter M.J., Cui J.Z., Chang T.S., Ma P., Maberley A.L., Ross W.H., White V.A., Samad A., Jia W., Hornan D., Matsubara J.A. Induction of proliferative vitreoretinopathy by a unique line of human retinal pigment epithelial cells. Can J Ophthalmol. 2002;37:211–220. doi: 10.1016/s0008-4182(02)80112-0. [DOI] [PubMed] [Google Scholar]
- 40.Ikuno Y., Kazlauskas A. An in vivo gene therapy approach for experimental proliferative vitreoretinopathy using the truncated platelet-derived growth factor alpha receptor. Invest Ophthalmol Vis Sci. 2002;43:2406–2411. [PubMed] [Google Scholar]
- 41.Fastenberg D.M., Diddie K.R., Sorgente N., Ryan S.J. A comparison of different cellular inocula in an experimental model of massive periretinal proliferation. Am J Ophthalmol. 1982;93:559–564. doi: 10.1016/s0002-9394(14)77369-6. [DOI] [PubMed] [Google Scholar]
- 42.Zhou B.P., Liao Y., Xia W., Zou Y., Spohn B., Hung M.C. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol. 2001;3:973–982. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]
- 43.Ogawara Y., Kishishita S., Obata T., Isazawa Y., Suzuki T., Tanaka K., Masuyama N., Gotoh Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem. 2002;277:21843–21850. doi: 10.1074/jbc.M109745200. [DOI] [PubMed] [Google Scholar]
- 44.Gottlieb T.M., Leal J.F., Seger R., Taya Y., Oren M. Cross-talk between Akt, p53 and Mdm2: possible implications for the regulation of apoptosis. Oncogene. 2002;21:1299–1303. doi: 10.1038/sj.onc.1205181. [DOI] [PubMed] [Google Scholar]
- 45.Haupt Y., Maya R., Kazaz A., Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 46.Druker B.J., Talpaz M., Resta D.J., Peng B., Buchdunger E., Ford J.M., Lydon N.B., Kantarjian H., Capdeville R., Ohno-Jones S., Sawyers C.L. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 47.Ikuno Y., Leong F.L., Kazlauskas A. Attenuation of experimental proliferative vitreoretinopathy by inhibiting the platelet-derived growth factor receptor. Invest Ophthalmol Vis Sci. 2000;41:3107–3116. [PubMed] [Google Scholar]
- 48.Lei H., Rheaume M.A., Kazlauskas A. Recent developments in our understanding of how platelet-derived growth factor (PDGF) and its receptors contribute to proliferative vitreoretinopathy. Exp Eye Res. 2010;90:376–381. doi: 10.1016/j.exer.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Giono L.E., Manfredi J.J. Mdm2 is required for inhibition of Cdk2 activity by p21, thereby contributing to p53-dependent cell cycle arrest. Mol Cell Biol. 2007;27:4166–4178. doi: 10.1128/MCB.01967-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mendrysa S.M., McElwee M.K., Michalowski J., O'Leary K.A., Young K.M., Perry M.E. Mdm2 is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol Cell Biol. 2003;23:462–472. doi: 10.1128/MCB.23.2.462-473.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mendrysa S.M., O'Leary K.A., McElwee M.K., Michalowski J., Eisenman R.N., Powell D.A., Perry M.E. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev. 2006;20:16–21. doi: 10.1101/gad.1378506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Loewer A., Batchelor E., Gaglia G., Lahav G. Basal dynamics of p53 reveal transcriptionally attenuated pulses in cycling cells. Cell. 2010;142:89–100. doi: 10.1016/j.cell.2010.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fastenberg D.M., Diddie K.R., Dorey K., Ryan S.J. The role of cellular proliferation in an experimental model of massive periretinal proliferation. Am J Ophthalmol. 1982;93:565–572. doi: 10.1016/s0002-9394(14)77370-2. [DOI] [PubMed] [Google Scholar]
- 54.Iotsova V., Stehelin D. Down-regulation of fibronectin gene expression by the p53 tumor suppressor protein. Cell Growth Differ. 1996;7:629–634. [PubMed] [Google Scholar]
- 55.Qiu J., Wang G., Hu J., Peng Q., Zheng Y. Id1-induced inhibition of p53 facilitates endothelial cell migration and tube formation by regulating the expression of beta1-integrin. Mol Cell Biochem. 2011;357:125–133. doi: 10.1007/s11010-011-0882-6. [DOI] [PubMed] [Google Scholar]
- 56.Morales S.A., Mareninov S., Coulam P., Wadehra M., Goodglick L., Braun J., Gordon L.K. Functional consequences of interactions between FAK and epithelial membrane protein 2 (EMP2) Invest Ophthalmol Vis Sci. 2009;50:4949–4956. doi: 10.1167/iovs.08-3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morales S.A., Mareninov S., Prasad P., Wadehra M., Braun J., Gordon L.K. Collagen gel contraction by ARPE-19 cells is mediated by a FAK-Src dependent pathway. Exp Eye Res. 2007;85:790–798. doi: 10.1016/j.exer.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 58.Morales S.A., Mareninov S., Wadehra M., Zhang L., Goodglick L., Braun J., Gordon L.K. FAK activation and the role of epithelial membrane protein 2 (EMP2) in collagen gel contraction. Invest Ophthalmol Vis Sci. 2009;50:462–469. doi: 10.1167/iovs.07-1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morales S.A., Telander D., Notterpek L., Wadehra M., Braun J., Gordon L.K. Rewiring integrin-mediated signaling and cellular response with the peripheral myelin protein 22 and epithelial membrane protein 2 components of the tetraspan web. Invest Ophthalmol Vis Sci. 2011;52:5465–5472. doi: 10.1167/iovs.10-6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Telander D.G., Morales S.A., Mareninov S., Forward K., Gordon L.K. Epithelial membrane protein-2 (EMP2) and experimental proliferative vitreoretinopathy (PVR) Curr Eye Res. 2011;36:546–552. doi: 10.3109/02713683.2011.561468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Levine A.J., Momand J., Finlay C.A. The p53 tumour suppressor gene. Nature. 1991;351:453–456. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Minimum effective dose and maximum tolerated dose of Nutlin-3. A: Western blot analysis to test levels of various proteins. RCFs are pretreated with the indicated concentrations of Nutlin-3 for 30 minutes, and then exposed to RV (diluted 1:3 in DMEM) for 2 hours. Cells are lysed, and the resulting lysates are subjected to Western blot analysis using the indicated antibodies. The data shown are representative of three independent experiments. B: Maximum tolerated dose analysis. RCFs are grown to near confluence in DMEM plus 10% FBS, and the medium is replaced with DMEM plus 5% FBS plus the indicated concentration of Nutlin-3. The medium is replaced every 24 hours, and the photographs are taken on day 3. The data shown are representative of two independent experiments. Scale bar = 50 μm.
Epiretinal membranes from rabbits subjected to the PVR protocol. Rabbits that undergo a gas vitrectomy are intravitreally injected with the indicated concentration of Nutlin-3 on days 0, 2, and 4. The rabbits undergo a fundus examination on days 1, 3, 5, 7, and 14. After the last fundus examination, the rabbits are euthanized, and the eyes are enucleated and fixed in 10% formalin. Sections are prepared, stained with H&E, and then photographed. Representative photographs of a Nutlin-3 (A) and a vehicle (B) injected eye are presented. Arrowheads and arrows, retina and epiretinal membrane, respectively. Scale bar = 50 μm. The lack of the retinal pigment epithelial cell layer is an artifact related to processing of the tissue.
Inhibiting PDGFRα kinase activity blocks RV-induced collagen contraction. The indicated RCFs (50,000 cells/mL) (A) or F cells (1 million cells/mL) (B) are subjected to a collagen contraction assay, as described in Materials and Methods. F cells are immortalized mouse embryo fibroblasts derived from PDGFR knockout mice that do not express either of the two PDGFR genes, and Fα and Fβ cells are F cells in which PDGFRα or PDGFRβ is re-expressed.21 After the collagen is solidified (90 minutes), DMEM or RV (diluted 1:3 in DMEM), supplemented with imatinib (10 μmol/L) or its vehicle, is added and replenished every 24 hours; the experiments are terminated on day 3. The panels below the bar graphs show photographs of representative gels; the data presented are representative of three independent experiments. The data are subjected to a paired t-test. The asterisk denotes a statistically significant difference. NS, not significant.
PDGFRα does more than suppress p53 to mediate RV-dependent contraction. A: Western blot analysis of knockdown RCFs. Lentiviruses are used to stably express shRNAs directed against GFP, PDGFRα, or p53 in primary RCFs. The resulting cell lysates are subjected to Western blot analysis using the indicated antibodies. The signal intensity is quantified and expressed as a ratio of the loading control (RasGAP). The data presented are representative of two independent experiments. B: Expression of PDGFRα potentiates RV-mediated suppression of p53. The cells in A are left resting or exposed to vitreous (diluted 1:3 in DMEM) from normal rabbits (RV) for 2 hours and lysed, and total cell lysates are subjected to Western blot analysis with the indicated antibodies. The signal intensity is quantified, and the data presented are representative of three independent experiments. C: RV-induced contraction of RCF-loaded collagen gels, which requires PDGFRα-mediated suppression of p53. The cells (50,000 cells/mL) in A are subjected to a collagen contraction assay. RV (diluted 1:3 in DMEM) is added on top of the gels on day 0 and replenished every 24 hours; the experiments are terminated on day 3. The panel below the bar graph shows photographs of representative gels; the data presented are representative of three independent experiments. The data are subjected to a paired t-test. The asterisk denotes a statistically significant difference. NS, not significant.
p53 is undetectable in human PVR membranes. Epiretinal membranes from patients with PVR who are subjected to p53 IHC. Although the positive control (rat colon cancer) demonstrates that this approach could readily detect p53 protein (B), p53 is undetectable in membranes from seven different patients, three of which are shown (D–F). The green-brown pigment is routinely observed in such samples, which contain many retinal pigment epithelial cells23; they are also present in the section of a PVR membrane that is stained with a nonimmune, isotype-matched control antibody (C). A: A section of rat colon cancer that serves as a negative control for B.