

Abstract
Evidence suggests a proinflammatory role of lysophosphatidic acid (LPA) in various pathologic abnormalities, including in the central nervous system. Herein, we describe LPA as an important mediator of inflammation after spinal cord injury (SCI) in zebrafish and mice. Furthermore, we describe a novel monoclonal blocking antibody raised against LPA that potently inhibits LPA's effect in vitro and in vivo. This antibody, B3, specifically binds LPA, prevents it from interacting with its complement of receptors, and blocks LPA's effects on the neuronal differentiation of human neural stem/progenitor cells, demonstrating its specificity toward LPA signaling. When administered systemically to mice subjected to SCI, B3 substantially reduced glial inflammation and neuronal death. B3-treated animals demonstrated significantly more neuronal survival upstream of the lesion site, with some functional improvement. This study describes the use of anti-LPA monoclonal antibody as a novel therapeutic approach for the treatment of SCI.
Spinal cord injury (SCI) often results in permanent damage at the site of the injury from the initial neurotrauma, and it is complicated by inflammatory and other processes that prevent neuronal regeneration and recovery by the central nervous system (CNS). Many approaches are under way to investigate mechanisms for improving neural regeneration and promoting functional recovery, including blockage of axonal growth inhibitory molecules, treatment with growth-promoting molecules, application of measures to reduce glial scarring, and the development of methods to limit diffuse inflammation and cell death. Thus, the key to an effective therapy after SCI is to identify factors that contribute to acute and secondary events that limit regeneration and functional recovery. Herein, we describe for the first time the role of lysophosphatidic acid (LPA) as a significant contributor to acute and secondary injury processes, and we describe a novel therapeutic antibody against LPA that may be useful as a treatment for SCI.
LPA is a bioactive lysophospholipid that is a collection of structural analogs with a single functional glycerol alcohol phosphate moiety esterified to a variety of fatty acyl hydrocarbons of varied lengths and degrees of saturation. LPA is detected in various body fluids, including serum and cerebrospinal fluid, and it is thought that LPA is an inflammatory and wound-healing mediator, released from activated platelets, astrocytes, and other cells participating in the inflammatory process.1 LPA is also known to induce the release of various proinflammatory cytokines from a variety of cells, including astrocytes.2
LPA mainly acts through binding to its specific G-protein–coupled receptors encoded by different genes: LPA1/Edg-2/rec.1.3/vzg-1/Gpcr26/Mrec1.3, LPA2/Edg-4, LPA3/Edg-7/RP4-678I3/HOFNH30, LPA4/P2Y9/GPR23, LPA5/GPR92, LPA6/P2Y5, GPR87, and P2Y10.1 In the developing nervous system, LPA targets most cell types and plays an important role during neurogenesis in establishing the cerebral cortex.1,3 In the adult mammalian nervous system, LPA1 is found in oligodendrocytes and Schwann cells,4–8 whereas LPA2-5 are weakly expressed.8–11 LPA and its producing enzyme autotaxin are present in the cerebrospinal fluid 12 and in the adult brain, including in the white matter, choroid plexus, and leptomeningeal cells.13–16 Furthermore, LPA can induce astrocyte reactivity in the mouse brain17 and neuropathic pain and demyelination in the spinal cord.18–20 Moreover, various studies showed that LPA can stimulate astrocytic proliferation,21 promote neuronal death,22 and mediate microglial activation23 and is cytotoxic to the neuromicrovascular endothelium.24 Considering the pleiotropic effects of LPA on many key CNS cell types, together with data showing localized up-regulation of LPA receptors after injury in mice and humans,8,25 it is likely that LPA regulates essential aspects of the cellular reorganization after neural trauma, such as reactive astrogliosis and neural degeneration. Thus, it is probable that LPA is a key player in regulating response to injury and, consequently, in modulating the outcome of CNS damage.
We used two animal models to examine the role of LPA in an established regenerative (zebrafish) SCI injury model and a nonregenerative (murine) model of SCI. We first examined the role of LPA in a zebrafish spinal cord transection model. This in vivo model regenerates damaged nerves and recovers full locomotor ability after SCI in a remarkably short period after injury.26,27 We then assessed the effect of LPA on inflammatory, glial, and neuronal cell activity and on neuronal regeneration in the zebrafish proregenerative model. Second, we used a well-characterized murine model of SCI to evaluate the role of LPA in a nonregenerative system. For this evaluation, we developed a novel and specific mouse monoclonal antibody (mAb) against LPA that blocks LPA's effects in vitro on neural stem/progenitor cells (NS/PCs) and in vivo by demonstrating neuroprotection and improving the functional outcomes of SCI in this murine model of SCI. Furthermore, we show the mechanism of LPA's effects in the CNS after SCI by assessing its effect on the various CNS cell types. Thus, using a regenerative model and a nonregenerative model, we demonstrated the proinflammatory role of LPA during SCI. This work highlights the importance of LPA modulation in neurotrauma and provides proof of concept for the blockage of LPA signaling to treat SCI. This work demonstrates that the anti-LPA mAb may be a useful therapeutic reagent for the treatment of SCI.
Materials and Methods
Ethics
All the experiments were approved by the human or animal research ethics committees of the University of Melbourne, St. Vincent's Hospital, and Monash University in accordance with the requirements of the National Health and Medical Research Council of Australia (Australian Code of Practice for the Care and Use of Animals for Scientific Purposes and the National Statement on Ethical Conduct in Human Research).
Reagents
Dilutions of LPA (Sigma-Aldrich, Castle Hill, Australia) were made in 0.1% fatty acid–free bovine serum albumin (BSA; final concentration, 0.01% BSA; Sigma-Aldrich). The murine anti-LPA mAb B3 and the isotype-matched control IgGB2 mAb were obtained from Lpath Inc. (San Diego, CA).
Zebrafish Strains
Two transgenic (Tg) lines were used: macrophage-expressed gene1 Tg(mpeg1:GFP) and neutrophils Tg(mpx:GFP)i114,3,28,29 GFAP promoter Tg(GFAP:GFP)mi200130 or motor neuron Islet1 promoter Tg(isl1:GFP)2rw012.31
Spinal Cord Lesion
Spinal cords of zebrafish were lesioned as described elsewhere.27,32 Briefly, adult fish (3 to 6 months old) were anesthetized in 0.033% tricaine methanesulfonate (MS-222) in fish tank water until respiratory movements of the opercula stopped (3 to 5 minutes). A longitudinal incision was made halfway between the dorsal fin and the operculum, corresponding to the eighth vertebra (∼5 mm caudal to the operculum) of the spinal cord. The initial incision was made through the muscle layer, and the vertebral column was exposed by holding the muscle tissue aside. Then, the vertebral column was completely transected using microscissors. A single injection of 10 μL of 5 μmol/L LPA in 0.1% BSA or 0.1% BSA alone (controls) was performed into the lesion site and around the muscle tissue immediately after spinal cord transection. The wound was sealed with a drop of 3M Vetbond tissue adhesive (3M, St. Paul, MN). The gills of the fish were flushed in a tank of fresh fish water by gently pulling the fish through the water. Fish resumed breathing within a few seconds.
Bromodeoxyuridine Application
Injections of 50 μL of i.p. bromodeoxyuridine (BrdU) (2.5 mg/mL; Sigma-Aldrich) were performed 0, 2, and 4 days after lesioning.
Locomotor Analysis
A scale from 1 to 5 was designed to assess fish motor function improvement at different time points after spinal cord transection as described previously.32
Zebrafish Tissue Preparation
To collect tissue after SCI, fish were deeply anesthetized with an overdose of 0.033% tricaine methanesulfonate (MS-222). The spinal cords were exposed and fixed for 2 hours in 4% paraformaldehyde (PFA) in PBS at room temperature, subsequently dissected out, postfixed for 2 to 3 hours at room temperature, followed by immersion in 30% sucrose in PBS overnight at 4°C before embedding in OCT. Spinal cords were cryostat sectioned at 20 μm thickness. Analyses were performed in each Tg strain at different time points after SCI (as indicated in Results). Time points were specifically chosen to be the most appropriate for each aspect investigated based on preliminary data not shown.
TUNEL Method
TUNEL staining was performed on the mpx:GFP:EGFP line 6 hours after SCI using an in situ cell death detection kit, TMR red (Roche Diagnostics GmbH, Mannheim, Germany), according to the manufacturer's protocol.
Immunohistochemical Analysis
Sections were labeled using standard immunohistochemical procedures to determine the expression and localization of BrdU. Sections were postfixed for 10 minutes in 4% PFA, and antigen retrieval was performed by incubating the sections for 15 minutes in 2 mol/L HCl, followed by blocking (PBS-TX containing 5% normal goat serum; Invitrogen, Carlsbad, CA) for 1 hour at room temperature. Mouse anti-BrdU (1:400; Roche Diagnostics) in blocking solution was incubated overnight at 4°C. After washing, sections were incubated for 2 hours at room temperature, with secondary antibodies diluted in blocking solution [goat anti-mouse Alexa Fluor 594 (1:1000; Molecular Probes Inc., Eugene, OR)].
RT-PCR of Spinal Cord Tissue
Approximately 0.5 g of brain, muscle, liver, and spinal cord tissues each were dissected from wild-type zebrafish, and RNA was extracted using TRI reagent (Sigma-Aldrich). Total RNA was reversed transcribed using the SuperScript III Reverse Transcriptase kit (Invitrogen), and the products were analyzed by RT-PCR. PCR products were amplified for 31 cycles and were separated on 2% agarose gel. β-Actin expression was evaluated as an internal control. The primer sequences used were LPAR133 and β-actin (forward: 5′-GCATTGCTGACCGTATGCAG-3′; reverse: 5′-GATCCACATCTGCTGGAAGGTGG-3′).
Probe Generation and in Situ Hybridization
A 1208-bp fragment of LPAR1 was amplified from mRNA templates with Platinum Taq DNA polymerase (Invitrogen) and was cloned into pGEM-T easy vector system (Promega Corp., Madison, WI). Primers used were as previously described.33 Plasmids were linearized, transcribed, and labeled using SP6 polymerase (Roche Diagnostics) and a DIG RNA labeling mix (Roche Diagnostics). In situ hybridization was performed by standard procedures on 30-μm sections. After staining, tissues were imaged using an Axio Imager Z1 compound microscope (Carl Zeiss MicroImaging GmbH, Jena, Germany).
Specificity of the Anti-LPA Antibody B3
The specificity of the murine anti-LPA IgGβ2 mAb B3 was determined by competition enzyme-linked immunosorbent assay (ELISA) using methods similar to those published for antibodies directed against sphingosine-1-phosphate.34 The 18:0 LPA coating material was diluted to 0.33 μg/mL in carbonate buffer (100 mmol/L NaHCO3, 33.6 mmol/L Na2CO3, pH 9.5). Plates were first coated with 100 μL per well of this coating solution and were incubated at 37°C for 1 hour. The plates were then washed four times with PBS (100 mmol/L Na2HPO4, 20 mmol/L KH2PO4, 27 mmol/L KCl, 1.37 mmol/L NaCl, pH 7.4) and were blocked with 150 μL per well PBS + 1% BSA + 0.1% Tween 20 for 1 hour at room temperature. The murine B3 IgGβ2 mAb was tested against lipids that are structurally and chemically similar to LPA at 5, 2.5, 1.25, 0.625, and 0.0 μmol/L. (Data in Table 1 are given for the 2.5-μmol/L level of competitive lipid and are expressed as a percentage of inhibition of binding). The antibody was diluted to 0.5 μg/mL in PBS + 0.1% Tween 20 and was combined with the lipid samples at a 1:3 ratio of antibody to sample on a nonbinding plate (usually 100 μL of mAb and 300 μL of samples/standards, enough to run in triplicate). The plates were then washed four times with PBS and were incubated for 1 hour at room temperature with 100 μL per well of the primary antibody combined with the samples. Next, the plates were washed four times with PBS and then were incubated for 1 hour at room temperature with 100 μL per well of horseradish peroxidase–conjugated goat anti-mouse antibody diluted 1:20,000 in PBS + 1% BSA + 0.1% Tween 20. Again, the plates were washed four times with PBS and were developed using 100 μL per well of 3,3′,5,5′-tetramethylbenzidine substrate at 4°C. After 8 minutes, the reaction was stopped with 1 mol/L H2SO4, 100 μL per well. The OD was measured at 450 nm using a Thermo Multiskan EX microplate photometer (Thermo Scientific, Rockford, IL). Raw data were transferred to GraphPad software version 5 (GraphPad Software Inc., San Diego, CA) for analysis. Values for percentage inhibition were calculated for each lipid using a Microsoft Excel spreadsheet (Microsoft Corp., Redmond, WA).
Table 1.
Competition ELISA
| Competitive lipid 2.5 μmol/L | B3 inhibition (%) |
|---|---|
| 1-(9Z,12Z-octadecadienoyl)-sn-glycero-3-phosphate (1-linoleoyl LPA, 18:2 LPA) | 71 |
| D-erythro-sphingosine-1-phosphate (S1P) | 4 |
| D-erythro-sphingosine (SPH) | 3 |
| D-erythro-sphingosine phosphocholine (lyso sphingomyelin, SPC) | 2 |
| 1-Stearoyl-2-hydroxy-sn-glycerol-3-phosphocholine (LPC) | −2 |
| 1-Stearoyl-2-hydroxy-sn-glycerol-3-phosphoethanolamine (LPE) | −2 |
| 1-Stearoyl-2-hydroxy-sn-glycero-3-[phosho-rac-1-glycerol)] (LPG) | 8 |
| 1-Oleoyl-2-hydroxy-sn-glycerol-3-phospho-L-serine (LPS) | 2 |
| 1-Olyoyl-2-Hydroxy-sn-glycero-3-phosphoinositol (LPI) | 3 |
| 1,2-Dipalmitoyl-sn-glycero-3-[phospho-L-serine] (DPPS) | 3 |
| 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) | 1 |
| 1,2-Dilinoleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (18:2 PG) | 0 |
| 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphate (16:0–18:1) (PA) | 0 |
| 1,2-Dieladoyl-sn-glycero-3-phosphoethanolamine | 6 |
| 1,2-Diundecanoyl-sn-glycero-3-phosphocholine | −1 |
| 1-Alkyl-2-acetoyl-sn-glycero-3-phosphocholine (PAF) | 4 |
| 1-Oleoyl-sn-glycero-2-3-cyclic-phosphate (cyclic LPA) | 16 |
| 1-Alkyl-2-hydroxy-sn-glycero-3-phosphocholine (Lyso PAF) | 6 |
| 1-Bromo-3(S)-hydroxy-4- (palmitoyloxy) butyl] phosphonate (BrP-LPA) | 6 |
| 1,2-Dioleoyl-sn-glycerol (DAG) | −5 |
| (R,R)−2,2′-bisdodecyl-LBPA | 2 |
| 2-Arachidonoyl glycerol (2-AG) | 7 |
| Glycerol phosphate | 3 |
2-AG, 2-arachidonoyl glycerol; 18;2 PG, 1,2-Dilinoleoyl-sn-Glycero-3-[Phospho-rac-(1-glycerol)]; BrP LPA, 1-Bromo-3(S)-hydroxy-4- (palmitoyloxy) butyl] phosphonate; Cyclic LPA, 1-Oleoyl-sn-Glycero-2-3-Cyclic-Phosphate; DAG, 1,2-dioleoyl-sn-glycerol; DPPS, 1,2-Dipalmitoyl-sn-Glycero-3-[Phospho-L-Serine]; Lyso PAF, 1-Alkyl-2-Hydroxy-sn-Glycero-3-Phosphocholine; LPC, 1-Stearoyl-2-Hydroxy-sn-Glycerol-3-Phosphocholine; LPE, 1-Stearoyl-2-Hydroxy-sn-Glycerol-3-Phosphoethanolamine; LPG, 1-Stearoyl-2-Hydroxy-sn-Glycero-3-[Phosho-rac-1-glycerol]; LPI, 1-Olyoyl-2-Hydroxy-sn-Glycero-3-Phosphoinositol; LPS, 1-Oleoyl-2-hydroxy-sn-glycerol-3-phospho-L-serine; PA, 1-Palmitoyl-2-Oleoyl-sn-Glycero-3-Phosphate (16:0-18:1) (phosphatidic acid); PAF, 1-Alkyl-2-Acetoyl-sn-Glycero-3-Phosphocholine; PC, 1,2-diundecanoyl-sn-glycero-3-phosphocholine; PE, 1,2-dieladoyl-sn-glycero-3-phosphoethanolamine; POPC, 1-Palmitoyl-2-Oleoyl-sn-Glycero-3-Phosphocholine; S1P, D-erythro-Sphingosine-1-Phosphate; SPC, D-erythro-Sphingosine Phosphocholine; SPH, D-erythro-Sphingosine.
LPA Receptor Signaling Assay
The ability of the murine anti-LPA antibody B3 to block LPA signaling via its receptor was studied in a series of assays using cells supplied by DiscoveRx (Freemont, CA). For the present studies, the DiscoveRx PathHunter CHO-K1 EDG2 or EDG4 or EDG7 β-arrestin cell lines were used to test LPA signaling to cells overexpressing these LPA receptors. DiscoverRX uses enzyme fragment complementation technology in which two weakly complementing fragments of the β-galactosidase enzyme are expressed in stable transfected cells. In this system, one fragment of the β-galactosidase, termed the enzyme acceptor, is fused to the C-terminus of the β-arrestin2. The complementing fragment of β-galactosidase, termed the ProLink tag (DiscoveRx), is expressed as a fusion protein with LPA1-3 at the C-terminus. On activation, the LPA receptor is phosphorylated, providing a binding site for β-arrestin. The interaction of β-arrestin and the LPA receptor forces the interaction of ProLink and enzyme acceptor, thus allowing complementation of the two fragments of β-galactosidase and the formation of functional enzyme capable of hydrolyzing substrate and generating a chemiluminescent signal. Complementation is driven by protein-protein interaction between arrestin–enzyme acceptor and ProLink-labeled LPA receptor. Cells were grown in culture following the vendor recommendations, and assays were run in a 96-well plate format. Assay plates were prepared by collecting cells in a sterile 50-mL conical tube using 5 mL of Cellstripper nonenzymatic cell dissociation buffer (Mediatech Inc., Oakland, CA). Cells were spun at 1100 rpm for 5 minutes, supernatant was removed, and cell pellet was resuspended in 5 mL of complete media with antibiotics. The cells were then counted using an automated cell counter and were plated at 20,000 cells per well (100 μL of total volume in each well) in 96-well white clear-bottom plates. Plates were incubated at 37°C in a humidified 5% CO2 atmosphere. After 24 hours, cells were starved in reduced serum media and incubated at 37°C for 24 hours. For the LPA binding experiments, a 100 μmol/L stock of 18:2 LPA (Avanti Polar Lipids, Alabaster, AL) was diluted 0 to 10 mmol/L in media containing 1 mg/mL of BSA, and for the antibody inhibition experiment, 200 nmol/L LPA for LPA1 receptor or 400 nmol/L LPA for LPA2 and 3-overexpresing cells was incubated with 0 to 2000 mg/mL anti-LPA antibody. The media from the 96-well plates containing the cells were removed, and 90 μL of either LPA or LPA plus anti-LPA mAb was added to each well. The plates were incubated at 37°C in a humidified 5% CO2 atmosphere for 90 minutes. Plates were then washed with 300 μL of media, and 100 μL of new media plus 50 μL of freshly made working detection reagent solution (DiscoverRx) was added to each well. The plates were incubated in the dark at room temperature for an additional 90 minutes. Finally, the plates were read on a standard luminescence plate reader. Data were fit to a four-parameter equation (GraphPad Prism; GraphPad Software) and were analyzed using GraphPad Prism 5 software. The standard relative light units were graphed using a four-parameter fit equation.
Neural Induction of Human Embryonic Stem Cells into NS/PCs
The human embryonic stem cell lines HES-3 (WiCell, Madison, WI) and ENVY (ES Cell International, Singapore) were used in this study. For each type of experiment, medium was changed every second day. Neural induction by noggin (500 ng/mL; R&D Systems, Minneapolis, MN) was performed as described.35 Noggin-treated cells were harvested after 14 days and were further subcultured as NS/PCs in suspension in neural basal medium together with basic fibroblast growth factor and epidermal growth factor (20 ng/mL each) as neurospheres.36 In the neurosphere formation assay, neurospheres were grown in the presence of various treatments for 5 to 7 days, and the number of neurospheres was counted in each condition. In the neural differentiation assay, after 2 weeks of growth in suspension culture, the neurospheres were plated as described onto laminin-coated 8-well glass chamber slides (BD Falcon; BD Biosciences, San Jose, CA) in neural basal medium lacking growth factors,36 were allowed to attach, and were incubated in the presence or absence of specified treatments for 3 to 5 days. Cells were then fixed with 4% PFA for 10 minutes, blocked in 10% fetal calf serum–PBS + Tween 20, and immunostained with a mouse anti-βΙΙΙ-tubulin antibody (1:1000; Millipore, Billerica, MA) and its appropriate conjugated secondary antibodies (Alexa Fluor 568 or 488, 1:1000; Molecular Probes-Invitrogen). The specificity of the staining was verified by the absence of staining in the negative control immunoglobulin fraction (Dako, Carpinteria, CA; data not shown). Frequency of neuron-forming spheres was measured by counting the number of spheres that showed neurons recognized by βΙΙΙ-tubulin staining.
Spinal Cord Lesions in Mice
Animal experiments were performed using adult male C57BL/6 mice. Mice were given a complete left hemisection at thoracic level T12, as previously described.37 To address the bias, reproducibility, and robustness of this study,38 we kept the injury homogenous and reproducible by excluding animals in which the spinal section was invading the contralateral side and in which weakness was observed in the right hind limb. This exclusion was performed blindly at the end of the first week in an unbiased and encoded manner. Approximately the same numbers of animals were discarded from different groups (isotype control versus treatment, ∼15% to 20%). Furthermore, at all times, the investigator was blinded to the nature of the drugs to be delivered. Decoding of the samples was performed only once data were quantified and analyzed.
The anti-LPA mAb,B3 or its IgGβ2 isotype-treated control (0.5 mg per mouse, 25 mg/kg) were encoded and randomly delivered subcutaneously 30 minutes after lesioning and then twice weekly for a maximum of 2 weeks. For short-term experiments, after 7 or 14 days, the animals were sacrificed and perfused with PBS, followed by 4% PFA; the spinal cords were removed, postfixed for 1 hour in 4% PFA, and then cryopreserved in 20% sucrose overnight at 4°C for frozen sections. Control tissues were taken from uninjured mice (sham).
For nerve regeneration and functional recovery, animals were injected for 2 weeks and were analyzed for up to 5 weeks.37,39 Tetramethylrhodamine dextran was used as an anterograde tracer to examine the extent of regeneration of lesioned lateral white matter tract axons or corticospinal tract axons. Tracing was performed 1 week before tissue collection. For lateral white matter tract axons, the mice were anesthetized and the spinal cord was exposed in the cervical region as described previously herein. The tracer was injected into the spinal cord at the level of the cervical enlargement, ipsilateral to the lesion (three injection points, 0.3 mL at 50 mg/mL each).37
Immunohistochemical Analysis in Murine Tissue
Tissue was cut into 20-μm sections using a cryostat and was processed for immunohistochemical analysis as described previously herein for zebrafish tissue. The primary antibodies used were mouse anti-NeuN (1:1000; Millipore), rabbit anti–glial fibrillary acidic protein (GFAP) (1:1000, Dako), mouse anti-GFAP (1:1000; Invitrogen), mouse anti–chondroitin sulfate proteoglycan (1:200; Sigma-Aldrich), rabbit anti–mitogen-activated protein kinase (MAPK) (1:1000; Cell Signaling Technology Inc., Beverly, MA), rabbit anti–Ki-67 (1:200; Thermo), rabbit antiactivated caspase-3 (1:200; BD Bioscience), and mouse antisynaptophysin (1:1000; Chemicon, Temecula, CA). The specificity of the staining was verified by the absence of staining in negative controls consisting of the appropriate negative control immunoglobulin fraction (data not shown).
Cell apoptosis was performed using a TUNEL kit as described previously herein for the zebrafish. DAPI was used to visualize cell nuclei (Sigma-Aldrich). The specificity of the staining was verified by the absence of staining in negative controls without the TdT enzyme. Appropriate conjugated secondary antibodies (Alexa Fluor 568 or 488; 1:1000; Molecular Probes) were used.
Cell Count
GFAP density was measured in a 200-μm2 grid at the lesion site (up to 400 μm distal from the center line of the lesion) using ImageJ software version 1.43u (NIH, Bethesda, MD). The GFAP density was averaged from ≥5 boxes per section and 10 sections per spinal cord, with the results being presented as the percentage of total area. To avoid bias choice of fields, the fields that were taken were 200 μm apart in any direction.
Cell proliferation was quantified by counting Ki-67–positive cells in a 200-μm2 grid at the lesion site (≤400 μm distal from the center line of the lesion). For neuronal counts, NeuN-positive cells were counted at two different distances from the lesion site (300 μm and 5 mm proximal to the lesion site) in a 200-μm2 field. Traced neurons were counted at four different distances from the lesion site (as specified in the graph). Synaptophysin density was measured around traced neurons at the lesion site using ImageJ software.
Microscopy
Sections were examined by bright field or fluorescence microscopy using an Axioplan Z1 epifluorescence microscope (Carl Zeiss MicroImaging GmbH). Photomicrographs (1300 × 1030 dpi) were obtained with 2.5× and 5× Plan-Neofluar objectives (Carl Zeiss MicroImaging GmbH) and were acquired as digital images using an AxioCam digital camera connected to AxioVision software version 4.4 (Carl Zeiss MicroImaging GmbH). Images were cropped and sized using Adobe Photoshop 11 and Illustrator 14 (Adobe Systems Inc., San Jose, CA). To confirm colocalization, optical sections of the sample were acquired using an Axoioplan Z1 microscope equipped with the ApoTome module and a 40× objective using AxioVision software.
Analysis of Signal Transduction
To examine signaling pathways (intact spinal cord, spinal cord after SCI with or without mAb treatment; the investigator was blinded to the nature of the drugs delivered), the spinal cord tissue–included lesion site ≤3 mm from each side was lysed 7 days after SCI with lysis buffer [50 mmol/L HEPES (pH 7.5), 150 mmol/L NaCl, 10% glycerol, 1% Triton X-100, 1 mmol/L EDTA, 1 mmol/L EGTA (pH 8), 1.5 mmol/L MgCl2, 2 mmol/L phenylmethylsulfonyl fluoride, 0.2 mmol/L Na3VO4] and a protein inhibitor cocktail (Roche Diagnostics). The lysates were then clarified by centrifugation, and protein levels were detected using Bradford assay (Bio-Rad Laboratories, Hercules, CA). For Western blot analysis, protein samples were mixed with one-quarter volume of 4× loading buffer (200 mmol/L Tris-HCl, pH 6.8, 8% SDS, 0.04% bromophenol blue, 40% glycerol, 400 mmol/L dithiothreitol) and heated for 5 minutes at 100°C. Proteins (30 μg per sample) were resolved by SDS-PAGE using 8% to 16% gradient polyacrylamide gels (Gradipore, Frenchs Forest, Australia) in running buffer (25 mmol/L Tris, 200 mmol/L glycine, 0.1% SDS) and were electrophoretically transferred onto a nitrocellulose membrane (0.45 μm; Bio-Rad Laboratories) in transfer buffer (25 mmol/L Tris, 192 mmol/L glycine, 20% methanol). Membranes were blocked for 2 hours in Tris-buffered saline (0.05 mol/L Tris, pH 7.5, 0.15 mol/L NaCl, 0.1% Tween 20) containing 6% skimmed milk powder and incubated with rabbit anti–p-MAPK (1:1000; Cell Signaling Technology Inc.) overnight, followed by a secondary antibody linked to horseradish peroxidase (HRP–anti-rabbit IgG, 1:1000; Cell Signaling Technology Inc.). Immunoreactive bands were detected using an enhanced chemiluminescence reagent (Pierce Biotechnology, Rockford, IL). Membranes were stripped using glycine solution (0.2 mol/L; pH 2) at 60°C for 10 minutes, followed by three washes in Tris-buffered saline with Tween and incubation with rabbit anti-MAPK (1:1000; Cell Signaling Technology Inc.). Alternatively, samples were loaded onto two different gels with the same protein amount, and each membrane was incubated with anti–p-MAPK or anti-MAPK Ab.
Rho GTPase activation was detected by using the rhotekin Rho-binding domain assay according to the manufacturer's instruction (Upstate Biotechnology, Lake Placid, NY) on spinal cord tissue lysates as described previously (350 μg per sample). Endogenous Rho-GTP was precipitated from the tissue lysates at 4°C for 1 hour using 30 μg of the rhotekin Rho-binding domain agarose for each sample. The beads were pelleted by centrifugation at 14,000 × g for 2 minutes, washed three times with the lysis buffer, and resuspended in 4× loading buffer. The eluted protein samples were resolved on 12% SDS gels and were analyzed by Western blot analysis. Rho was detected using rabbit anti-Rho (1:1000; Cell Signaling Technology Inc.).
Quantitative analysis of detected bands was performed by densitometric analysis of scanned blots using ImageJ software. Statistical comparison was made against adjusted relative densities of triplicate loaded samples using GraphPad Prism software.
Behavioral Analyses
The investigator was blinded to the nature of the treatments. Ability to walk on a horizontal was performed as previously described.37,39 The ability to walk on a horizontal wire grid (1.2 × 1.2-cm grid spaces; 35 × 45-cm total area) was determined to assess their ability to control movement in the left hind limb. The mice were tested 1 to 5 weeks after SCI, once a week. Each mouse was allowed to walk freely around the grid for 2 minutes before testing and then was tested for 3 minutes. When the left hind limb paw protruded entirely through the grid, with all the toes and the heel extending below the wire surface, this was counted as a misstep. The total number of steps taken with the left hind limb was also counted. The results are expressed as the percentage of accurate footsteps.
Open-field locomotion score was evaluated by using the modified Basso-Beattie-Bresnahan (mBBB) scoring system of 20 points.40 The mice were placed in an open field on a nonslippery surface (46 × 57 cm). In each testing session, the mice were observed individually for 3 minutes by the observer. The mice were tested once per week every week for 5 weeks. Decoding of the samples was performed only after the data were quantified and analyzed.
In the data presented herein, one animal in the cohort “B3−” died and 2 “B3−” animals and 2 “isotype-treated control” animals were removed because of double paralysis.
Statistical Analysis
All the sets of experiments were performed at least three times in triplicates (n refers to the number of independent experiments performed on different cell cultures/animals). All the data are expressed as mean ± SEM as indicated. Significance of the differences was evaluated using either two-tailed t-tests with 95% confidence intervals in case of only two variables to compare or the one-way analysis of variance followed by the Tukey test for multiple comparisons, with α = 0.001. Statistical significance was established at P < 0.05, P < 0.01, and P < 0.001. For behavioral analysis, fish locomotor analysis, mBBB score, and grid walking, a nonparametric Mann-Whitney test was used, with α set to 5% (P < 0.05 and P < 0.01).
Results
LPA Mediates Inflammatory Responses and Glial Cell Proliferation After SCI in the Zebrafish Danio rerio
Contrary to what is observed in mammals, fish regenerate their spinal cord after full transection within a few weeks after injury, making the zebrafish a good model for neuronal regeneration.26,27,32 Accordingly, we examined the effect of local LPA administration after full spinal cord transection in the zebrafish to determine the different cellular responses in vivo to LPA at the lesion site. Taking advantage of various zebrafish Tg lines, we tracked key cellular responses to injury and to LPA administration. Leukocytes, glia, and neurons were analyzed in Tg fish expressing the GFP transgene under the control of the macrophage lineage promoter macrophage expressed gene1 Tg(mpeg1:GFP), the neutrophil lineage promoter myelopeoxidase Tg(mpx:GFP)i114,3,28,29 the GFAP promoter Tg(GFAP:GFP)mi2001,30 or the motor neuron Islet1 promoter Tg(isl1:GFP)2rw012.31 RT-PCR of the adult fish brain and spinal cord revealed that LPA1 is highly expressed in the adult fish CNS (Figure 1A). Based on morphologic features and location, in situ hybridization showed that LPA1 is expressed on large neuronal-like cells along the spinal cord and glia-like cells located along the midline and central canal of the spinal cord (Figure 1, B and C). We next tested the responses to injured animals after LPA treatment (see Materials and Methods). Six hours after SCI, LPA-treated animals displayed substantially more apoptotic cells, assessed by TUNEL staining at the lesion site, and more neutrophils (Figure 1, D–F). Three days after complete spinal cord transection, macrophage infiltration was observed at the lesion site, and some macrophage/microglia activation was also observed distal from the lesion site (Figure 1G). Significantly more cells expressing the macrophage/microglia marker mpeg1 were activated in the injured spinal cords of LPA-treated zebrafish, although at the lesion site, a substantial increase in macrophage infiltration was not seen (Figure 1, G–M). These results suggest that LPA promotes inflammation and neuronal cell death during the acute phase of neurotrauma.
Figure 1.

LPA promotes inflammation and cell death in zebrafish after SCI. A: LPA1 expressed in the brain and spinal cord of adult zebrafish. B, brain; L, liver; M, muscle; SC, spinal cord; NC, negative control consisting of no cDNA in the PCR. B and C: In intact adult zebrafish spinal cord, in situ hybridization shows LPA1 expression in large neuronal cells along the spinal cord (arrowheads) and in glial cells along the midline in spinal cord tissue (arrows). Six hours after SCI in the mpx:GFP line, there is little infiltration of neutrophils into the control fish and some TUNEL-labeled cells (D); however, in LPA-treated animals, increased neutrophil infiltration and neuronal apoptosis was demonstrated by TUNEL staining (E). F: Quantitation of neutrophils at the lesion site reveals a significant increase in LPA treatment versus control (Con). Results are given as mean ± SEM (n = 5 in each group). *P < 0.001 by two-tailed t-test, 95% confidence. G: Three days after SCI in the mpeg1:GFP line demonstrating infiltration of mpeg1-expressing cells in the lesion site (box enlargement; H) and their presence distal from the lesion site (box enlargement; I). J: LPA treatment increased the presence of mpeg1-expressing cells distal to the lesion site (box enlargement; K), with similar macrophage infiltration (box enlargement; L). M: Quantitation of macrophages/microglia shows a significant increase in LPA-treated versus control (Con) spinal cords. Results are mean ± SEM (n = 8 in each group) *P < 0.001 by two-tailed t-test, 95% confidence. Scale bars: 50 μm (B, C, H, I, K, and L); 100 μm (D and E); 300 μm (G and J).
The longer-term, secondary effects of LPA were then studied in animals subjected to SCI. Five days after SCI, LPA administration increased glial cell proliferation at the central canal (Figure 2A, B, and E), and these cells also demonstrated increased levels of GFAP expression compared with injection of vehicle alone (Figure 2, C and D), suggesting that LPA promotes glial cell proliferation and reactivity in the late, secondary phases of neurotrauma. In addition, 10 days after SCI, islet1-expressing neurites were observed sprouting at the lesion site, and quantitation of the number of neurite projections 200 μm from the lesion site revealed substantially more neurites in control compared with LPA treatment (Figure 2F). Moreover, 3 weeks after SCI, neurites were observed regenerating through the lesion site in control animals, which is in contrast to LPA-treated fish, which displayed reduced neurite sprouting (Figure 2G). Overall locomotor recovery of LPA-treated fish was worse, although this did not reach significance at this time point (Figure 2H). This suggests that the regeneration process still occurs to some extent in LPA-treated zebrafish. Collectively, these results suggest that LPA increases inflammation and glial cell proliferation and reduces neurite sprouting in the zebrafish model of regeneration after SCI.
Figure 2.

LPA increases glia proliferation and decreases neurite sprouting in zebrafish after SCI. Five days after SCI in the GFAP:GFP line, BrdU-positive glial cells proliferate at the central canal and accumulate at the edges of the lesion site (A) compared with LPA-treated fish (B). GFAP expression at the edges of lesion in control fish (C) compared with more abundant GFAP expression in LPA-treated animals (D). E: Quantitation of BrdU-positive cells reveals an increase with LPA treatment. Results are given as mean ± SEM (n = 10 in each group). *P < 0.001 by two-tailed t-test, 95% confidence). F: Three weeks after SCI, the Islet1:GFP line shows a significant decrease in neurite number at the lesion site in LPA-treated fish compared with controls (Con). Results are given as mean ± SEM (n = 8 for each genotype). ***P < 0.001 by two-tailed t-test, 95% confidence). G: Neurite sprouting in the islet1:EGFP line around the lesion site in control treatment compared with reduced neurite number from both sides of the lesion site in LPA treatment. H: Locomotor recovery assays show motor function 3 weeks after SCI (n = 7 animals from each group) NS, not significant. Scale bars: 100 μm (A, B, and G); 50 μm (C and D).
Since the exogenous LPA application in the zebrafish model suggested that LPA may have an inhibitory role in regeneration after neurotrauma, thereby influencing inflammatory cell infiltration and activation, glial cell proliferation, and neuronal sprouting inhibition, we next used the murine model of SCI to investigate the role of endogenous LPA generation during neurotrauma and to determine whether intervention in LPA signaling could be a useful therapeutic strategy in the treatment of SCI. Consequently, an anti-LPA mAb was developed as a therapeutic molecular sponge to neutralize LPA in a murine model of SCI. The specificity of the mAb against LPA was initially demonstrated.
Blockage of LPA's Effect by the Specific Anti-LPA mAb
The availability of highly specific mAbs directed against LPA offers a unique tool to probe the role of LPA in cell signaling and to demonstrate use of the antibody as a potential therapeutic. Anti-LPA mAbs specifically bind LPA and prevent the ligand from interacting with its complement of receptors. Table 1 shows a competition ELISA where the murine anti-LPA mAb B3 was tested for its ability to recognize 18:2 LPA by comparison with a variety of other bioactive lipids delivered at the high concentration of 2.5 μmol/L. The table shows that at this concentration of 18:2 LPA as the competing lipid, the binding of B3 to the plate was inhibited by 71%. In contrast, structurally related bioactive lipids, such as sphingosine-1-phosphate, platelet activating factor, and sphingosine, were not recognized by the B3 mAb. In addition, lysophosphatidyl choline, the immediate precursor of LPA generation by autotaxin, was not recognized by the B3 mAb, along with other potential precursors, such as phosphatidic acid or glycerol-3-phosphate. Structural phospholipids also did not cross-react, and neither did the autotaxin inhibitors cyclic LPA and bromomethylene phosphonate–LPA.
Having demonstrated that the B3 mAb was specific for LPA, it was then studied for its ability to block specific LPA receptor signaling. For these experiments, the CHO-K1 EDG2 (LPA1)–, CHO-K1 EDG4 (LPA2)–, and CHO-K1 EDG7 (LPA3)–β-arrestin clones (PathHunter) generated by DiscoveRx were used in very sensitive assays where the readout was a luminescence signal associated with G-protein–coupled receptors. Figure 3, A, C, and E, shows the dose-dependent effect of 18:2 LPA to activate the LPA1/EDG2-, LPA2/EDG4-, or LPA3/EDG7-overexpressing cells with EC50 values of ∼82, 293, and 837 nmol/L LPA, respectively. For all three receptors, Figure 3, B, D, and F, demonstrates the dose-dependent ability of B3 to completely knock down LPA signaling to baseline levels of luminescence. Using data from cells expressing the highest-affinity receptor, LPA1/EDG2 (Figure 3, A and D), the EC50 of LPA required to activate the receptor was 82 nmol/L, whereas B3 blocked LPA signaling with an ID50 of ∼5 μg/mL. This represents an antibody concentration of 30 nmol/L, which is stoichiometrically sufficient to neutralize the LPA (200 nmol/L) present in the assay (ie, twice the EC50 for LPA). Similar results were observed when B3 was tested for its ability to neutralize LPA in cells expressing the less sensitive LPA receptors, LPA2/EDG4 and LPA3/EDG7 (Figure 3, D and F).
Figure 3.
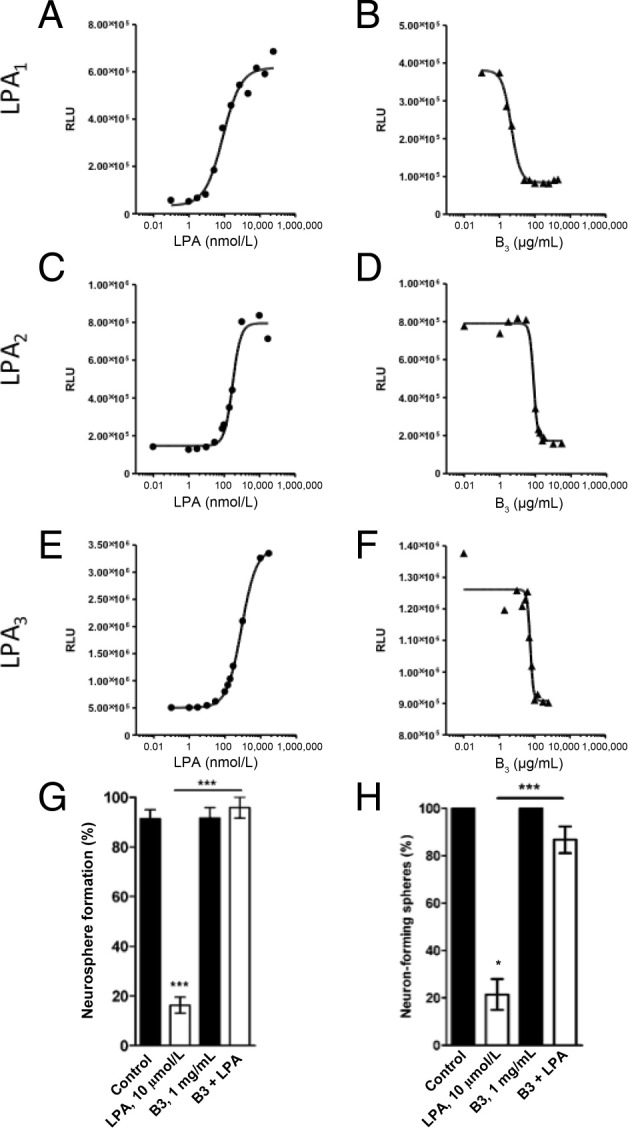
Anti-LPA mAb (B3) blocks LPA signaling in LPA1, 2, or 3 overexpressing CHO-K1 cells and in human NS/PCs. LPA activates signaling via LPA receptors LPA1 (A), LPA2 (C), or LPA3 (E). CHO-K1 cells were plated at 2.0 × 104 cells per well and were incubated at 37°C/5% CO2. After 24 hours, plates were starved with reduced serum media at 37°C for another 24 hours. The LPA standard curve was prepared by titrating 18:2 LPA (0 to 10 μmol/L) in media containing 1 mg/mL of BSA onto the plate and was analyzed as described in Materials and Methods. B, D, and F: B3 blocks LPA signaling by inhibiting LPA binding to it receptors. Media containing 200 nmol/L (for LPA1/EDG2) or 400 nmol/L (for LPA2/EDG4 and LPA3/EDG7) 18:2 LPA and increasing concentrations of B3 antibody (0 to 2000 μg/mL) were added to the corresponding CHO-K1 cells (B, D, and F, respectively) and processed as described for the LPA signaling experiment. RLU, relative light unit. Frequency of neurosphere formation (G) and neuronal differentiation (neuron-forming neurospheres) (H) in the presence or absence (control) of LPA (10 μmol/L) and/or B3 (1 mg/mL). Data are expressed as the mean ± SEM from at least three independent experiments. ***P < 0.001 by one-way analysis of variance followed by the Tukey test, α = 0.001.
Having determined the potency and specificity of B3 to block LPA binding to LPA1-3 receptors in the DiscoveRx cells, the ability of B3 to block LPA-induced biological effects was then assessed in vitro using human embryonic stem cell–derived NS/PCs. These cells were chosen as a more biologically relevant cell assay readout of the anti-LPA mAb potency, as we previously reported that LPA dose dependently inhibits the neural differentiation of human embryonic stem cell–derived NS/PCs by having effects on neurosphere formation and neuronal differentiation.41 We also previously reported that these cells express LPA1-5 mRNAs.41 We tested the potency of B3 to block LPA's effects in two different cell assays: inhibition of neurosphere formation and inhibition of NS/PC differentiation toward neurons. As shown in Figure 3, G and H, the sole application of B3 did not significantly modify neurosphere formation frequency or neuronal differentiation. As expected, exogenous LPA addition substantially and significantly inhibited neurosphere formation. Coincubation of LPA with B3 abolished LPA's effect on neuronal differentiation and neurosphere formation. Altogether, these cell-based assays demonstrate the strong potency of B3 to block LPA's biological effects in cells, including human NS/PCs.
Anti-LPA mAbs Decrease Astrogliosis and Microglial Activation in the in Vivo Mouse Model of SCI
Having demonstrated the potency of the murine anti-LPA mAb B3 to block the effects of LPA in cell assays, the ability of B3 to block LPA effects in vivo was then tested in a mouse model of SCI. These in vivo studies consisted of treating hemisectioned animals s.c. twice a week for 1 to 2 weeks with either isotype control or B3 (0.5 mg per mouse, ∼25 mg/kg, in a blinded manner) after SCI. At both time points, a reduction in astrocyte reactivity was observed after anti-LPA mAb treatment. Densitometry revealed a significant decrease in GFAP expression by astrocytes around the lesion site in B3-treated mice compared with isotype-treated control mice 1 week (Figure 4, A, B, D, E, and G) and 2 weeks (data not shown) after injury. In addition, this reduced glial response resulted in a marked reduction in glial scarring as assessed by immunostaining for chondroitin sulfate proteoglycan, a key component of the glial scar (Figure 4, C and F). These results also demonstrated a significant reduction in cell proliferation at the lesion site, as assessed by quantification of the proliferation marker Ki-67 (Figure 4H). Most of the cells were colabeled with GFAP, suggesting that most of the proliferating cells at the lesion site at this time point were astrocytes (Figure 4H). These results suggest that treatment with B3 reduces the astrocytic response after SCI and attenuates reactive gliosis.
Figure 4.

Anti-LPA mAb (B3) reduces glial scar after SCI. Immunostaining at the injury site of mouse spinal cords 1 week after SCI shows a dense network of astrocytes expressing high levels of GFAP (A and B), which is reduced after B3 treatment (D and E). C: Immunostaining for chondroitin sulfate proteoglycans 2 weeks after lesioning revealed that the scar was diminished in B3-treated mice (F). G: Quantitation of GFAP density around the lesion site showed a significant decrease with B3 treatment. Results are the mean ± SEM percentage of GFAP staining density in the field (n = 7 in each group). *P < 0.001 by two-tailed t-test, 95% confidence). Most proliferating cells at the lesion site are GFAP positive. H: Quantitation of Ki-67–positive cells at the lesion site showed a significant decrease with B3 treatment. Results are the mean ± SEM number of Ki-67–positive cells in the field (n = 7 in each group). *P < 0.001 by two-tailed t-test, 95% confidence). I: p-MAPK levels were reduced on astrocytes at the lesion site in B3-treated mice. Western blot analysis of spinal cord tissue 1 week after injury showed a significant reduction in p-MAPK signaling (n = 7) (J) and Rho-GTP levels (n = 4) (K) in B3-treated mice. Data are expressed as the mean ± SEM from at least three independent animals. *P < 0.05, **P < 0.01 by one-way analysis of variance followed by the Tukey test, α = 0.001. Scale bars: 200 μm (A, C, D, and F); 50 μm (B, E, H, and I).
To understand more precisely the signaling pathways involved in the LPA effect after SCI, we assessed levels of the extracellular signal-regulated kinase 1 and 2 phosphorylation (p-MAPK) in the spinal cord of sham or hemisectioned animals that received either B3 treatment or its isotype control for 1 week (Figure 4, I, and J). Western blot analysis indicated high and sustained levels of extracellular signal-regulated kinase 1 and 2 phosphorylation in injured spinal cords compared with sham spinal cords (Figure 4J). Moreover, treatment with the anti-LPA mAb strongly reduced p-MAPK down to control levels, suggesting that endogenous LPA induces extracellular signal-regulated kinase 1 and 2 phosphorylation after SCI (Figure 4J). These data were confirmed by immunostaining of the spinal cords, which indicated colocalization of p-MAPK and GFAP, demonstrating that LPA-dependent extracellular signal-regulated kinase 1 and 2 signaling in the tissue likely resulted from astrocyte activity (Figure 4I). In addition, B3 treatment also decreased Rho activation (Figure 4K), a major regulator of cytoskeletal changes.42 Activation of Rho in reactive astrocytes and in neuronal growth cone collapse has been reported after SCI.37,43,44 Furthermore, CD11b immunostaining and density quantitation also revealed that B3 significantly reduced microglial cell activation near the lesion site compared with isotype-treated control (Figure 5), which may suggest that LPA contributes to the proinflammatory response at the lesion site.
Figure 5.
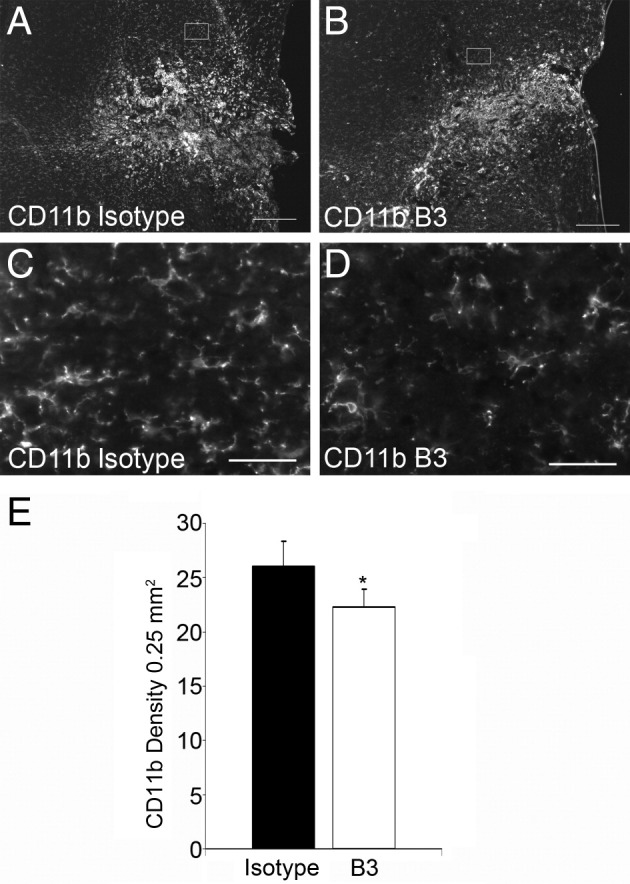
Anti-LPA mAb (B3) reduces microglia activation around the lesion site. One week after SCI, CD11b immunostaining in isotype-treated control mice (A, and C) compared with reduced levels in B3-treated mice (B and D). E: Quantitative analysis of the reactive microglia marker CD11b from 200-mm2 areas adjacent to the lesion site. Results are the mean ± SEM (n = 7 in each group). *P < 0.001 by two-tailed t-test, 95% confidence). Scale bar s: 100 μm (A and B); 50 μm (C and D).
Anti-LPA mAb Is Neuroprotective and Decreases Neuronal Apoptosis Near the Lesion Site After SCI in the Mouse
In the mouse 1 week after SCI, treatment with B3 dramatically reduced apoptosis at the lesion site, as revealed by TUNEL assay (Figure 6, A–D). Moreover, TUNEL staining colocalized with NeuN in isotype-treated controls (Figure 6, E–G), suggesting that B3 had an antiapoptotic effect on neurons by virtue of neutralizing the proapoptotic LPA produced at the injury site. This observation is further supported by decreased activation of caspase-3 at the lesion site in B3-treated animals compared with isotype-treated control mice (Figure 6, H and I); furthermore, caspase-3 colocalized with NeuN (Figure 6, J–L). Moreover, these data indicate a statistically higher number of NeuN-positive cells at the lesion site in B3-treated animals compared with isotype-treated controls (Figure 6M), suggesting increased neuronal survival of 51%, from a mean ± SEM of 7.68% ± 2.2% to 11.6% ± 2.2% cells per field (Figure 6M). Thus, these data demonstrate that B3 is neuroprotective after SCI.
Figure 6.

Anti-LPA mAb (B3) is neuroprotective by reducing apoptotic neuronal death after SCI. One week after SCI, apoptotic cells were observed at the lesion site in isotype-treated control mice (A and B; box enlargement) compared with reduced TUNEL staining in B3 treatment (C and D; box enlargement). E–G: Colabeling of TUNEL staining with the neuronal marker NeuN shows apoptotic neurons (arrowheads). Immunostaining for active caspase-3 increased in control (H) compared with B3 (I) treatment and colocalized with NeuN (J–L). M: Quantitation of number of neuronal cells at the lesion site and 5 mm proximal (upstream of the lesion site) reveals that in B3 treatment, the number of neuronal cells at the lesion site was significantly higher than in controls. Results are the mean ± SEM of NeuN-positive cells in the field (n = 7 in each group). *P < 0.001 by two-tailed t-test, 95% confidence). Scale bars: 100 μm (A, B, H, and I); 50 μm (C–G and J–L).
Anti-LPA mAbs Promote Recovery After SCI in the Mouse
As B3 decreased astrogliosis and neuronal cell death 1 to 2 weeks after SCI, we next examined whether this treatment would promote a favorable environment for neurons to regenerate. Eight weeks after injury, axonal regeneration through the lesion site was not observed in any isotype mAb–treated control animals but was observed only in a few animals treated with B3 for 2 weeks, hence suggesting that B3's effect is not mainly through regeneration (data not shown). We observed a significantly higher number of tetramethylrhodamine dextran–traced neurons 500 μm to 1 mm proximal to the lesion site in all B3-treated mice compared with isotype-treated control animals (Figure 7, A–C). Moreover, these were accompanied by a significant increase in synapse density in the area around traced neurons, as determined by synaptophysin immunostaining (Figure 7, D–F).
Figure 7.

Anti-LPA mAb (B3) increases neuronal survival and synaptic densities at the lesion site. A–C: Eight weeks after SCI, in B3-treated mice, significantly more traced neurons (arrowheads) are observed in the areas adjacent and proximal to the lesion site (n = 7) compared with controls (n = 8). D–F: Quantitation of synaptic density around the traced neuronal cells shows significantly more synapses in B3-treated mice compared with isotype-treated control mice. TMRD, tetramethylrhodamine dextran. Results are mean ± SEM cell number or density in the field. *P < 0.001 by two-tailed t-test, 95% confidence. Scale bars: 100 μm (B and C); 25 μm (E and F).
As a critical consequence of the neuroprotective and proregenerative effects of the anti-LPA mAb in vivo, injured mice were assessed for functional recovery after a 2-week antibody treatment, as determined by use of the left hind limb in the grid walking assay and the mBBB scale. As seen in Figure 8A, anti-LPA B3 treatment resulted in a significant improvement in the ability to walk on a grid 3 weeks after SCI. SCI mice receiving B3 treatment made far fewer foot falls and showed greater weight support with the affected left hind limb (Figure 8A). B3-treated mice also showed significant improvements in function by 4 weeks after SCI, as assessed by the modified open-field behavior test using the mBBB scale (Figure 8B).40 These data demonstrate the relevance of anti-LPA mAb treatment for improving key behavioral outcomes after neurotrauma.
Figure 8.
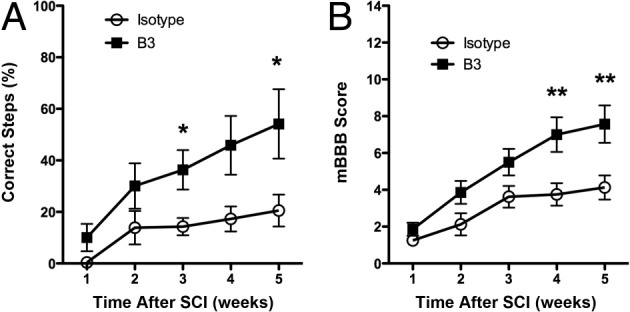
Anti-LPA mAb (B3) improves motor function after SCI. A: Walking on a grid was significantly improved after 2 weeks of treatment with B3. Results are given as mean ± SEM. *P < 0.05 by nonparametric Mann-Whitney test, α = 5%). B: The mBBB score was measured up to 5 weeks after SCI in isotype-treated control (n = 8) and B3-treated (n = 7) mice. Results are given as mean ± SEM. **P < 0.01 by nonparametric Mann-Whitney test, α = 5%).
Discussion
Herein we demonstrate for the first time the role of LPA in promoting neuronal cell death and reactive gliosis and inhibiting neuronal regeneration after SCI using murine and zebrafish models of SCI. We also demonstrate the neuroprotective and proregenerative effects of a therapeutic anti-LPA antibody and the ability of antibody therapy to improve functional and behavioral outcomes.
Using the zebrafish model of regeneration after neurotrauma and taking advantage of several Tg zebrafish lines expressing markers for key cellular components of neurotrauma, we demonstrated the ability of LPA to inhibit the normal regenerative response after SCI. In particular, LPA mediates neuronal death, microglial activation, astroglial proliferation, and GFAP up-regulation and, over the long term, inhibits neurite sprouting in injured zebrafish.
Using the murine SCI model, these data using a specific anti-LPA mAb strongly indicate that LPA produced endogenously after neurotrauma inhibits SCI regeneration by increasing gliosis and promoting neuronal death. The specific mAb raised against LPA, B3, inhibited the effects of LPA in vitro and in vivo. The anti-LPA mAb not only validates the role of LPA in neurotrauma but also suggests that LPA is causally involved in nearly all the pathologic consequences of SCI, including gliosis, nerve cell death, inflammatory responses, and the inhibition of neuroregeneration. However, it remains to be determined whether these consequences are due to pleiotropic effects of LPA or to a sole cellular effect leading to broader consequences. Treatment of SCI mice with the anti-LPA mAb significantly reduced glial scar formation and inflammatory responses while substantially increasing neuronal survival. The observed increasing density of synapses around neuronal cells proximal to the lesion site after mAb treatment may suggest that neuronal activity was also improved compared with isotype-treated control mice. All these lead to motor function improvement with mAb treatment. All the mice exhibited increased neuronal cell survival and increased synaptic density at the rostral part of the lesion site, suggesting that the antibody promoted neuronal survival rather than regeneration. After spinal hemisection, a gradual return of locomotor function occurs in mice45,46 and is thought to be due to activation of central pattern generators.47 Due to the neuronal prosurvival effects of anti-LPA mAb treatment, neurons innervating the hip (found in the region of the hemisection) improve significantly beyond normal recovery. The survival effect of the antibody on neurons explains the improvement of B3 treatment on locomotor hip movement and weight support as measured by the behavioral readouts used in this study and, ultimately, improves locomotor function.
These data in zebrafish and mice support the concept that LPA promotes activation of microglial cells, which, in turn, might also regulate LPA de novo synthesis.1 It has been demonstrated previously in the adult mouse spinal cord that LPA is synthesized by microglial cells in their early phase of activation and that this LPA is likely responsible for neuropathic pain.48 Consistent with this report, we demonstrate that anti-LPA mAb treatment significantly reduced microglial activation after SCI. Although outside the scope of the present study, one could speculate that the anti-LPA mAb blocks de novo synthesis of LPA by microglia, hence reducing their activation by disrupting the autocrine and paracrine actions of LPA on microglia.
These data also suggest that the anti-LPA mAb treatment–induced improvements are likely to be mediated by the decrease in Rho-GTP and p-MAPK activation pathways that mediate glial cell proliferation and differentiation, resulting in scarring. In addition, the Rho pathway has already been described as the main pathway used by LPA to induce growth cone collapse1 and is inhibited partially by antibody treatment.
The present study with the anti-LPA mAb provides in vivo support of previous work suggesting that LPA plays an active role in blocking the regenerative response by inhibiting the neuronal differentiation of NS/PCs1,49 as antibody treatment promoted axonal sprouting after neurotrauma in SCI mice. Consistent with the in vivo effects of the antibody, the in vitro data obtained on human embryonic stem cell–derived NS/PCs demonstrated the ability of the anti-LPA mAb to counter negative effects of LPA on NS/PCs. Thus, the present data also indicate a potential role of LPA mAbs to modify the outcome of neurogenesis after inflammation or injury by reducing inhibition of NS/PC neuronal differentiation by LPA.
We demonstrate for the first time that blocking LPA signaling may be a useful and novel therapeutic strategy for SCI. Targeting the ligand with the anti-LPA mAbs may offer some advantages as a therapeutic strategy in the treatment of neurotrauma.1 Lpath Inc. has now humanized B3 and performed affinity maturation for eventual use in clinical trials. The murine mAb was used in the mouse model for proof of concept and to avoid complicating results due to potential immunogenicity and inflammatory effects of using a human antibody in a mouse.
It must be appreciated that this was a proof-of-concept study intended to demonstrate a role of LPA in SCI and needs to be followed up with other studies where the dosing regimen is fully explored as per the dose level and therapeutic window. Regarding the therapeutic window, we would not move forward in preclinical development in investigational new drug (IND)-enabling studies unless we can demonstrate that our antibodies can be efficacious when given in a therapeutically relevant timeframe after injury, as well as a relevant route of administration (i.v. versus s.c. or even i.t.). Moreover, once the dose level and therapeutic window are optimized, we will test the humanized mAbs to make sure that a dosing regimen is not complicated by a mouse-against-human antibody immunogenicity or by inflammatory responses. Although this was a proof-of-concept study, one can argue that the exposure of the animal to mAb portends efficacy in a clinically relevant timeframe, as the pharmacodynamics of our mAb in mice demonstrates that maximum exposure in blood after s.c. injection does not occur until 24 hours after the first s.c. injection. Thus, the therapeutic benefit seen in this study extends well into what could be a clinically relevant timeframe.
A related limitation of this study is the observation that the extent of neuroprotection seems to fall off beyond 1 mm from the injury site. One would expect that optimizing the therapeutic regimen in future planned studies will extend this anatomical limitation. Regardless of the anatomical limitation, substantial and significant improvements in functional behavior were observed.
Taken together, the present work suggests that anti-LPA mAbs could be a useful approach in treating SCI and may have advantages over other approaches intended to intervene in the LPA signaling pathway. Targeting the enzymes of the LPA synthetic pathways with enzyme inhibitors or LPA analogs presents significant challenges considering that the metabolic pathway of LPA is complex and that multiple parallel pathways for LPA synthesis exist.50 Similarly, the receptor-specific antagonist approach presents pitfalls as most cell types express more than one LPA receptor subtype and the pattern of receptor expression can change during the progression of disease. Because LPA receptors share signaling pathways, the activities of these receptors might be redundant, and blockage of one receptor may be insufficient to fully silence the pathway. Similarly, blocking even a principal pathway for LPA synthesis, such as targeting autotaxin, would not totally eliminate extracellular LPA levels as other routes of LPA synthesis exist. In light of these considerations, the neutralization of the ligand itself may be a simple and effective approach for the development of an LPA-based therapeutic.
Acknowledgments
We thank Amanda Rixon and the staff of the Experimental Medical and Surgical Research Unit at St. Vincent's Hospital (Melbourne, Australia) for assistance with animal work and care.
Footnotes
Supported by the National Health and Medical Research Council of Australia project (grant 454723) and Career Development Award Fellowship (A.P.), a Transport Accident Commission project grant (A.P.), NIH grant 1R43CA132395-01A2 (R.M.), the Victorian State Government's Department of Innovation, Industry, and Regional Development's Operational Infrastructure Support Program, and an Australian Development Scholarship from the Australian government (AusAID)(F.F.). The Australian Regenerative Medicine Institute is supported by grants from the State Government of Victoria and the Australian government.
Disclosures: R.A.S. and R.M. are shareholders in and inventors for Lpath Inc., R.A.S. is a consultant to and has stock in Lpath Inc., R.M. and K.M. are employees of Lpath Inc., and R.A.S. and A.P. are inventors on patents related to the work.
References
- 1.Frisca F., Sabbadini R.A., Goldshmit Y., Pebay A. Biological effects of lysophosphatidic acid in the nervous system. Int Rev Cell Mol Biol. 2012;296:273–322. doi: 10.1016/B978-0-12-394307-1.00005-9. [DOI] [PubMed] [Google Scholar]
- 2.Lin M.-E., Herr D.R., Chun J. Lysophosphatidic acid (LPA) receptors: signaling properties and disease relevance. Prostaglandins Other Lipid Mediat. 2010;91:130–138. doi: 10.1016/j.prostaglandins.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fukushima N. LPA in neural cell development. J Cell Biochem. 2004;92:993–1003. doi: 10.1002/jcb.20093. [DOI] [PubMed] [Google Scholar]
- 4.Moller T., Musante D.B., Ransom B.R. Lysophosphatidic acid-induced calcium signals in cultured rat oligodendrocytes. Neuroreport. 1999;10:2929–2932. doi: 10.1097/00001756-199909290-00010. [DOI] [PubMed] [Google Scholar]
- 5.Weiner J.A., Chun J. Schwann cell survival mediated by the signaling phospholipid lysophosphatidic acid. Proc Natl Acad Sci U S A. 1999;96:5233–5238. doi: 10.1073/pnas.96.9.5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiner J.A., Hecht J.H., Chun J. Lysophosphatidic acid receptor gene vzg-1/lpA1/edg-2 is expressed by mature oligodendrocytes during myelination in the postnatal murine brain. J Comp neurol. 1998;398:587–598. [PubMed] [Google Scholar]
- 7.Stankoff B., Barron S., Allard J., Barbin G., Noel F., Aigrot M.S., Premont J., Sokoloff P., Zalc B., Lubetzki C. Oligodendroglial expression of Edg-2 receptor: developmental analysis and pharmacological responses to lysophosphatidic acid. Mol Cell Neurosci. 2002;20:415–428. doi: 10.1006/mcne.2002.1129. [DOI] [PubMed] [Google Scholar]
- 8.Frugier T., Crombie D., Conquest A., Tjhong F., Taylor C., Kulkarni T., McLean C., Pebay A. Modulation of LPA receptor expression in the human brain following neurotrauma. Cell Mol Neurobiol. 2011;31:569–577. doi: 10.1007/s10571-011-9650-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Contos J.J., Chun J. The mouse lp(A3)/Edg7 lysophosphatidic acid receptor gene: genomic structure, chromosomal localization, and expression pattern. Gene. 2001;267:243–253. doi: 10.1016/s0378-1119(01)00410-3. [DOI] [PubMed] [Google Scholar]
- 10.Lee C.W., Rivera R., Dubin A.E., Chun J. LPA(4)/GPR23 is a lysophosphatidic acid (LPA) receptor utilizing G(s)-: G(q)/G(i)-mediated calcium signaling and G(12/13)-mediated Rho activation. J Biol Chem. 2007;282:4310–4317. doi: 10.1074/jbc.M610826200. [DOI] [PubMed] [Google Scholar]
- 11.Kotarsky K., Boketoft A., Bristulf J., Nilsson N.E., Norberg A., Hansson S., Owman C., Sillard R., Leeb-Lundberg L.M., Olde B. Lysophosphatidic acid binds to and activates GPR92, a G protein-coupled receptor highly expressed in gastrointestinal lymphocytes. J Pharmacol Exp Ther. 2006;318:619–628. doi: 10.1124/jpet.105.098848. [DOI] [PubMed] [Google Scholar]
- 12.Sato K., Malchinkhuu E., Muraki T., Ishikawa K., Hayashi K., Tosaka M., Mochiduki A., Inoue K., Tomura H., Mogi C., Nochi H., Tamoto K., Okajima F. Identification of autotaxin as a neurite retraction-inducing factor of PC12 cells in cerebrospinal fluid and its possible sources. J Neurochem. 2005;92:904–914. doi: 10.1111/j.1471-4159.2004.02933.x. [DOI] [PubMed] [Google Scholar]
- 13.Narita M., Goji J., Nakamura H., Sano K. Molecular cloning, expression, and localization of a brain-specific phosphodiesterase I/nucleotide pyrophosphatase (PD-I alpha) from rat brain. J Biol Chem. 1994;269:28235–28242. [PubMed] [Google Scholar]
- 14.Fuss B., Baba H., Phan T., Tuohy V.K., Macklin W.B. Phosphodiesterase I, a novel adhesion molecule and/or cytokine involved in oligodendrocyte function. J Neurosci. 1997;17:9095–9103. doi: 10.1523/JNEUROSCI.17-23-09095.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Savaskan N.E., Rocha L., Kotter M.R., Baer A., Lubec G., van Meeteren L.A., Kishi Y., Aoki J., Moolenaar W.H., Nitsch R., Brauer A.U. Autotaxin (NPP-2) in the brain: cell type-specific expression and regulation during development and after neurotrauma. Cell Mol Life Sci. 2007;64:230–243. doi: 10.1007/s00018-006-6412-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dennis J., Nogaroli L., Fuss B. Phosphodiesterase-Ialpha/autotaxin (PD-Ialpha/ATX): a multifunctional protein involved in central nervous system development and disease. J Neurosci Res. 2005;82:737–742. doi: 10.1002/jnr.20686. [DOI] [PubMed] [Google Scholar]
- 17.Sorensen S.D., Nicole O., Peavy R.D., Montoya L.M., Lee C.J., Murphy T.J., Traynelis S.F., Hepler J.R. Common signaling pathways link activation of murine PAR-1, LPA, and S1P receptors to proliferation of astrocytes. Mol Pharmacol. 2003;64:1199–1209. doi: 10.1124/mol.64.5.1199. [DOI] [PubMed] [Google Scholar]
- 18.Elmes S.J., Millns P.J., Smart D., Kendall D.A., Chapman V. Evidence for biological effects of exogenous LPA on rat primary afferent and spinal cord neurons. Brain Res. 2004;1022:205–213. doi: 10.1016/j.brainres.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 19.Fujita R., Kiguchi N., Ueda H. LPA-mediated demyelination in ex vivo culture of dorsal root. Neurochem Int. 2007;50:351–355. doi: 10.1016/j.neuint.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Ueda H. Molecular mechanisms of neuropathic pain-phenotypic switch and initiation mechanisms. Pharmacol Ther. 2006;109:57–77. doi: 10.1016/j.pharmthera.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 21.Keller J.N., Steiner M.R., Holtsberg F.W., Mattson M.P., Steiner S.M. Lysophosphatidic acid-induced proliferation-related signals in astrocytes. J Neurochem. 1997;69:1073–1084. doi: 10.1046/j.1471-4159.1997.69031073.x. [DOI] [PubMed] [Google Scholar]
- 22.Holtsberg F.W., Steiner M.R., Keller J.N., Mark R.J., Mattson M.P., Steiner S.M. Lysophosphatidic acid induces necrosis and apoptosis in hippocampal neurons. J Neurochem. 1998;70:66–76. doi: 10.1046/j.1471-4159.1998.70010066.x. [DOI] [PubMed] [Google Scholar]
- 23.Moller T., Contos J.J., Musante D.B., Chun J., Ransom B.R. Expression and function of lysophosphatidic acid receptors in cultured rodent microglial cells. J Biol Chem. 2001;276:25946–25952. doi: 10.1074/jbc.M102691200. [DOI] [PubMed] [Google Scholar]
- 24.Brault S., Gobeil F., Jr, Fortier A., Honore J.C., Joyal J.S., Sapieha P.S., Kooli A., Martin E., Hardy P., Ribeiro-da-Silva A., Peri K., Lachapelle P., Varma D., Chemtob S. Lysophosphatidic acid induces endothelial cell death by modulating the redox environment. Am J Physiol. 2007;292:R1174–R1183. doi: 10.1152/ajpregu.00619.2006. [DOI] [PubMed] [Google Scholar]
- 25.Goldshmit Y., Munro K., Leong S.Y., Pebay A., Turnley A.M. LPA receptor expression in the central nervous system in health and following injury. Cell Tissue Res. 2010;341:23–32. doi: 10.1007/s00441-010-0977-5. [DOI] [PubMed] [Google Scholar]
- 26.Becker C.G., Lieberoth B.C., Morellini F., Feldner J., Becker T., Schachner M. L1.1 is involved in spinal cord regeneration in adult zebrafish. J Neurosci. 2004;24:7837–7842. doi: 10.1523/JNEUROSCI.2420-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Becker T., Wullimann M.F., Becker C.G., Bernhardt R.R., Schachner M. Axonal regrowth after spinal cord transection in adult zebrafish. J Comp Neurol. 1997;377:577–595. doi: 10.1002/(sici)1096-9861(19970127)377:4<577::aid-cne8>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 28.Ellett F., Pase L., Hayman J.W., Andrianopoulos A., Lieschke G.J. mpeg1 promoter transgenes direct macrophage-lineage expression in zebrafish. Blood. 2011;117:e49–e56. doi: 10.1182/blood-2010-10-314120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Renshaw S.A., Loynes C.A., Trushell D.M., Elworthy S., Ingham P.W., Whyte M.K. A transgenic zebrafish model of neutrophilic inflammation. Blood. 2006;108:3976–3978. doi: 10.1182/blood-2006-05-024075. [DOI] [PubMed] [Google Scholar]
- 30.Bernardos R.L., Raymond P.A. GFAP transgenic zebrafish. Gene Expr Patterns. 2006;6:1007–1013. doi: 10.1016/j.modgep.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 31.Uemura O., Okada Y., Ando H., Guedj M., Higashijima S., Shimazaki T., Chino N., Okano H., Okamoto H. Comparative functional genomics revealed conservation and diversification of three enhancers of the isl1 gene for motor and sensory neuron-specific expression. Dev Biol. 2005;278:587–606. doi: 10.1016/j.ydbio.2004.11.031. [DOI] [PubMed] [Google Scholar]
- 32.Goldshmit Y., Sztal T., Hall T., Jusuf P.R., Nguyen-Chi M., Currie P.D. Fgf-dependent glial cell bridges facilitate spinal cord regeneration in zebrafish. J Neurosci. 2012;32:7477–7492. doi: 10.1523/JNEUROSCI.0758-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee S.-J., Chan T.-H., Chen T.-C., Liao B.-K., Hwang P.-P., Lee H. LPA1 is essential for lymphatic vessel development in zebrafish. FASEB J. 2008;22:3706–3715. doi: 10.1096/fj.08-106088. [DOI] [PubMed] [Google Scholar]
- 34.O'Brien N., Jones S.T., Williams D.G., Cunningham H.B., Moreno K., Visentin B., Gentile A., Vekich J., Shestowsky W., Hiraiwa M., Matteo R., Cavalli A., Grotjahn D., Grant M., Hansen G., Campbell M.A., Sabbadini R. Production and characterization of monoclonal anti-sphingosine-1-phosphate antibodies. J Lipid Res. 2009;50:2245–2257. doi: 10.1194/jlr.M900048-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pera M.F., Andrade J., Houssami S., Reubinoff B., Trounson A., Stanley E.G., Oostwaard D.W., Mummery C. Regulation of human embryonic stem cell differentiation by BMP-2 and its antagonist noggin. J Cell Sci. 2004;117:1269–1280. doi: 10.1242/jcs.00970. [DOI] [PubMed] [Google Scholar]
- 36.Reubinoff B.E., Itsykson P., Turetsky T., Pera M.F., Reinhartz E., Itzik A., Ben-Hur T. Neural progenitors from human embryonic stem cells. Nat Biotechnol. 2001;19:1134–1140. doi: 10.1038/nbt1201-1134. [DOI] [PubMed] [Google Scholar]
- 37.Goldshmit Y., Galea M.P., Wise G., Bartlett P.F., Turnley A.M. Axonal regeneration and lack of astrocytic gliosis in EphA4-deficient mice. J Neurosci. 2004;24:10064–10073. doi: 10.1523/JNEUROSCI.2981-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steward O., Popovich P.G., Dietrich W.D., Kleitman N. Replication and reproducibility in spinal cord injury research. Exp Neurol. 2012;233:597–605. doi: 10.1016/j.expneurol.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 39.Goldshmit Y., Spanevello M.D., Tajouri S., Li L., Rogers F., Pearse M., Galea M., Bartlett P.F., Boyd A.W., Turnley A.M. EphA4 blockers promote axonal regeneration and functional recovery following spinal cord injury in mice. PLoS One. 2011;6:e24636. doi: 10.1371/journal.pone.0024636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y., Oskouian R.J., Day Y.J., Kern J.A., Linden J. Optimization of a mouse locomotor rating system to evaluate compression-induced spinal cord injury: correlation of locomotor and morphological injury indices. J Neurosurg Spine. 2006;4:165–173. doi: 10.3171/spi.2006.4.2.165. [DOI] [PubMed] [Google Scholar]
- 41.Dottori M., Leung J., Turnley A.M., Pebay A. Lysophosphatidic acid inhibits neuronal differentiation of neural stem/progenitor cells derived from human embryonic stem cells. Stem Cells. 2008;26:1146–1154. doi: 10.1634/stemcells.2007-1118. [DOI] [PubMed] [Google Scholar]
- 42.Hall A. Rho GT: pases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 43.Wahl S., Barth H., Ciossek T., Aktories K., Mueller B.K. Ephrin-A5 induces collapse of growth cones by activating Rho and Rho kinase. J Cell Biol. 2000;149:263–270. doi: 10.1083/jcb.149.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dubreuil C.I., Winton M.J., McKerracher L. Rho activation patterns after spinal cord injury and the role of activated Rho in apoptosis in the central nervous system. J Cell Biol. 2003;162:233–243. doi: 10.1083/jcb.200301080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steward O., Schauwecker P.E., Guth L., Zhang Z., Fujiki M., Inman D., Wrathall J., Kempermann G., Gage F.H., Saatman K.E., Raghupathi R., McIntosh T. Genetic approaches to neurotrauma research: opportunities and potential pitfalls of murine models. Exp Neurol. 1999;157:19–42. doi: 10.1006/exnr.1999.7040. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Z., Guth L., Steward O. Mechanisms of motor recovery after subtotal spinal cord injury: insights from the study of mice carrying a mutation (WldS) that delays cellular responses to injury. Exp Neurol. 1998;149:221–229. doi: 10.1006/exnr.1997.6717. [DOI] [PubMed] [Google Scholar]
- 47.Juvin L., Le Gal J.P., Simmers J., Morin D. Cervicolumbar coordination in mammalian quadrupedal locomotion: role of spinal thoracic circuitry and limb sensory inputs. J Neurosci. 2012;32:953–965. doi: 10.1523/JNEUROSCI.4640-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma L., Nagai J., Ueda H. Microglial activation mediates de novo lysophosphatidic acid production in a model of neuropathic pain. J Neurochem. 2010;115:643–653. doi: 10.1111/j.1471-4159.2010.06955.x. [DOI] [PubMed] [Google Scholar]
- 49.Pitson S.M., Pébay A. Regulation of stem cell pluripotency and neural differentiation by lysophospholipids. Neurosignals. 2009;17:242–254. doi: 10.1159/000231891. [DOI] [PubMed] [Google Scholar]
- 50.Pebay A., Bonder C.S., Pitson S.M. Stem cell regulation by lysophospholipids. Prostaglandins Other Lipid Mediat. 2007;84:83–97. doi: 10.1016/j.prostaglandins.2007.08.004. [DOI] [PubMed] [Google Scholar]