

Abstract
Mcl-1, an anti-apoptotic member of the Bcl-2 protein family, is overexpressed in a broad range of human cancers and plays a critical role in conferring resistance to chemotherapy. In the course of screening a natural product-like library of sesquiterpenoid analogs, we identified substituted hexahydronaphthalenes that showed activity against the Mcl-1/BimBH3 interaction in vitro. Here, we describe the synthesis of a small library of analogs and their biological evaluation. The most potent inhibitor in the series (19) exhibits an IC50 of 8.3 μM by ELISA and disrupts the interaction between endogenously expressed Mcl-1 and Bim in cultured MDA-MB-468 breast cancer cells.
Keywords: Mcl-1, Bcl-2, Protein-protein interactions, Small molecule inhibitors, Anticancer agents
Bcl-2 family proteins regulate apoptosis through their influence on mitochondrial outer membrane (MOM) permeability and the release of cell death factors such as cytochrome c in response to cellular stress.1–3 Bax and Bak are pro-apoptotic multidomain (BH1-BH4) family members necessary for apoptosis and are directly involved in binding to the MOM.4 Other pro-apoptotic Bcl-2 proteins such as Bim, Bid, Bad, Bik, Puma, and Noxa contain only a single BH3 domain and indirectly modulate MOM permeability upstream of Bax and Bak.5 The anti-apoptotic family members Bcl-2, Bcl-xL, Bcl-w, Mcl-1, and Bfl-1 exert their influence by heterodimerizing with these pro-apoptotic substrates.6–8
Mcl-1 is over-expressed in greater than 50% of hepatocellular carcinomas,9 pancreatic adenocarcinomas,10 cervical cancers,11 non-Hodgkin’s lymphomas,12 and non-small cell lung cancers.13 Mcl-1 is known to compensate for the loss of Bcl-2 or Bcl-xL activity induced by selective antagonists14,15 and is especially well suited to provide protection from apoptosis due to its relatively short half-life (between 0.5 and 3 hours).16 Mcl-1 is thus a critical survival factor in a variety of human tumors and has emerged as a promising target for small molecule inhibitors.
In our search for new antagonists of anti-apoptotic Bcl-2 proteins, we screened a small in-house library of natural product-like scaffolds for activity against the Mcl-1/BimBH3 interaction. This resulted in the identification of hexahydronaphthalene 1 (Figure 1) as hit compound for further investigation. Here, we describe the synthesis of analogs of 1 and their biological evaluation as novel Mcl-1 antagonists.
Figure 1.

Structure of hexahydronaphthanlene 1 and analogs.
The hexahydronaphthalene core structure was prepared via Diels-Alder reaction between dihydrobezaldehyde derivative 317 and (E)-trimethyl((3-methylbuta-1,3-dien-1-yl)oxy)silane under thermal conditions (Scheme 1).18 Treatment of the intermediate cycloaddition adduct with TBAF in THF promoted silyl ether cleavage and concomitant alkene migration to give core structure 4 as a single diastereomer. Extensive 2D NMR experiments established the structural connectivity of the product while relative stereochemistry was confirmed by X-ray diffraction of reduced derivative 9.18 Presumably, treatment of the intermediate Diels-Alder adduct with TBAF promotes retro-aldol ring opening and equilibration to the more stable α-OH isomer. While the precise mechanism of the concomitant alkene migration is unknown, this process seems to occur during the retro-aldol reaction and ultimately gives rise to a lower energy didehydrodecalin. The relative stereochemistry of the hexahydronaphthalene scaffold was confirmed by X-ray diffraction of diol derivative 5.
Scheme 1.

Synthesis of hexahydronaphthalene scaffold 5.
Elaboration of 4 into the desired compounds was achieved through silylation of the secondary alcohol followed by aldehyde reduction and conversion to the methoxymethyl ether 7 (Scheme 2). To assess the impact of the amide substituent of 1 on biological activity, we prepared truncated analogue 8 via silyl ether cleavage and reaction of the resulting alcohol with cyclohexylisocyanate. Hydrolysis of 7 followed by dialkylation afforded analogs 10 and 11. Condensation of 9 with various amines and subsequent carbamoylation gave a series of diversely substituted hexahydronaphthalenes for biological evaluation.
Scheme 2.

Analog synthesis.
All newly synthesized compounds were tested for their ability to block the interaction between GST-Mcl-1 and the BimBH3 domain in an ELISA assay19,20 at a single concentration (25 μM). A peptide corresponding to the BimBH3 helix was used as a reference inhibitor and average percent inhibition was calculated from three separate experiments. As shown in Table 1, initial lead compound 1 exhibited >50% inhibition of Mcl-1 at 25 μM. Truncations at either end of the hexahydronaphthalene core as in 5, 9, and 12 resulted in diminished activity, as did the introduction of a more polar morpholine group in place of the isobutenyl amide as in 22. Various hydrophobic substitutions were well tolerated, with the exception of the smaller cyclopentyl and isopropyl carbamates in 17 and 18. The 3-chlorophenyl carbamate derivative (19) exhibited slightly enhanced activity. Compound 10, which is readily accessible from the core scaffold via dialkylation, also showed significant inhibition at 25 μM.
Table 1.
Inhibition of the GST-Mcl-1/BimBH3 interaction by ELISA at 25 μM (mean given with standard deviation, n= 3).
| Compound | % inhibition | Compound | % inhibition |
|---|---|---|---|
 1 |
53.8 ± 5.1 |
 18 |
31.2 ± 7.1 |
 8 |
34.8 ± 8.4 |
 19 |
74.3 ± 6.0 |
 12 |
17.1 ± 1.8 |
 20 |
40.3 ± 8.4 |
 21 |
53.9 ± 3.7 |
 10 |
64.9 ± 4.7 |
 22 |
26.5 ± 5.9 |
 11 |
31.2 ± 1.0 |
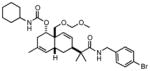 23 |
64.4 ± 5.9 |
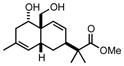 5 |
25.0 ± 2.6 |
 16 |
61.8 ± 10.6 |
 9 |
18.2 ± 6.0 |
 17 |
19.7 ± 4.0 |
We then selected the three most active compounds from single dose testing for determination of IC50 values. Figure 2 depicts the ELISA dose response curves for compounds 10, 19, and 23 over a range of concentrations between 0.1 and 100 μM. Each compound exhibited dose-dependant inhibition of Mcl-1 in the micromolar range. The most potent compound, 19, showed an IC50 of 8.3 μM in vitro.
Figure 2.

ELISA Mcl-1/BimBH3 dose-response curves for 10, 19, and 23 (95% confidence intervals in parentheses).
We next determined whether the most potent inhibitor in vitro (19) is able to enter human cancer cells, reach its target and disrupt the Mcl-1/Bim interaction. To this end, we treated MDA-MB-468 human breast cancer cells that ectopically express Bcl-xL and Bim with either vehicle V (0.1% DMSO) or 19 at 25 or 50 μM, lysed the cells, immunoprecipitated Mcl-1 from the lysates and immunoblotted with Bim as described by us previously.21 As a positive control we used BH3M6, a substituted terphenyl derivative that has previously been shown to disrupt the interaction of various anti-apoptotic Bcl-2 family proteins with Bim in whole cells.21 TPC, an unsubstituted analog of BH3M6 with no appreciable in vitro activity was also included as a negative control. Figure 3 shows that in vehicle-treated MDA-MB-468 cells Bim co-immunoprecipitated with Mcl-1. Similarly, cells treated with negative control TPC also show complex formation between Mcl-1 and Bim. In contrast, 19 inhibited the interaction between Mcl-1 and Bim at both 25 and 50 μM. Compound 19 had similar potency to BH3M6 at both concentrations.
Figure 3.

Co-immunoprecipitation of Mcl-1 (with Bim) from MDA-MB-468 cells. (A) Samples compared in the same gel for each protein and (B) densitometric analysis of inhibition.
Compound 19 also exhibited approximately 80% growth inhibition toward MDA-MB-468 cells above 100 μM by MTT assay (see Supplementary Materials). However, increased concentrations did not result in greater cytotoxicity. Poor solubility in culture medium beyond 100 μM precluded an accurate determination of GI50 values.
To gain structural insight into the possible binding interaction between 19 and Mcl-1, we performed computational docking experiments using the GLIDE protocol. Since 19 was produced as a racemic mixture, we docked both enantiomers to the X-ray crystal structure of human Mcl-1 (derived from PDB code 2NL9).22 Figure 4 depicts the top scoring docking poses for each antipode, both of which bind to the hydrophobic cleft occupied by the BH3 helical domains of pro-apoptotic partners. Notably, the two large hydrophobic groups of the 1R,4aS,6R,8aS enantiomer (isobutenyl and 3-chlorophenyl) bind to the sites on Mcl-1 that normally accommodate the Leu62 (i+4) and Phe69 (i+11) side chains in human Bim (Figure 4A). While the 3-chlorophenyl substituent in the 1S,4aR,6S,8aR enantiomer also appears to mimic the Phe69 residue, the isobutenyl substituent resides well outside of the Leu62 pocket (Figure 4B). In both docked structures, the methoxymethyl ether substitutent of 19 makes extensive stabilizing contacts with Asn260 and Arg263 in Mcl-1. Furthermore, this functional group overlays well with the carboxy side chain of Asp67 (i+9) in human Bim.
Figure 4.

Top-scoring poses for (A) (1R,4aS,6R,8aS)-19 and (B) (1S,4aR,6S,8aR)-19 docked to human Mcl-1 (PDB code 2NL9) using GLIDE.
In summary, we have described the synthesis and preliminary SAR for a series of hexahydronapthalenes that disrupt the Mcl-1/BimBH3 interaction in vitro. The most potent inhibitor in the series (19) exhibits an IC50 of 8.3 μM by ELISA. Compound 19 also disrupts the interaction between endogenously expressed Mcl-1 and Bim in intact MDA-MB-468 breast cancer cells. Computational docking suggests that 19 interacts with the BH3-binding hydrophobic cleft in human Mcl-1. Efforts toward the synthesis of enantiopure 19 and other analogs for evaluation against a wider panel of anti-apoptotic Bcl-2 family proteins are currently underway.
Supplementary Material
Acknowledgments
This research was supported by the James and Esther King Biomedical Research Program, Florida Department of Health (Grant 1BN03 to JRD) and the National Institutes of Health (Grant P01CA118210 to SMS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Crystallographic data (excluding structure factors) for compound 5 has been deposited with the Cambridge Crystallographic Data Centre as supplementary CCDC publication number 834136. Experimental procedures, complete spectral data, and copies of NMR spectra for final compounds.
Supplementary data associated with this article can be found, in the online version, at doi:xxxxxxxx..
References and notes
- 1.Kelly PN, Strasser A. Cell Death Diff. 2011;18:1414. doi: 10.1038/cdd.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Korsmeyer SJ. Cancer Res. 1999;59:1693s. [PubMed] [Google Scholar]
- 3.Youle RJ, Strasser A. Nat Rev Mol Cell Biol. 2008;9:47. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 4.Wei MC, Zong WX, Cheng EHY, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Science. 2001;292:727. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adams JM, Cory S. Oncogene. 2007;26:1324. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Willis SN, Adams JM. Curr Opin Cell Biol. 2005;17:617. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green DR. Cancer Cell. 2006;9:328. doi: 10.1016/j.ccr.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 8.Ewings KE, Wiggins CM, Cook SJ. Cell Cycle. 2007;6:2236. doi: 10.4161/cc.6.18.4728. [DOI] [PubMed] [Google Scholar]
- 9.Sieghart W, Losert D, Strommer S, Cejka D, Schmid K, Rasoul-Rockenschaub S, Bodingbauer M, Crevenna R, Monia BP, Peck-Radosavljevic M, Wacheck V. J Hepatol. 2006;44:151. doi: 10.1016/j.jhep.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 10.Miyamoto Y, Hosotani R, Wada M, Lee JU, Koshiba T, Fujimoto K, Tsuji S, Nakajima S, Doi R, Kato M, Shimada Y, Imamura M. Oncology. 1999;56:73. doi: 10.1159/000011933. [DOI] [PubMed] [Google Scholar]
- 11.Chung TK, Cheung TH, Lo WK, Yim SF, Yu MY, Krajewski S, Reed JC, Wong YF. Cancer Lett. 2002;180:63. doi: 10.1016/s0304-3835(01)00842-4. [DOI] [PubMed] [Google Scholar]
- 12.Michels J, O’Neill JW, Dallman CL, Mouzakiti A, Habens F, Brimmell M, Zhang KYJ, Craig RW, Marcusson EG, Johnson PWM, Packham G. Oncogene. 2004;23:4818. doi: 10.1038/sj.onc.1207648. [DOI] [PubMed] [Google Scholar]
- 13.Song LX, Coppola D, Livingston S, Cress D, Haura EB. Cancer Biol Ther. 2005;4:267. doi: 10.4161/cbt.4.3.1496. [DOI] [PubMed] [Google Scholar]
- 14.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. Nature. 2005;435:677. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 15.Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi YX, Sneed T, Verhaegen M, Soengas M, Ruvolo VR, McQueen T, Schober WD, Watt JC, Jiffar T, Ling X, Marini FC, Harris D, Dietrich M, Estrov Z, McCubrey J, May WS, Reed JC, Andreeff M. Cancer Cell. 2006;10:375. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 16.Zhou P, Levy NB, Xie H, Qian L, Lee CY, Gascoyne RD, Craig RW. Blood. 2001;97:3902. doi: 10.1182/blood.v97.12.3902. [DOI] [PubMed] [Google Scholar]
- 17.Saito S, Sone T, Murase M, Yamamoto H. J Am Chem Soc. 2000;122:10216. [Google Scholar]
- 18.Kim YB, Del Valle JR. Tetrahedron Lett. 2011;52:6396. [Google Scholar]
- 19.Tang G, Yang CY, Nikolovska-Coleska Z, Guo J, Qiu S, Wang R, Gao W, Wang G, Stuckey J, Krajewski K, Jiang S, Roller PP, Wang S. J Med Chem. 2007;50:1723. doi: 10.1021/jm061400l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doi K, Li R, Sung SS, Wu H, Liu Y, Manieri W, Krishnegowda G, Awwad A, Dewey A, Liu X, Amin S, Cheng C, Qin Y, Schonbrunn E, Daughdrill G, Loughran TP, Jr, Sebti S, Wang HG. J Biol Chem. 2012;287:10224. doi: 10.1074/jbc.M111.334532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kazi A, Sun J, Doi K, Sung SS, Takahashi Y, Yin H, Rodriguez JM, Becerril J, Berndt N, Hamilton AD, Wang HG, Sebti SM. J Biol Chem. 2011;286:9382. doi: 10.1074/jbc.M110.203638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Czabotar PE, Lee EF, van Delft MF, Day CL, Smith BJ, Huang DC, Fairlie WD, Hinds MG, Colman PM. Proc Natl Acad Sci USA. 2007;104:6217. doi: 10.1073/pnas.0701297104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.