

Abstract
Background
Lysis of endocytic organelles is a necessary step in many cellular delivery methodologies. This is achieved efficiently in the photochemical internalization approach but the cell death that accompanies this process remains a problem.
Methods
We investigate the mechanisms of cell death that accompanies photochemical internalization of the fluorescent peptide TMR-TAT.
Results
TMR-TAT kills cells after endocytosis and light irradiation. The lysis of endocytic organelles by TMR-TAT causes a rapid increase in the concentration of calcium in the cytosol. TMR-TAT co-localizes with endocytic organelles containing calcium prior to irradiation and photochemical internalization leads to the release of the lumenal content of these organelles. Ruthenium red and cyclosporin A, inhibitors of calcium import in mitochondria and of the mitochondria permeability transition pore, inhibit cell death.
Conclusions
TMR-TAT mediated photochemical internalization leads to a disruption of calcium homeostasis. The subsequent import of calcium in mitochondria is a causative factor of the cell death that accompanies photochemical internalization.
General Significance
Understanding how the lysis of endocytic organelles affects cellular physiology and causes cell death is crucial to the development of optimal delivery methodologies.
Keywords: Photochemical Internalization, TAT, Cell-penetrating peptide, Calcium
1. Introduction
Many methods used to deliver macromolecules into cells employ the endocytic pathway as a route of cellular entry. Macromolecules need, however, to escape from endosomes in order to reach cytosolic targets or other intracellular organelles. Efficient endosomal release is therefore necessary to achieve successful delivery in most applications [1]. Accordingly, different strategies have been used to enable the release of trapped molecules from endosomes. A challenge consists in mediating the lysis of endosomes without damaging other cellular membranes so as to prevent cell death. Interestingly, this can be achieved by using a combination of photosensitizing compounds and irradiation with visible light as demonstrated with the photochemical internalization (PCI) methodology developed by Berg and co-workers [2]. PCI has been successfully used to transfer proteins or RNA into cells both in vitro and in vivo [3–4]. PCI is based on the use of phthalocyanine or porphyrin derivatives that localize inside endosomes and disrupt endosomal membranes specifically at illuminated sites [5]. More recently, several investigators recognized that similar photo-endosomolytic effects could be achieved with the cell-penetrating peptides (CPPs) TAT or polyarginines labeled with various fluorophores. Matsushita and co-workers have, for instance, shown that irradiation of FITC-R11-p53 at 480 nm could cause the release of the protein from endosomes [6]. Ferrer and co-workers have reported similar light-triggered endosomal release with peptides containing the polyarginine peptide R7 labeled with fluorescein or AlexaFluor633 [7]. Ohtsuki and co-workers have shown that TatU1A, a protein fusion between TAT and the RNA binding protein U1A, could form complexes with RNA molecules and that these complexes accumulate inside endosomes when incubated with live cells. Attachment of fluorophores such as Alexa546 or Cy3 at the C-terminus of TatU1A however enables the release of the RNA into the cytosolic space of cells upon light irradiation [8–11]. Light-dependent delivery of the RNA leads to the silencing of target genes and the authors have therefore demonstrated that this approach can be used for the spatial and temporal photo-activation of RNA interference.
While PCI is a potentially powerful approach for therapeutic applications and for cell biology assays, several reports suggest that cell viability can be greatly reduced post irradiation. Schiffelers and co-workers have for instance shown that 95% of cells incubated with 1 μg/mL of TPPS2a, a sulfonated meso-tetraphenylporphine commonly used for PCI, are killed 24 h after a 90 sec of exposure to 13 mW/cm2 of light [12]. Berg and co-workers demonstrated that TPPS2a localizes in lysosomes and that irradiation causes significant cytotoxicity [13]. Irradiation mediates the release of hydrolytic lysosomal enzymes into the cytosol but, interestingly, it was found that the released lysosomal enzymes were photochemically inactivated or inhibited by cytosolic inhibitors. These studies did not identify the cause of cell death but on the other hand suggested that cells could survive partial lysosomal disruption [13]. We have recently reported that TAT labeled with tetramethylrhodamine, TMR-TAT, also kills cells readily upon light irradiation during PCI experiments [14]. TMR-TAT efficiently escapes from endosomes on a millisecond time scale when using an epifluorescence microscope as light source. The fluorescent peptide is also photolytic toward biological membranes such as the plasma membrane of red blood cells [14]. These effects involve the targeting of TMR by TAT to cellular membranes and the formation of singlet oxygen within membranes. TMR-TAT is not cytotoxic in the dark but PCI leads to rapid cell death. In particular, in our hands, endosomal release of TMR-TAT could not be achieved without observing cellular killing in the form of blebbing and permeabilization of the plasma membrane [14].
In this report, our goal is to investigate the mechanism of cell-death involved in TAT-mediated PCI (TM-PCI). Our rationale is that identification of the possible causes of cell-death resulting from TM-PCI could aid in developing analogs or delivery protocols that can promote endosomal release rapidly and efficiently while avoiding cell killing.
2. Material and methods
2.1. Materials used
Cell culture reagents and the calcium probes fluo-4 AM, fura-2 AM, and fura-2 were purchased from Invitrogen. Peptide synthesis reagents were from Novabiochem. All other reagents were from Sigma. HeLa, COS-7 and NIH 3T3 were obtained from ATCC. Cerulean-Lamp1 was a kind gift from Dr. Jennifer Lippincott-Schwartz (Cell Biology and Metabolism Program, Eunice Kennedy Shriver National Institute of Child Health and Human Development). pTagCFP-mito vector was bought from Evrogen. Whole blood was purchased from Gulf Coast Regional Blood Center (Houston, TX).
2.2. Peptide synthesis
TMR-TAT (TMR-GRKKRRQRRRG-NH2) and the protease-resistant TMR-riTAT were synthesized by solid phase peptide synthesis technique as previously described [14]. Purified and lyophilized TMR-TAT was dissolved in water to obtain a 1 mM stock solution and diluted in PBS or cell culture media to desired concentrations for experiments.
TMR-S-S-TAT synthesis. The synthesis of 2-(2-pyridyldithio)ethylamine hydrochloride (PDA-HCl) was conducted as previously reported [15]. EDCI (0.22 g, 1.2 mmol) was added to a solution of 5(6)-carboxytetramethylrhodamine (0.30 g, 0.70 mmol), PDA-HCl (0.12 g, 0.54 mmol), HOBt (0.18 g, 1.2 mmol), and TEA (0.20 mL, 1.4 mmol) in DMF (4 mL) at 0 °C. The reaction solution was stirred under N2 at 0 °C for 30 min, and then warmed to room temperature. After reacting overnight at room temperature, DMF was dried under vacuum. The resulting crude material was purified by silica gel chromatography (DCM:MeOH = 6:1) to give the mixture of two TMR-PDA isomers (0.22 g, 68%). Mass spectrometry analysis (MALDI-TOF): calculated for C32H30N4O4S2 598.2, observed 599.4 (M+H)+.
Cys-TAT (CGRKKRRQRRRG-NH2) was synthesized by adding Fmoc-Cys (StBu)-OH to the N-terminus of the TAT peptide assembled on the solid phase as Fmoc-Gly-Arg (Pbf)-Lys (Boc)-Lys (Boc)-Arg (Pbf)-Arg (Pbf)-Gln-Arg (Pbf)-Arg (Pbf)-Arg (Pbf)-Gly-Rink amide MBHA resin. The resin was deprotected with 20% piperidine to remove Fmoc and the Cys side chain was deprotected by treatment with tributylphosphine in dimethylformamide/dichloromethane (1:3:3 by volume). Cleavage of the peptide from the resin was then performed by treatment with trifluoroacetic acid/triisopropylsilane/ethanedithiol/water (92.5:2.5:2.5:2.5 by volume). The peptide was purified by semi-preparative HPLC and analyzed by mass spectrometry (calculated mass: 1555.9, observed: 1558.2 (M+H)+). The N-terminal cysteine of Cys-TAT was reacted with TMR-PDA in Tris buffer at pH 8.3 for 30 min. The product, TMR-S-S-TAT was purified by semi-preparative HPLC. Mass spectrometry analysis (MALDI-TOF): calculated mass: 2044.4, observed mass: 2045.7 (M+H)+.
2.3. Cell-based assays
Cells were cultured in Dulbecco’s Minimum Essential Media (DMEM) supplemented with 10% fetal bovine serum (FBS) and maintained at 37°C in a humidified environment with 5% CO2. Cells were plated in 8-well dishes so that the cells were 80% confluent after 24 h. Cells were washed 3 times with PBS or Leibovitz’s L-15 medium, incubated with 3 μM TMR-TAT at 37°C for 1 h, washed 3 times with PBS supplemented with 2 mg/mL of heparin. Cells were the incubated in L-15 and placed on the microscope stage at 37°C for imaging. For experiments involving calcium depletion, MEM Eagle Joklik modification supplemented with 2 g/L of sodium bicarbonate but lacking calcium was used. Cells with a damaged plasma membrane were detected using 5 μM cell-impermeable DNA stains SYTOX® Blue or SYTOX® Green after photosensitization. Ruthenium red was used from 1 mM stock solutions in water and diluted to 5 μM for incubation with cells. Cyclosporin A was used from 1 mM stock solutions in DMSO and diluted to 20 μM for incubation with cells. For transfection and expression of a fluorescent mitochondrial marker, the pTagCFP-mito plasmid was mixed with Lipofectamine LTX reagent to form a complex in opti-MEM media. The DNA complex was added to previously seeded HeLa cells (60% confluent) on an 8-well dish and cells were kept at 37°C for 18 hr. The transfection media was then removed and replaced with L-15 media prior to performing experiments.
2.4. Photo-hemolysis assay
Cells were centrifuged for 10 min at 1500g to separate the erythrocytes from the plasma supernatant. Erythrocyte pellets were resuspended in PBS and the process was repeated four times to remove plasma and buffy coat. The erythrocytes (50% volume in PBS) were diluted in PBS to a final concentration of 0.1 %. Peptides and erythrocytes were mixed in PBS and added to a 384 well plate. Cells were incubated with the peptide for 15 minutes and allowed to settle to the bottom of the dish prior to imaging to get a layer of cells at the focal plane. The photo-hemolytic activity of TMR-TAT or TMR-S-S-TAT was assessed by irradiation of the peptide and cell mixtures at 560 nm on the microscope (RFP channel). The number of lysed cells was counted after irradiation by using bright field images in which intact erythrocytes have a dark contrast while lysed ghosts do not. The data reported represents the averages and the corresponding standard deviations of five experiments for which a minimum of 500 cells were examined.
2.5. Fluorescence Microscopy Imaging
Multi-well dishes with 80% cell confluency in each well were placed on an inverted epifluorescence microscope (Model IX81, Olympus, Center Valley, PA). The microscope is equipped with a heating stage which was maintained at 37°C. The microscope is configured with a spinning disk unit to perform both confocal and wide-field fluorescence microscopy. Images were captured with a Rolera-MGI Plus back-illuminated EMCCD camera (Qimaging, Surrey, BC, Canada). Imaging was performed using bright field imaging and the fluorescence filter sets: RFP (Ex = 560±20 nm/Em= 630±35 nm), FITC (Ex = 488±10 nm/Em= 520±20 nm) and CFP (Ex = 436±10 nm/Em= 480±20 nm). The excitation light was from a 100W mercury lamp (Leeds Precision Instruments # L202 Osram) passed through the filter cubes and 20, 40, or 100X objectives. Neutral density filters (ND 1, 2, 3 and 4 on the instrument corresponds to 100, 25, 12.5 and 5% transmittance) and different exposure times were used to adjust the dose of light given to the cells. The bright field and fluorescence intensities of HeLa cells were measured with the SlideBook 4.2 software (Olympus, Center Valley, PA). Irradiances at the specimen were 100 mW/cm2 when no neutral density filter and no objective were present in the light path (and, for instance, 5 mW/cm2 when ND4 was inserted). Irradiances were measured using a monochromic photometer (model 840-c, Newport, Irvine, CA). Irradiation area has a diameter of 1.3 cm without objective but the light beam is focused into an area of 3×10−3 cm2 by the 100X objective. Irradiances can therefore be approximated to be at 21 or 420 W/cm2 with ND4 or ND1, respectively. To determine the percentage of cells with a compromised plasma membrane after light irradiation, cells were incubated with SYTOX® Green and Hoechst. SYTOX® Green is cell-impermeable and only stains cells with a compromised plasma membrane while Hoechst stains all cells. Cells were imaged with a 40X objective. Ten to twenty images were acquired in the green and blue channels for each experiment. The total number of cells in a given image was determined from the blue channel image (Hoechst) by counting the number of blue nuclei present. The number of dead cells was determined by identifying cells containing a green fluorescent nucleus stained by SYTOX® green. Cell viability was determined by establishing a ratio of dead cells/total number of cells for each sample (at least 1000 cells were counted in each experiment and each experiment was repeated 3 times).
2.6. Calcium imaging and calibration
The calcium indicators fluo-4-AM and fura-2 were used from 1 mM DMSO stock solutions and diluted to 5 μM in media for experiments. Fluo-4 AM was incubated with cells for 1 h. After incubation, cells were washed with fresh medium and imaged 20 min later to allow intracellular cleavage of the acetoxymethyl esters. The maximum fluorescence intensity of fluo-4 (Fmax) was determined in cellulo by addition of the calcium ionophore ionomycin (1μM) in L-15 supplemented with 2 mM calcium chloride. The fluorescence intensity of fluo-4 in the absence of calcium (Fmin) was determined by addition of EGTA (10 mM) to cells incubated in L-15. Quantification of emitted fluorescence was calculated by taking the mean fluorescence intensity of the nucleus of a cell using the Slidebook software. The approximate calcium concentrations reported in the text were calculated from the formula [Ca2+]=Kd*(F−Fmin)/(Fmax−F) [16]. Ratiometric imaging of fura-2 was performed on a Stallion digital imaging workstation with a c-apochromat 63X/1.2 W objective. Excitation was performed at 340 nm and 380 nm with rapid switching (< 2 msec) between excitation wavelengths. Emission was collected at 505 nm. Calibration experiments were performed in vitro on the microscope using fura-2 solutions buffered at pH 7.2 and pH 5.5 in 20 μm flat capillary tubes (vitrotubes 5002-050, Vitrocom). The fluorescence signals from the 380 and 340 nm excitation images, F380 and F340, were measured in each case in the absence or presence of 2 mM calcium chloride. Alternatively, in cellulo calibration experiments were performed as described for fluo-4 by using the cell-permeable fura-2 analog fura-2 AM. Quantification of F380 and F340 was calculated by taking the mean fluorescence intensity of a region of interest (ROI) using the Slidebook software after subtraction of the fluorescence background obtained from a ROI outside the cell. The ratio of F340/F380 signals, R, were calculated for each set of images (Rmin and Rmax corresponding to the ratios in the absence or presence of saturating amount of calcium, respectively) and the calcium concentrations reported in the text were calculated from the formula [Ca2+]=Kd*[(R−Rmin)/(Rmax−R)]*(F380max/F380min) as previously described [16]. The apparent Kd of fura-2 used in our calculations were 130 nM at pH 7.2 and 1188 nM at pH 5.5, as previously established [17].
2.7. Microinjection of TMR-TAT into Live Cells
HeLa cells were cultured on 35 mm plates (P35G-1.5-7-C-grid, MatTek Corp., Ashland, MA). Cells were washed and incubated with Leibovitz’s L-15 Medium and placed on the microscope. Femtoliter aliquots of TMR-TAT (10 μM) were directly injected into the cytoplasm of live HeLa cells using an InjectMan NI2 micromanipulator equipped with a FemtoJet microinjector (Eppendorf, Westbury, NY). The microinjected cells were imaged immediately after microinjection. Irradiation was performed in the microscope as described for TM-PCI.
3. Results
3.1. Microinjection of TMR-TAT followed by irradiation does not cause membrane blebbing or cell death
TMR-TAT is photolytic towards endosomes but also towards other biological membranes such as the plasma membrane of red blood cells [14]. We were therefore interested in testing the hypothesis that TMR-TAT might kill cells after escaping from endosomes by causing photolytic damage to other intracellular organelles. In order to determine whether TMR-TAT induces a phototoxic response when present in the cytosolic space of cells, TMR-TAT was directly microinjected into the cytoplasm of HeLa cells (Figure 1). Cells were then irradiated under conditions similar to that used for PCI. The cell impermeable DNA stain SYTOX® Blue was added to the media in order to assess the permeability of the plasma membrane. A PCI experiment for which TMR-TAT was incubated with cells to permit accumulation inside endocytic organelles was carried out in parallel. In both experiments, irradiation of the cells was performed directly on the microscope by using the RFP channel to excite TMR (Ex = 560nm). In the PCI experiment, TMR-TAT escaped from endosomes and distributed in the cytosol and nucleus of cells upon brief irradiation (Figure 1). As previously reported, PCI was accompanied by rapid plasma membrane blebbing and staining with SYTOX® Blue, indicative of plasma membrane disruption and cell killing [14]. In contrast, when similar irradiation conditions were used, cells containing microinjected TMR-TAT did not show loss of plasma membrane permeability or blebbing. In order to compare the amount of TMR-TAT microinjected inside cells to that of TMR-TAT released into the cell after PCI, the whole cell fluorescence intensities of cells post-PCI as well as the microinjected cells were measured. Based on this analysis, the amount of TMR-TAT present in the cytosol of microinjected cells was equal or greater than that obtained after PCI (please note the greater TMR fluorescence intensity for the cells microinjected in Figure 1; the contrast is kept constant between the different images). Microinjected TMR-TAT therefore, appears to be overall less phototoxic than TMR-TAT released from endosomes.
Figure 1. Irradiation of endocytosed TMR-TAT kills cell while irradiation of microinjected TMR-TAT does not.

A) HeLa cells were incubated with TMR-TAT (3 μM), washed, and irradiated at 560 ± 20 nm through a 100 X objective on a wide-field microscope. The time displayed on the images represents the total light exposure time. The fluorescence signal of TMR-TAT (TMR channel) initially shows a punctate distribution consistent with the compound being present within endocytic organelles. Brief irradiation at 560 nm (TMR channel) causes TMR-TAT to redistribute throughout the cells. This is accompanied by blebbing of the plasma membrane (visible in the bright field image) and permeabilization of the plasma membrane, as seen by staining of the cells with SYTOX Blue. The TMR and SYTOX BLUE fluorescence signals are represented as inverted monochrome (black = fluorescent signal, white = no signal). B) TMR-TAT was microinjected into the cell and irradiated at 560 nm immediately. Irradiation had no apparent effect on the cells under conditions where the peptide is more concentrated inside cells and the light dose is more intense than in the incubated sample. Identical data were obtained with the protease resistant TMR-riTAT (not shown).
3.2. TMR-S-S-TAT kills cells as efficiently as TMR-TAT
We have previously shown that the attachment of TMR to TAT is necessary to mediate photo-induced lysis [14]. We therefore reasoned that, if TMR-TAT exerts phototoxic effects after it escapes from endosomes, the phototoxicity of the compound could be abolished by introducing a linker between TMR and TAT that would cleave upon entry of the peptide into the cytosolic space of irradiated cells. With this principle in mind, TMR and TAT were attached to one another through a disulfide bridge to generate TMR-S-S-TAT. Disulfide bonds are cleaved within seconds in the reducing environment of the cytosol but not inside endosomes of HeLa cells [18]. TMR-S-S-TAT was as efficient as TMR-TAT at inducing the lysis of red blood cells (Figure 2A). Addition of the cytosolic reducing agent glutathione (GSH) however, abolished the photolytic effect of TMR-S-S-TAT but not that of TMR-TAT. These results are therefore consistent with the idea that TMR-S-S-TAT is inactivated by reduction. Like TMR-TAT, TMR-S-S-TAT was endocytosed by cells (Figure S2) and released from endosomes upon irradiation with light (Figure 2B). Interestingly, TMR-S-S-TAT led to cell-death as readily as TMR-TAT. Cell-death was in particular not reduced when PCI irradiation was performed at a slow frequency (every 10 s) so as to allow sufficient time for TMR-S-S-TAT to be reduced in the cytosol in between small bursts of endosomal release (100 ms excitation, Figure S1). Overall, these results suggest that TMR-TAT does not cause the cell-death observed during PCI by causing photolytic damage in the cytosolic space of cells.
Figure 2. TMR-S-S-TAT loses its photolytic activity upon reduction but displays the same cell killing activity as TMR-TAT.

A) Peptide-mediated lysis of red blood cells as a function of exposure time (irradiation at 560 nm). TMR-TAT and TMR-S-S-TAT (3 μM each) display similar photo-hemolytic activity. The activity of TMR-TAT is unaffected by the reducing agent glutathione (GSH, 5 mM) but that of TMR-S-S-TAT is abolished. B) Irradiation of endocytosed TMR-S-S-TAT results in plasma membrane blebbing and permeabilization in a manner similar to TMR-TAT. Note that the TMR signal does not show the nucleoli staining typical of TAT after irradiation (see Figure 1). The TMR signal is also weak, presumably because the reduced fluorophore is able to diffuse out of the permeabilized cell more readily than TMR-TAT.
3.3. TMR-TAT endosomal release is accompanied by an increase in cytosolic [Ca2+]
The previous results suggested that cell death observed during TM-PCI might be a direct result of the photo-induced lysis of endocytic organelles. In particular, TM-PCI might induce the release of toxic molecules into cells other than the PCI agent itself. While the PCI-mediated release of lysosomal hydrolytic enzymes might impact cell viability, the rapidity with which cell death occurs prompted us to test other potential causes of cell death. In particular, it has been established that an increase in the concentration of calcium in the cytosol can cause cell death [19]. Interestingly, early endosomes, late endosomes and lysosomes are thought to contain calcium at high concentrations [20–23]. We therefore hypothesized that TMR-TAT mediated PCI could trigger cell death by releasing pools of calcium from endosomes and lysosomes. To test this idea, the fluorescent calcium indicator, fluo-4 (Kd ~ 345 nM) was used to detect the increase of cytosolic calcium during TM-PCI [16, 24]. As shown in Figure 3, TMR-TAT endosomal release was accompanied by a burst increase in the cytosolic calcium levels. This increase in fluorescence is consistent with the calcium indicator being saturated and indicates that the [Ca2+]cyt increases from less than 100 nM to over 1 μM after PCI. The L-15 medium used for incubation in these assays initially contained calcium at a physiological concentration of 1.3 mM. Calcium might therefore enter the cell from the extracellular milieu during PCI. To test this hypothesis, the experiment was repeated with a medium formulated without calcium and supplemented with the calcium chelator EGTA. This medium was used to wash the cells after incubation with TMR-TAT and during light irradiation and microscopy. PCI was performed immediately after incubating the cells with the calcium-free medium so as to create a condition where calcium was removed from the extracellular milieu without causing the depletion of the intracellular pools of calcium. PCI performed under these conditions led to an increase in fluorescence of fluo-4 only slightly reduced in comparison to that observed when calcium is present in the medium (Figure 3B). Moreover, plasma membrane blebbing and permeabilization also accompanied the PCI procedure in calcium-free media. It should therefore be noted that permeabilization presumably accounts for the decrease in signal observed after 4 s of irradiation. Plasma membrane permeabilization might also contribute to the smaller signal observed for TM-PCI performed in the absence of extracellular calcium. Together, these results suggest that [Ca2+]cyt increases greatly during PCI and that the calcium released into the cytosol comes from an intracellular pool rather than from the extracellular milieu.
Figure 3. TM-PCI causes an increase in the cytosolic concentration of calcium.
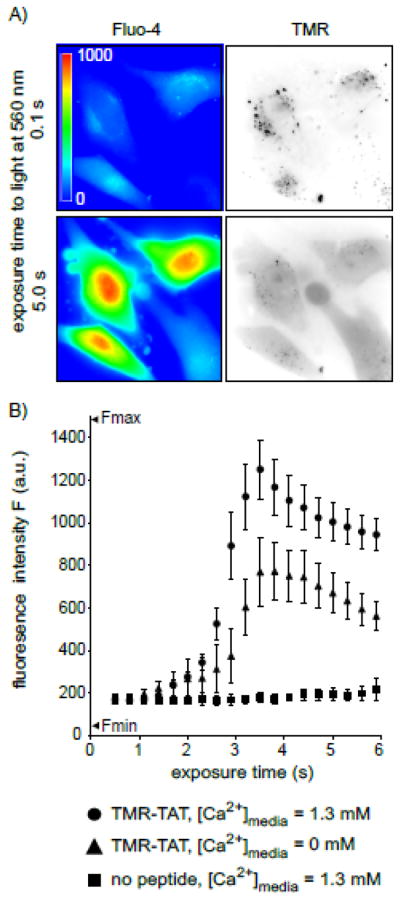
A) Images of HeLa cells incubated with TMR-TAT and the calcium indicator fluo-4AM. The fluo-4 images are pseudocolored based on the fluorescence intensity of the probe. Images representing the distribution of TMR-TAT before and after light irradiation are presented as inverted monochromes. B) Intensity of the fluo-4 signal as a function of light exposure time (irradiation at 560 nm). Cells were incubated with fluo-4AM alone, or with TMR-TAT (3 μM) in L-15 containing 1.3 mM CaCl2. Cells were then placed in fresh medium containing 0 or 1.3 mM CaCl2. Cells were irradiated immediately so as to prevent depletion of calcium intracellular stores for the cells lacking calcium in the extracellular milieu.
3.4. Import of calcium in mitochondria is a causative factor of TM-PCI mediated cell killing
Following the initial burst of fluorescence of fluo-4, we observed that the signal of the calcium indicator localized at continuous tubular structures (Figure S3) corresponding to mitochondria (Figure S4). A link between mitochondrial calcium and cell death has been clearly established in the literature and we therefore decided to test whether calcium import into the mitochondria could account for the rapid cell death observed. To test this hypothesis, the inhibitor of mitochondrial calcium import ruthenium red (RuRed) was used [25–27]. Mitochondrial calcium is also known to cause opening of the permeability transition pore (PTP), an event that can lead to apoptosis. PTP opening can be inhibited by cyclosporin A [28–30]. TMR-TAT mediated PCI was therefore performed with cells incubated with RuRed or cyclosporin A to assess the relationship between Ca2+ mitochondrial import and cell death (Figure 4). Cells treated with RuRed or cyclosporin A showed a burst of fluo-4 fluorescence similar to that observed in an untreated control sample, indicating that these inhibitors do not affect the cytosolic flux of Ca2+ (Figure 4B). However, RuRed and cyclosporin A inhibited the rapid plasma membrane blebbing and permeabilization obtained during TM-PCI. These data suggest that calcium import into mitochondria and opening of the PTP are causative factors to the rapid cell death observed during the TM-PCI.
Figure 4. Ruthenium red (RuRed) and cyclosporin A inhibit cell killing by TM-PCI.

A) HeLa cells were incubated with TMR-TAT (3 μM) in the absence or presence of RuRed (5 μM) or cyclosporin A (40 μM). Cells were irradiated at 560 nm for 3 sec and cell killing was measured by counting the number of cells stained by SYTOX Blue 30 min after irradiation. B) Images of a similar experiment performed with cells incubated with fluo-4AM (5 μM). Images of the fluo-4 signal before (insert) and after irradiation are pseudo-colored based on the intensity of the fluorescence signal. Presence of RuRed or cyclosporin A does not affect the increase of [Ca2+]cyt observed upon irradiation. However, membrane blebbing and permeabilization are observed when cells are incubated with TMR-TAT alone but not when RuRed or cyclosporin A are added.
3.5. TMR-TAT co-localizes with endocytic organelles containing calcium ions and TM-PCI causes the release of their lumenal content
The previous results establish that calcium is released into the cytosol of cells during TM-PCI. In order to test whether calcium is actually released from endocytic organelles during TM-PCI, calcium depletion experiments were performed. Cells were incubated with TMR-TAT in media in which calcium was chelated by EGTA (Figure 5). Because EGTA is cell impermeable, it is expected that EGTA would be endocytosed along with TMR-TAT and chelate calcium in the lumen of endocytic organelles. After incubation, the medium was replaced with L-15 containing 1.3 mM calcium so as to not deplete other intracellular calcium stores. TM-PCI led to endosomal release of TMR-TAT without causing an increase in fluo-4 fluorescence in the cytosol of cells (TMR-TAT uptake and endosomal release was similar to the conditions lacking EGTA, Figure 5B), These results are consistent with the notion that calcium present within endocytic organelles is involved in the increase in [Ca2+]cyt observed during TM-PCI. In addition, a quantitative cell viability assay was performed to establish that cells incubated with EGTA did not die upon light irradiation (Figure S5C), further confirming that the release of calcium in the cytosol of cells is mediating cell death. Next, we tested whether TMR-TAT would localize inside late endosomes and lysosomes. This is because high concentrations of calcium have already been detected in these organelles [20–23]. For these experiments, cells were transfected with Cerulean-LAMP1, a marker of late endosomes and lysosomes [31]. As shown in Figure 6A, TMR-TAT co-localizes with these organelles prior to light irradiation. Upon irradiation, the TMR-TAT signal vanishes from Cerulean-LAMP1 positive organelles (as shown in Figure 6A) and is distributed into the cytosol (as previously shown in Figure 1). Importantly, the TMR signal is still present in some organelles, thereby indicating that the loss of signal observed is not due to photobleaching. These data therefore suggest that TMR-TAT accumulates in organelles known to contain calcium and that TMR-TAT can escape from these organelles upon irradiation. To further confirm these results, we tested whether endocytic organelles containing TMR-TAT also contain calcium. This was determined by incubating cells with TMR-TAT and with the cell-impermeable calcium indicator fura-2. Fura-2 has a maximum of excitation at 380 nm in its unbound form (emission is at 510 nm). This maximum of excitation shifts to 340 nm when fura-2 binds calcium and, given a Kd for the binding of fura-2 to calcium, one can use ratiometric analysis of the signals obtained after excitation at 340 and 380 nm to estimate the concentration of calcium present in a cell [16]. TMR-TAT and fura-2 were incubated together in L-15 medium for 1 hour and cells were imaged immediately after incubation. As shown in Figure 6B, TMR-TAT and fura-2 co-localized within endocytic organelles. Ratiometric imaging of the fura-2 signals indicated that the 340/380nm ratio was 1.9±0.3 on average (30 organelles examined in 5 cells). This, in turn, would correspond to a [Ca2+]endo of 470 nM at pH 7.2 or 3.9 μM at pH 5.5. Overall, these results suggest that calcium is present inside endocytic organelles containing TMR-TAT. Next, we tested whether TM-PCI causes the release of the luminal content of endosomes. TMR-TAT was co-incubated with the pinocytosis marker 3 kDa Dextran-Rhodamine Green (Dx-RG) (Figure 6C). Prior to PCI, the fluorescence signals of TMR-TAT and Dx-RG co-localized in a punctate distribution consistent with the molecules being trapped inside endocytic organelles. Upon light irradiation, the signals of both TMR-TAT and Dx-RG redistributed homogeneously throughout the cell. These results therefore suggest that TMR-TAT is able to cause the release of molecules trapped in the lumen of endocytic organelles. Overall, these results indicate that TMR-TAT localizes within endocytic organelles that contain calcium in their lumen and that light irradiation can cause the release of the lumenal content of these organelles into the cytosol of cells.
Figure 5. Co-incubation of TMR-TAT with the cell-impermeable calcium chelator EGTA inhibits cell death induced by TM-PCI.

A) HeLa cells were incubated with TMR-TAT (3 μM) in media containing calcium (1.3 mM) with or without EGTA (2 mM) for 30 min. Cells were then washed and imaged in media containing calcium (1.3 mM) to avoid calcium depletion of intracellular organelles. As EGTA is cell-impermeable, it is expected to accumulate inside endocytic organelles with TMR-TAT. Fluo-4 imaging indicates that TMR-PCI did not cause a significant increase in fluo-4 signal as compared to the control without EGTA present. B) Fluorescence imaging of cells incubated with EGTA and TMR-TAT after exposure to light for 6 sec. The intensity of the fluorescence signals of TMR-TAT inside cells indicate that incubation with EGTA does not cause a significant reduction in the amount of TMR-TAT endocytosed and released into the cytosol after light irradiation. C) Quantitative analysis of cell killing that accompanies TM-PCI in cells incubated without or with EGTA. A SYTOX® Green exclusion assay was used to distinguish live cells (stained by Hoechst, pseudo-colored purple) from dead cells (stained by SYTOX® Green, pseudo-colored green). The histogram represents the fraction of cells stained by SYTOX® Green before and after light irradiation. The average values of three experiments and the corresponding standard deviations are presented along with two representative SYTOX® Green and Hoechst overlay microscopy images.
Figure 6. TMR-TAT co-localizes with endocytic organelles that contain calcium and TM-PCI causes the release of the content of endocytic organelles.

A) Microscopy images showing that TMR-TAT (pseudo-colored red) co-localizes with intracellular organelles expressing Cerulean-LAMP1 (pseudo-colored green). Upon irradiation, the TMR-TAT signal decreases from certain organelles as indicated by the white arrows. B) TMR-TAT co-localizes with endocytic organelles that contain calcium. HeLa cells were incubated with TMR-TAT and the cell-impermeant calcium probe fura-2. Ratiometric imaging of fura-2 was performed by measuring the emission of the probe at 505 nm after excitation at 340 nm or 380 nm. C) TM-PCI causes the release of the pinocytosis marker 3 kDa Dextran Rhodamine green (Dx-RG). Representative images of HeLa cells incubated with TMR-TAT and RG-Dx before and after light irradiation at 560 nm. The overlay image of the TMR (pseudo-colored red) and RG (pseudo-colored green) shows a punctate distribution for both species prior to irradiation (a yellow color being indicative of co-localization). After irradiation, the fluorescent signals of TMR-TAT and Dx-RG are distributed throughout the cell. This does not happen if TMR-TAT is not present (not shown).
4. Discussion
PCI is an attractive approach to deliver molecules into cells. Light provides spatial and temporal control over the delivery process and endosomal lysis is efficient. It has been well appreciated, however, that this approach is potentially toxic to cells. The photosensitizers typically used for PCI are related to photodynamic therapy (PDT) agents, a treatment modality for which the goal is to actually kill cells with light [5]. As in PDT, high doses of light during PCI induce cell apoptosis and necrosis [2, 12–13]. PCI experiments are therefore typically performed at lower light doses that do not cause a significant reduction in cell viability. However, lower doses of light might lead to a reduction in the efficiency with which endosomal release is achieved. In practice, optimal conditions therefore need to be found to achieve delivery while maintaining cell viability. In our hands, however, PCI mediated by TMR-TAT could not be achieved without cell killing being observed. The cell killing is rapid with plasma membrane blebbing and loss of permeability appearing within seconds. We were therefore interested in identifying the mechanisms associated with cell death in order to potentially circumvent this problem. Our initial hypothesis was that the photolytic peptide might lyse intracellular organelles after escaping from endosomes. Light irradiation of TMR-TAT directly microinjected into the cytosol of cells however, did not reproduce the cell killing observed during PCI. TMR-S-S-TAT was also used to test this hypothesis. TMR-S-S-TAT is a compound that mimics the activity of TMR-TAT inside endosomes but that loses its photolytic activity upon reduction of the disulfide bond that links TMR to TAT. We have shown that the reduction of such compounds takes place within seconds upon entry in the cytosol of cells [18]. We therefore anticipated that, if TMR-TAT causes cell death because of a photo-induced activity in the cytosol, cell death would be abolished with TMR-S-S-TAT. However, TMR-S-S-TAT produces a similar killing as TMR-TAT. Together, these results suggest that TMR-TAT is not significantly phototoxic in the cytosol of cells and under the irradiation conditions used for PCI. Instead, the endosomal release process itself appears to be involved in the cell killing.
TMR-TAT might lyse late endosomes or lysosomes during PCI. One can therefore envision that PCI is accompanied by the release of hydrolytic enzymes from these organelles. This is a phenomenon that has been appreciated in the PDT field as being a potential causative factor for the phototoxic activity of various PDT photosensitizers [13, 32–34]. Yet, there is usually a significant delay between lysosomal lysis and observable cell death. Because the cell death observed during TM-PCI is rapid, we wondered whether cell death could be caused by toxic molecules other than hydrolytic enzymes. Based on the well-documented link between calcium and cell death, the involvement of calcium during PCI was investigated.
The concentration of calcium is typically low in the cytosol (100 nM) of cells. The flux of calcium into the cytosol and an increase in [Ca2+]cyt have been linked to apoptosis and necrosis [19]. It has also been well-established that an increase in [Ca2+]cyt can cause the accumulation of calcium in mitochondria by transport through a low affinity uniporter [25, 35]. The resulting increase of [Ca2+] in the mitochondrial matrix causes the opening of the permeability transition pore (PTP) [19, 36]. Opening of the PTP can then result in the escape of mitochondrial matrix components, including the pro-apoptotic factor cytochrome c, into the cytosol. In our assays, TM-PCI caused a large increase in the fluorescence signal of the calcium indicator fluo-4. This fluorescence increase was observed when calcium was removed from the extracellular milieu immediately prior to irradiation of the cells. When cells where incubated in a media lacking calcium for more than 20 minutes to allow the calcium depletion of intracellular stores, the increase in fluorescence of fluo-4 was abolished. Taken together, these data indicate that calcium is released from intracellular stores during PCI. In addition, ruthenium red and cyclosporin A, inhibitors of the mitochondrial calcium uniporter and of PTP opening, respectively, led to an inhibition of the cell-killing observed during PCI even though the increase in [Ca2+]cyt was still observable. These results therefore suggest that the plasma membrane blebbing and loss of permeability observed during PCI are caused by the release of calcium into the cytosol of cells followed by the transport of calcium into mitochondria and PTP opening (see Figure 7 for a proposed model).
Figure 7. Proposed model for the increase of cytosolic calcium upon TM-PCI and for the induction of cell death.
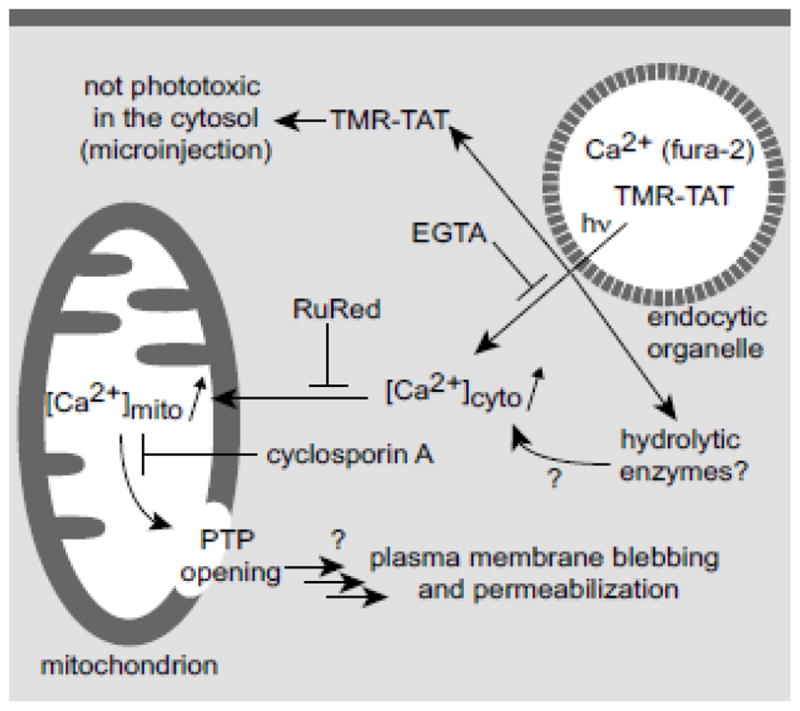
Experimental evidence for some aspects of this model is provided in parentheses and the action of various inhibitors is indicated. Areas that remain to be tested are highlighted with question marks.
Intracellular stores of calcium include the endoplasmic reticulum and mitochondria [19]. Interestingly, endocytic organelles also contain high concentrations of calcium [20–23]. Cells internalize calcium within endocytic vesicles by pinocytosis of calcium present in the extracellular milieu [22]. The concentration of calcium drops to low micromolar concentration in early endosomes but several reports suggest that it rises to higher levels in late endosomes and lysosomes [21, 23]. Christensen and coworkers have for instance measured that [Ca2+] is 600 μM in the lysosomes of macrophages [21]. To test whether calcium is released from endocytic organelles during TM-PCI, we first performed a calcium depletion experiment. The increase in fluo-4 fluorescence was abolished when calcium was chelated by EGTA in the incubation medium and in endocytic organelles. While these results are consistent with the model proposed in Figure 7, these experiments have, however, a caveat. Indeed, it is not possible to exclude the possibility that unbound EGTA might be released from endosomes during TM-PCI and that it might then chelate calcium ions released from other intracellular stores. Other experiments were therefore performed to test the hypothesis that calcium is released from endocytic organelles.
We first tested whether TMR-TAT co-localizes with endocytic organelles that contain Ca2+. Co-localization of TMR-TAT with late endosomes and lysosomes was demonstrated by expression of the fluorescent marker Cerulean-LAMP1. Irradiation of late endosomes or lysosomes labeled with both LAMP1 and TMR-TAT led to the local dissipation of the TMR-TAT signal, indicating that the peptide escapes from these organelles upon PCI. In addition, co-localization experiments between TMR-TAT and the endocytosed calcium indicator fura-2 were also performed. In principle, ratiometric imaging of fura-2 can be used to determine the concentration of calcium in a particular milieu. However, a complication arises when investigating calcium present in the endocytic pathway. This is because the pH of endocytic organelles is acidic and the Kd of fura-2 increases at acidic pH [17]. In addition, the calcium-free form of fura-2 can potentially interact with proteins more preferentially at acidic pH [37]. Together, these issues make the precise determination of calcium concentration within endosomes a technical challenge. However, we reasoned that, since both caveats lead to an underestimation of the actual concentration of calcium in endosomes (both problems diminish the ability of fura-2 to bind to calcium ions), a lower limit for [Ca2+]endo can nonetheless be determined. This lower limit was determined to be 470 nM, the concentration of calcium that would be present given a pH of 7.2. Considering that the pH of the endocytic organelles observed are likely to be acidic, the calcium concentration should be significantly higher (for instance, 3.9 μM at pH 5.5, a pH expected inside late endosomes). Overall, our results indicated that calcium is present in the lumen of endocytic organelles also containing TMR-TAT (Figure 6). Finally, TM-PCI causes the release of the content of endocytic organelles, as demonstrated by the release of Dx-RG. Taken together, these results suggest that TM-PCI causes the release of Ca2+ from endocytic organelles. As indicated in Figure 7, it is however interesting to note that our data do not reveal whether the increase in [Ca2+]cyto detected originates solely from Ca2+ escaping from the endocytic pathway. As a matter of fact, it is possible that the release of endocytic calcium might trigger the release of more calcium from other organelles such as the endoplasmic reticulum [19, 38]. Alternatively, other released molecules such as hydrolytic enzymes (but not TMR-TAT itself) might cause an increase in cytosolic calcium indirectly [19]. Whether this is the case will be the object of future investigations.
Overall, our data support a model in which the flux of calcium in the cytosol during TM-PCI is a cause of cell photo-killing. Our results also highlight how efficient and rapid disruption of the endocytic pathway might be problematic not only during PCI but also for other delivery methods relying on endosomal escape. Interestingly, crotamine, a cell-penetrating peptide derived from the venom of the rattlesnake Crotalus durissus terrificus, causes cell death in a manner dependent on the disruption of lysosomes and on rapid intracellular calcium release [39]. While other endosomolytic agents have been used to deliver macromolecules into cells without affecting cell viability, the release of calcium from endosomal and lysosomal organelles could be a factor contributing to the toxicity associated with certain delivery procedures. Paradoxically, a goal in cellular delivery is often to make endosomal escape as efficient as possible [1]. This is based on the principle that a greater biological effect can be achieved if more of a delivered macromolecule can escape from endosomes. Yet, our results suggest that the efficient lysis of endocytic organelles leads to calcium-induced cell death. This therefore highlights that a cell might remain viable during delivery only if endosomal lysis is relatively inefficient. Interestingly, it is also possible that TM-PCI causes cell death not because of the efficiency of endosomal escape per se, but because of the rapidity of this process. In particular, a small increase in [Ca2+]cyt can be tolerated by a cell [19]. Calcium homeostasis can also be quickly reestablished by the various calcium pumps and channels present in the cell [19]. It is therefore possible that slowing down the rate of endosomal release might allow the cell to cope with the calcium entering the cytosol. In the context of PCI, this might mean that low doses of light might be preferable to intense bursts of irradiation. This could for instance explain why extensive cell killing is not observed in reports of TM-PCI performed on laser-scanning confocal microscope (e.g. an intense laser is used but a particular endosome is irradiated only briefly with long time intervals between scans) [6–7]. Overall, it is clear that understanding the interplay between endosomal lysis and cell viability in greater details is required to permit the development of delivery procedures that have a minimum impact on cellular physiology.
Supplementary Material
HIGHLIGHTS.
TMR-TAT mediated PCI is a promising delivery method but accompanies cell death
TMR-TAT co-localizes with calcium containing endocytic vesicles in cells
Endosomal escape of TMR-TAT with light increases cytosolic calcium concentration
Calcium flux into the cytosol is followed by import of calcium into mitochondria
Release of calcium from endocytic vesicles is a causative factor of the cell death
Acknowledgments
We thank Professor Donald Pettigrew for his help and valuable discussions. Ratiometric imaging was done at the Image Analysis Laboratory of the College of Veterinary medicine and Biomedical Sciences at Texas A&M University. This work was supported by Award Number R01GM087227 and R01GM087981 from the National Institute of General Medical Sciences, the Norman Ackerman Advanced Research Program, and the Robert A. Welch foundation (Grant A-1769).
Abbreviations
- TMR
carboxytetramethylrhodamine
- CPP
cell-penetrating peptide
- TAT
trans-activator of transcription
- TM-PCI
TMR-TAT mediated photochemical internalization
- RBC
red blood cell
- PDT
photodynamic therapy
- DMF
dimethylformamide
- DCM
dichloromethane
- TEA
triethylamine
- PDA
2-(2-pyridyldithio)ethylamine
- HOBt
N-hydroxybenzotriazole
- PBS
phosphate-buffered saline
- Dx-RG
dextran-rhodamine green
- RuRed
ruthenium red
- EGTA
ethylene glycol tetraacetic acid
- PTP
permeability transition pore
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Varkouhi AK, Scholte M, Storm G, Haisma HJ. Endosomal escape pathways for delivery of biologicals. J Control Release. 2011;151:220–228. doi: 10.1016/j.jconrel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Berg K, Selbo PK, Prasmickaite L, Tjelle TE, Sandvig K, Moan J, Gaudernack G, Fodstad O, Kjolsrud S, Anholt H, Rodal GH, Rodal SK, Hogset A. Photochemical internalization: a novel technology for delivery of macromolecules into cytosol. Cancer Res. 1999;59:1180–1183. [PubMed] [Google Scholar]
- 3.Berg K, Weyergang A, Prasmickaite L, Bonsted A, Hogset A, Strand MT, Wagner E, Selbo PK. Photochemical internalization (PCI): a technology for drug delivery. Methods Mol Biol. 2010;635:133–145. doi: 10.1007/978-1-60761-697-9_10. [DOI] [PubMed] [Google Scholar]
- 4.Berg K, Berstad M, Prasmickaite L, Weyergang A, Selbo PK, Hedfors I, Hogset A. Photochemical internalization: a new tool for gene and oligonucleotide delivery. Top Curr Chem. 2010;296:251–281. doi: 10.1007/128_2010_63. [DOI] [PubMed] [Google Scholar]
- 5.Berg K, Selbo PK, Weyergang A, Dietze A, Prasmickaite L, Bonsted A, Engesaeter BO, Angell-Petersen E, Warloe T, Frandsen N, Hogset A. Porphyrin-related photosensitizers for cancer imaging and therapeutic applications. J Microsc. 2005;218:133–147. doi: 10.1111/j.1365-2818.2005.01471.x. [DOI] [PubMed] [Google Scholar]
- 6.Matsushita M, Noguchi H, Lu YF, Tomizawa K, Michiue H, Li ST, Hirose K, Bonner-Weir S, Matsui H. Photo-acceleration of protein release from endosome in the protein transduction system. FEBS Lett. 2004;572:221–226. doi: 10.1016/j.febslet.2004.07.033. [DOI] [PubMed] [Google Scholar]
- 7.Maiolo JR, 3rd, Ottinger EA, Ferrer M. Specific redistribution of cell-penetrating peptides from endosomes to the cytoplasm and nucleus upon laser illumination. J Am Chem Soc. 2004;126:15376–15377. doi: 10.1021/ja044867z. [DOI] [PubMed] [Google Scholar]
- 8.Endoh T, Sisido M, Ohtsuki T. Photo inducible RNA interference using cell permeable protein carrier. Nucleic Acids Symp Ser (Oxf) 2007:127–128. doi: 10.1093/nass/nrm064. [DOI] [PubMed] [Google Scholar]
- 9.Matsushita-Ishiodori Y, Kuwabara R, Sakakoshi H, Endoh T, Ohtsuki T. Photosensitizing carrier proteins for photoinducible RNA interference. Bioconjug Chem. 2011;22:2222–2226. doi: 10.1021/bc200095a. [DOI] [PubMed] [Google Scholar]
- 10.Endoh T, Ohtsuki T. Cellular siRNA delivery using TatU1A and photo-induced RNA interference. Methods Mol Biol. 2010;623:271–281. doi: 10.1007/978-1-60761-588-0_17. [DOI] [PubMed] [Google Scholar]
- 11.Endoh T, Sisido M, Ohtsuki T. Spatial regulation of specific gene expression through photoactivation of RNAi. J Control Release. 2009;137:241–245. doi: 10.1016/j.jconrel.2009.04.015. [DOI] [PubMed] [Google Scholar]
- 12.Oliveira S, Fretz MM, Hogset A, Storm G, Schiffelers RM. Photochemical internalization enhances silencing of epidermal growth factor receptor through improved endosomal escape of siRNA. Biochim Biophys Acta. 2007;1768:1211–1217. doi: 10.1016/j.bbamem.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 13.Berg K, Moan J. Lysosomes as photochemical targets. Int J Cancer. 1994;59:814–822. doi: 10.1002/ijc.2910590618. [DOI] [PubMed] [Google Scholar]
- 14.Srinivasan D, Muthukrishnan N, Johnson GA, Erazo-Oliveras A, Lim J, Simanek EE, Pellois JP. Conjugation to the cell-penetrating peptide TAT potentiates the photodynamic effect of carboxytetramethylrhodamine. PLoS ONE. 2011;6:e17732. doi: 10.1371/journal.pone.0017732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ebright YW, Chen Y, Pendergrast PS, Ebright RH. Incorporation of an EDTA-metal complex at a rationally selected site within a protein: application to EDTA-iron DNA affinity cleaving with catabolite gene activator protein (CAP) and Cro. Biochemistry. 1992;31:10664–10670. doi: 10.1021/bi00159a004. [DOI] [PubMed] [Google Scholar]
- 16.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 17.Lattanzio FA., Jr The effects of pH and temperature on fluorescent calcium indicators as determined with Chelex-100 and EDTA buffer systems. Biochem Biophys Res Commun. 1990;171:102–108. doi: 10.1016/0006-291x(90)91362-v. [DOI] [PubMed] [Google Scholar]
- 18.Lee YJ, Datta S, Pellois JP. Real-time fluorescence detection of protein transduction into live cells. J Am Chem Soc. 2008;130:2398–2399. doi: 10.1021/ja7102026. [DOI] [PubMed] [Google Scholar]
- 19.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 20.Scott CC, Gruenberg J. Ion flux and the function of endosomes and lysosomes: pH is just the start: the flux of ions across endosomal membranes influences endosome function not only through regulation of the luminal pH. Bioessays. 2011;33:103–110. doi: 10.1002/bies.201000108. [DOI] [PubMed] [Google Scholar]
- 21.Christensen KA, Myers JT, Swanson JA. pH-dependent regulation of lysosomal calcium in macrophages. J Cell Sci. 2002;115:599–607. doi: 10.1242/jcs.115.3.599. [DOI] [PubMed] [Google Scholar]
- 22.Gerasimenko JV, Tepikin AV, Petersen OH, Gerasimenko OV. Calcium uptake via endocytosis with rapid release from acidifying endosomes. Curr Biol. 1998;8:1335–1338. doi: 10.1016/s0960-9822(07)00565-9. [DOI] [PubMed] [Google Scholar]
- 23.Haller T, Dietl P, Deetjen P, Volkl H. The lysosomal compartment as intracellular calcium store in MDCK cells: a possible involvement in InsP3-mediated Ca2+ release. Cell Calcium. 1996;19:157–165. doi: 10.1016/s0143-4160(96)90084-6. [DOI] [PubMed] [Google Scholar]
- 24.Gee KR, Brown KA, Chen WN, Bishop-Stewart J, Gray D, Johnson I. Chemical and physiological characterization of fluo-4 Ca(2+)-indicator dyes. Cell Calcium. 2000;27:97–106. doi: 10.1054/ceca.1999.0095. [DOI] [PubMed] [Google Scholar]
- 25.Gunter TE, Buntinas L, Sparagna G, Eliseev R, Gunter K. Mitochondrial calcium transport: mechanisms and functions. Cell Calcium. 2000;28:285–296. doi: 10.1054/ceca.2000.0168. [DOI] [PubMed] [Google Scholar]
- 26.Rossi CS, Vasington FD, Carafoli E. The effect of ruthenium red on the uptake and release of Ca 2+ by mitochondria. Biochem Biophys Res Commun. 1973;50:846–852. doi: 10.1016/0006-291x(73)91322-3. [DOI] [PubMed] [Google Scholar]
- 27.Reed KC, Bygrave FL. The inhibition of mitochondrial calcium transport by lanthanides and ruthenium red. Biochem J. 1974;140:143–155. doi: 10.1042/bj1400143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fournier N, Ducet G, Crevat A. Action of cyclosporine on mitochondrial calcium fluxes. J Bioenerg Biomembr. 1987;19:297–303. doi: 10.1007/BF00762419. [DOI] [PubMed] [Google Scholar]
- 29.Broekemeier KM, Dempsey ME, Pfeiffer DR. Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J Biol Chem. 1989;264:7826–7830. [PubMed] [Google Scholar]
- 30.Szabo I, Zoratti M. The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J Biol Chem. 1991;266:3376–3379. [PubMed] [Google Scholar]
- 31.van der Goot FG, Gruenberg J. Intra-endosomal membrane traffic. Trends Cell Biol. 2006;16:514–521. doi: 10.1016/j.tcb.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 32.Allison AC, Magnus IA, Young MR. Role of lysosomes and of cell membranes in photosensitization. Nature. 1966;209:874–878. doi: 10.1038/209874a0. [DOI] [PubMed] [Google Scholar]
- 33.Woodburn KW, Fan Q, Miles DR, Kessel D, Luo Y, Young SW. Localization and efficacy analysis of the phototherapeutic lutetium texaphyrin (PCI-0123) in the murine EMT6 sarcoma model. Photochem Photobiol. 1997;65:410–415. doi: 10.1111/j.1751-1097.1997.tb08579.x. [DOI] [PubMed] [Google Scholar]
- 34.O’Connor AE, Gallagher WM, Byrne AT. Porphyrin and nonporphyrin photosensitizers in oncology: preclinical and clinical advances in photodynamic therapy. Photochem Photobiol. 2009;85:1053–1074. doi: 10.1111/j.1751-1097.2009.00585.x. [DOI] [PubMed] [Google Scholar]
- 35.Kruman, Mattson MP. Pivotal role of mitochondrial calcium uptake in neural cell apoptosis and necrosis. J Neurochem. 1999;72:529–540. doi: 10.1046/j.1471-4159.1999.0720529.x. [DOI] [PubMed] [Google Scholar]
- 36.Baumgartner HK, Gerasimenko JV, Thorne C, Ferdek P, Pozzan T, Tepikin AV, Petersen OH, Sutton R, Watson AJ, Gerasimenko OV. Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J Biol Chem. 2009;284:20796–20803. doi: 10.1074/jbc.M109.025353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bancel F, Salmon JM, Vigo J, Vo-Dinh T, Viallet P. Investigation of noncalcium interactions of fura-2 by classical and synchronous fluorescence spectroscopy. Anal Biochem. 1992;204:231–238. doi: 10.1016/0003-2697(92)90232-v. [DOI] [PubMed] [Google Scholar]
- 38.Alonso MT, Chamero P, Villalobos C, Garcia-Sancho J. Fura-2 antagonises calcium-induced calcium release. Cell Calcium. 2003;33:27–35. doi: 10.1016/s0143-4160(02)00179-3. [DOI] [PubMed] [Google Scholar]
- 39.Nascimento FD, Sancey L, Pereira A, Rome C, Oliveira V, Oliveira EB, Nader HB, Yamane T, Kerkis I, Tersariol IL, Coll JL, Hayashi MA. The Natural Cell-Penetrating Peptide Crotamine Targets Tumor Tissue in Vivo and Triggers a Lethal Calcium-Dependent Pathway in Cultured Cells. Mol Pharm. 2011 doi: 10.1021/mp2000605. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.