

Abstract
Purpose
We tested the combination of a tumor lysate vaccine with a panel of costimulatory molecules to identify an immunotherapeutic approach capable of curing established murine gliomas.
Experimental Design
Glioma-bearing mice were primed with a tumor lysate vaccine, followed by systemic administration of the following costimulatory ligands: OX40L, CD80, 4-1BBL, and GITRL which were fused to the Fc portion of human immunoglobulin. Lymphocytes and mRNA were purified from the brain tumor site for immune monitoring studies. Numerous variations of the vaccine and Fc-OX40L regimen were tested alone or in combination with temozolomide.
Results
Lysate vaccinations combined with Fc-OX40L led to the best overall survival, yielding cure rates of 50–100% depending on the timing, regimen, and combination with temozolomide. Cured mice that were rechallenged with glioma cells rejected the challenge, demonstrating immunological memory. Lymphocytes isolated from the draining lymph nodes of vaccine/Fc-OX40L-treated mice had superior tumoricidal function relative to all other groups. Vaccine/Fc-OX40L-treated mice exhibited a significant increase in proliferation of brain infiltrating CD4 and CD8 T cells, as indicated by Ki67 staining. Fc-OX40L had single agent activity in transplanted and spontaneous glioma models and the pattern of inflammatory gene expression in the tumor predicted the degree of therapeutic response.
Conclusions
These data demonstrate that Fc-OX40L has unique and potent activity against experimental gliomas and warrants further testing.
Keywords: Glioblastoma, immunotherapy, OX40L, costimulatory molecules, temozolomide
Introduction
Glioblastoma multiforme (GBM) is a lethal brain tumor with a median survival of 15–19 months (1). There is an urgent need for more targeted and effective therapies. Recent studies have shown vaccines to be a promising adjuvant therapy for GBM (reviewed in (2)). Major challenges in the field include the immunologically specialized nature of the brain and the immune suppression documented in GBM patients (3–5). Vaccines (peptide, dendritic cell (DC), and lysate-based) have been used to overcome inadequate natural antigen presentation by increasing the frequency of tumor-reactive T cells. When tumor antigen is presented to T cells in the absence of proper levels of costimulation, functionally anergic cells can be primed that have poor tumoricidal function. T cell costimulation occurs secondary to T cell receptor (TCR) recognition of antigen by MHC presentation. One of the first costimulatory signals occurs through the CD28 receptor, which is constitutively expressed on naïve T cells, recognizing its ligands, CD80 or CD86. Members of the tumor necrosis factor receptor superfamily are upregulated on T cells following this initial activation. These receptors include glucocorticoid-induced TNFR-related protein (GITR), 4-1BB, and OX40 (6). Engagement of these receptors by their corresponding ligands leads to enhanced T cell survival and differentiation into effector and memory cells.
Prior studies have shown that it is possible to break anergy by delivering agonist antibodies or recombinant ligands that activate costimulatory receptors present on T cells (reviewed in (7)). Blockade of inhibitory checkpoints such as Cytotoxic T-Lymphocyte Antigen 4 (CLTA4) on T cells is another strategy that has gained attention. The recent FDA approval of ipilimumab (a CTLA4 antagonist) for the treatment of metastatic melanoma is a first in class success that has invigorated the field of immunotherapy (8). However, ipilimumab is not specific to tumor-reactive T cells, resulting in severe and long-lasting adverse events in an appreciable fraction of patients (9); this has led some to challenge the rationale for its use (10). Indeed, many questions remain unresolved about the optimal use of these novel immunomodulatory drugs. In the case of drugs that activate T cell costimulatory receptors, what are the relative potencies of agonist antibodies versus recombinant activating ligands? How should such drugs be combined with the standard of care chemotherapy or experimental vaccines? The details of combination therapy, such as dose, timing, and frequency of administration are important considerations that can be preliminarily addressed in preclinical models. Laying this basic foundation of knowledge will enable rational clinical trial design.
We previously characterized immunomodulatory proteins consisting of the C-terminus of the Fc portion of immunoglobulin fused to costimulatory ligands: CD80, GITRL, OX40L, or 4-1BBL (11–14). Comparative studies of an anti-OX40 agonist antibody with Fc-OX40L demonstrated Fc-OX40L was far more potent on a per molecule basis (13), establishing the rationale to develop this class of Fc-ligand fusion proteins. In this prior work, varying the dose and frequency of each drug led to the development of optimized single-agent treatment regimens. These regimens were capable of triggering tumor-reactive T cells and tumor regression in murine models of colon and renal cell carcinoma (11–14). Nonetheless, such single agent approaches rely on endogenous tumor-reactive T cells as targets, which may be too limiting, justifying the investigation of vaccines that increase the frequency of responder T cells. We previously demonstrated the efficacy of a tumor cell lysate /toll-like receptor (TLR) agonist vaccine to treat mice bearing intracranial gliomas. The vaccine consisting of tumor cell lysate and CpG, a TLR 9 agonist, cured 50% of glioma-bearing mice through a mechanism requiring CD4 and CD8 T cells (15). However, when this tumor cell vaccine was prepared in standard tissue culture conditions (e.g., atmospheric 21% oxygen) and modeled at clinically scalable antigen doses, the efficacy of vaccination was abolished despite evidence of immune priming (16). One goal of this study was to determine if additional costimulation was necessary to achieve efficacy following vaccination with lysate/CpG. To address this we screened a panel of Fc-ligand fusion proteins in combination with the tumor lysate/CpG vaccine and identified Fc-OX40L as the most potent, demonstrating both single agent activity and synergy with the CpG/lysate vaccine. The optimal frequency, timing, and combination of Fc-OX40L with vaccination were established in mice bearing implanted GL261 gliomas. The efficacy of the optimized regimen was generalizable to other vaccine adjuvants (PolyICLC, a TLR3 agonist), other tumor models (breast carcinoma), and effective in combination with the standard of care chemotherapy for GBM, temozolomide. Comparative studies in two glioma models revealed inflammatory gene expression signatures that were associated with response rates to Fc-OX40L as a single agent. Collectively, these studies provide a wealth of information that can be exploited when considering the design of clinical trials.
Materials and Methods
Animal Models and Cell Lines
The culture conditions for GL261-Luc and EMT6 have been described elsewhere (15, 17). All animals were maintained in a specific pathogen free facility in accordance with the University of Minnesota Institutional Animal Care and Use Committee (IACUC) guidelines. For the implanted glioma model, seven-week-old C57BL/6 (B6) mice were purchased from Jackson Laboratory. Gliomas were established by intracranial inoculation of 15,000 GL261-Luc cells in 1 μL of Hank’s balanced salt solution (HBSS) (Gibco) to syngeneic B6 mice. Animals were anesthetized with a ketamine/xylazine cocktail (54.7 mg/mL ketamine and 9.26 mg/mL xylazine) prior to surgery. Cells were implanted into the right striatum at coordinates 2.5 mm lateral, 0.5 mm anterior from the bregma, and 3 mm ventral from the surface of the brain (15). Cells were delivered over 5 minutes at a rate of 0.2 μL/min. Tumor implantation was confirmed by bioluminescence imaging 3 days following inoculation; animals received 100 μL Luciferin (Gold Biotechnology) by intraperitoneal (IP) injection and were imaged with an IVIS50 system (Caliper Life Sciences). The de novo glioma model was induced by transposon-mediated gene transfer, as we have previously described (18). The following plasmids were injected into the brains of B6 mice at 1–2 days following birth, pT2/C-Luc//PGK-SB13 (0.07μg), pT/CAGGS-NRASV12 (0.14 μg), and pT2/shP53/GFP4/mPDGF (0.14 μg). Mice that became moribund where humanely euthanized.
The breast carcinoma model was induced in BALB/c mice that were purchased from Taconic. Breast carcinoma was induced by injecting 4 ×106 EMT6 cells in 100 μL of HBSS cells into the upper right mammary fat pad of syngeneic BALB/c mice while under sedation from isoflurane gas (17). Tumor burden was determined with calipers by measuring the greatest longitudinal diameter (length) and greatest transverse diameter (width). When a measurement in either direction reached 20 mm the animal was humanely euthanized.
Activity Assay
Splenocytes from naïve C57BL/6 mice were isolated and enriched for mononuclear cells by density gradient centrifugation. Cells were cultured at 3×106 cells/mL in anti-mouse CD3-coated [clone 145-2C11, 3mg/mL (eBioscience)] 96-well tissue culture plates in complete medium in the indicated concentrations of murine costimulatory ligand fusion proteins, vehicle control, or CD3/CD28 beads (positive control, Invitrogen). Cytokines were measured in 3-day culture supernatants using BD CBA Flex sets for mouse IL-2 and IFNγ, per the manufacturer’s instructions. Samples were run on a BD LSRII flow cytometer using FACSDiva software and analyzed using FCAP ARRAY software.
Quantitative RT-PCR for immune-related gene expression
Total RNA was isolated from tumor-bearing brain or control tissue (normal brain) using Qiagen’s RNeasy mini kit (Valencia, CA) and DNase treated using Turbo DNase (Applied Biosystems, Foster City, CA) per manufacturer’s instructions. For quantitative RT-PCR, 100 ng RNA was amplified with gene specific primers using one-step Power SYBR green RNA-to-Ct kit (Applied Biosystems) and run in an MX3000P Stratagene thermocycler. Data were acquired and analyzed using MxPro software (Stratagene). Samples were run in triplicate and gene expression was normalized to the housekeeping gene GAPDH. Primer sequences were obtained from the NIH qRT-PCR database [http://primerdepot.nci.nih.gov] and synthesized by the USC Microchemical Core Facility.
Vaccine Production and Delivery
Cells were collected and washed 3 times with phosphate buffered saline (PBS), resuspended in PBS and flash frozen with liquid nitrogen. Cell lysis was induced by 5 cycles of freezing in liquid nitrogen and thawing in a 37°C water bath, vortexing after each round. Cell death was verified by trypan blue dye exclusion. Lysates were stored at −80°C until use. Protein concentration was determined using Pierce BCA Assay kit (Thermo Scientific). Purified CpG 1826, an unmethylated oligodeoxynucleotide (ODN) sequence (5′-tccatgacgttcctgacgtt-3′) with a full phosphorothioate backbone was obtained from Integrated DNA Technologies (IDT, Coralville, IA), and verified to be endotoxin free. CpG 1826 was resuspended in 1× TE at a concentration of 10 mg/mL and stored at −80°C until use. PolyICLC was provided by Oncovir Inc. The final vaccine was generated by combining 65 μg of cell lysate with 50 μg CpG 1826 or 10 μg PolyICLC in a 100μL final volume (adjusted with saline). Animals received 100 μL of vaccine or saline by intradermal (i.d.) injection above the shoulders.
Costimulatory Fusion Proteins Production and Delivery
Costimulatory fusion proteins were genetically engineered, biochemically characterized, and functionally validated as described previously (11–14). Previously published optimal doses for each costimulatory fusion protein were used: Fc-4-1BBL (150 μg/dose), CD80-Fc (40μg/dose), Fc-GITRL (25 μg/dose), and Fc-OX40L (50 μg/dose). Each costimulatory fusion protein was brought to a final volume of 100 μL per dose with PBS and delivered by IP injection.
CTL Assay
GL261 cells were labeled with 10μM/mL CFSE (Invitrogen) for 15 minutes then thoroughly washed with PBS. Cervical lymph nodes were harvested, dissociated by passing through a 70 μm filter, and incubated with CFSE labeled GL261 cells for 6 hours. Following incubation, cells were stained with 7-AAD (ImmunoChemistry Technologies) and analyzed by flow cytometry. The percentage of CFSE labeled target cells that incorporated 7-AAD was plotted as the percent lysis.
Flow Cytometry
Cervical lymph nodes were passed through a 70μm filter to generate a single cell suspension. To harvest brain-infiltrating lymphocytes (BIL), mice were first perfused with PBS to flush the capillaries. The brains were removed and minced into very fine (< 1mm) pieces with a razor, passed through a 70μm filter, and subjected to a two layer Percoll gradient (70% and 30%) and centrifuged at 800×g for 30 minutes. The lymphocyte interface was harvested by aspiration. Cells were counted using trypan blue dye exclusion and a hemocytometer. For these studies antibodies against CD3 (clone 17A2), CD4 (clone GK1.5), CD8 (clone 53–67) (ebioscience), and Ki67 (clone B56) (BD Pharminogen) were used for phenotypic analysis. Intracellular Ki67 staining was achieved by using the ebioscience Foxp3/transcription factor intracellular staining kit according to the manufacturer’s instructions. A BD Bioscience FACSCanto was used for analysis of cells and data were analyzed using Flowjo software (Tree Star). The total number of stained cells was determined by multiplying the percentage of stained cells by the total number of viable cells determined previously by trypan blue dye exclusion.
Temozolomide Administration
Temozolomide (TMZ) was purchased from Toronto Research Chemicals as powder and resuspended in PBS at a final concentration of 10 mg/mL. A dose of 50 mg/kg was administered by oral gavage for 5 days starting on day 7 following inoculation.
Histopathology and Immunohistochemistry
Tissues were fixed in 10% neutral buffered formalin, routinely processed into paraffin using standard histology techniques, sectioned at a thickness of 4 μm, stained with hematoxylin and eosin, and evaluated by light microscopy. For immunohistochemistry (IHC) preparations, 4 μm formalin-fixed, paraffin-embedded sections of tissue were deparaffinized and rehydrated, followed by antigen retrieval (using 10 mM Citrate buffer pH 6.0 or Tris EDTA pH 9.0) in a steamer prior to IHC procedures performed on a Dako Autostainer. IHC for was performed for CD3 (Clone CD3–12, Serotech), B220 (Clone RA3-6B2, BD Pharmingen) and Pax5 (Santa Cruz Biotechnology, catalog number SC-1974). Detection of bound primary antibody was achieved using the appropriate Rat-HRP or Goat-HRP Polymer Detection systems (Biocare Medical), with DAB as the chromogen. All slides were evaluated by a board certified veterinary pathologist (MGO). Confocal microscopy was performed at the University of Minnesota Imaging Center, St. Paul Campus.
Statistical Analyses
Statistical comparisons for lymphocyte counts were made by a Mann-Whitney (non-parametric) test. Expression of immune-related genes was compared using a student’s t test. Differences in animal survival were evaluated by log-rank test. Differences in mean cytokine secretion among groups was evaluated for significance by ANOVA followed by Dunnett’s test for pairwise comparisons. All tests were performed with Prism 4 software (Graph Pad Software, Inc). P values <0.05 were considered statistically significant.
Results
Comparative Efficacy of Combination Vaccine with Costimulatory Ligands
The efficacy of vaccination with lysate/CpG was tested alone and in combination with Fc-ligand fusion proteins. Mice bearing GL261 gliomas were primed intradermally by three vaccinations (days 3, 7, and 10), followed by daily IP injection of Fc-ligand on days 17–21 following tumor challenge. The rationale for this timing was to first prime tumor-reactive T cells, then provide a sustained (five consecutive days) co-stimulatory signal. Vaccine alone or the combination of vaccine with CD80-Fc or Fc-4-1BBL did not enhance survival compared to saline controls (Fig. 1A–C). The vaccine/Fc-GITRL combination resulted in a statistically significant, albeit incremental, increase in survival without long-term survivors (Fig. 1D). The vaccine/Fc-OX40L combination resulted in a significant increase in survival and 50% long-term survivors (Fig. 1E). In long-term survivors, the kinetics of complete tumor regression varied from 24–45 days after inoculation as determined by bioluminescence imaging (BLI) (Supplementary Fig. S1A). In order to assay for immunological memory, long-term survivors and naïve controls were intracerebrally challenged with GL261 in the contralateral hemisphere relative to initial challenge. No additional treatment was administered. All cured mice rejected rechallenge within 8 days (Supplementary Fig. S1B) and survived long-term, whereas all naïve animals succumbed to tumor burden (Fig. 1F).
Figure 1. Comparative efficacy of combination vaccine with costimulatory ligand fusion proteins.
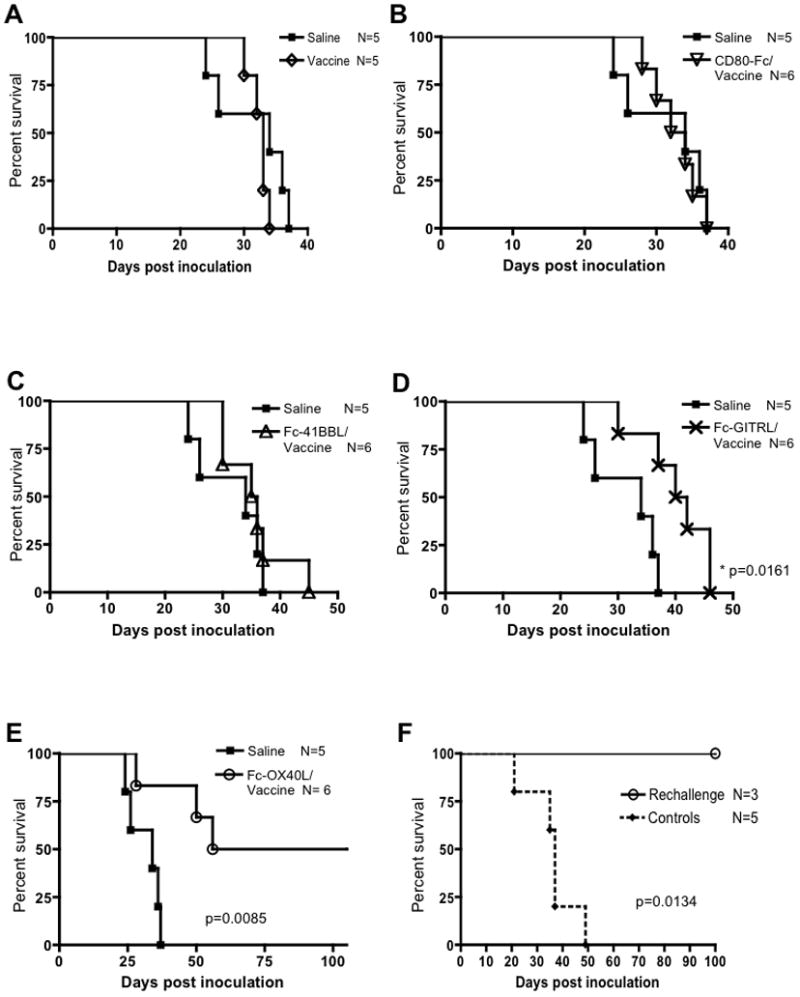
Animals were inoculated with 15,000 GL261-Fluc cells on day 0. Vaccine, lysate/CpG, was administered i.d. on days 3, 7, and 10. On day 17 animals received one of four costimulatory molecules (CD80, 4-1BBL, GITRL, or OX40L) as Fc fusion proteins or PBS control for five consecutive days. Overall survival compared to saline controls was documented for vaccine alone (A), CD80-Fc/vaccine (B), Fc-4-1BBL/vaccine (C), Fc-GITRL/vaccine (D), and Fc-OX40L/vaccine (E). This experiment was repeated twice, once at optimized doses (shown) and once at equivalent doses (data not shown) with similar results. Fc-OX40L/vaccine survival has been repeated at least 2 additional times with similar results. (F) 100 days post inoculation the vaccine/Fc-OX40L-treated survivors were rechalleged with GL261cells in the contralateral hemisphere. In addition, naïve control animals were also inoculated in the same manner. This experiment was performed once.
This initial screen was carried out using previously determined optimal doses of each Fc-ligand fusion protein (range 25–150 μg/dose). For a more direct comparison, the identical screen was repeated by administering the Fc-fusion proteins or an agonist OX40 antibody (OX86) at the same dose of 50 μg. Using this more equivalent dosing scheme, only mice treated with vaccine/Fc-OX40L survived long-term (data not shown), confirming our initial experiments and the superior potency of Fc-OX40L to OX86 that was reported previously in other tumor models (13). The difference in therapeutic potency of these molecules was not due to inactivate preparations because they all had an equivalent ability to enhance IL-2 elaboration in primary splenocytes cultures stimulated with a CD3 agonist antibody (Supplementary Fig. S2A). Furthermore, IFNγ production was enhanced upon stimulation with each of the reagents, except CD80-Fc (Supplementary Fig. S2B). The unique ability of the vaccine/Fc-OX40L combination therapy to cause tumor regression and establish immunological memory warranted further mechanistic studies.
T cell response correlates with survival outcome
In order to understand how the various Fc-ligands altered immune responses, animals were treated as described in Figure 1 and euthanized on day 25. This time point was four days following the final Fc-ligand injection and was chosen because it was when initial tumor regression was noted by BLI (Supplementary Fig. S1). Lymphocytes from the brain and brain draining lymph node (DLN) were quantified and assayed for tumoricidal function. There was a two-to-three fold increase in the total number of cells in the DLN of vaccine/Fc-OX40L-treated animals compared to all other groups except Fc-GIRTL/vaccine (Fig. 2A). The difference between the Fc-GIRTL/vaccine and vaccine/Fc-OX40L groups was not statistically significant (p = 0.15). However, only cells isolated from the DLN of vaccine/Fc-OX40L-treated animals had appreciable tumoricidal function in ex vivo CTL assays (Fig. 2B). These experiments revealed quantitative and functional differences in lymphocytes in the DLN that correlated with outcome; Fc-GIRTL/vaccine followed by vaccine/Fc-OX40L had increasingly superior survival as shown in Fig. 1, whereas tumoricidal function was unique to vaccine/Fc-OX40L.
Figure 2. Superior priming in the draining lymph nodes of vaccine/Fc-OX40L-treated mice.
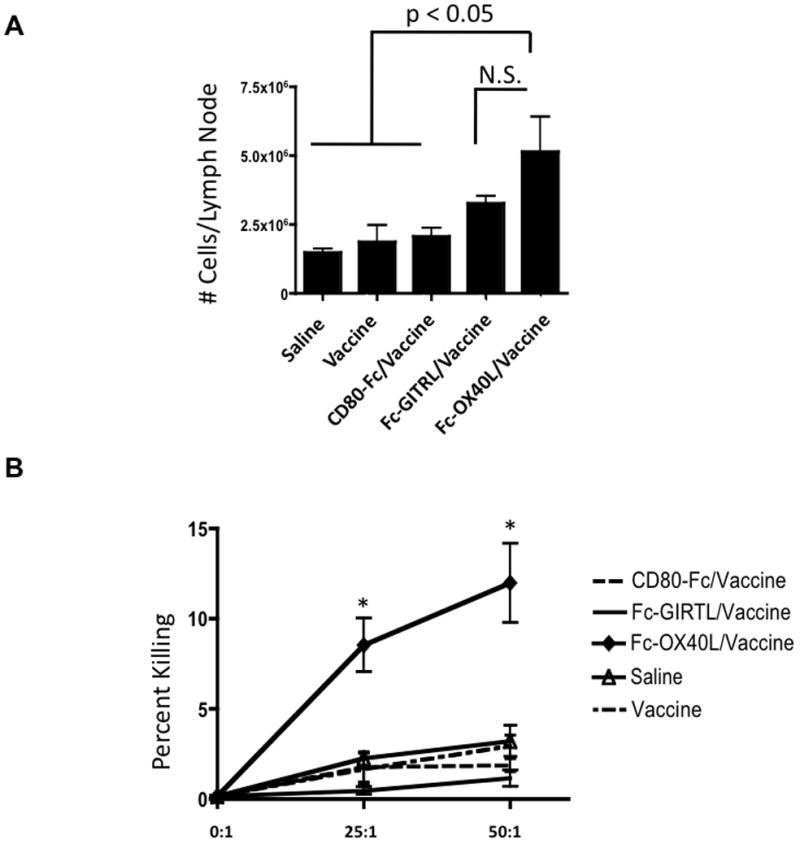
In order to understand the effect that the costimulatory molecules have on the immune system, animals were treated with the reagents as described in Figure 1 and euthanized on day 25. Lymphocytes from the cervical lymph nodes (draining LN) were collected, counted, and assayed for tumoricidal function in CTL assays. (A) Aggregate data showing cell counts in each treatment group. (B) Cell killing plotted as percent lysis. This experiment was performed once. Statistical significance is indicated as p < 0.05 (*), or not significant (N.S.). Mean values are shown −/+ SEM; n=4–5/group.
Flow cytometry analyses demonstrated that animals in the vaccine/Fc-OX40L group had an increase in the percentage of CD4 and CD8 T cells at the tumor site (Fig. 3A). However, when absolute numbers were analyzed only 3/5 mice treated with vaccine/Fc-OX40L had increased numbers of CD4 and CD8 T cells at the tumor site (Fig. 3A). Due to the high animal-to-animal variability, this difference was statistically insignificant, with the exception of brain infiltrating CD4 T cells in two cases. Notably, the percentage of vaccine/Fc-OX40L-treated mice with increased tumor infiltrating T cells (~60%) correlated with the long-term term survival rate of 50%. A more robust assay was Ki67 staining, a marker of proliferation. There was a significant increase in the frequency of CD4 and CD8 brain infiltrating T cells that were mitotically active in vaccine/Fc-OX40L-treated mice (Fig. 3B). The significance of this finding is unclear, but it may represent an intrinsic difference in the proliferative competence of T cells, consistent with previous studies that demonstrated OX40 agonists can break T cell anergy in tumor models (19). All together, these experiments established a link between complete response rate, priming in the DLN, tumoricidal function, and tumor infiltration of T cells that was unique to the vaccine/Fc-OX40L combination therapy. Further studies were then undertaken to systematically dissect the contribution of Fc-OX40L and the vaccine and optimize combination therapies.
Figure 3. Analysis of brain infiltrating lymphocytes.

Glioma-bearing animals were treated as described in Figure 1 and euthanized on day 25. Lymphocytes were isolated from the brains and analyzed by flow cytometry. (A) The percentage and total numbers of CD4 and CD8 T cells are plotted. (B) The percentage of proliferating CD4 and CD8 T cells was determined by Ki67 staining. This experiment was performed once. Statistical significance is indicated as p < 0.05 (*), or not significant (N.S.). Mean values are shown −/+ SEM; n=4–5/group.
Determination of the optimal treatment strategy
The potency of Fc-OX40L as a single agent was examined in glioma-bearing animals. We treated well-established tumors, beginning treatment on day 17. Impressively, Fc-OX40L was capable of extending survival of tumor-bearing animals and curing 20% (1/5) (Fig. 4A, Regimen 1). In a further attempt to optimize complete response rates, vaccination was given concurrently with Fc-OX40L on days 7, 10, and 13 followed by 5 daily doses of Fc-OX40L alone (days 15–19). This was reproducibly the most potent treatment, with 70% cure rates (5/7) (Fig. 4B, Regimen 2). Variations of this treatment were tested to determine the contribution of vaccine and Fc-OX40L. Fc-OX40L alone given early (days 7, 10, and 13) (Regimen 3) had a weak therapeutic effect unless given in combination with vaccine (Regimen 4) (Fig. 4A, B). The efficacy of Fc-OX40L given late (days 15–19; Regimen 5) was improved if vaccination was administered on days 7, 10, and 13 (Regimen 6) (Fig. 4A, B). We focused on the timing and frequency used in regimen 2 in subsequent studies due to the greatest therapeutic index of this regimen.
Figure 4. Optimization of Fc-OX40L Treatment.

(A) The efficacy of Fc-OX40L monotherapy was established at different time points following GL261 inoculation. (B) Treatment with vaccine (lysate plus CpG) and Fc-OX40L was optimized by varying the treatment schedule as indicated. Data represents one experiment; Regimen 2 has been repeated at least three additional times with similar results.
Contribution of vaccine components (antigen vs. adjuvant) with Fc-OX40L
The components of the vaccine/Fc-OX40L treatment were broken down to determine which is necessary for maximum effect. Lysate, CpG, or the combination “vaccine” had no effect on survival (Supplementary Fig. S3A). Both lysate/Fc-OX40L and lysate/CpG/Fc-OX40L were able to significantly extend survival with 50–60% cure rates (Supplementary Fig. S3B). Therefore, a TLR agonist is dispensable to achieve complete responses. However, the addition of a TLR agonist (PolyICLC or CpG) to the regimen was associated with a delay in the time of initial death of the first 20–40% of animals compared to lysate/Fc-OX40L (Supplementary Fig. S3B–C). In clinical oncology, the closest analogy would be to describe the contribution of the TLR agonist as increasing progression-free survival. Additionally, a single animal in the lysate/Fc-OX40L treatment group that was determined to be a complete responder through BLI subsequently died due to tumor recurrence at day 158, a phenomenon never observed in the lysate/CpG/Fc-OX40L-treated animals (Supplementary Fig. S3B). Thus, inclusion of a TLR agonist in the treatment cocktail may increase the durability of complete responses. Increased survival caused by inclusion of the TLR agonist (to lysate/Fc-OX40L) never reached statistical significance compared to lysate/Fc-OX40L alone, but was reproducible and generalizable to both TLR3 (PoyICLC) and TLR9 (CpG). Unexpectedly, CpG or PolyICLC given without lysate antigen but with Fc-OX40L had no effect on survival (Supplementary Fig. S3B–C). Hence, apparently TLR agonists administered i.d. near the tumor DLN diminished the efficacy of Fc-OX40L administered IP as a single agent. This finding reveals the importance of co-delivering a source of antigen with TLR ligand adjuvant in settings where Fc-OX40L is administered systemically. Although the mechanisms of how these combinations are promoting or antagonizing therapeutic efficacy are unclear, the combination of vaccine (TLR agonist plus lysate) with Fc-OX40L consistently yielded highest survival rates. This was a robust finding in the GL261 glioma model and extended to other models; notably the vaccine/Fc-OX40L regimen was curative in 5/5 mice bearing established EMT6 breast carcinoma (Supplementary Fig. S4).
Complete responses using temozolomide combination therapy associated with secondary malignancies
TMZ is a DNA alkylating chemotherapy that is part of the current standard of care for GBM (20). Any investigational treatment will likely be delivered in combination with TMZ in the clinic. For that reason we studied the compatibility of the two treatment strategies. We attempted to model a likely scenario in newly diagnosed GBM patients, which historically have received TMZ prior to vaccine immunotherapy (21–23), by administering vaccine or Fc-OX40L alone and in combination with TMZ. TMZ was administered by oral gavage for five consecutive days beginning on day 7 post GL261 inoculation. Vaccination was performed on day 12 and 19, followed by 5 daily doses of Fc-OX40L beginning on day 19. When TMZ was given alone or in combination with vaccine, survival was significantly enhanced, although there were no long-term survivors (Fig. 5A). The survival of glioma bearing mice was not significantly different between the TMZ/Fc-OX40L and vaccine/Fc-OX40L groups; yielding 20–40% long-term survivor rates (Fig. 5B). Strikingly, the addition of TMZ to vaccine/Fc-OX40L resulted in 100% complete tumor regression by day 50 after GL261 challenge (Fig. 5B). However, 80% of the mice cured by the TMZ/vaccine/Fc-OX40L combination developed lymphoid neoplasia involving multiple organs, killing 80% (4/5) with a latency of 82– 378 days (depicted in Fig. 5B). These animals had enlarged spleens, lymph nodes, and livers. Microscopic examination revealed massive infiltration of these organs and variable involvement of other tissues (e.g., lungs, skeletal muscle, pancreas, kidneys) by neoplastic medium to large round cells with a high mitotic index (up to or exceeding 3–6 mitotic figures per high powered field, 40× objective). Immunohistochemistry of the liver revealed that the neoplastic cells were CD3 positive but B220 and Pax5 negative in 2 animals, consistent with T cell lymphosarcoma (Supplementary Fig. S5A). In the other 2 animals, the neoplastic cells were variably CD3, B220 and Pax5 positive (Supplementary Fig. S5B). Confocal microscopy confirmed that the CD3 and B220 positive cells in these animals comprised distinct cell populations, and this was interpreted to be lymphosarcoma involving both T- and B-cell lineages in these animals (Supplementary Fig. S5C). Glioma progression was the cause of death in all other animals in this experiment with the exception of a single mouse in the TMZ/Fc-OX40L group that died with evidence of microscopic disease on day 170 (Fig. 5B). In all other experiments glioma progression was also the cause of death, as documented by neurologic deterioration and histological analysis of treated brains, except for two mice in the lysate/Fc-OX40L group (Supplementary Fig S3B) that died of unknown causes (data not shown).
Figure 5. Combination therapy with TMZ is efficacious but associated with secondary malignancy.

TMZ was administered via oral gavage for 5 days beginning on day 7 post GL261 inoculation. Vaccinations occurred on days 12 and 19, followed by 5 daily doses of Fc-OX40L beginning on day 19. BLI plots are shown below corresponding survival data in each panel, whereby each line represents the tumor-burden of an individual mouse over time. (A) TMZ given alone was compared to saline and TMZ with vaccination. (B) The survival effect of the combinations of Fc-OX40L with vaccine, TMZ, and vaccine/TMZ were compared in glioma-bearing animals. “L” indicates death due to lymphoid neoplasia rather than glioma burden. This experiment was performed once.
Analysis of single agent Fc-OX40L in transplanted and spontaneous glioma models
It remained unclear how generalizable these data were to autochthonous glioma models. We therefore investigated the efficacy of Fc-OX40L in a de novo glioma model that is induced by oncogene transfer into neonatal mice as previously described (18). Plasmids encoding three oncogenes and a firefly luciferase reporter were delivered intracerebrally to transform endogenous brain cells within 1–2 days of birth. Beginning at the time of weaning, animals were imaged to identify when tumor formation occurred. For lack of an adequate cell line to use for lysate generation (equivalent to autologous as in the GL261 system), Fc-OX40L was administered as a single agent. Treatment began when tumor was first detected by BLI as depicted in Fig. 6A. Fc-OX40L monotherapy caused a statistically significant increase in survival, although this difference was incremental and there were no long-term survivors (Fig. 6B). This finding was in contrast to Fc-OX40L monotherapy in the GL261 model, which consistently cured 20–25% of mice (even those bearing 17-day established tumors), prompting us to further investigate the microenvironment of both tumor systems.
Figure 6. Differential survival and inflammatory gene expression patterns between implanted and spontaneous glioma models.

(A) Schematic illustrating spontaneous tumor model and treatment schedule. (B) Survival of glioma-bearing mice following Fc-OX40L monotherapy. Seven mice were treated individually and data pooled. (C) qRT-PCR analysis was performed on tumors collected from GL261 implanted and plasmid oncogene induced (spontaneous) animals. Gene expression was normalized to the housekeeping gene GAPDH. 3–4 animals per group were analyzed in triplicate; statistical significance is indicated as p < 0.05 (*), mean values are shown −/+ SEM.
Gene expression profiling has proven useful in identifying differences between tumors and has lead to the further classification of gliomas beyond the histological standards to a molecular subtype (24). In an attempt to understand the molecular differences between the two glioma models, we conducted a quantitative RT-PCR analysis of 30 immune-related genes in brains from both glioma models and normal B6 mice. Relative to normal brain, both tumor models exhibited log-fold increases in CD11c expression, a DC marker, and arginase-1 (Fig. 6C). The arginase enzyme is released by myeloid derived suppressor cells, locally depletes L-arginine, and consequently impairs T cell proliferation and survival (25). Response to costimulation may be dependent on which costimulatory molecules are absent endogenously. Interestingly, OX40L was the only co-stimulatory ligand of those tested in our models that had lower expression in the tumor relative to the normal brain in either model. The GL261 and spontaneous models differed in several important ways. GL261 tumors exhibited a 15-fold increase in IFN-γ expression compared to the spontaneous model, and consistent with this expressed more PD1-L, which is induced by IFNγ (26). In addition, CD3 (a T cell marker) was more highly expressed in the GL261 model, whereas the NK cell marker CD49b was more highly expressed in the spontaneous model (Fig. 6C); although these were not statistically different they were trending toward significance (CD49b p=0.071, CD3 p=0.088).
Discussion
Recent clinical trials employing peptide vaccines or lysate-pulsed DC vaccines have shown promising results in GBM patients (21–23). These studies have documented immunologic and radiographic responses in select patients, and favorable survival rates that await validation in randomized clinical trials. However, the majority of patients treated by vaccination ultimately experienced recurrence and succumbed to disease, highlighting the need for more potent immunotherapy and the induction of memory cells. Recombinant costimulatory ligand-Fc fusion proteins offer an attractive adjuvant to vaccine immunotherapy. To this end, the purpose of this study was to screen a panel of Fc-ligands using doses previously optimized in other tumor models (11–14). Fc-OX40L was clearly the most efficacious in the GL261 vaccination model. We are cautious to avoid over-interpreting these data; this does not mean that one Fc-ligand is universally better than the others, considering that the dose, timing in relation to vaccination, and biologic activity of each agent was not systematically optimized prior to the screen that identified Fc-OX40L as our lead candidate. It is likely that the efficacy of each drug could be significantly changed by altering the temporal relationship to TCR signaling following vaccination. Nonetheless, this study is the first head-to-head screen using Fc-ligand fusion proteins, and the first to clearly demonstrate the therapeutic potency of the combination of vaccine/Fc-OX40L in a brain tumor model.
The reproducibility of our data and that of others using OX40 agonists strongly suggests that OX40 agonists warrant clinical testing in a variety of cancers. The vaccine/Fc-OX40L combination was generalizable to various TLR agonists (TLR9 or TLR3) and other tumor models (GL261 glioma or EMT6 breast carcinoma), yielding cure rates of 50–100% depending on the model and adjuvant used. To our knowledge, no immunotherapy has improved survival in an implanted brain tumor model beyond day 10 (27), whereas Fc-OX40L improved survival of mice treated starting 17 days after tumor challenge (GL261), and in a spontaneous model as a single agent. We noted a curious finding following Fc-OX40L monotherapy: that treating earlier relative to tumor implantation progressively decreased the median survival (compare regimen 1, 3, and 5 in Fig. 4A). This is the opposite of conventional wisdom, which would argue immunotherapy works better on smaller tumors. We speculate that this is due to a delay in natural antigen presentation caused by tumor cell death, leading to more responder T cells in animals with advanced tumors. Indeed, the appeal of cancer vaccines is to increase the frequency of tumor-reactive T cells. Although vaccination did not alter T cell priming measured in the DLN on day 25 (15 days after last vaccine; Fig. 2A), vaccination primed equivalent expansion of CD4 and CD8 T cells in the DLN at day 15 relative to the vaccine/Fc-OX40L combination (Supplementary Fig. S6). Consistent with a model of vaccination increasing tumor-reactive T cell frequency, vaccination combined with Fc-OX40L synergistically increased survival rates in the GL261 model relative to Fc-OX40L monotherapy. We speculate that an important effect of Fc-OX40L is rescuing T cell anergy, as shown by enhancing tumoricidal function (Fig. 2B). This hypothesis is consistent with other reports that have studied the OX40/anergy axis in greater detail (19, 28). In addition, Fc-OX40L has been shown to activate tumor vasculature and tumor-associated DCs, revealing the complexity of response to this drug (29). A significant percentage of infiltrating T cells expressed the proliferation marker Ki67 in the vaccine/Fc-OX40L group, supporting the notion that this treatment yields a qualitative difference in T cell response (Fig. 3). Restimulation by antigen presenting cells (APCs) can induce proliferation in responding T cells, which has been documented to occur at the tumor site, in addition to the lymph nodes (30). It has also been documented that Ki67 has a half life of 60–90 minutes and therefore it is also possible that these cells have been stimulated in the lymph nodes and recently migrated to the brain (31). In either situation, it appears that the infiltrating T cells in the vaccine/Fc-OX40L group have recently experienced a productive encounter with an APC and this is a likely explanation for the corresponding increase in tumoricidal function.
The naturally occurring immune response to cancer warrants detailed consideration because there is evidence it may predict therapeutic response. Untreated GL261 tumors had an inflammatory phenotype, dominated by T cells and strong IFNγ expression, whereas the spontaneous glioma model had higher IL-6 expression and was NK cell dominated (Fig. 6). This is intriguing because Fc-OX40L monotherapy consistently cured 20–25% of mice in the GL261 model, but only incrementally increased survival in the spontaneous model. These results are reminiscent of those reported by Prins et al. in GBM patients treated by lysate-pulsed DCs and TLR7 or TLR3 agonists (23). There are at least three molecular subclasses of GBM based on microarray signature: mesenchymal, proneural, and proliferative, where mesenchymal is associated with inflammation and has the worst outcome (24). The study by Prins et al. preliminarily demonstrated an increase in survival of vaccinated patients of the mesenchymal compared to the proneural subclass. Although this study was not powered to detect clinical efficacy, it offers a provocative hypothesis consistent with our data: that the pre-existing immune response and inflammatory milieu might be used to predict responders to specific immunotherapy protocols. This paradigm is quintessentially personalized medicine and warrants testing in prospectively designed, randomized clinical trials. The 30-gene immune phenotyping panel used in this study is a simplified assay relative to microarray that could be useful in such efforts. We are currently evaluating these ideas by conducting a randomized study of vaccine/Fc-OX40L immunotherapy in pet dogs with naturally occurring glioma.
The high incidence of secondary malignancy in TMZ-treated animals warrants attention. It is possible that TMZ-exposed T cells are generally more susceptible to transformation in B6 mice, rather than requiring a specific Fc-OX40L/TMZ combination. Further investigation into the effect of TMZ on secondary malignancies in the context of immunotherapy is beyond the scope of this report, but demands caution. We are currently investigating delaying TMZ until after vaccine/Fc-OX40L is completed in dogs with naturally occurring glioma in an attempt to address the theoretical risk of accelerated secondary malignancy by priming TMZ-exposed T cells. The vaccine trials conducted in GBM patients to date have not reported an elevated risk of secondary malignancy, although the caveat is that most treated patients died before meaningful long-term follow up was obtained. As novel immunotherapies are approved that significantly increase patient survival, the risk of secondary malignancy in heavily pre-treated patients may become increasingly important.
In summary, these preclinical studies have laid the foundation for the clinical development of Fc-OX40L for the treatment of GBM and other cancers. The combination of Fc-ligand fusion proteins with vaccines is an appealing strategy worth pursuing. Such combination therapies may represent an important leap forward in the field of cancer immunotherapy.
Supplementary Material
Translational Relevance.
Protein biologics that activate immunostimulatory pathways or inhibit immunoregulatory pathways are entering the clinic at an unprecedented rate. Comparative studies of this class of agents, given alone or in combination with vaccines and chemotherapy, should inform rational clinical trial design. We performed a screen in glioma-bearing mice to identify a lead candidate for further optimization with the following costimulatory ligands: OX40L, CD80, 4-1BBL, and GITRL. OX40L proved to be most efficacious and synergized with vaccines to cure well-established tumors. The combination with temozolomide, the standard of care chemotherapy for glioma patients, was very efficacious but was associated with secondary malignancies, warranting caution and further study. Inflammatory gene expression signatures in different glioma models provided insight as to how the pre-existing immune response at the time of treatment may predict responders and non-responders. Together, these studies reveal exciting prospects and challenges in applying this class of immune modifiers to clinical trials.
Acknowledgments
This work was supported in part by NIH grants R01 CA154345, R01 CA160782, the American Cancer Society grant RSG-09-189-01-LIB, and generous support from the Minnesota Medical Foundation, the Hedberg Family Foundation, and the Children’s Cancer Research Fund to JRO. We acknowledge the University of Minnesota Masonic Cancer Center Comparative Pathology Shared Resource for histology and immunohistochemistry preparations, and thank Josh Parker for preparation of Supplementary Figure S5. We thank Nicole Feng for assistance with qRT-PCR.
References
- 1.Grossman SA, Ye X, Piantadosi S, Desideri S, Nabors LB, Rosenfeld M, et al. Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide in research studies in the United States. Clin Cancer Res. 2010;16:2443–9. doi: 10.1158/1078-0432.CCR-09-3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finocchiaro G, Pellegatta S. Immunotherapy for glioma: getting closer to the clinical arena? Curr Opin Neurol. 2011;24:641–7. doi: 10.1097/WCO.0b013e32834cbb17. [DOI] [PubMed] [Google Scholar]
- 3.Okada H, Kohanbash G, Zhu X, Kastenhuber ER, Hoji A, Ueda R, et al. Immunotherapeutic approaches for glioma. Crit Rev Immunol. 2009;29:1–42. doi: 10.1615/critrevimmunol.v29.i1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitchell DA, Fecci PE, Sampson JH. Immunotherapy of malignant brain tumors. Immunol Rev. 2008;222:70–100. doi: 10.1111/j.1600-065X.2008.00603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gustafson MP, Lin Y, New KC, Bulur PA, O’Neill BP, Gastineau DA, et al. Systemic immune suppression in glioblastoma: the interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncol. 2010;12:631–44. doi: 10.1093/neuonc/noq001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pardee AD, Wesa AK, Storkus WJ. Integrating costimulatory agonists to optimize immune-based cancer therapies. Immunotherapy. 2009;1:249–64. doi: 10.2217/1750743X.1.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–9. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer. 2011;11:805–12. doi: 10.1038/nrc3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bakacs T, Mehrishi JN, Moss RW. Ipilimumab (Yervoy) and the TGN1412 catastrophe. Immunobiology. 2012;217:583–9. doi: 10.1016/j.imbio.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 11.Hu P, Arias RS, Sadun RE, Nien YC, Zhang N, Sabzevari H, et al. Construction and preclinical characterization of Fc-mGITRL for the immunotherapy of cancer. Clin Cancer Res. 2008;14:579–88. doi: 10.1158/1078-0432.CCR-07-0940. [DOI] [PubMed] [Google Scholar]
- 12.Liu A, Hu P, Khawli LA, Epstein AL. Combination B7-Fc fusion protein treatment and Treg cell depletion therapy. Clin Cancer Res. 2005;11:8492–502. doi: 10.1158/1078-0432.CCR-05-1411. [DOI] [PubMed] [Google Scholar]
- 13.Sadun RE, Hsu WE, Zhang N, Nien YC, Bergfeld SA, Sabzevari H, et al. Fc-mOX40L fusion protein produces complete remission and enhanced survival in 2 murine tumor models. J Immunother. 2008;31:235–45. doi: 10.1097/CJI.0b013e31816a88e0. [DOI] [PubMed] [Google Scholar]
- 14.Zhang N, Sadun RE, Arias RS, Flanagan ML, Sachsman SM, Nien YC, et al. Targeted and untargeted CD137L fusion proteins for the immunotherapy of experimental solid tumors. Clin Cancer Res. 2007;13:2758–67. doi: 10.1158/1078-0432.CCR-06-2343. [DOI] [PubMed] [Google Scholar]
- 15.Wu A, Oh S, Gharagozlou S, Vedi RN, Ericson K, Low WC, et al. In vivo vaccination with tumor cell lysate plus CpG oligodeoxynucleotides eradicates murine glioblastoma. J Immunother. 2007;30:789–97. doi: 10.1097/CJI.0b013e318155a0f6. [DOI] [PubMed] [Google Scholar]
- 16.Olin MR, Andersen BM, Zellmer DM, Grogan PT, Popescu FE, Xiong Z, et al. Superior efficacy of tumor cell vaccines grown in physiologic oxygen. Clin Cancer Res. 2010;16:4800–8. doi: 10.1158/1078-0432.CCR-10-1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiong Z, Gharagozlou S, Vengco I, Chen W, Ohlfest JR. Effective CpG immunotherapy of breast carcinoma prevents but fails to eradicate established brain metastasis. Clin Cancer Res. 2008;14:5484–93. doi: 10.1158/1078-0432.CCR-07-4139. [DOI] [PubMed] [Google Scholar]
- 18.Wiesner SM, Decker SA, Larson JD, Ericson K, Forster C, Gallardo JL, et al. De novo induction of genetically engineered brain tumors in mice using plasmid DNA. Cancer Res. 2009;69:431–9. doi: 10.1158/0008-5472.CAN-08-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Redmond WL, Gough MJ, Weinberg AD. Ligation of the OX40 co-stimulatory receptor reverses self-Ag and tumor-induced CD8 T-cell anergy in vivo. Eur J Immunol. 2009;39:2184–94. doi: 10.1002/eji.200939348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 21.Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 2011;29:330–6. doi: 10.1200/JCO.2010.30.7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. 2011;13:324–33. doi: 10.1093/neuonc/noq157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011;17:1603–15. doi: 10.1158/1078-0432.CCR-10-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer cell. 2006;9:157–73. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 25.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5:641–54. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 26.Chen J, Feng Y, Lu L, Wang H, Dai L, Li Y, et al. Interferon-gamma-induced PD-L1 surface expression on human oral squamous carcinoma via PKD2 signal pathway. Immunobiology. 2012;217:385–93. doi: 10.1016/j.imbio.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 27.Ali S, King GD, Curtin JF, Candolfi M, Xiong W, Liu C, et al. Combined immunostimulation and conditional cytotoxic gene therapy provide long-term survival in a large glioma model. Cancer research. 2005;65:7194–204. doi: 10.1158/0008-5472.CAN-04-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirschhorn-Cymerman D, Rizzuto GA, Merghoub T, Cohen AD, Avogadri F, Lesokhin AM, et al. OX40 engagement and chemotherapy combination provides potent antitumor immunity with concomitant regulatory T cell apoptosis. The Journal of experimental medicine. 2009;206:1103–16. doi: 10.1084/jem.20082205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pardee AD, McCurry D, Alber S, Hu P, Epstein AL, Storkus WJ. A therapeutic OX40 agonist dynamically alters dendritic, endothelial, and T cell subsets within the established tumor microenvironment. Cancer Res. 2010;70:9041–52. doi: 10.1158/0008-5472.CAN-10-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masson F, Calzascia T, Di Berardino-Besson W, de Tribolet N, Dietrich PY, Walker PR. Brain microenvironment promotes the final functional maturation of tumor-specific effector CD8+ T cells. J Immunol. 2007;179:845–53. doi: 10.4049/jimmunol.179.2.845. [DOI] [PubMed] [Google Scholar]
- 31.Brown DC, Gatter KC. Ki67 protein: the immaculate deception? Histopathology. 2002;40:2–11. doi: 10.1046/j.1365-2559.2002.01343.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.