

Abstract
Background
Expression microarrays have evolved into a powerful tool with great potential for clinical application and therefore reliability of data is essential. RNA amplification is used when the amount of starting material is scarce, as is frequently the case with clinical samples. Purification steps are critical in RNA amplification and labelling protocols, and there is a lack of sufficient data to validate and optimise the process.
Results
Here the purification steps involved in the protocol for indirect labelling of amplified RNA are evaluated and the experimentally determined best method for each step with respect to yield, purity, size distribution of the transcripts, and dye coupling is used to generate targets tested in replicate hybridisations. DNase treatment of diluted total RNA samples followed by phenol extraction is the optimal way to remove genomic DNA contamination. Purification of double-stranded cDNA is best achieved by phenol extraction followed by isopropanol precipitation at room temperature. Extraction with guanidinium-phenol and Lithium Chloride precipitation are the optimal methods for purification of amplified RNA and labelled aRNA respectively.
Conclusion
This protocol provides targets that generate highly reproducible microarray data with good representation of transcripts across the size spectrum and a coefficient of repeatability significantly better than that reported previously.
Background
Expression microarrays have shown great potential for clinical application [1,2], and therefore it is critical that data is reproducible. Clinical specimens frequently contain small amounts of RNA and amplification is required to obtain sufficient material for expression analysis. RNA amplification by T7-polymerase is commonly used to generate cDNA or amplified RNA (aRNA) for direct and indirect labelling reactions [3-6].
Nucleic acid purification and recovery steps have a critical impact on the quality of the labelled targets for microarray experiments. Although a variety of methods have been applied for the purification steps [5-8], there has not been a systematic evaluation to optimize these methods. We present here a comprehensive study of all purification steps involved in the process of indirect labeling of aRNA (Figure 1), to generate anti-sense targets applicable on both cDNA and oligonucleotide arrays [9].
Figure 1.
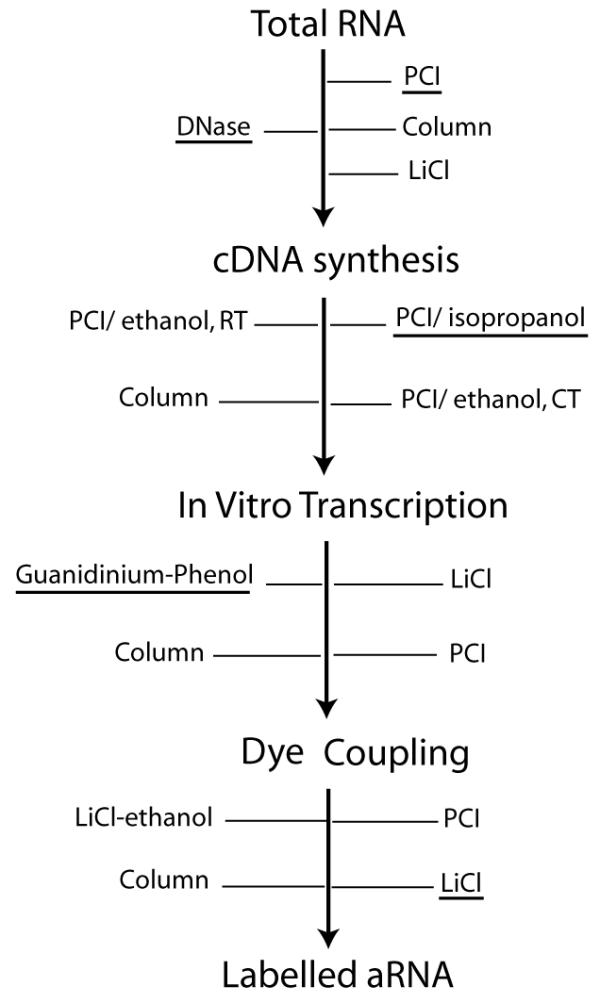
Purification steps in indirect aRNA labelling. Methods evaluated are indicated for each step. Experimentally determined optimal methods are underlined. DNase – DNase I treatment; PCI – Phenol:Chloroform:Isoamyl alcohol; LiCl – Lithium Chloride; Ethanol at RT – ethanol at room temperature; Ethanol at CT – ethanol with cold incubation; LiCl-ethanol – Lithium Chloride/ethanol precipitation.
Results and Discussion
Effect of genomic DNA carry over on RNA amplification
To evaluate the optimal method for removal of genomic DNA contamination from RNA samples, total RNA from each of the five cell lines was treated using one of four methods: Qiagen RNeasy minikit columns (with on-column DNase digestion); DNase treatment followed by 2.5 M LiCl precipitation, and DNase treatment (using two different RNA concentrations, D5 and D20) followed by PCI extraction.
The efficiency of each method was first assessed by agarose gel and Agilent Bioanalyzer. Agarose gel was sufficient to demonstrate that specimens purified by the LiCl method had a high level of genomic DNA contamination (Figure 2A). Using the Agilent Bioanalyzer revealed that both the column method and D20 samples had genomic DNA carry over, manifested as a small shoulder after the 28S band (Figure 2B). To further evaluate the effect of genomic DNA on downstream reactions, RNA samples processed with each method were amplified and the products of amplification precipitated with LiCl to preserve high molecular weight species. D5 samples generated amplification products without heavy molecular weight genomic bands, whereas the column method showed genomic bands (Figure 2C). These findings were consistent in the five cell lines studied. Contamination with shorter genomic fragments could not be ruled out with these methods.
Figure 2.

Effect of genomic DNA contamination in total RNA. (A) 1% agarose gel of purified MCF-7 total RNA samples. L-1 kb ladder (Invitrogen); Col. – column purified RNA; D20 – DNase treated/PCI extracted (RNA concentration – 20 μg/100 μl); D5 – DNase treated/PCI extracted (RNA concentration – 5 μg/100 μl); LiCl – DNase treated/Lithium Chloride purified. (B) Agilent Bioanalyzer image of MCF-7 total RNA sample purified using column method. Arrow pointing at shoulder after 28S band indicating genomic DNA carry over. (C) 1% agarose/formamide denaturing gel of MCF-7 aRNA. L1 – 6000 RNA ladder (Ambion); L2-1 Kb ladder (Invitrogen). (D) Absorption at 260 nm of nucleic acid products derived from the 4 total RNA purification methods. 2 μg of total RNA from each of the five cell lines was amplified with and without reverse transcriptase being added to the cDNA synthesis reaction (with RT and no RT respectively). C – column; Li – LiCl precipitation; D5/D20 – as in A. Cell lines included MCF-7, ZR-75-1-1, OCUB-M, Cal51, and HCT-1187.
Finally, to quantify the effect of genomic DNA contamination cDNA synthesis was conducted without the addition of reverse transcriptase, but keeping all the downstream steps for RNA amplification, including DNA polymerase. In these circumstances cDNA synthesis cannot occur and absorptions at 260 nm of the amplification products reflect genomic DNA contamination and not aRNA. D5 samples showed minimal absorptions at 260 nm, whereas other samples had significant amounts of nucleic acid (Figure 2D). The experiments were also conducted with the inclusion of reverse transcriptase as a control.
Application of DNase I (2 Units/1 μg of total RNA) to diluted samples followed by phenol extraction is the most effective method for removal of genomic DNA. Dilution leads to easier access of DNase to genomic DNA and probably allows more efficient phenol extraction due to lower viscosity of the sample. Genomic DNA contamination could be a potential problem when quantifying the amplification products and may interfere with the downstream reactions.
cDNA purification affects transcript representation
To evaluate the effect of cDNA clean up on amplified products, total RNA samples from the five cell lines studied were processed with the optimized method (D5/PCI) and 2 μg of total RNA from each cell line used for cDNA synthesis. For cDNA purification 4 methods were tested: column, PCI/ethanol at RT, PCI/ethanol at CT, and PCI/isopropanol. Each purified sample was used for T7-amplification followed by recovery with 2.5 M LiCl precipitation. PCI/isopropanol showed the highest overall yield for both RNA samples of good quality (28S/18S atio of 1.9) and of poor quality (28S/18S ratio of 1.4) (p < 0.01, Table 1). Column and PCI/ethanol at RT provided high yields only for samples with 28S/18S ratio of 1.9.
Table 1.
Yield of RNA amplification generated from different cDNA purification methods.*
| Method | 28S/18S = 1.9 | 28S/18S = 1.4 |
| column | 18 μg ± 2 | 7 μg ± 1 |
| ethanol at RT | 17 μg ± 2 | 10 μg ± 1 |
| isopropanol | 19 μg ± 3 | 14 μg ± 2 |
| ethanol at CT | 14 μg ± 2 | 12 μg ± 1 |
*RNA from MCF-7 and HCC-1187 (28S/18S = 1.9) and OCUB-M (28S/18S = 1.4) were used and reactions were repeated five times. Values represent averages with standard errors.
To evaluate the effect of cDNA purification methods on aRNA pattern, fluorescence absorptions were measured using the Agilent Bioanalyzer at 100-nucleotide (nt), 1000 nt, and 6000 nt data points (representing short, medium, and large size transcripts). For each total RNA isolate, one amplification reaction was tested with each cDNA purification method and fluorescence absorptions for different methods were compared using the mean values obtained from the five cell lines (Figure 3A). The isopropanol method provided better recovery of both short and large size transcripts. Ethanol at RT was less efficient for small transcripts and the opposite was observed for ethanol at CT. Columns resulted in lower recovery for both small and large transcript sizes. Medium sized transcripts were recovered with similar efficiency by all 4 methods. The difference in preserving smaller transcripts was more evident for poor quality RNA samples (OCUB-M with 28S/18S of 1.4) which showed significantly higher fluorescence absorption at 100 nt with PCI/isopropanol (Figure 3B) compared to PCI/ethanol at RT (Figure 3C). The difference was less marked for other cell lines (data not shown). The better overall yield and the ability to preserve both long and short transcripts, suggests that PCI extraction with isopropanol recovery is the method of choice for cDNA purification.
Figure 3.

Analysis of aRNA generated after different cDNA purification methods. (A) Fluorescence absorptions at 100 nucleotides (100 nt), 1000 nucleotides (1000 nt), and 6000 nucleotides (6000 nt) data points. The results were obtained by plotting the mean fluorescence absorption values for the five cell lines using the different cDNA purification methods. cDNA purification methods: Col. – Column; E-R – ethanol at room temperature; IS – Isopropanol; E-C – ethanol at cold temperature. (B) Pattern of amplified RNA from OCUB-M cell line using cDNA purified by isopropanol method. First peak-absorption at about 100 nucleotides; second peak-absorption at 1000 nucleotides. (C) Pattern of amplified RNA from OCUB-M cell line using cDNA purified by ethanol at room temperature method.
Guanidinium-phenol extraction is the optimal method to purify aRNA
To evaluate aRNA purification methods, 3 μg of total RNA from each cell line was amplified in twelve replicates using the optimized total RNA and cDNA purification methods described above. aRNA purification was then performed in triplicate with each of the following 4 methods: column, LiCl, PCI, and guanidinium. Guanidinium isothiocyanate (or guanidinium containing compounds such as TRI reagent) was added to phenol extraction to test whether the improved denaturing ability resulted in more effective purification. Purified aRNA products were assessed with respect to yield, purity (260/280 ratio), and pattern of aRNA. The mean values obtained with different methods for the five cell lines were compared (Figure 4A). Columns had the lowest yield but showed optimal purity. Both guanidinium and LiCl resulted in good yield and purity. The yield was higher with PCI but aRNA purity was suboptimal (1.7). These results show that phenol extraction alone could not efficiently purify aRNA probably due to the high concentration of proteins such as spermidine in the reaction buffer. The addition of guanidinium salt with its chaotropic and denaturing effects [10], improved the purification without compromising yield.
Figure 4.

Analysis of aRNA purification. (A) Amplified RNA yields and 260/280 ratios with 4 methods of purification. Col – column; PCI – phenol/chlorform/isoamyl alchohol; LiCl – 2.5 M LiCl; G-P – guanidinium-phenol. (B) 1% denaturing agarose/formamide gel of MCF-7 aRNA purified using column (C) Agilent Bioanalyzer analysis of aRNA (from MCF-7) purified by G-P. 6000 nano marker from Ambion (blue) superimposed on the RNA trace. (D) Plot of aRNA yield in μg for different starting total RNA quantities. C1 – OCUB-M with Col.; L1 – OCUB-M with LiCl; G1 – OCUB-M with G-P; C2 – MCF-7 with Col.; L2 – MCF-7 with LiCl; G2 – MCF-7 with G-P.
To evaluate the pattern of amplification, aRNA samples of all five cell lines were analysed on both denaturing agarose gels and the Agilent Bioanalyzer. Column-purified samples showed a smear ranging from 400 to 6000 bp (figure 4B), guanidinium purified samples showed a smear from 100 to over 6000 nt (figure 4C), and samples purified with LiCl had preservation of larger products with a variable recovery of transcripts less than 200 bases (data not shown). These data indicate that the guanidinium method is optimal for aRNA purification.
The best method for aRNA recovery is dependent on total RNA quality
To evaluate the effect of total RNA quality on recovery of aRNA, different amounts of total RNA (2, 3, and 5 μg) from a cell line with poor quality RNA (OCUB-M: 28S/18S = 1.4) and from a cell line with good quality RNA (MCF-7: 28S/18S = 1.9) were tested. Amplification reactions were performed in fifteen replicates for each starting quantity of RNA from MCF-7 and OCUB-M. Five replicate products from each starting quantity were precipitated with either 2.5 M LiCl, column, or guanidinium methods (figure 4D). The guanidinium method provided the best aRNA recovery with OCUB-M (p #60; 0.01), showing that samples with lower 28S/18S ratios can be reliably recovered by this method. The yield of aRNA was significantly higher with MCF-7 (p < 0.001) compared to OCUB-M. Furthermore, LiCl and guanidinium methods provided better yields in the MCF-7 samples compared to the column method (p < 0.01). The LiCl was a robust method for the recovery of MCF-7 aRNA products but not for OCUB-M samples.
RNA samples with low 28S/18S ratios have reduced amplification efficiency and contain shorter transcripts. Columns result in size exclusion of short aRNA products and have therefore limited ability for recovery in lower quality RNA samples. LiCl has a variable yield for short transcripts and cannot efficiently precipitate RNA in low concentrations (data not shown); therefore it is not reliable for samples with low 28S/18S ratios. The guanidinium method performs consistently for samples with different qualities of starting RNA and is therefore the best method for aRNA purification.
Removal of protein impurities is essential for aRNA labelling reaction
To study the effect of aRNA purity (260/280 ratio) on coupling efficiency, labelling reactions of MCF-7 aRNA samples with 260/280 ratios from 1.5 to 2 were performed (Figure 5A). Cy5 labelling was done in triplicate for each different ratio using 10 μg of aRNA and purified by LiCl precipitation. Coupling efficiency was measured using the Agilent Bioanalyzer by obtaining mean ratios of coupled to total Cy5 fluorescence readings. The 320/650 ratio (Cy5) was also determined using the Nanodrop device. The results showed a positive correlation between aRNA purity and coupling efficiency (Figure 5A). Samples with 260/280 of less than 1.8 had coupling ratios below 0.5 (Figure 5B), but with the increase of 260/280 to above 1.8 coupling efficiency improved to over 0.9 (Figure 5C). Furthermore, Cy5-labelled products with 320/650 ratios over 0.1 (Figure 5D) showed a lower coupling efficiency (p < 0.01) compared to products with ratios equal or less than 0.1 (Figure 5E). The experiments were also performed with Cal51 aRNA samples with similar results (data not shown).
Figure 5.

Effect of aRNA purity and labelled-aRNA purification. (A) Graph showing coupling efficiency (C) and 320/650 (B) values for six MCF-7 aRNA samples with different 260/280 ratios. (B) Agilent Bioanalyzer pattern of MCF-7 Cy5-labelled target using an aRNA with 260/280 ratio of 1.6. Sharp spikes represent uncoupled Cy5 dye. (C) Same as B for aRNA with 260/280 ratio of 2 and coupling efficiency near 1. (D) NanoDrop® ND-1000 Spectrophotometer absorptions at 320 and 650 nm wavelengths of a MCF-7 Cy5-labelled target. 320/650 ratio-0.6. (E) Same as in d (different sample). 320/650 ratio-0.09. (F) Recovery rates (R) and coupling efficiencies (C) for different labelled-aRNA purification methods. Data is presented for MCF-7 cell line and measurements represent an average of three separate reactions. Li-ETOH – LiCl-ethanol; PCI – phenol/chloroform/isoamyl alcohol.
These data demonstrate that purification of aRNA is critical for obtaining an efficient labelling reaction. The presence of protein impurities measured by a 260/280 < 1.8 inhibits dye coupling most likely by competing with aminoallyl groups for esterification with Cy dyes. Measurement at 320 nm indicates background absorption and in the past 320/260 ratios have been used to assess the purity of nucleic acids [11]. Measurements at 650 and 550 nm are for Cy5 and Cy3 dyes respectively [12,13]. We applied 320/650 and 320/550 ratios to estimate the insoluble by-products for Cy5 and Cy3 coupling reactions, which serve as measures for the purity of labelled products.
A correlation coefficient of 0.95 (n = 14) between 320/550 and 320/650 ratios was noted in each sample set suggesting that 320/550 ratios can be used to assess Cy3 coupling reactions. Background at 320 nm may represent dye particles or other insoluble by-products of the coupling reaction and at times these particles can also be seen on Agilent Bioanalyzer as a slow moving peak (data not shown). Since 320/550 and 320/650 values can be easily measured using a spectrophotometer device, they provide a fast and cost effective method for evaluating the quality of labelled aRNA products.
LiCl is the optimal method for purification of labelled aRNA
The recovery rates and free-dye removal of labelled aRNA were evaluated by four different purification methods (column, LiCl-ethanol, PCI, and LiCl) using 10 μg of guanidinium-purified aRNA from MCF-7 cell line. Since LiCl is not an effective precipitant for low concentrations of RNA, it is important not to dilute the samples at this stage (we use 10 μg of starting aRNA and apply 2.5 M final LiCl concentration from 7.5 M LiCl working stock). For each purification method five replicates of Cy3 and Cy5 labelling were tested. The ratios of recovered labelled aRNA to starting aRNA were measured. Mean recovery ratios for combined Cy3 and Cy5-labelled aRNAs were compared and coupling efficiency of Cy5 (see above) determined as an indicator for free-dye removal using Agilent Bioanalyzer (Figure 5F). LiCl had the best overall performance with a recovery rate of 0.6 and coupling efficiency of 0.95.
Optimised purification protocol generates reproducible expression data
To evaluate hybridisation efficiency, labelled-aRNA targets from Cal51 and ZR-75-1-1 cell lines were generated using the experimentally determined optimal purification steps (underlined in Figure 1). For each cell line RNA sample two separate amplification reactions were carried out independently using the optimised method. After labelling of each amplified RNA with Cy3 and Cy5, the reactions were purified with the LiCl method and used in hybridisation experiments. With a total of four slides, a balanced dye-reversal experiment was carried out by hybridising Cy5-labelled targets from each cell line against Cy3-labelled targets of the other cell line using one set of amplified products for each two slides. This generated a total of two technical replicate slides (same dye order) and two dye-reversals slides (opposite dye order). Since each slide contained an internal replicate, the data generated after hybridisation included a total of four internal replicates, eight technical replicates, and sixteen dye-reversal combinations. Data were normalised using the SMA package and analysed using R program (Figure 6). Correlation coefficients in figure 6 represent each pair of replicate and dye-reversal data set with mean values of 0.85 (± 0.05, n = 4) for internal replicates, 0.8 (± 0.01, n = 8) for technical replicates, and 0.63 (± 0.02, n = 16) for dye reversal pairs. The average A value (median of log2 intensities for two dye channels) across slides was 8.7 and 85% of the spots (an average of 11,500/13,000 per each slide) had measurable signals. These data suggest that the optimised method described here can generate reproducible microarray results with good signal intensities and provide hybridisation for the majority of spotted cDNA probes indicating a diverse range of transcript representation.
Figure 6.

Scatter plots and correlation coefficients for each microarray experiment pair. Cal51 versus ZR-75-1-1 cell lines. Rep1 and Rep2 represent the replicates within each slide (internal replicates). Slides d94/d97 and d96/d98, have the same dye orders. Slides d94/d97 are dye reversal experiments for slides d96/d98.
Transcript representation and repeatability
Two housekeeping genes with extreme size distributions were selected to test their representation in aRNA by RT-PCR: as an example of a small transcript the R38b sno gene with a cDNA size of only 86 bases and as an example of long transcript human guanine nucleotide exchange factor p532 gene with a cDNA size of 15,164 bases. The presence of both transcripts in aRNA was confirmed (Figure 7A,7B). Total RNA was also tested to confirm that the transcripts were present (data not shown). PCR reactions with no cDNA were used as negative controls in all experiments.
Figure 7.

Representation of small and large transcripts in aRNA generated by optimised purification protocol. (A) 2.5% agarose gel of R38b RT-PCR product using HCC-1187 aRNA as template. (B) 0.7% agarose gel of p532 RT-PCR product using HCC-1187 aRNA as template. (C) Box-plot for A values (average log2 intensity) of three transcript size categories (Long: > 7,000 bases, Medium: 500–7,000 bases, Short: <500 bases). (D) Coefficients of repeatability (CR) as described by Jenssen et al [14], are demonstrated in 8 replicate data sets for three transcript size categories.
The size representation of the transcripts was also tested globally using the array data. The A value (median of log2 intensity) was categorized for long transcripts >7000 nt (n = 239), short transcripts <500 nt (n = 38), and medium transcripts 500–7,000 nt (n= 5,375). The three size categories had very similar A values signifying equal representation independent of the size (Figure 7C). It should be noted that the amplified products were hybridized without fragmentation and that longer transcripts could potentially mark the corresponding features with more label than shorter transcripts, which can lead to an overestimation of A values for the longer transcripts.
To further evaluate the quality of the microarray data, the coefficient of repeatability (CR) was determined as described by Jenssen et al [14]. Repeatability of M values across the eight replicates showed a median of 0.16 for all three size categories (Figure 7D). These results are significantly better when compared with published CR values of five landmark microarray studies ranging from 0.518 to 1.101 [see ref. [14]]. Although the presented method was not directly compared to the other purification techniques in terms of reproducibility and repeatability, the better CR values compared to the published studies in addition to the improved transcript representation and dye coupling support the contention that the protocol described here is an improvement over currently used methods.
A major concern during the purification steps is the exclusion of transcripts based on their size, which can potentially lead to selection bias in subsequent expression microarray analysis. RT-PCR in addition to analysis of A and CR values demonstrated good representation for various size transcripts using the purification protocol described here.
Conclusions
This manuscript describes a rigorous evaluation of purification methods involved in RNA amplification and labelling. The proposed purification protocol (see Figure 1, underlined methods) provides good yield, purity, coupling efficiency and preservation of different size transcripts. It is also cost effective when compared with methods using multiple column steps, and provides labelled targets for microarray hybridisation with an optimal coefficient of repeatability.
Methods
Samples and total RNA extraction
Total RNA was isolated from five breast cancer cell lines (MCF-7, HCC-1187, Cal51, ZR-75-1-1, and OCUB-M) using TRI-reagent (Sigma) following the manufacturer's recommendations.
Purification of total RNA and genomic DNA removal
The following were tested:
Column method, using RNeasy mini-kit (Qiagen) with on-column DNase I treatment, following the manufacturer's instructions.
Non-column methods, using DNase I treatment followed by a clean-up step. For DNase treatment 2 units of DNase I (Roche Applied Sciences) were used per μg of total RNA at 37°C for 30 minutes. The reaction was tested on total RNA dilutions of 20 μg/100 μl and 5 μg/μl (D20 and D5 respectively). Two clean-up methods were evaluated:
1- Lithium Chloride (LiCl) precipitation. D20 samples were purified using a final concentration of 2.5 M LiCl. After incubation at -20°C for 2 hours the sample was centrifuged (16,000 g) at 4°C for 20 minutes (min). The pellet was then washed with 70% ethanol before drying.
2- Extraction with phenol:chloroform:isoamyl alcohol (25:24:1, pH: 5.2, PCI). D20 and D5 samples were mixed with one volume of PCI in a Phase-Lock-Gel™ (PLG) tube (Heavy Gel, Eppendorf). After mixing, the tube was centrifuged at room temperature (RT) for 5 min. The aqueous phase was transferred to a new PLG tube and a second extraction was done using chloroform. The aqueous phase was mixed with 100% ethanol and 0.1 volumes of 7.5 M NH4Acetate, incubated at -20°C from 2 hours to overnight (ON), followed by washing with 70% ethanol.
RNA amplification
cDNA synthesis
First strand cDNA was synthesized using 1 to 5 μg of total RNA (see below). RNA was mixed with 1 μl of T7-oligo (dT) primer (100 ng/μl, Ambion) in nuclease-free water to a total volume of 8 μl and added to EndoFree RT™ enzyme (Ambion) in a 21 μl reaction following the instruction manual. The reaction was incubated at 50°C for 2 hours.
Second strand cDNA was synthesized by mixing the first strand reaction with 95 μl nuclease-free water, 15 μl KOD XL Buffer (Novagen®), 15 μl of dNTPs (final concentration 0.2 mM), 1 μl RNase H (10 U/μl, Ambion), and 3 μl of KOD XL polymerase (Novagen®). Incubation was at 37°C for 5 min, followed by 94°C for 2 min, 65°C for 1 min and 75°C for 30 min. The reaction was stopped by 15 μl of 0.1 M NaOH/2 mM EDTA and incubated at 65°C for 10 min and neutralized by 15 μl of 0.1 M HCl.
Reactions carried out without reverse transcriptase were used as negative controls.
Purification of cDNA
cDNA was purified using the following methods:
1) cDNA clean-up column (DNA clear™ kit, Ambion) using the manufacturer's instructions.
2) PCI (pH:8.2) extraction with isopropanol precipitation at room temperature (Isopropanol method): reaction volume was adjusted to 200 μl with nuclease-free water, mixed with 200 μl of PCI and transferred to a PLG tube. After centrifugation (12,000 g) at RT for 5 min, the aqueous phase was transferred to a fresh PLG tube and a separate chloroform extraction was carried out. The final aqueous phase was precipitated using 1 μl of linear acrylamide (0.1 μg/μl, Ambion), 0.5 volumes of 7.5 M NH4Acetate and two volumes of isopropanol. The mixture was incubated at RT for 10 min and centrifuged (12,000 g) at RT for 20 min. The pellet was washed with 500 μl of 75% ethanol, centrifuged for 5 min, dried, and re-suspended in nuclease free water.
3) PCI extraction with ethanol precipitation at room temperature (ethanol at RT): Ethanol was replaced for isopropanol after PCI extraction and sample was immediately centrifuged (Modified from Zhao et al, [8]).
4) PCI extraction with cold ethanol precipitation (ethanol at CT): After PCI extraction, 0.1 volumes of 7.5 M NH4Acetate were added to the aqueous phase and mixed with 2.5 volumes of pre-chilled 100% ethanol. The mixture was incubated at -20°C for 2 hours and centrifuged at 4°C for 20 min, followed by washing with pre-chilled 70% ethanol and re-suspension.
In vitro transcription to generate amplified RNA
aRNA was generated by T7 MEGAscript™ kit (Ambion) with incorporation of aminoallyl-UTP (aa-UTP) in the process. 15 μl of purified cDNA was mixed with 3 μl of aaUTP solution (50 mM, Ambion), 12 μl of ATP, CTP, GTP mix (25 mM), 2 μl of UTP solution (75 mM), 4 μl of T7 10 × reaction buffer and 4 μl of T7 enzyme mix. A ratio of 1:1 for aaUTP: UTP was used [9]. The reaction mix was incubated at 37°C for 14 hours followed by treatment with 2 μl of DNase I (2 U/μl) at 37°C for 30 minutes.
Purification of amplified RNA
The following methods were used to purify aRNA:
1) Column purification with RNeasy kit (Qiagen) following the manufacturer's instruction.
2) PCI extraction (pH: 5.2): an equal volume of PCI was added to aRNA and transferred to the PLG tube as described above. After two rounds of PCI extraction, a separate chloroform extraction step was carried out followed by precipitation with NH4Acetate and ethanol at -20°C.
3) LiCl precipitation: aRNA was precipitated with a final concentration of 2.5 M LiCl. After cold incubation at -20°C for 2 hours, the sample was precipitated and washed as described above.
4) Guanidinium Isothiocyanate-phenol or TRI-reagent™ purification (guanidinium method). After addition of 100 μl of 4 M guanidinium isothiocyanate to the aRNA sample, it was purified using the PLG tubes and phenol as described by the manufacturer (PLG manual). Alternatively 1 ml of TRI-reagent™ (Sigma) was added to each aRNA sample, mixed well and transferred to a PLG tube. After adding 200 μl of chloroform, the solution was mixed by shaking, incubated at RT for 2 min and centrifuged (12,000 g) at 4°C for 20 min. The aqueous phase was then transferred to a new PLG tube and mixed with 600 μl of chloroform. After centrifuging (12,000 g) at 4°C for 10 min, the aqueous phase was transferred to a 1.5 ml tube and precipitated by adding 1 μl of linear acrylamide (0.1 μg/μl, Ambion), 0.1 volumes of 3 M NaAcetate and an equal volume of isopropanol followed by incubation at -20°C ON. The centrifuge and washing steps were carried out as described previously for PCI extraction.
Labelling of amplified RNA
Coupling reaction
Aminoallyl modified-aRNA (aa-aRNA) was coupled with monoreactive Cy3 and Cy5 dyes (Amersham). One vial of dye was dissolved in 40 μl of dimethylsulfoxide (DMSO) and divided into aliquots of 4 μl and dried by speed vacuum. To 10 μg of aa-aRNA in 6.7 μl of nuclease-free water, 10 μl of DMSO, and 3.3 μl of 0.3 M NaHCO3 (pH: 9) were added. The mixture was immediately transferred to Cy3 or Cy5 dried dyes and mixed by pipetting. Coupling reactions were carried out for 1 hour in the dark followed by quenching with 4.5 μl of 4 M hydroxylamine for 15 minutes.
Purification of labelled aRNA
Labelled targets were cleaned-up by the following methods:
1) Column purification with Qiagen RNA columns.
2) LiCl-Ethanol precipitation was carried out by adding 0.1 volumes of 4 M LiCl and 2.5 volumes of pre-chilled 100% ethanol. The mix was incubated at -20°C for 2 hours and centrifuged (12,000 g) at 4°C for 20 min followed by washing with 500 μl of pre-chilled 70% ethanol and re-spinning at 12,000 g for 5 min. The pellet was then air-dried and re-suspended in nuclease-free water.
3) PCI extraction (pH: 5.2): One round of PCI extraction followed by precipitation.
4) LiCl precipitation. To each reaction 12.5 μl of 7.5 M LiCl was added (2.5 M final concentration). The mixture was incubated at -20°C overnight followed by precipitation as described before.
Assessment of RNA quality
The NanoDrop® ND-1000 Spectrophotometer (NanoDrop Technologies) was used to determine: 260/280 ratio, to assess total RNA and aRNA purity; 320/550 and 320/650 ratios, to evaluate Cy3 and Cy5-labelled aRNAs respectively.
Quality of total RNA and patterns of amplified or labelled aRNA were evaluated using the Agilent-2100 Bioanalyzer with the RNA 6000 Nano Lapchip® kit (Agilent Technologies) and also by 1% denaturing agarose/formamide gels. Coupling efficiency for Cy5 dyes was assessed using the Agilent Bioanalyzer.
Hybridisation of cDNA microarrays
Expression microarrays containing 6528 pairs of duplicate cDNA spots were used (Cancer Research UK DNA Microarray Facility at the Institute of Cancer Research; CR-UK DMF Human 6.5 k genome-wide array).
Labelled targets from two cell lines, Cal51 and ZR-75-1-1, were generated using the optimized purification protocol. A total of 4 hybridizations were done: two slides were used with the same dye combination (replicates) and two slides with reversal of the dyes (dye reversal).
For each hybridization 2 μg of each Cy3 and Cy5-labelled targets (corresponding to 110–130 pmols of dye) were used. Hybridisation was performed as described http://www.crcdmf.icr.ac.uk with minor modifications. In brief, the volume was adjusted to 15 μl with nuclease-free water, to which 15 μl of pre-warmed (37°C) Amersham Hybridisation Buffer (Amersham Biosciences), 30 μl of deionised formamide, and 1 μl of Poly-dA (10 μg/μl, Amersham Biosciences) were added. After mixing, the samples were denatured at 92°C for 2 min and centrifuged at 12,000 g for 5 min. Slides were placed in Glass Array Hybridisation Cassettes (Ambion), targets were applied and cover slips fitted. Hybridization was carried out at 42°C overnight in a waterbath.
Washing was done in 2XSSC, 0.2%SDS at 42°C for 30 min, 2XSSC, 0.1%SDS at 42°C for 30 min, and 0.1XSSC, 0.1%SDS at RT for 10 min. Slides were then plunged ten times in 0.1XSSC to remove extra SDS with subsequent washes in 0.1XSSC two times for 2 minutes and once for one minute. Subsequently they were washed in 0.01XSCC for 15 seconds and submerged quickly in 96% ethanol followed by spin-drying at 500 rpm for 5 min.
Scanning, feature extraction and analysis
Slides were scanned using the ScanArray® 4000 microarray analysis system (Packard BioChip Technologies). Feature extraction was done using ScanArray Express software (Packard BioChip Technologies) and spots with high background were flagged manually. Data was transferred as tab delimited text files and analyzed using R mathematical program http://cran.us.r-project.org and Statistics for Microarray Analysis package (SMA), http://stat-www.berkeley.edu/users/terry/zarray/Software/smacode.html. Student's t-test and Chi-Square statistics were used for analysis of parametric and non-parametric factors respectively.
Bioinformatics
The longest annotated transcript for each of the analysed genes was computationally determined after combining the information available in RefSeq [http://www.ncbi.nlm.nih.gov/RefSeq/ and http://ncbi.nlm.nih.gov/books/bookres.fcgi/handbook/ch18d1.pdf, The NCBI handbook, Chapter 18. The Reference Sequence (RefSeq) Project] and Ensembl (Human v.16.33.1, http://www.ensembl.org) databases [15]. The search was automated by using BioPerl http://www.bioperl.org and Ensembl Perl modules on a Linux platform [16].
Reverse Transcription-PCR of selected transcripts
RT-PCR was done using RNA from cell line HCC-1187 to amplify a 50 base pair (bp) fragment of R38bsno and a 7130 bp fragment of Human guanine nucleotide exchange factor (p532) located 1 kb from the 3' end of the cDNA.
Reverse transcription was done using either 5 μg of total RNA with 1 μl of oligo-dT (16) primer (Roche Applied Biosciences) or 2 μg of aRNA with 25 pmols of gene specific reverse primer. The volume was adjusted to 9 μl with nuclease-free water, incubated at 70°C for 3 min and cooled on ice for 2 min. The following were added to the primed RNA: 4 μl of first strand buffer (BD Biosciences Clontech), 2 μl of 0.1 M DTT, 1 μl of RNase Inhibitor, 2 μl of 10 mM dNTPs, and 2 μl Powerscript Reverse™ Transcriptase (BD Biosciences Clontech). The reaction was incubated at 42°C for 2 hours, heat inactivated for 15 min at 70°C, and treated with 1 μl of Ribonuclease H (Promega) at 37°C for 30 min.
The primers used for PCR amplification were (p532 primers designed as described [17]):
R38bForward-5'GCTGAGTCCATGATGATTTC3'
R38bReverse-5'GCCTTTCTTTGCCTTCAGAC3'
p532Forward-5'AACTCACGGCAGTGGAGGGAAAG3'
p532Reverse-5'TGCTGTTCTGGTTGTTGGGGCTA3'
PCR conditions were:
For R38b-2 μl of reverse transcription product, 8 μl of MgCl2 (25 mM), 4 μl of 10 × PCR Buffer2 (Promega), 2 μl of 10 mM dNTPs, 1 pmol of each of the primers, and 1 μl of AmpliTaq polymerase (Promega). The volume was adjusted to 50 μl and thermal cycling carried out for 1 min at 95°C, 45 seconds at 51°C, 1 min at 72°C for 30 cycles with a 10 min extension period at 72°C in the last cycle. As a negative control, cDNA was excluded from the PCR reaction. The product was analysed on a 2.5% agarose gel.
For p532-2 μl of reverse transcription product, 2 μl of each of the primers (5 pmol/μl), and KOD XL Polymerase (Novagen®) following the product instructions. Thermal cycling was carried out for 10 seconds at 94°C and 7.1 minutes at 68°C for 35 cycles, followed by 10 minutes at 72°C. Negative control included a PCR reaction without cDNA. The PCR product was analysed on a 0.7% agarose gel.
Authors' contributions
AN planned the study, carried out the experiments, and drafted the manuscript. AAA performed the statistical analysis and participated in study design. NLB-M performed bioinformatics analysis. SA supervised bioinformatics analysis. JDB participated in study design, drafting the manuscript and supervised statistical analysis. CC supervised study design and experiments, drafting the manuscript, and carried out final editing. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
NLB-M is the recipient of a Praxis XXI doctoral fellowship from FCT, Ministry of Science, Portugal; AAA is the recipient of MRC Clinical Training and Sackler Fellowships. Research in the Cancer Genomics Program is funded by grants from Cancer Research UK.
Contributor Information
Ali Naderi, Email: an258@cam.ac.uk.
Ahmed A Ahmed, Email: aaa42@cam.ac.uk.
Nuno L Barbosa-Morais, Email: nlbmb2@cam.ac.uk.
Samuel Aparicio, Email: saparici@hgmp.mrc.ac.uk.
James D Brenton, Email: jdb1003@cam.ac.uk.
Carlos Caldas, Email: cc234@cam.ac.uk.
References
- Van De Vijver MJ, He YD, Van'T Veer LJ, Dai H, Hart AAM, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Van Gelder RN, Von Zastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc Natl Acad Sci USA. 1990;87:1663–1667. doi: 10.1073/pnas.87.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van't Veer LJ, Dal H, Van de Vijver MJ, He YD, Hart AAM, Mao M, Peterse HL, Van der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Wang E, Miller LD, Ohnmacht GA, Liu ET, Marincola FM. High-fidelity mRNA amplification for gene profiling. Nat Biotechnol. 2000;18:457–459. doi: 10.1038/74546. [DOI] [PubMed] [Google Scholar]
- Hughes TR, Mao M, Jones AR, Buchard J, Marton MJ, Shannon KW, Lefkowitz SM, Ziman M, Schelter JM, Meyer MR, et al. Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat Biotechnol. 2001;19:342–347. doi: 10.1038/86730. [DOI] [PubMed] [Google Scholar]
- Hu L, Wang J, Baggerly K, Wang H, Fuller GN, Hamilton SR, Coombes KR, Zhang W. Obtaining reliable information from minute amounts of RNA using cDNA microarrays. BMC Genomics. 2002;3:16. doi: 10.1186/1471-2164-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Hastie T, Whitfield ML, Borresen-Dale AL, Jeffery SS. Optimization and evaluation of T7 based RNA linear amplification protocols for cDNA microarray analysis. BMC Genomics. 2002;3:31. doi: 10.1186/1471-2164-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 'T Hoen PAC, De Kort F, Van Ommen GJB, Den Dunnen JT. Fluorescent labelling of cRNA for microarray applications. Nucleic Acids Res. 2003;31:e20. doi: 10.1093/nar/gng020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A, Carmenate T, Delgado M, Gonzalez S. Recombinant Opc meningococcal protein, folded in vitro, elicits bactericidal antibodies after immunization. Vaccine. 1997;15:751–8. doi: 10.1016/S0264-410X(96)00198-3. [DOI] [PubMed] [Google Scholar]
- Winfrey MR, Rott MA, Wortman AT. Molecular Biology for the Laboratory. NJ: Prentice Hall; 1997. Unravelling DNA; pp. 1–2. Laboratory 8 Addendum. [Google Scholar]
- Ferri GL, Isola J, Berger P, Giro G. Direct eye visualization of Cy5 fluorescence for immunohistochemistry and in situ hybridization. J Histochem Cytochem. 2000;48:437–44. doi: 10.1177/002215540004800314. [DOI] [PubMed] [Google Scholar]
- Kimura E, Aoki S, Kikuta E, Koike T. A macrocyclic Zinc (II) fluorophore as a detector of apoptosis. Proc Natl Acad Sci USA. 2003;100:3731–6. doi: 10.1073/pnas.0637275100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenssen TK, Langaas M, Kuo WP, Smith-Sorensen B, Myklebost O, Hovig E. Analysis of repeatability in spotted cDNA micoarrays. Nucleic Acid Res. 2002;30:3235–3244. doi: 10.1093/nar/gkf441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clamp M, Andrews D, Barker D, Bevan P, Cameron G, Chen L, Clark L, Cox T, Cuff J, Curwen V, et al. Ensembl 2002: accommodating comparative genomics. Nucleic Acids Res. 2003;31:38–42. doi: 10.1093/nar/gkg083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, Fuellen G, Gilbert JGR, Kork I, Lapp H, et al. The Bioperl Toolkit: Perl modules for the life sciences. Genome Res. 2002;12:1161–8. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Fockler C, Barnes WM, Higuchi R. Effective amplification of long targets from cloned inserts and human genomic DNA. Proc Natl Acad Sci USA. 1994;91:5695–5699. doi: 10.1073/pnas.91.12.5695. [DOI] [PMC free article] [PubMed] [Google Scholar]