

Abstract
In Salmonella enterica, the protein acetyltransferase (Pat) enzyme is part of the sirtuin-dependent acylation/deacylation system (SDPADS) that modulates the activity of several proteins via the acylation of lysine residues critical to their activities. Pat is a ~98 kDa protein that, with two distinct domains, an N-terminal acyl-CoA synthetase (NDP-forming) domain (~700 aa) and a C-terminal acetyltransferase domain (~160 aa), with homology to proteins of the Gcn5-related N-acetyltransferase (GNAT) superfamily. Although the role of the GNAT-like domain is likely responsible for the catalytic activity of Pat, the role of the N-terminal domain remains unclear. Here we report the use of positive selection for identification of residues critical for Pat enzyme activity. This approach revealed seven residues that, when changed, resulted in drastic loss of Pat activity in vitro which caused a discernable loss-of-function phenotype. Five of the seven residues were located in the N-terminal region of Pat and two were located in the GNAT-like domain. Each single-amino-acid variant had a circular dichroism spectrum that differed from that of the wild-type Pat protein, suggesting that loss of enzymatic activity in the mutant proteins was likely due to an inability to acquire its biologically active fold.
Keywords: Post-translational modification, Protein acetylation, Lysine acylation, Protein acetyltransferases, Metabolic regulation, Acetate metabolism
1. Introduction
Nε-lysine (Nε-Lys) acetylation plays a critical role in the regulation of protein function in all forms of life (Jones and O’Connor, 2011; Thao and Escalante-Semerena, 2011). Although the role of Nε-Lys acetylation has been extensively studied in eukaryotes, our understanding of the role of this posttranslational modification in prokaryotic cell physiology is limited. Recent proteomic studies have proposed that Nε-Lys acetylation may be as extensively used in prokaryotes as it is in eukaryotes ( Wang et al., 2010; Yu et al., 2008; Zhang et al., 2009). In one of these studies, the S. enterica protein acetyltransferase (Pat) enzyme was specifically implicated in the modification of several metabolic enzymes, namely, glyceraldehyde phosphate dehydrogenase (GapA), isocitrate lyase (AceA) and isocitrate dehydrogenase kinase/phosphatase (AceK) (Wang et al., 2010).
Pat belongs to the Gcn5-related N-acetyltransferase (GNAT) superfamily of acyltransferases (pfam00583) that is conserved in all domains of life. GNATs are best described as enzymes that use acyl-coenzyme A as a donor for acylation of Lys residues of proteins and small molecules Neuwald and Landsman, 1997; Vetting et al., 2005). These enzymes belong to a larger superfamily of lysine acetyltransferases [LATs (Yang, 2004) or KATs (Allis et al., 2007) (formerly known as histone acetyltransferases (HATs) (Kouzarides, 1999; Neuwald and Landsman, 1997)
In S. enterica, Pat inactivates acetyl-CoA synthetase (Acs; AMP-forming; EC 6.2.1.1; (Starai and Escalante-Semerena, 2004)) by acetylating residue Lys609, and the NAD+-dependent CobB sirtuin-like deacetylase reactivates Acs by deacetylation of AcsAc (Fig. 1) (Starai et al., 2002 ;Starai et al., 2003, 2004). Acs is a ubiquitous enzyme important for the conversion of acetate to acetyl-coenzyme A (Ac-CoA), a necessary step for its utilization as carbon and energy source (Starai and Escalante-Semerena, 2004).
Fig. 1. Posttranslational control of Acs activity by acetylation/deacetylation.
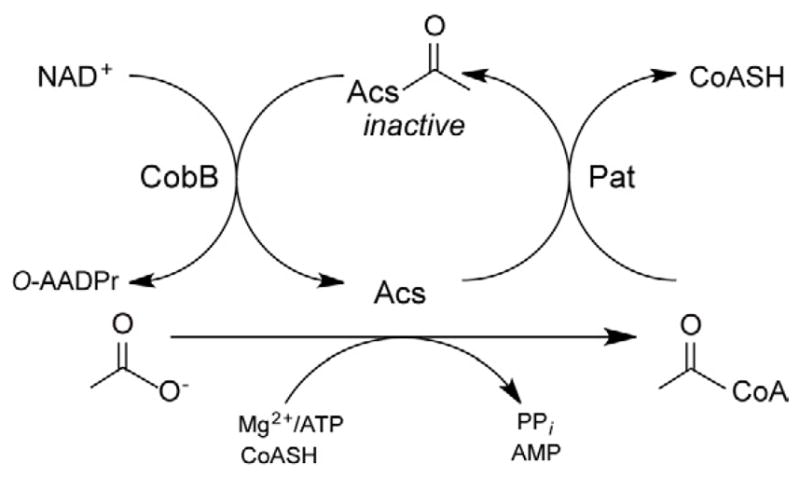
Acs is inactivated by Ac-CoA-dependent Pat acetylation. AcsAc is reactivated by NAD+-dependent CobB deacetylation. O-AADPr, O-Acetyl-ADP-ribose. Figure adapted from (Starai and Escalante-Semerena, 2004).
Acs was the first enzyme known to be under Nε-Lys acetylation control (Starai et al., 2002). This mechanism of control of Acs activity was later expanded to mammalian Acs enzymes (Hallows et al., 2006 ;Schwer et al., 2006). Other proteins reported to be modified by Pat include propionyl-coenzyme A synthetase (PrpE), which was shown to be inactivated by Pat-dependent propionylation (Garrity et al., 2007). More recently, Pat was shown to acetylate a critical Lys residue of the E. coli RcsB transcriptional regulator, with concomitant inhibition of the DNA binding activity of the protein (Thao et al., 2010). The latter was the first report of Nε-Lys acetylation control of the activity of a bacterial regulatory protein.
Pat is a multidomain protein that appears to have two distinct domains, an N-terminal domain that has similarity to members of the NDP-forming acyl-CoA synthetase family (COG1042) and a C-terminal domain that has similarity to GNATs (Fig. 2A). Notably, Pat lacks the catalytic histidine residue needed for NDP-forming acyl-CoA synthetase activity and cannot catalyze synthesis of Ac-CoA from acetate, ATP or CoA (Starai and Escalante-Semerena, 2004).
Fig. 2. Residues critical for Pat activity identified from loss-of-function mutations.

A. Pat appears to be a multidomain protein that has a predicted acetyltransferase fold belonging to the GNAT superfamily (CDD-search (Marchler-Bauer et al., 2009)). Seven residues (eight amino acid changes) were identified by localized mutagenesis as being important for pat function. B. Growth behavior on 10 mM acetate. In the presence of a functional allele of pat, a cobB strain has a severe growth defect (top left box). Upon inactivation of pat, growth is restored (bottom left box). All eight mutant alleles restored growth on acetate, i.e. they behaved as non-functional alleles of pat. C. Western blot analysis showed that variant Pat proteins were stable, ruling out the possibility that the observed effect was due to the absence of Pat.
Although initial biochemical characterization of the S. enterica Pat enzyme was recently reported (Thao and Escalante-Semerena, 2011), the contribution of the N-terminal domain to Pat function has not been explored. From the above-mentioned studies, we learned that Pat has two distinct Ac-CoA-binding sites, that Ac-CoA binding to Pat is positively cooperative in nature, that Pat oligomerizes into a tetramer upon Ac-CoA binding in vitro and that the N-terminal domain is required for oligomerization and activity (Thao and Escalante-Semerena, 2011).
The goal of the work reported here was to identify residues of S. enterica Pat that were critical for activity, such that substitutions at those positions would abolish enzyme activity. Single amino acid substitutions at seven positions dramatically reduced Pat activity. Five of these amino acid changes occurred across the N-terminal domain of the protein. Pat variants were isolated and their activities were assessed in vitro. Seven of the 8 Pat variants studied lost >94% of their activity; the severe loss of activity was attributed to incorrect folding, as detected by circular dichroism spectroscopy.
2. Materials and methods
2.1. Localized mutagenesis of pat
Point mutations were introduced into pat using a localized mutagenesis protocol described elsewhere (Hong and Ames, 1971). In such a procedure, the high-frequency-of-transduction, generalized transducing bacteriophage P22 mutant (HT 105/1, int201) was grown on a strain containing a target gene (Davis et al., 1980; Schmieger and Backhaus, 1973) and the phage lysate was mutagenized using hydroxylamine. We mutagenized strain JE6579, which carried a yfiD::kan+ mutation near pat+ (Table 1). The absence of YfiD did not affect acetate utilization under the conditions tested.
Table 1.
Strains and plasmids used in this study.
| Strainsa | ||||
|---|---|---|---|---|
|
| ||||
| Strain number | Genotype | Source | ||
|
| ||||
| TR6583 | metE205 ara-9 | K. Sanderson via J. Roth | ||
|
| ||||
| Derivatives of TR6583
| ||||
| JE6579 | yfiD181::kan+ | Lab collection | ||
| JE6318 | cobB1176::Tn10d(Tc) pta103::cat+ | Lab collection | ||
| JE13928 | cobB1176::Tn10d(Tc) pta103::cat+pat2 ::kan+ | |||
| JE12412 | cobB1176::Tn10d(Tc) pta103::cat+yfiD181 ::kan+pat5 | |||
| JE12413 | cobB1176::Tn10d(Tc) pta103::cat+yfiD181 ::kan+pat6 | |||
| JE12414 | cobB1176::Tn10d(Tc) pta103::cat+yfiD181 ::kan+pat7 | |||
| JE12415 | cobB1176::Tn10d(Tc) pta103::cat+yfiD181 ::kan+pat8 | |||
| JE12416 | cobB1176::Tn10d(Tc) pta103::cat+yfiD181 ::kan+pat9 | |||
| JE12417 | cobB1176::Tn10d(Tc) pta103::cat+yfiD181 ::kan+pat10 | |||
| JE12418 | cobB1176::Tn10d(Tc) pta103::cat+yfiD181 ::kan+pat11 | |||
| JE12419 | cobB1176::Tn10d(Tc) pta103::cat+yfiD181 ::kan+pat12 | |||
| JE9314 | E. coli C41(DE3) yfiQ11::kan+ | Lab collection | ||
|
| ||||
| Plasmids | ||||
|
| ||||
| pTEVb | pat Allele | Mutation | Primersc | |
|
| ||||
| pPAT9 | pat5 | D165N (gac – aac) | 5′-gtccaatactattcttaactgggcgcaacagcg -3′ | |
| 5′-cgctgttgcgcccagttaagaatagtattggac -3′ | ||||
| pPAT10 | pat6 | R170H (cgt – cat) | 5′-gactgggcgcaacagcatgaaatgggcttttc -3′ | |
| 5′-gaaaagcccatttcatgctgttgcgcccagtc -3′ | ||||
| pPAT20 | pat7 | A220T (gcc – acc) | 5′-gttttgtttccaccgcccgtagcg -3′ | |
| 5′-cgctacgggcggtggaaacaaaac -3′ | ||||
| pPAT11 | pat8 | R450W (cgg – tgg) | 5′-ccagaagcaactgtgggaaacgccagcgttg -3′ | |
| 5′-caacgctggcgtttcccacagttgcttctgg -3′ | ||||
| pPAT12 | pat9 | D592N (gat – aat) | 5′-gtggtggtcgagcacaatccggtgtttg -3′ | |
| 5′-caaacaccggattgtgctcgaccaccac -3′ | ||||
| pPAT13 | pat10 | A780T (gcc – acc) | 5′-ctacgatcgagaaatgacctttgtggccgtg -3′ | |
| 5′-cacggccacaaaggtcatttctcgatcgtag -3′ | ||||
| pPAT14 | pat11 | A780V (gcc – gtc) | 5′-ctacgatcgagaaatggtctttgtggccgtg -3′ | |
| 5′-cacggccacaaagaccatttctcgatcgtag -3′ | ||||
| pPAT15 | pat12 | A811 V (gcc – gtc) | 5′-gtagatgccgaatttgtcgtattggtgcgttc -3′ | |
| 5′-gaacgcaccaatacgacaaattcggcatctac -3′ | ||||
All strains are derivatives of S. enterica sv. Typhimurium LT2
Plasmids derived from pTEV cloning vector pKLD66 (Rocco et al., 2008) for overproduction and purification of products.
Primers used to introduce the amino acid substitution. Nucleotide changes are underscored.
A hydroxylamine-mutagenized P22 lysate grown on strain JE6579 was crossed with strain JE6318 (metE ara cobB::Tn10d(Tc) pta::cat+; Table 1). The pta::cat+ allele was needed in the recipient strain to abolish acetate assimilation via the acetate kinase (AckA)/phosphotransacetylase (Pta) pathway, thus eliminating all background growth on acetate.
Inheritance of the yfiD::kan+ marker was selected for on nutrient broth (NB, Difco) + kanamycin (Sigma, 25 μg/ml) plates. The resulting kanamycin-resistant (Kmr) transductants were replica-printed onto minimal acetate plates [no-carbon E (NCE)] (Berkowitz et al., 1968) supplemented with 10 mM acetate to select for strains that inherited null alleles of pat and thus could grow. We note that the minimal medium plates used in these experiments did not contain kanamycin; instead, the selective pressure we applied was growth.
Kmr transductants that grew on minimal acetate plates were freed of phage (Chan et al., 1972) and P22 lysates of each of the strains were used to reconstruct the original mutant strains using the cob pat strain (JE6318) as recipient. Reconstructed strains that were resistant to kanamycin and grew on minimal acetate plates were used in subsequent studies. All growth conditions were performed at 37°C. The nature of the point mutations in pat was identified by DNA sequencing using BigDye® Terminator v3.1 (Applied Biosystems) protocols; the reactions were resolved and analyzed at the University of Wisconsin Biotechnology Center.
2.2. Growth analysis of strains carrying null alleles of pat
A 2-μl aliquot of an overnight culture grown in nutrient broth was used to inoculate 198 μl of 10 mM acetate medium (NCE + acetate, pH 7). Growth was monitored at OD650 over a 60-h time period at 37°C with shaking in a 96-well microtiter plate using an ELx808 Ultra microplate reader (Bio-Tek Instruments). Data were obtained from three independent experiments from individual cultures done in triplicate for each strain.
2.3. Antibody preparation and western blot analysis
Untagged wild-type Pat protein was used to elicit rabbit polyclonal antibodies (Harlan Laboratories). An overnight culture of each strain was used to inoculate 150 ml of nutrient broth in 500 ml flasks at a 1:100 starter culture to media ratio. The cultures were grown at 37°C to a cell density of OD650 of 0.7, 50-ml cultures were harvested by centrifugation at 1,825 × g for 45 min at 4°C and then re-suspended in 1.0 ml of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (50 mM, pH 7.5) containing lysozyme (1 mg/ml), DNAse I (25 μg/ml) and phenylmethylsulfonyl fluoride (PMSF, 0.5 mM). Cells were lysed by sonication for two 1 min intervals using a Heat Systems Ultrasonics sonicator (Model W-10) at setting 3 on ice. Cell debris was removed by centrifugation at 16,000 × g for 15 min at 4°C and 800 μg of soluble extract resolved by 12% SDS-PAGE (Laemmli, 1970). Binding of α-Pat antibodies to blots was visualized using alkaline-phosphatase-conjugated goat α-rabbit immunoglobulin G (ThermoFisher) and NBT/BCIP chemistry. Bands were detected using a Fotodyne Digital Imaging system and TotalLab v2005 software. The experiment was performed in duplicate using two independent cultures.
2.4. Construction of pat overexpression plasmids
Strains and plasmids used in this study are listed in Table 1. The 2661-bp pat gene of S. enterica sv. Typhimurium LT2 was inserted into plasmid pKLD66 (Rocco et al., 2008) using KpnI and HindIII sites as reported previously (Thao et al., 2010). The resulting overexpression plasmid, pPAT8, directs the synthesis of wild-type Pat protein with an N-terminal hexahistidine-maltose binding protein (His6-MBP) tag cleavable by recombinant tobacco etch virus (rTEV) protease (Blommel and Fox, 2007; Parks et al., 1994) Plasmid pPAT8 was used as template for site-directed mutagenesis using the QuikChange XL kit (Stratagene). Primers for generation of plasmids for overexpression of the mutant alleles of pat are listed in Table 1. DNA sequencing was used to verify the presence of null pat alleles on plasmids constructed during this work.
2.5. Overproduction and purification of Acs and Pat proteins
2.5.1. Acs
Wild-type Acs protein was isolated as described using chitin column chromatography (NEB) (Starai et al., 2002), stored in HEPES buffer (50 mM, pH 7.5) containing NaCl (150 mM) and glycerol (20% v/v) and drop-frozen in liquid nitrogen prior to storage at −80°C.
2.5.2. Pat proteins
His6-MBP tagged wild-type and variant Pat proteins were purified by a two-step process at 4°C similarly to a previously described method (Thao et al., 2010). Briefly, plasmids carrying individual pat alleles were transformed into JE9314 (E. coli C41(DE3) yfiQ::kan+; Table 1) and 5 ml of an overnight culture was used to inoculate 500 ml of lysogeny broth (LB) medium (1.0% tryptone, 0.5% yeast extract, 0.5% NaCl) containing ampicillin (200 μg/ml). Cultures were grown at 37°C with shaking to an OD600 of 0.6 prior to induction of pat expression by isopropyl-β-D-thiogalactopyranoside (IPTG, 1 mM); induced cultures were shaken overnight at 28°C. Cells were harvested by centrifugation at 8,394 × g for 15 min at 4°C.
2.5.3. Purification step 1
Cells were resuspended in 10 ml of resuspension buffer [binding buffer A (Hepes buffer (20 mM, pH 7.5), NaCl (500 mM) and imidazole (20 mM)) plus lysozyme (1 mg/ml), DNAse I (25 μg/ml) and phenylmethanesulfonyl fluoride (PMSF, 0.5 mM)]. Cells were lysed by sonication for 2 min (30 s, 50% duty) on ice using a 550 Sonic Dismembrator (Fisher Scientific) at setting 4. Clarified cell lysates were obtained after centrifugation for 45 min at 4°C at 43,667 × g followed by filtration of the supernatant through a 0.2 μm filter (Millipore). Samples were loaded onto 1 ml Ni-NTA Superflow resin (Qiagen) pre-equilibrated with binding buffer A, and TEV-cleavable His6-MBP tagged Pat protein eluted with elution buffer [HEPES buffer (20 mM, pH 7.5), NaCl (500 mM) and imidazole (500 mM)] following a wash step with the same buffer system containing a lower amount of imidazole (40 mM).
To cleave the tag, His7-TEV protease (rTEV) was purified as described (Blommel and Fox, 2007) and cleavage of tagged Pat proteins was performed as follows. rTEV was added to the eluted protein supplemented with dithiothreitol (DTT, 1 mM) at a 1:50 protease:tagged mg protein ratio and the mixture was incubated at room temperature for 3 h.
2.5.4. Purification step 2
The protein mixture was dialyzed against binding buffer B (HEPES buffer (20 mM, pH 7.5), NaCl (500 mM) and tris(2-carboxyethyl)phosphine hydrochloride (TCEP, 0.5 mM)) plus ethylenediaminetetra-acetic acid (EDTA, 0.5 mM). Prior to loading onto the 1 ml Ni-NTA Superflow resin, DTT and EDTA were extensively dialyzed away at 4°C in binding buffer B. Protein was eluted off the column using a 40 mM imidazole wash step, followed by 500 mM imidazole, which allowed us to separate tagged from untagged protein. Untagged Pat protein eluted in the flow step. Pat proteins were stored in Hepes buffer (50 mM, pH 7.5) containing NaCl (150 mM) and glycerol (50% v/v), and stored at 4°C.
2.6. Pat in vitro activity assays
Reaction mixtures (total volume = 30 μl) contained Pat (3 μM), Acs (5 μM), [14C, C-1]-Ac-CoA (25 μM) and tris(2-carboxyethyl)phosphine (TCEP, 1 mM) in 50 mM Hepes (pH 7.5) buffer. Reactions were incubated at 37°C for 30 min, followed by quenching with 6 μl 6X SDS-PAGE loading buffer and heating at 95°C for 2 min. A 6-μl sample (25 pmol of Acs protein) from each quenched reaction was resolved in a 12% SDS-PA gel, the gel was dried and exposed to a phosphor screen for 24 h. Phosphor images were obtained and analyzed using a Typhoon Trio Variable Mode Imager and ImageQuant v5.2 software (GE Healthcare). A no-enzyme reaction was used for background subtraction. Reactions were performed in triplicate.
2.7. Circular dichroism (CD) studies
Circular dichroism spectroscopy was performed on an Aviv Model 202SF circular dichroism spectrometer at the Biophysics Instrumentation Facility at the University of Wisconsin-Madison. Far-UV CD spectra were obtained at 4°C from protein samples (0.15 mg/ml) in 20 mM sodium phosphate buffer pH 7.4 containing NaCl (70 mM) and glycerol (5%, v/v). The spectra were obtained in a 0.1-cm path length cuvette every 0.5 nm with a spectral band width of 1 nm and 5 s averaging time. Data were converted to molar ellipticity units by using the mean residue mass of 110.40 Da.
3. Results and discussion
3.1. In vivo strategy for identification of Pat residues critical for function
To identify residues important for Pat activity in vivo, we exploited the fact that inactivation of pat restores growth of a CobB (deacetylase)-deficient strain on 10 mM acetate. We previously showed that inactivation of pat in a cobB strain restored growth on 10 mM acetate because Acs was no longer inactivated by acetylation (Starai and Escalante-Semerena, 2004).
From Fig. 1, one can infer that in a CobB-deficient strain, Acs would remain acetylated due to the absence of CobB deacetylase activity. As a result, acetylated (i.e. inactive) Acs would not convert acetate to Ac-CoA and growth would be arrested (Starai et al., 2004); a control experiment confirmed this prediction (Fig. 2B, left panel, top box).
Placement of a selectable marker near the pat gene (a kanamycin insertion in yfiD) allowed us to mobilize mutant pat alleles into genetic backgrounds of interest by crossing the mutagenized P22 phage lysate with strain JE6318 (cob pta). Under the conditions of our search, we isolated Kmr transductants that grew on 10 mM acetate. Using this approach, we identified 35 pat alleles, each containing a single nucleotide change. In all cases, loss of Pat function was observed. Twenty-seven of the above-mentioned changes introduced premature stop codons throughout the pat gene (data not shown). Notably, a termination codon (amber, TAG) introduced by a C→T transition at nucleotide 2590 indicated that the last 23 amino acids of the GNAT domain are crucial to enzyme activity and stability.
Eight mutant pat alleles contained single-nucleotide changes that resulted in variants of Pat with single amino acid substitutions (Fig. 2A); all eight Pat variants were further characterized. Five of these variants (i.e. PatD165N, PatR170H, PatA220T, PatR450W and PatD592N) had two notable features. Firstly, all substitutions were located within the N-terminal region, outside the GNAT domain of Pat, and secondly, they had reduced acetyltransferase activity (Fig. 2A,B). We also identified two residues within the GNAT domain (A780, A811) which, when changed to A780T/V or A811V, abolished the acetyltransferase activity of Pat (Fig. 2A,B).
Of note, no amino acid change was found in the predicted interdomain region (Fig. 2A), and only one of the residues critical to Pat activity, i.e. D592, was conserved in its respective domain family (Sánchez et al., 2000). All proteins encoded by the mutant alleles of pat were stable, as assessed by western blot analysis (Fig. 2C) indicating that the loss of function was not due to the absence of the mutant protein.
3.2. The enzymatic activity of Pat variants is dramatically reduced
We overproduced, isolated and quantified in vitro the enzymatic activity of each of the Pat variants. The activity of seven variants was reduced >94% relative to the activity of the wild-type protein, with one variant (PatR170H) being about one third as active as wild-type Pat (Fig. 3). Of note, none of the variants including the PatR170H variant had sufficient activity in vivo to raise the level of acetylated Acs (inactive) to the point where growth on 10 mM acetate would be prevented.
Fig. 3. In vitro activity assays of Pat variants show loss in enzyme activity.

A) The variant Pat proteins containing single amino acid changes were incubated with Acs protein and [14C, C-1]-Ac-CoA. B) The amount of radioactive label transferred to Acs by the variants was plotted as percent of that transferred by wild-type Pat and are reported as mean ± s.d. from experiments performed in triplicate.
3.3. The single amino acid substitutions in the Pat variants result in substantial conformational changes that compromise the activity of the enzyme
We used circular dichroism spectroscopy to investigate the effect of these substitutions on the conformation of Pat. As shown in Fig. 4, native Pat has a double minima at 208 nm and 222 nm characteristic of α-helical proteins (Kelly et al., 2005). The α-helical content of Pat upon mutation of the identified residues was decreased in all of the proteins to varying degrees (Table 2), suggesting that the loss of enzymatic activity was likely due to incorrect protein folding. Not surprisingly, attempts to produce larger amounts of mutant protein (from >1 L cultures, or at protein concentrations >0.3 mg/ml) was problematic, since the mutant proteins were prone to aggregate-forming higher-order oligomers eluting in the void volume in gel filtration experiments (data not shown). We were unable to perform circular dichroism spectroscopy studies at temperatures above 4°C due to the instability of the variants and wild-type Pat, a problem that was likely compounded by the necessity for minimal buffer conditions when using this method.
Fig. 4. CD spectra of wild-type and mutant Pat proteins.

Far UV spectra of wild-type (black tracing) and mutant Pat enzymes show the proteins have different secondary structures, suggesting that loss of catalytic activity in the mutant proteins was likely due to an inability to acquire the correct structural fold.
Table 2.
Helical content of native Pat and its mutantsa.
| WT | D165N | R170H | A220T | R450W | D592N | A780T | A780V | A811V |
|---|---|---|---|---|---|---|---|---|
| 20.6 % | 10.9 % | 19.4 % | 16.4 % | 9.9 % | 12.1 % | 11.7 % | 16.9 % | 17.9 % |
Based on the 222 nm values. (Chen et al., 1974).
Clearly, the substitutions we identified further destabilized an already inherently unstable wild-type Pat protein (Gasteiger et al., 2003; Guruprasad et al. 1990). Tag removal by rTEV treatment led to visible aggregation; however, the protein did not completely fall out of solution, but the addition of NaCl (0.5 M) and TCEP (0.5 mM) was needed to maintain wild-type Pat and its variants in solution during the purification process. The addition of glycerol (20% v/v) was necessary to prevent wild-type Pat from aggregating, especially at higher concentrations; this, however, did not help maintain the mutant proteins in solution.
3.4. Potential roles for the residues of Pat identified from this work
Although we cannot yet assign function to the seven amino acids identified by this study as important for Pat activity, sequence analysis suggests roles for at least three of the residues. As mentioned above, only D592 is conserved in its respective superfamily domain (acyl-CoA synthetases; ACS (NDP-forming)) (Sánchez et al., 2000). This residue is located within a region of the protein that has been proposed to be involved in nucleotide binding, as shown by tryptic digestion and mass spectrometry for E.coli succinyl-coenzyme A synthetase (SCS) (Joyce et al., 1999, Joyce et al., 2000). Many residues important for SCS activity are conserved in the ACS (NDP-forming) superfamily; however, the domain organization of the members appears to be modular (Sánchez et al., 2000). There are several notable differences between S. enterica Pat and SCS. For example, the α- and β-subunits of SCS are fused in Pat, and within the β-subunit of Pat the domains appear to be in reverse order (Sánchez et al., 2000). Because of this, and the fact that a direct interaction has not been shown even though structures of SCS have been reported (Fraser et al., 1999; Wolodko et al. 1994), the role of this residue is speculative.
The other two residues, A780 and A811, are likely important since they are located within the core GNAT domain. The structures of many GNATs have been solved, revealing a conserved fold important for Ac-CoA binding and catalysis despite structure-based alignments showing low pairwise sequence identity (Vetting et al., 2005) (Fig. 5). Although neither of these residues is conserved, A780 falls in strand β2 and A811 falls in strand β4 (Fig. 5), two structural features that constitute the core Ac-CoA binding fold of GNATs (Dyda et al., 2000; Vetting et al., 2005). It is likely that mutations at both of these residues resulted in changes in folding or conformation. The different CD-spectra we obtained from these variant Pats as compared to wild-type Pat support this idea (Fig. 4).
Fig. 5. Secondary structure prediction of S. enterica Pat.

The prediction of secondary structures was carried out by using PSIPRED (Jones, 1999). Residues identified as being important for pat function and enzyme activity are highlighted with black background and yellow bold typeface font. Secondary structure elements (designated β0 to β6) are associated with the core GNAT fold (Vetting et al., 2005). Secondary structures are shown in purple (alpha helices) and red (β sheets).
Interestingly, the glutamate that acts as the general base catalyst of yeast GCN5 (yGCN5), Glu173 (Langer et al., 2001; Tanner et al., 1999), which resides within the core GNAT domain (Fig. 5), is also conserved in Pat (Fig. 6A). Site-directed mutation of this residue in Pat, Glu809, from a glutamate to glutamine (GAA → CAG), which has a similar side chain structure but no acidic group required for it to function catalytically, resulted in over 96% loss in activity in vitro (Fig. 6B). Although, the E809Q change had a drastic effect on protein folding (Fig. 6C), the variant was not defective in vivo (the mutation on the chromosome was introduced as described (Blank et al., 2011); data not shown). This result is unlike the eight-mutant non-functional pat alleles that restored growth on acetate in the cob-defective strain (Fig. 2B)). These data suggest that Glu809 does not behave as a catalytic residue. However, it is important to note that there have been examples where a general base catalyst appeared unnecessary for catalysis (see review (Vetting et al., 2005)). The yeast Hpa2 GNAT has no obvious catalytic residue that could function as a general base, and it was proposed that a net positive electrostatic potential of the active site might assist in deprotonation of the amino group of the lysine residue (Angus-Hill et al., 1999).
Fig. 6. A putative active-site glutamate is present in Salmonella Pat, but is likely not important for catalysis.

A. The putative general base involved in histone acetylation, Glu173 in yeast GCN5, is conserved in Pat, Glu809 (highlighted in black with asterisk). The alignment was generated with reference to the SCOP superfamily of acyl-CoA N-acetyltransferases (SCOP 55729, (Gough and Chothia, 2002)) and Dyda, et al. (Dyda et al., 2000). The region designated motif A (indicated by box) corresponds to the four (motifs C, D, A, B) (Neuwald and Landsman, 1997), which are hallmarks of GNATs. Motif A is the longest and most conserved motif, encompassing a region crucial for Ac-CoA binding (Dyda et al., 2000). B. Glu809 in the predicted Ac-CoA binding domain appears critical for acetyltransferase activity in vitro, but the severe loss in Pat activity was likely due to significant structural changes upon mutation from Glu-to-Gln at residue 809, as observed from circular dichroism studies (C).
3.5. Conclusions
In this work, we contrived a genetic selection for the identification of residues that are critical for S. enterica Pat function. Using this method we identified seven out of 886 residues in the protein that, when changed, resulted in the enzyme losing >95% of its activity. It was unexpected to find that <1% of the residues in Pat (7/886) were critical for function. These results show that demanding growth of a CobB deacetylase-deficient strain on 10 mM acetate is a strong positive selection for null pat alleles. Collectively, the data suggest that changes in these residues lead to substantial conformational changes and protein instability, indicating that the critical nature of each of the alluded side chains is for proper enzyme folding and that, although other substitutions may be tolerated, those reported in this work are not. These findings, in addition to recent biochemical and thermodynamic analyses of the wild-type Pat enzyme, advance our understanding of Pat, a multidomain, oligomeric GNAT used by the cell to control metabolism.
Acknowledgments
This work was supported by USPHS grant R01-GM62203 to J.C.E.-S.
S.T. was supported in part by the USPHS Molecular Biosciences Training Grant T32-GM07215, and NRSA Predoctoral Fellowship F31-GM083668. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Sandy Thao, Email: sthaovue@gmail.com.
Jorge C. Escalante-Semerena, Email: escalante@bact.wisc.edu.
References
- Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, Pillus L, Reinberg D, et al. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–636. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Angus-Hill ML, Dutnall RN, Tafrov ST, Sternglanz R, Ramakrishnan V. Crystal structure of the histone acetyltransferase Hpa2: A tetrameric member of the Gcn5-related N-acetyltransferase superfamily. J Mol Biol. 1999;294:1311–1325. doi: 10.1006/jmbi.1999.3338. [DOI] [PubMed] [Google Scholar]
- Berkowitz D, Hushon JM, Whitfield HJ, Jr, Roth J, Ames BN. Procedure for identifying nonsense mutations. J Bacteriol. 1968;96:215–220. doi: 10.1128/jb.96.1.215-220.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank K, Hensel M, Gerlach RG. Rapid and highly efficient method for scarless mutagenesis within the Salmonella enterica chromosome. PLoS ONE. 2011;6:e15763. doi: 10.1371/journal.pone.0015763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blommel PG, Fox BG. A combined approach to improving large-scale production of tobacco etch virus protease. Protein Expr Purif. 2007;55:53–68. doi: 10.1016/j.pep.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RK, Botstein D, Watanabe T, Ogata Y. Specialized transduction of tetracycline resistance by phage P22 in Salmonella typhimurium. II. Properties of a high-frequency-transducing lysate. Virology. 1972;50:883–898. doi: 10.1016/0042-6822(72)90442-4. [DOI] [PubMed] [Google Scholar]
- Chen YH, Yang JT, Chau KH. Determination of the helix and beta form of proteins in aqueous solution by circular dichroism. Biochemistry. 1974;13:3350–3359. doi: 10.1021/bi00713a027. [DOI] [PubMed] [Google Scholar]
- Davis RW, Botstein D, Roth JR. A manual for genetic engineering: advanced bacterial genetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1980. [Google Scholar]
- Dyda F, Klein DC, Hickman AB. GCN5-related N-acetyltransferases: a structural overview. Annu Rev Biophy Biomol Struc. 2000;29:81–103. doi: 10.1146/annurev.biophys.29.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser ME, James MN, Bridger WA, Wolodko WT. A detailed structural description of Escherichia coli succinyl-CoA synthetase. J Mol Biol. 1999;285:1633–1653. doi: 10.1006/jmbi.1998.2324. [DOI] [PubMed] [Google Scholar]
- Garrity J, Gardner JG, Hawse W, Wolberger C, Escalante-Semerena JC. N-lysine propionylation controls the activity of propionyl-CoA synthetase. J Biol Chem. 2007;282:30239–30245. doi: 10.1074/jbc.M704409200. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough J, Chothia C. SUPERFAMILY: HMMs representing all proteins of known structure. SCOP sequence searches, alignments and genome assignments. Nucleic Acids Res. 2002;30:268–272. doi: 10.1093/nar/30.1.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruprasad K, Reddy BV, Pandit MW. Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. 1990;4:155–161. doi: 10.1093/protein/4.2.155. [DOI] [PubMed] [Google Scholar]
- Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci U S A. 2006;103:10230–10235. doi: 10.1073/pnas.0604392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JS, Ames BN. Localized mutagenesis of any specific small region of the bacterial chromosome. Proc Natl Acad Sci U S A. 1971;68:3158–3162. doi: 10.1073/pnas.68.12.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- Jones JD, O’Connor CD. Protein acetylation in prokaryotes. Proteomics. 2011 doi: 10.1002/pmic.201000812. [DOI] [PubMed] [Google Scholar]
- Joyce MA, Fraser ME, Brownie ER, James MN, Bridger WA, Wolodko WT. Probing the nucleotide-binding site of Escherichia coli succinyl-CoA synthetase. Biochemistry. 1999;38:7273–7283. doi: 10.1021/bi990527s. [DOI] [PubMed] [Google Scholar]
- Joyce MA, Fraser ME, James MN, Bridger WA, Wolodko WT. ADP-binding site of Escherichia coli succinyl-CoA synthetase revealed by x-ray crystallography. Biochemistry. 2000;39:17–25. doi: 10.1021/bi991696f. [DOI] [PubMed] [Google Scholar]
- Kelly SM, Jess TJ, Price NC. How to study proteins by circular dichroism. Biochim Biophys Acta. 2005;1751:119–139. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Histone acetylases and deacetylases in cell proliferation. Curr Opin Genet Dev. 1999;9:40–48. doi: 10.1016/s0959-437x(99)80006-9. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Langer MR, Tanner KG, Denu JM. Mutational analysis of conserved residues in the GCN5 family of histone acetyltransferases. J Biol Chem. 2001;276:31321–31331. doi: 10.1074/jbc.M103839200. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, et al. CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009;37:D205–210. doi: 10.1093/nar/gkn845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwald AF, Landsman D. GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem Sci. 1997;22:154–155. doi: 10.1016/s0968-0004(97)01034-7. [DOI] [PubMed] [Google Scholar]
- Parks TD, Leuther KK, Howard ED, Johnston SA, Dougherty WG. Release of proteins and peptides from fusion proteins using a recombinant plant virus proteinase. Anal Biochem. 1994;216:413–417. doi: 10.1006/abio.1994.1060. [DOI] [PubMed] [Google Scholar]
- Rocco CJ, Dennison KL, Klenchin VA, Rayment I, Escalante-Semerena JC. Construction and use of new cloning vectors for the rapid isolation of recombinant proteins from Escherichia coli. Plasmid. 2008;59:231–237. doi: 10.1016/j.plasmid.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez LB, Galperin MY, Muller M. Acetyl-CoA synthetase from the amitochondriate eukaryote Giardia lamblia belongs to the newly recognized superfamily of acyl-CoA synthetases (Nucleoside diphosphate-forming) J Biol Chem. 2000;275:5794–5803. doi: 10.1074/jbc.275.8.5794. [DOI] [PubMed] [Google Scholar]
- Schmieger H, Backhaus H. The origin of DNA in transducing particles in P22-mutants with increased transduction-frequencies (HT-mutants) Mol Gen Genet. 1973;120:181–190. doi: 10.1007/BF00267246. [DOI] [PubMed] [Google Scholar]
- Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A. 2006;103:10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starai VJ, Celic I, Cole RN, Boeke JD, Escalante-Semerena JC. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science. 2002;298:2390–2392. doi: 10.1126/science.1077650. [DOI] [PubMed] [Google Scholar]
- Starai VJ, Escalante-Semerena JC. Acetyl-coenzyme A synthetase (AMP forming) Cell Mol Life Sci. 2004;61:2020–2030. doi: 10.1007/s00018-004-3448-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starai VJ, Escalante-Semerena JC. Identification of the protein acetyltransferase (Pat) enzyme that acetylates acetyl-CoA synthetase in Salmonella enterica. J Mol Biol. 2004;340:1005–1012. doi: 10.1016/j.jmb.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Starai VJ, Takahashi H, Boeke JD, Escalante-Semerena JC. Short-chain fatty acid activation by acyl-coenzyme A synthetases requires SIR2 protein function in Salmonella enterica and Saccharomyces cerevisiae. Genetics. 2003;163:545–555. doi: 10.1093/genetics/163.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starai VJ, Takahashi H, Boeke JD, Escalante-Semerena JC. A link between transcription and intermediary metabolism: a role for Sir2 in the control of acetyl-coenzyme A synthetase. Curr Opin Microbiol. 2004;7:115–119. doi: 10.1016/j.mib.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Tanner KG, Trievel RC, Kuo MH, Howard RM, Berger SL, Allis CD, Marmorstein R, Denu JM. Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. J Biol Chem. 1999;274:18157–18160. doi: 10.1074/jbc.274.26.18157. [DOI] [PubMed] [Google Scholar]
- Thao S, Chen CS, Zhu H, Escalante-Semerena JC. N(epsilon)-Lysine acetylation of a bacterial transcription factor inhibits its DNA-binding activity. PLoS ONE. 2010;5:e15123. doi: 10.1371/journal.pone.0015123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thao S, Escalante-Semerena JC. Biochemical and thermodynamic analyses of Salmonella enterica Pat, a multidomain, multimeric N(epsilon)-lysine acetyltransferase involved in carbon and energy metabolism. mBio. 2011;2:e00216–00211. doi: 10.1128/mBio.00216-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thao S, Escalante-Semerena JC. Control of protein function by reversible N(epsilon)-lysine acetylation in bacteria. Curr Opin Microbiol. 2011;14:200–204. doi: 10.1016/j.mib.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetting MW, Carvalho LPSd, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys. 2005;433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolodko WT, Fraser ME, James MN, Bridger WA. The crystal structure of succinyl-CoA synthetase from Escherichia coli at 2.5-A resolution. J Biol Chem. 1994;269:10883–10890. doi: 10.2210/pdb1scu/pdb. [DOI] [PubMed] [Google Scholar]
- Yang XJ. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. 2004;32:959–976. doi: 10.1093/nar/gkh252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu BJ, Kim JA, Moon JH, Ryu SE, Pan JG. The diversity of lysine-acetylated proteins in Escherichia coli. J Microbiol Biotechnol. 2008;18:1529–1536. [PubMed] [Google Scholar]
- Zhang J, Sprung R, Pei J, Tan X, Kim S, Zhu H, Liu CF, Grishin NV, et al. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol Cell Proteomics. 2009;8:215–225. doi: 10.1074/mcp.M800187-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]