

Abstract
The mammalian target of rapamycin (mTOR) signaling pathway in neurons integrates a variety of extracellular signals to produce appropriate translational responses. mTOR signaling is hyperactive in neurological syndromes in both humans and mouse models that are characterized by epilepsy, autism, and cognitive disturbances. In addition, rapamycin, a clinically important immunosuppressant, is a specific and potent inhibitor of mTOR signaling. While mTOR is known to regulate growth and synaptic plasticity of glutamatergic neurons, its effects on basic parameters of synaptic transmission are less well studied, and its role in regulating GABAergic transmission is unexplored. We therefore performed an electrophysiological and morphological comparison of glutamatergic and GABAergic neurons in which mTOR signaling was either increased by loss of the repressor Pten or decreased by treatment with rapamycin. We found that hyperactive mTOR signaling increased evoked synaptic responses in both glutamatergic and GABAergic neurons by ∼50%, due to an increase in the number of synaptic vesicles available for release, the number of synapses formed, and the miniature event size. Prolonged (72 h) rapamycin treatment prevented these abnormalities and also decreased synaptic transmission in wild-type glutamatergic, but not GABAergic, neurons. Further analyses suggested that hyperactivation of the mTOR pathway also impairs presynaptic function, possibly by interfering with vesicle fusion. Despite this presynaptic impairment, the net effect of Pten loss is enhanced synaptic transmission in both GABAergic and glutamatergic neurons, which has numerous implications, depending on where in the brain mutations of an mTOR suppressor gene occur.
Introduction
Fast synaptic transmission is the most basic process of information transfer in the CNS. Changes in fundamental aspects of synaptic transmission, such as the amount of neurotransmitter a neuron releases and the postsynaptic response it elicits, can have profound effects on the function of neurons, circuits, and the organism as a whole. There is increasing evidence that defects in synaptic transmission lead to or play a role in many neurodevelopmental disorders (Zoghbi, 2003; van Spronsen and Hoogenraad, 2010; Waites and Garner, 2011). Physiological studies of basic synaptic transmission of disease models are therefore crucial to further our understanding of how molecular abnormalities might translate into functional deficits.
Improper regulation of the mammalian target of rapamycin (mTOR) signaling pathway by several relevant proteins causes distinct neurological syndromes in both humans and animals, characterized by epilepsy, autism, and malformations of the CNS (Backman et al., 2001; Kwon et al., 2001; Meikle et al., 2007; Orlova et al., 2010). The mTOR pathway is a ubiquitous signaling cascade that integrates extracellular stimuli such as growth factors, nutrient availability, and synaptic input to control translation machinery. In neurons, mTOR signaling regulates soma size, dendrite and axon growth, and spine density and structure (Kwon et al., 2001, 2006; Jaworski et al., 2005; Tavazoie et al., 2005; Fraser et al., 2008), and properly balanced mTOR signaling is necessary for both learning and memory and various forms of long-term potentiation and long-term depression (Costa-Mattioli et al., 2009; Hoeffer and Klann, 2010). Electrophysiological studies of glutamatergic synaptic transmission in slice preparations with hyperactive mTOR signaling have found increases in both mEPSC frequency and mEPSC amplitude, which were attributed to increases in dendrite length and/or spine density (Tavazoie et al., 2005; Jurado et al., 2010; Bateup et al., 2011; Luikart et al., 2011; Xiong et al., 2012).
Despite the clear effect hyperactive mTOR signaling has on the overall growth of glutamatergic neurons, there are few data on how mTOR signaling regulates neurotransmitter release machinery or GABAergic neurotransmission and growth. We therefore decided to compare the primary effects of altered mTOR signaling on synaptic transmission in both glutamatergic and GABAergic neurons by characterizing autaptic cultures of neurons in which mTOR activity was increased by loss of the negative regulator Pten or decreased by treatment with the mTOR inhibitor rapamycin. We found that mTOR bidirectionally regulates synaptic strength of single glutamatergic neurons through changes in synapse number, synaptic vesicle number, and mEPSC amplitude. In GABAergic neurons, increased mTOR signaling also increased synaptic strength through increases in synapse number, synaptic vesicle number, and mIPSC amplitude; however, decreased mTOR signaling did not reduce inhibitory transmission. In addition, we found that alterations in mTOR signaling regulate the rate constant for spontaneous vesicle fusion. These results pinpoint specific functional consequences of altered mTOR signaling and suggest a previously unappreciated role for altered GABAergic transmission in the pathology of mTOR related neurodevelopmental disorders.
Materials and Methods
Mice and cell culture.
PtenloxP/loxP; Gfap-Cre mice were described previously (Backman et al., 2001; Kwon et al., 2001; Ljungberg et al., 2009). Animal housing and use were in compliance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and were approved by the institutional animal care committee at Baylor College of Medicine. Microisland cultures of postnatal day 1 (P1) striatal and hippocampal dentate granule neurons were prepared according to published procedures (Reim et al., 2001; Xue et al., 2008; Rost et al., 2010). Microislands were made by coating collagen (0.7 mg ml−1) and poly-d-lysine (0.1 mg ml−1) on coverslips with a custom-built stamp to achieve uniform size (200 μm diameter). Astrocytes were grown on microislands for 1 week before plating of neurons. The hippocampus or striatum was removed from P1 mice of either sex, and the dentate gyrus was then microdissected from the CA3/CA1 region. Neurons were then digested with papain (Worthington) and plated on astrocytes derived from wild-type neonatal cortex tissue at a density of 2000–3000 neurons per 35 mm dish and grown in a chemically defined medium (Neurobasal-A medium supplemented with Glutamax and B-27; Invitrogen). Under these conditions, a single neuron on an astrocyte microisland forms recurrent synapses (autapses) (Bekkers and Stevens, 1991).
Electrophysiology.
The standard extracellular solution contained the following (in mm): 140 NaCl, 2.4 KCl, 10 HEPES, 10 glucose, 4 MgCl2, and 2 CaCl2, pH 7.3 (305 mOsm). The internal solution contained the following: 136 mm KCl, 17.8 mm HEPES, 1 mm EGTA, 0.6 mm MgCl2, 4 mm ATP, 0.3 mm GTP, 12 mm creatine phosphate, and 50 U/ml phosphocreatine kinase. These concentrations set the chloride reversal potential high enough that GABA receptor-mediated synaptic responses resulted in an inward current. All experiments were performed at room temperature (23–24°C). Whole-cell recordings were performed on neurons from control and experimental groups in parallel on the same day in vitro (days 9–14 in vitro) and held at −70 mV unless noted otherwise. Dentate granule neurons were identified with the mGluR2/3 agonist l-CCG I [(2S,1′S,2′S)-2-(carboxycyclopropyl)glycines] (Tocris Bioscience) (Rost et al., 2010). Only neurons whose EPSC peak amplitudes were reduced by >60% were considered granule neurons. For rapamycin treatment experiments, rapamycin (LC Laboratories) was dissolved in DMSO at a concentration of 1 mm and then further diluted in culture media to 100 nm.
For voltage-clamp experiments, action potential (AP)-evoked EPSCs or IPSCs were triggered by a 2 ms somatic depolarization to 0 mV. The shape of the evoked response and antagonists (either 3 mm kynurenic acid or 20 μm bicuculline; Tocris Bioscience) were applied to verify glutamatergic or GABAergic identities. Neurons were stimulated at 0.2 Hz (for EPSCs) or 0.1 Hz (for IPSCs) in standard external solution to measure basal evoked synaptic responses. Membrane capacitance measurements were obtained from the membrane test function in pClamp. Readily releasable pool (RRP) size was determined by measuring the charge transfer of the transient synaptic current induced by a 4 s application of hypertonic sucrose solution directly onto the neuron (Rosenmund and Stevens, 1996). The number of synaptic vesicles in the RRP of each neuron recorded was then obtained by dividing the sucrose charge by the charge of the average miniature event. To obtain Pvr, the basal evoked synaptic responses and the response to the hypertonic sucrose solution were recorded successively from the same neuron. The evoked response was integrated to calculate the charge transfer. Pvr was calculated as the ratio of evoked response charge to RRP charge. Short-term plasticity was examined either by evoking 50 synaptic responses at 10 Hz or 2 responses with a 20 ms interval in standard external solution for glutamatergic neurons. For GABAergic neurons, 5 Hz and 100 ms were used.
For current-clamp experiments, bias current was injected to achieve a resting membrane potential of −70 mV, and kynurenic acid or bicuculline were applied to block synaptic responses. Input resistance and membrane time constant were calculated from the steady state and charging transient, respectively, of voltage responses to 1 s, 20 pA hyperpolarizing current steps. Membrane capacitance was calculated by dividing the time constant by the input resistance. APs were evoked with 1 s, 20 pA depolarizing current steps. AP threshold was defined as Vm at the inflection point of the rising phase of the AP. AP amplitude was defined as the difference in Vm between the peak and threshold. AHP amplitude was defined as the difference in Vm between the peak AHP and AP threshold.
Data were analyzed offline with AxoGraph X 1.0 (AxoGraph Scientific) and KaleidaGraph (Synergy Software). Values for analysis were always pooled from at least two independent cultures. Normality was tested by the Kolmogorov–Smirnov test. Statistical significances were tested by using Student's t test for two groups or one-way ANOVA with Newman–Keuls post-test for more than two groups with normally distributed data sets, or the nonparametric Mann–Whitney test. For values reported as normalized, the average value of the control group was calculated for each day and then used to normalize individual neuron values from each group.
Miniature event analysis.
Miniature synaptic potentials were recorded for 60–80 s with either 3 mm kynurenic acid or 20 μm bicuculline applied for 2 s of every 10 s for background noise subtraction and to verify the identity (glutamatergic or GABAergic) of the neuron under study. For some neurons, 500 nm tetrodotoxin (TTX) was applied; however, there was no difference in the amplitude or frequency of events with or without TTX. For each cell, data were filtered at 1 kHz and analyzed using template-based miniature event detection algorithms implemented in analysis software AxoGraph X 1.0. The threshold for detection was set at three times the baseline SD from a template of 0.5 ms rise time and 3 ms decay for glutamatergic events and 0.5 ms rise and 25 ms decay for GABAergic events. The rate constant for spontaneous vesicle fusion (k) was calculated for each neuron with the first-order reaction equation r = k [A], where r is the observed minifrequency, and A is the number of synaptic vesicles in the RRP.
Virus production.
The preparation of lentiviral particles expressing cre-RFP fusion protein was done as described previously (Lois et al., 2002). Briefly, HEK293T cells were cotransfected with 8 μg shuttle vector F(SYN)UGW-RBN bearing cDNA for cre-RFP, and the mixed helper plasmids pCMVdR8.9 and pVSV-G (5 μg each) with Fugene 6 transfection reagent (Roche Diagnostics). After 48 h, the cell culture supernatant was collected, and cell debris was removed by filtration with a 0.45 μm polyethersulfone membrane (Pall Life Sciences). Aliquots of the filtrate were flash frozen in liquid nitrogen and stored at −80°C. Estimation of the titer was done on mass cultures of wild-type hippocampal neurons. For infection of the neurons for experiments, 100 μl of the viral solution (4 × 106 U/ml) was used 18–24 h after plating.
Immunocytochemistry and analysis.
After electrophysiology experiments were completed, the culture medium was removed, and neurons were rinsed with PBS and fixed in 4% paraformaldehyde for 30 min, and then washed three times with PBS again. Samples were incubated in blocking buffer (5% normal horse serum, 0.1% Triton X-100 in PBS) at room temperature for 1 h. Primary antibodies diluted in blocking buffer were applied overnight at 4°C. The following antibody dilutions were used: rabbit anti-phospho-S6 Ser 240/244 (1:1000; Cell Signaling Technology), mouse anti-microtubule-associated protein 2 (MAP2; 1:2000; Millipore Bioscience Research Reagents), rabbit anti-MAP2 (1:2000; Millipore Bioscience Research Reagents), rabbit Prox-1, (1:1000; Millipore Bioscience Research Reagents), guinea pig anti-VGLUT2 (1: 5000; Synaptic Systems), and rabbit anti-vesicular GABA transporter (VGAT; 1:2000; Synaptic Systems). After three washes for 5 min in PBS, secondary goat antibodies coupled to/conjugated with Alexa 488, 555, or 647 (Invitrogen) were diluted 1:1000 in blocking buffer and applied for 1 h at room temperature. All images were obtained using the Zeiss AxioImager M1 system with 40× Zeiss objectives and AxioVision software.
For neuron reconstruction and analysis, images were acquired using equal exposure times and processed using the National Institutes of Health ImageJ software (http://rsbweb.nih.gov/ij/). A threshold macro was applied to the VGLUT or VGAT channel to identify synapses. Neuron cell bodies, dendrites, and synapses were drawn using Neurolucida software (MicroBrightField). Quantification of the traced images was conducted with Neuroexplorer software (MicroBrightField). Raw values were then exported to KaleidaGraph (Synergy Software) for further analysis.
Results
Loss of Pten increases the magnitude of glutamatergic and GABAergic evoked synaptic responses
To identify specific roles for mTOR signaling in the regulation of glutamatergic and GABAergic synaptic transmission, we used the autaptic culture system, which allows the precise quantification of multiple parameters of presynaptic and postsynaptic function. To investigate glutamatergic transmission, we dissected and cultured the dentate gyrus of either PtenloxP/loxP; Gfap-Cre mice (in which Pten is absent from dentate granule and other restricted neuronal populations, but not astrocytes, described previously) (Backman et al., 2001; Kwon et al., 2001; Ljungberg et al., 2009) or PtenloxP/loxP mice and their wild-type littermates. Cultures made from PtenloxP/loxP and wild-type littermates were infected with a lentivirus expressing a cre-RFP fusion protein after one day in vitro. The results from either the conditional knock out mouse or in vitro viral infections were indistinguishable, and the data presented combine the results from both methods. To investigate GABAergic transmission, we dissected the striatum of PtenloxP/loxP mice and wild-type littermates and again infected with the lentivirus expressing cre-RFP. To ensure we had loss of Pten and hyperactivation of the mTOR signaling pathway, we immunostained for the phosphorylated ribosomal protein S6 (pS6) and observed a strong increase in pS6 fluorescent intensity in Pten-cre neurons compared to both wild-type neurons infected with cre-RFP (Fig. 1A) and uninfected PtenloxP/loxP neurons. We then performed whole-cell voltage-clamp or current-clamp analysis of single neurons.
Figure 1.
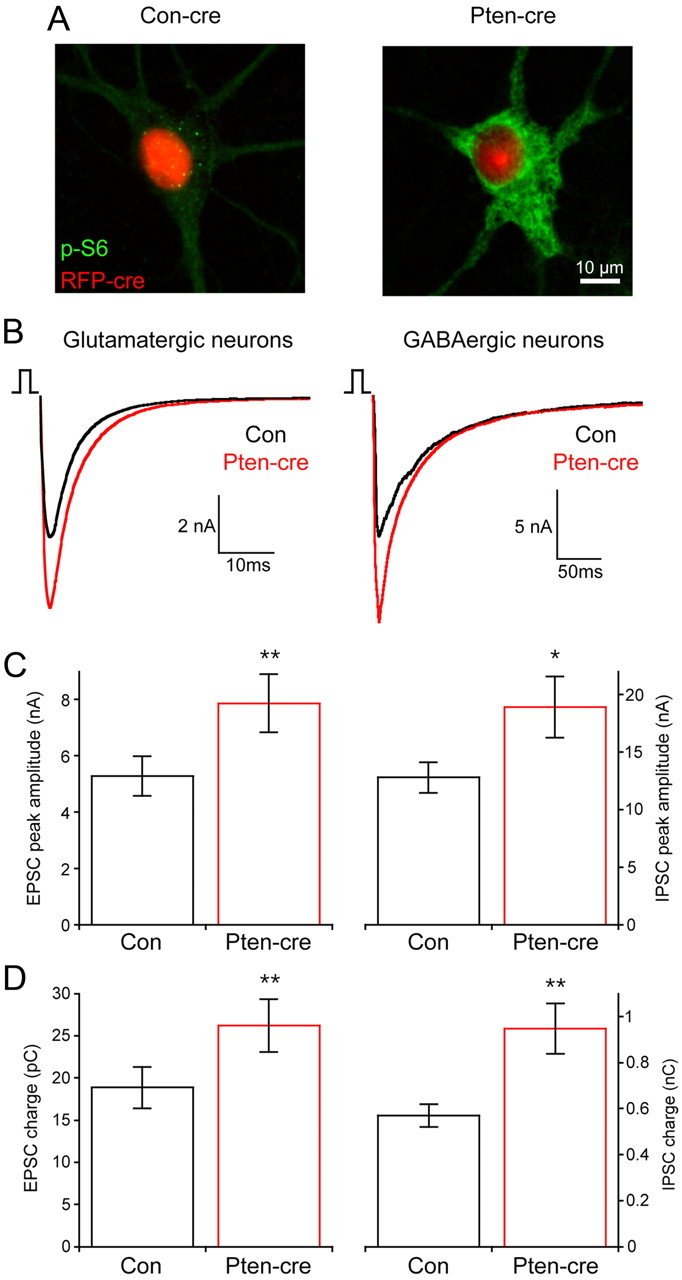
Loss of Pten increases synaptic transmission in glutamatergic and GABAergic neurons. A, Representative images from wild-type (left) and PtenloxP/loxP (right) neurons infected with a cre-RFP-expressing lentivirus showing the fluorescence from the RFP fusion protein (red) and the immunoreactivity against phospho-S6 (green). B, Representative traces showing 2 ms depolarization evoked synaptic responses from control (black traces) and Pten-cre (red traces) glutamatergic (left traces) and GABAergic (right traces) neurons. GABAergic responses are inward currents due to the Cl− concentration of the internal solution. C, Peak amplitudes of evoked EPSCs from glutamatergic (left) and evoked IPSCs from GABAergic (right) neurons. D, The charge contained in the evoked EPSCs of glutamatergic (left) and evoked IPSCs of GABAergic (right) neurons. All values are presented as mean ± SEM. *p ≤ 0.05; **p ≤ 0.01.
As a starting point for our functional analysis, we examined action potential-evoked synaptic currents under voltage clamp. The peak EPSC amplitudes of glutamatergic hippocampal granule neurons lacking Pten (7.87 ± 1.0 nA; n = 43) were significantly larger than those of control neurons (5.29 ± 0.7 nA; n = 48; p = 0.01.) (Fig. 1B,C, left). The charge contained in the EPSC of Pten-cre neurons was also significantly increased (Fig. 1D, left bars). The time constant of decay of the synchronous component of the synaptic current was not significantly different in control (5.05 ± 0.22 ms; n = 23) and Pten-cre (4.99 ± 0.23 ms; n = 25) glutamatergic neurons.
IPSCs from Pten-cre striatal neurons (18.9 ± 2.7 nA; n = 55) were also significantly larger in peak amplitude than control neurons (12.8 ± 1.3 nA; n = 57; p = 0.04) (Fig. 1B,C, right panels), as was the mean IPSC charge (Fig. 1D, right). The decay time constant of the GABAergic synaptic current was not significantly different in control (24.6 ± 2.3 ms; n = 27) and Pten-cre (27.6 ± 2.5 ms; n = 27) neurons. The magnitude of the increase in the IPSC was equivalent to the increase seen in the EPSC, about 50%. Thus, Pten regulates the magnitude of synaptic transmission at a single cell level in both glutamatergic and GABAergic neurons, and to a similar extent in both cell types.
We also performed a current-clamp analysis of Pten-cre and control glutamatergic and GABAergic neurons to evaluate changes in passive membrane properties and action potential dynamics (Table 1). Both glutamatergic and GABAergic Pten-cre neurons had significantly greater membrane capacitance (Cm) than control, indicating an increase in cell surface area and providing further evidence for hyperactive mTOR signaling. We also found that the action potential half-width was significantly longer in both glutamatergic and GABAergic neurons lacking Pten.
Table 1.
Passive membrane and action potential properties of Pten-cre neurons
| Glutamatergic neurons |
GABAergic neurons |
|||
|---|---|---|---|---|
| Control (n = 21) | Pten-cre (n = 22) | Control (n = 23) | Pten-cre (n = 24) | |
| Vrest (in mV) | −56 ± 2.1 | −58.1 ± 2.2 | −51.7 ± 2.1 | −56.1 ± 2.4 |
| Rinput (in MΩ) | 438 ± 44 | 356 ± 42 | 618 ± 85 | 551 ± 47 |
| Time constant (in ms) | 23.2 ± 2.8 | 22.7 ± 1.9 | 33.3 ± 5.2 | 41.1 ± 4.3 |
| Cm (in pF) | 59.8 ± 4.6 | 72.8 ± 6.5* | 53.3 ± 5.9 | 76.0 ± 6.2** |
| AP threshold (in mV) | −29.2 ± 1.1 | −30.7 ± 2.1 | −33.4 ± 1.5 | −32.6 ± 1.1 |
| AP amplitude (in mV) | 93.2 ± 3.2 | 82.9 ± 5.2 | 79.5 ± 2.5 | 76.2 ± 2.7 |
| AP half-width (in ms) | 1.12 ± 0.05 | 1.48 ± 0.13* | 1.49 ± 0.07 | 1.77 ± 0.11* |
| AP rise max slope (in mV/ms) | 212 ± 14 | 161 ± 21 | 116 ± 8 | 100 ± 9 |
| AP fall max slope (in mV/ms) | −89 ± 6 | −69 ± 8 | −60 ± 5 | −49 ± 5 |
| AHP amplitude (in mV) | −21.4 ± 1.7 | −19.4 ± 1.9 | −13.7 ± 1.9 | −14.1 ± 1.7 |
Measurements are shown as mean ± SEM.
*p ≤ 0.05;
**p ≤ 0.01 compared to control with unpaired t test or Mann–Whitney test.
An increase in quantal size partially accounts for the increase in evoked release
The magnitude of the evoked response depends on the postsynaptic response to the release of an individual vesicle (quantal size), the number of fusion competent vesicles (RRP), and the probability that a fusion competent vesicle is released in response to an action potential (vesicular release probability, Pvr) (del Castillo and Katz, 1954; Reim et al., 2001; Stevens, 2003). To determine which of these parameters underlies the increase in evoked release of Pten-cre neurons, we first analyzed spontaneous release in neurons lacking Pten to see if changes in mEPSC and mIPSC size were responsible. We found that the mean peak mEPSC amplitude of Pten-cre neurons (46.07 ± 2.3 pA; n = 50) was significantly greater than controls (35.8 ± 2.4 pA; n = 46; p ≤ 0.001) (Fig. 2A,C, left), as was the mEPSC charge (Fig. 2D, left). The decay time constant of glutamatergic miniature events was not significantly different between control and Pten-cre neurons (3.19 ± 0.1 ms, n = 27 for control vs 3.24 ± 0.1 ms, n = 26 for Pten-cre). The mIPSC peak amplitude (43.8 ± 2.3 pA, n = 57 for control vs 53.2 ± 2.7 pA, n = 61 for Pten-cre; p = 0.01) and charge were also increased in GABAergic Pten-cre neurons compared to control (Fig. 2B–D). The decay time constant of GABAergic miniature events was not significantly different between control and Pten-cre neurons (24.7 ± 0.9 ms, n = 22 for control vs 23.4 ± 0.9 ms, n = 22 for Pten-cre).
Figure 2.
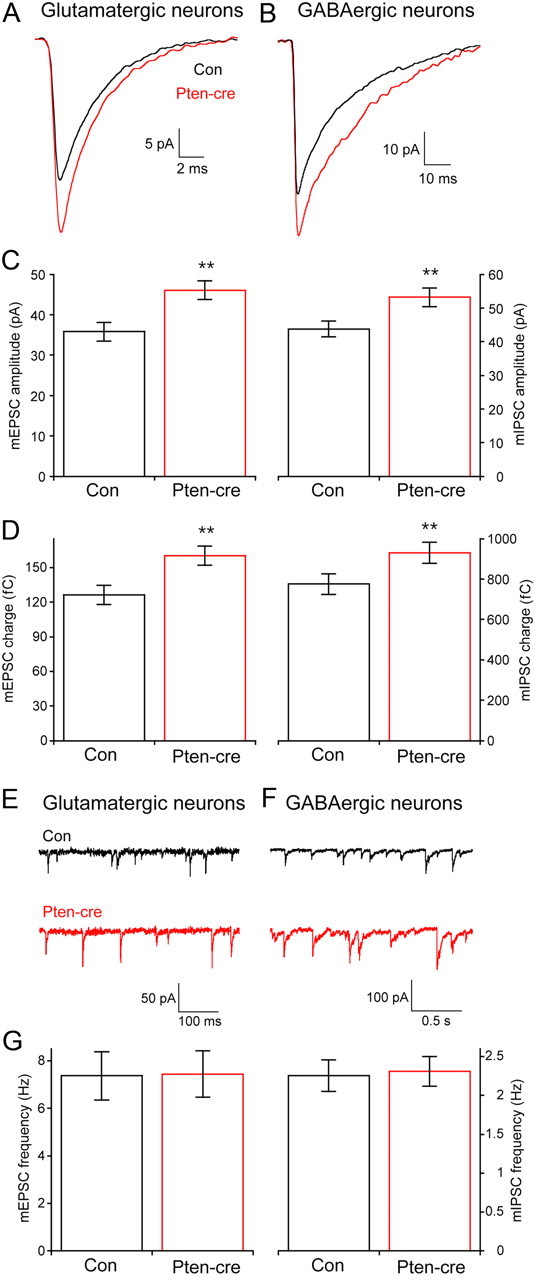
Loss of Pten increases the amplitude but not the frequency of glutamatergic and GABAergic miniature events. A, Representative traces showing the average of mEPSCs collected from one control (black trace) and one Pten-cre (red trace) glutamatergic neuron. B, Representative traces showing the average of mIPSCs collected from one control (black trace) and one Pten-cre (red trace) GABAergic neuron. C, Peak amplitudes of mEPSCs from glutamatergic (left) and mIPSCs from GABAergic (right) neurons. D, The charge contained in the mEPSCs of glutamatergic (left bars) and mIPSCs of GABAergic (right bars) neurons. E, F, Representative traces showing control (black traces) and Pten-cre (red traces) miniature postsynaptic current activity from glutamatergic (E) and GABAergic (F) neurons. G, Bar graph showing no significant difference in the miniature event frequency between control and Pten-cre neurons in glutamatergic (left bars) and GABAergic (left bars) neurons. All values are presented as mean ± SEM. **p ≤ 0.01.
The frequency of miniature events is not different between Pten-cre and control neurons
The magnitude of the increase in the miniature amplitude and charge was similar in glutamatergic and GABAergic neurons, ∼25%. It was, however, smaller than the increase in the evoked response in both cases, indicating that an increase in either the Pvr or the number of vesicles in the RRP must also contribute to the increase in the evoked EPSC and IPSC size. Normally, an increase in the number of synaptic vesicles in the RRP will cause an increase in the miniature event frequency, and previous studies have shown an increase in minifrequency as a result of hyperactive mTOR signaling in postsynaptic neurons (Tavazoie et al., 2005; Bateup et al., 2011; Luikart et al., 2011; Xiong et al., 2012) Unexpectedly, we found no difference in either the mEPSC or the mIPSC frequency between control and Pten-cre neurons (Fig. 2E–G). Control experiments showed that the lack of an effect was not due to an inability of the assay to detect higher frequencies, as a 10 Hz train of depolarizations increased the mEPSC frequency approximately threefold in both control and Pten-cre neurons (data not shown). The lack of an effect of Pten loss on the minifrequency indicates two possibilities: (1) there is no increase in the RRP of Pten-cre neurons, which predicts an increase in Pvr, or (2) there is an increase in the RRP that does not lead to an increase in minifrequency because loss of Pten also in some way impairs spontaneous vesicle fusion.
Loss of Pten increases the size of the RRP of vesicles
The number of synaptic vesicles in the RRP can be directly quantified in autaptic neurons by applying a 4 s pulse of hypertonic sucrose to the neuron, which induces the exocytosis of all of a neuron's fusion competent vesicles (Rosenmund and Stevens, 1996; Reim et al., 2001). The integral of the transient current during sucrose application represents the total charge contained in the RRP, and the total number of vesicles in the RRP can then be calculated by dividing the total charge by the average charge of the miniature events from each neuron. We found that the RRP charge of glutamatergic neurons lacking Pten was increased nearly twofold (Fig. 3A,C, left), while the RRP charge of GABAergic neurons was increased by 65% (Fig. 3B,C, right). As a result, the mean number of synaptic vesicles contained in the RRP of Pten-cre glutamatergic neurons (13852 ± 1605 vesicles; n = 34) was significantly greater than the number in control neurons (8867 ± 906 vesicles; n = 35; p ≤ 0.01), and the mean number of synaptic vesicles contained in the RRP of Pten-cre GABAergic neurons (5137 ± 493 vesicles; n = 56) was significantly greater than the number in their controls (3911 ± 365 vesicles; n = 54; p ≤ 0.05) (Fig. 3D). These results indicate that the combined effect of an increase in the size of miniature events and an increase in the number of synaptic vesicles in the RRP are sufficient to account for the magnitude of the increase in the evoked response seen in Pten-cre glutamatergic and GABAergic neurons (Fig. 1C–E).
Figure 3.
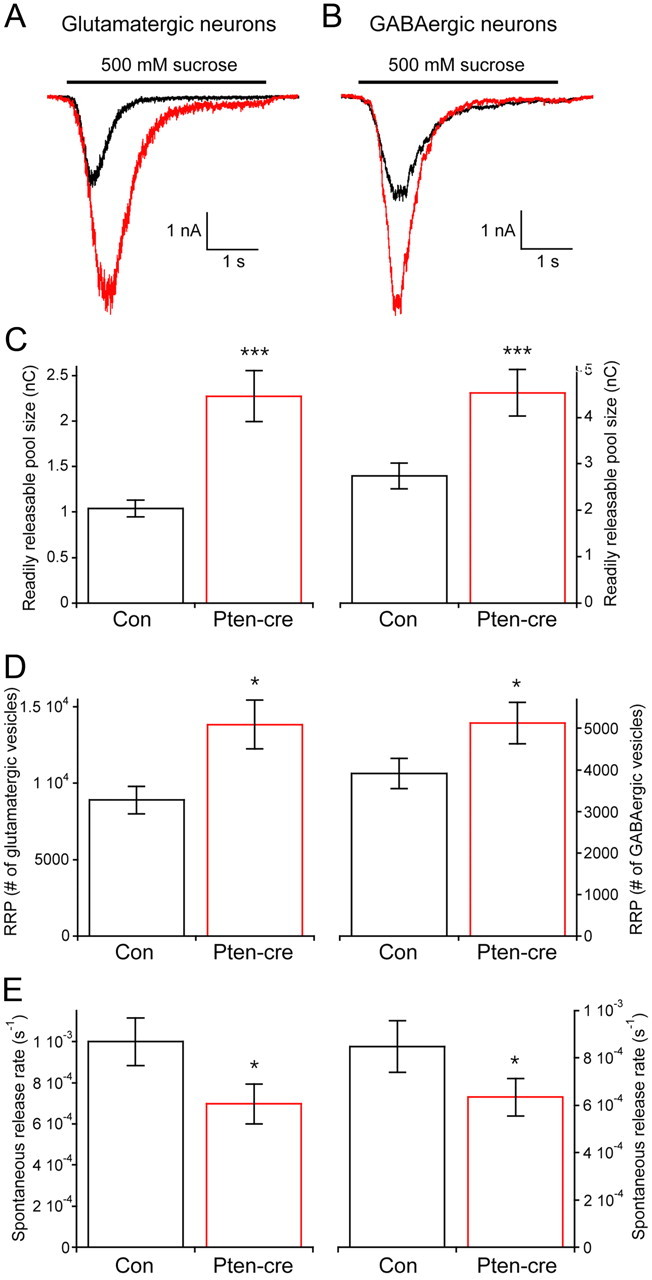
Loss of Pten increases the number of synaptic vesicles in the RRP. A, Representative traces of the response of glutamatergic control (black trace) and Pten-cre (red trace) neurons to a 4 s pulse of 500 mm sucrose. B, Representative traces of the response of GABAergic control (black trace) and Pten-cre (red trace) neurons to a 4 s pulse of 500 mm sucrose. C, The charge contained in the transient current induced by sucrose application in glutamatergic (left) and GABAergic (right) neurons. D, The number of synaptic vesicles contained in the RRP of glutamatergic (left) and GABAergic (right) neurons. E, The rate constant of spontaneous release of synaptic vesicles from glutamatergic (left) and GABAergic (right) neurons. All values are presented as mean ± SEM. *p ≤ 0.05; ***p ≤ 0.001.
Loss of Pten decreases the spontaneous release rate constant
An increase in the number of vesicles in the RRP often leads to an increase in both evoked and spontaneous release, because there is an increased number of synaptic vesicles available for fusion, either in response to an action potential or spontaneously (Murthy et al., 2001; Tyler and Pozzo-Miller, 2001; Habets and Borst, 2005). Although we detected an increase in evoked release, we found no difference in spontaneous release, as shown in Figure 2G. This suggests that loss of Pten may inhibit the rate constant of spontaneous vesicle fusion. To test this, we calculated the spontaneous vesicular release rate constant, or the rate at which an individual synaptic vesicle fuses with the plasma membrane in the absence of stimulation, by dividing the miniature event frequency by the number of vesicles in the RRP for each neuron (see Materials and Methods). We found that Pten-cre neurons had lower spontaneous release rate constants than control neurons (Fig. 3E). This indicates that, although Pten-cre neurons have more synaptic vesicles in their RRP, each one of these vesicles is less likely to fuse spontaneously than a vesicle from a control neuron.
Effects on Pvr and short-term plasticity
Next, we tested whether loss of Pten affected the probability of evoked vesicle fusion (Pvr) by dividing the number of vesicles released in response to action potential stimulation by the number of vesicles in the RRP. The Pvr was not significantly different between Pten-cre and control in glutamatergic or GABAergic neurons (Fig. 4A), indicating that the effect of Pten loss on vesicle fusion may be specific to spontaneous fusion. Next, we measured paired-pulse ratios (PPR) as an alternative measure of presynaptic efficiency (Fig. 4B–D). In glutamatergic neurons, loss of Pten caused a small, but significant, increase in the PPR at a 20 ms interstimulus interval (ISI; 1.13 ± 0.04, n = 49 for control vs 1.26 ± 0.04, n = 49 for Pten-cre; p = 0.04). PPR in GABAergic neurons was also significantly increased at a 100 ms ISI (0.89 ± 0.06, n = 33 for control vs 1.05 ± 0.07, n = 31 for Pten-cre; p = 0.04). Finally, we measured the response of Pten-cre neurons to high-frequency stimulation. In glutamatergic neurons, a 10 Hz train resulted in slightly larger responses in Pten-cre neurons than control neurons after 50 stimulations (Fig. 4E) (0.97 ± 0.05, n = 64 for control vs 1.23 ± 0.09, n = 66 for Pten-cre; p = 0.02), while in GABAergic neurons there was no difference after 50 stimulations in response to a 5 Hz train (Fig. 4F) (0.33 ± 0.04, n = 52 for control vs 0.34 ± 0.03, n = 52 for Pten-cre). Two- way ANOVA also detected a significant effect of genotype for glutamatergic neurons (p = 0.01), but not for GABAergic neurons (p = 0.74). Thus, loss of Pten caused changes in short-term plasticity consistent with an inhibition of evoked release efficiency, although we detected no difference in the Pvr.
Figure 4.
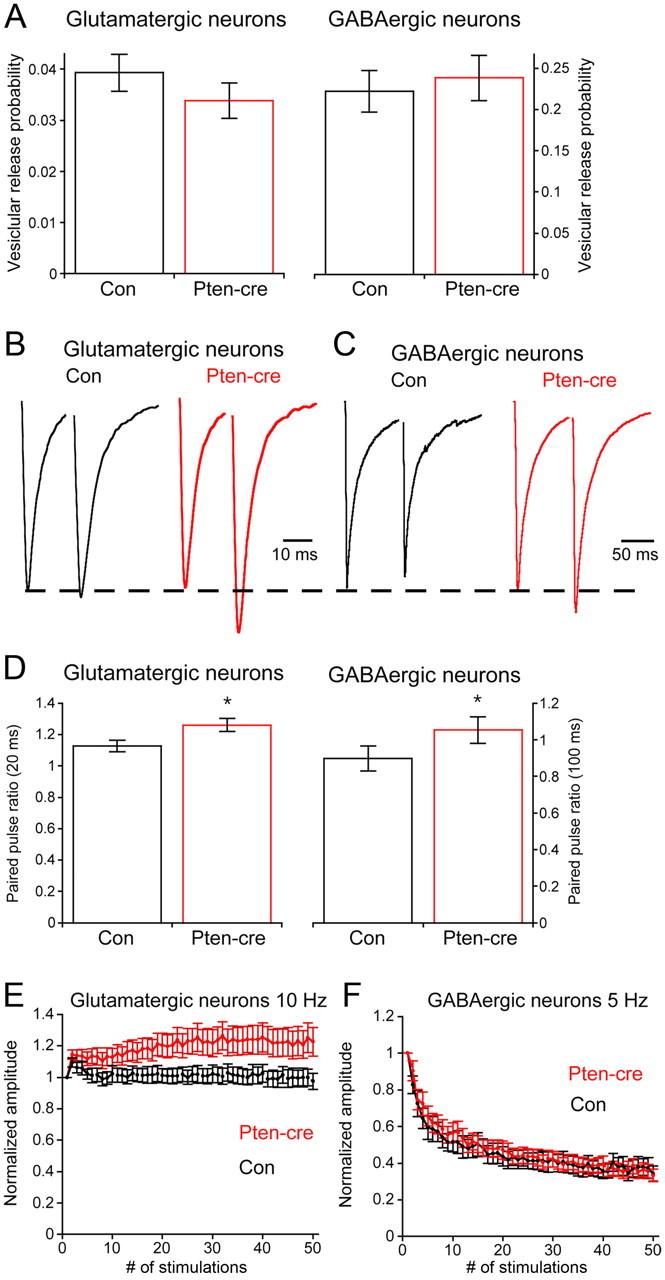
Presynaptic neurotransmitter release efficiency in Pten-cre neurons. A, The probability that an individual synaptic vesicle fuses in response to a 2 ms depolarization (Pvr) in control (black) and Pten-cre (red) neurons. B, Representative traces of the response of control (black trace) and Pten-cre (red trace) glutamatergic neurons to paired 2 ms depolarizations separated by 20 ms. C, Representative traces of the response of control (black trace) and Pten-cre (red trace) GABAergic neurons to paired 2 ms depolarizations separated by 100 ms. D, Paired pulse ratios in control (black) and Pten-cre (red) neurons. E, Line plot of the responses of control (black line) and Pten-cre (red line) glutamatergic neurons to 10 Hz stimulation. Values are normalized to the peak amplitude of the first response in the train. F, Line plot of the responses of control (black line) and Pten-cre (red line) GABAergic neurons to 5 Hz stimulation. Values are normalized to the peak amplitude of the first response in the train. All values are presented as mean ± SEM. *p ≤ 0.05.
The increase in the RRP is likely due to an increase in synapse number concomitant with longer dendritic arbors
An increase in the RRP can be caused by either an increase in the number of synapses or an increase in the number of docked synaptic vesicles per synapse. Since loss of Pten is known to affect cell growth by enhancing mTOR signaling, and our Cm measurements indicated increased surface area (Table 1), we analyzed immunofluorescent images of single neurons to determine whether there was an increase in cell size and total number of synapses in cells lacking this regulatory protein. For glutamatergic neurons, we used VGLUT1 and MAP2 antibodies, while for GABAergic neurons we used VGAT and MAP2 (Fig. 5A). Granule neuron identity was confirmed by positive Prox 1 staining. The soma and dendritic trees of the neurons were then traced, and VGLUT1- and VGAT-positive punctae counted. Representative tracings are shown in Figure 5B. The soma area was increased by loss of Pten in glutamatergic neurons (161.3 ± 20 μm2, n = 24 for control vs 227.1 ± 36 μm2, n = 23 for Pten-cre; p = 0.02), and there was a 60% increase in the number of VGLUT1 punctae per neuron (208.3 ± 26, n = 12 for control vs 333.3 ± 38, n = 12 for Pten-cre; p = 0.01) (Fig. 5C, left bars), a change nearly identical in magnitude to the increase in the number of vesicles in the RRP as measured by electrophysiology (Fig. 3D). The number of VGAT punctae was also significantly increased (Fig. 5C, right bars). Next, we quantified the total dendritic length to determine if the increase in the number of synapses was due to an increase in dendritic length, synapse density, or both. We found that total dendritic length was greater in both glutamatergic and GABAergic neurons (Fig. 5D), and that the increase was accompanied by an increase in the number of dendritic branch points in glutamatergic neurons (12.1 ± 0.9 branch points per neuron for control, n = 24 vs 17.5 ± 1.2 for Pten-cre, n = 23; p = 0.01) and GABAergic neurons (13.0 ± 1.8 branch points per neuron for control, n = 22 vs 19.0 ± 1.6 for Pten-cre, n = 19; p = 0.02). Finally, we calculated the synapse density. The synapse density was significantly increased in glutamatergic neurons, while GABAergic neurons showed a slight, nonsignificant increase (Fig. 5E). Thus, the increase in the number of vesicles in the RRP of glutamatergic and GABAergic Pten-cre neurons is likely due to an increase in the number of synaptic connections and not an increase in the number of synaptic vesicles per synapse (but see Discussion). This increase in connectivity is likely a result of increased dendritic area available for synapse formation and perhaps also an increase in synapse density, at least in glutamatergic neurons.
Figure 5.

Loss of Pten increases the length of dendrites and number of glutamatergic and GABAergic synapses. A, Representative images from control and Pten-cre glutamatergic (left images) and GABAergic (right images) neurons showing immunoreactivity for MAP2 (green) and either VGLUT1 or VGAT (red). B, Representative tracings of the control (top) and Pten-cre (bottom) GABAergic images shown in A. C, The number of VGLUT1 (left) and VGAT (right) punctae per each control (black) and Pten-cre (red) neuron. D, The total length of dendrites from control (black bars) and Pten-cre (red bars) glutamatergic (left) and GABAergic (right) neurons. E, The number of VGLUT1 (left) and VGAT (right) punctae per micron of dendritic length in each control (black) and Pten-cre (red) neuron. All values are presented as mean ± SEM. *p ≤ 0.05; **p ≤ 0.01.
Expression of loss of Pten phenotypes requires signaling through mTOR
Rapamycin is a widely used and recognized specific inhibitor of mTOR signaling. If the changes in Pten-cre neurons are due to increased mTOR activity, then rapamycin should block the effects of Pten loss (Heitman et al., 1991; Sarbassov et al., 2006; Zhou et al., 2009). We therefore treated Pten-cre neurons with 100 nm rapamycin at 6 DIV and then waited 72 h before electrophysiological analysis. We compared these rapamycin-treated neurons to both vehicle-treated Pten-cre and vehicle-treated control neurons. We found that after 72 h rapamycin treatment the number of vesicles in the RRP were nearly identical to control levels in both glutamatergic and GABAergic Pten-cre neurons (Fig. 6A–D). Rapamycin treatment also decreased both the mEPSC and mIPSC amplitudes of Pten-cre neurons to control levels (Fig. 6E–H).
Figure 6.
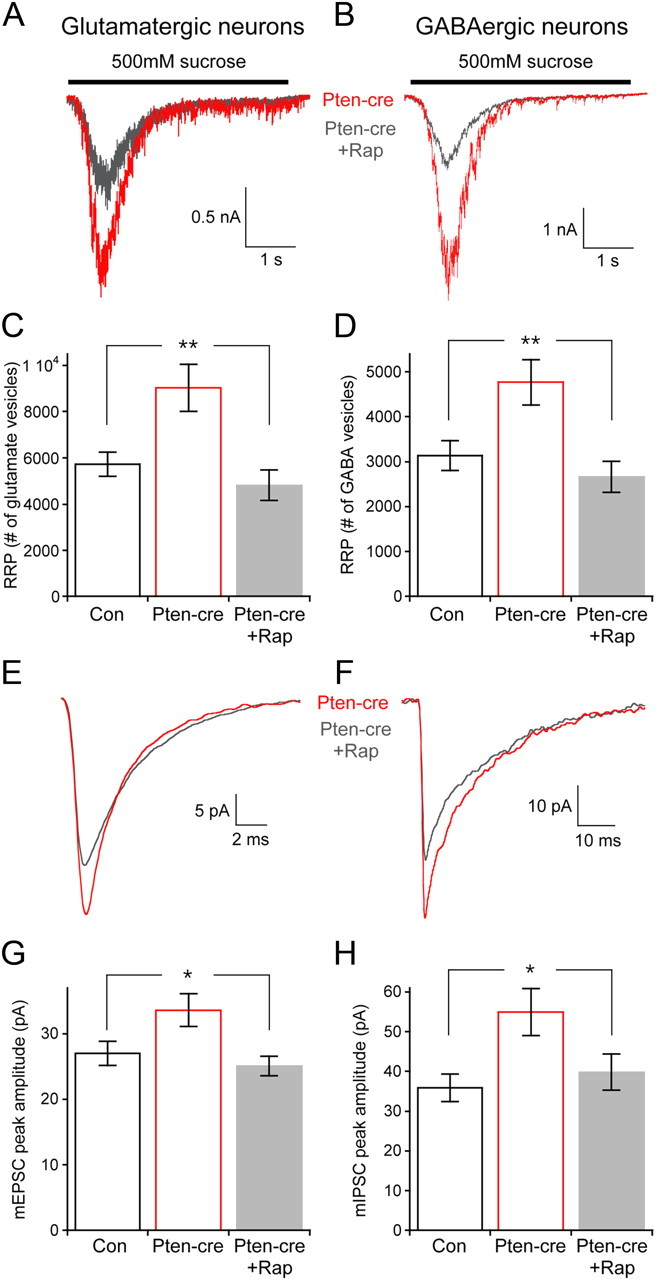
Seventy-two hour rapamycin treatment prevents the increase in RRP size and miniamplitude caused by Pten loss. A, Representative traces of the response of glutamatergic Pten-cre vehicle-treated (red trace) and 100 nm rapamycin-treated (gray trace) neurons to a 4 s pulse of 500 mm sucrose. B, Representative traces of the response of GABAergic Pten-cre vehicle-treated (red trace) and 100 nm rapamycin-treated (gray trace) neurons to a 4 s pulse of 500 mm sucrose. C, The number of synaptic vesicles contained in the RRP of glutamatergic vehicle-treated control, vehicle-treated Pten-cre, and rapamycin-treated Pten-cre neurons. D, The number of synaptic vesicles contained in the RRP of GABAergic vehicle-treated control, vehicle-treated Pten-cre, and rapamycin-treated Pten-cre neurons. E, Representative traces showing the average of mEPSCs collected from one vehicle-treated Pten-cre (red trace) and one rapamycin-treated Pten-cre (gray trace) glutamatergic neuron. F, Representative traces showing the average of mIPSCs collected from one vehicle-treated Pten-cre (red trace) and one rapamycin-treated Pten-cre (gray trace) GABAergic neuron. G, mEPSC peak amplitudes from vehicle-treated control, vehicle-treated Pten-cre, and rapamycin-treated Pten-cre glutamatergic neurons. H, mIPSC peak amplitudes from vehicle-treated control, vehicle-treated Pten-cre, and rapamycin-treated Pten-cre GABAergic neurons. All statistical tests are one-way ANOVA with a Student–Newman–Keuls post-test. All values are presented as mean ± SEM. *p ≤ 0.05; **p ≤ 0.01.
Next, we analyzed images of single neurons from vehicle-treated control, vehicle-treated Pten-cre, and rapamycin-treated Pten-cre neurons to test for an effect of rapamycin on cell size, synapse number, and dendritic length. First, we confirmed that rapamycin reduced mTOR hyperactivity in Pten-cre neurons by phospho-S6 immunocytochemistry (Fig. 7A). We then immunostained for MAP2 and either VGLUT1 or VGAT. We found that the soma size, number of VGLUT1-positive punctae per neuron, and total dendritic length of rapamycin-treated Pten-cre glutamatergic neurons were nearly identical to control neurons and significantly less than vehicle-treated Pten-cre neurons (Fig. 7B,C). The soma size, number of VGAT-positive punctae per neuron, and total dendritic length of 72 h rapamycin-treated Pten-cre GABAergic neurons were also indistinguishable from control neurons and significantly less than vehicle-treated Pten-cre neurons (Fig. 7D,E).
Figure 7.
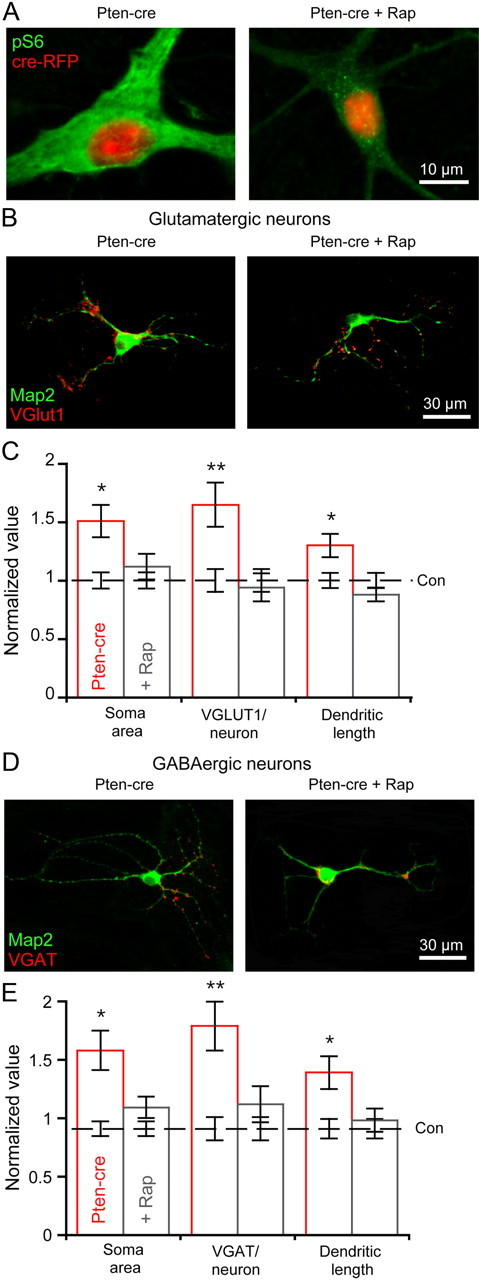
Seventy-two hour rapamycin treatment normalizes dendritic length and synapse number. A, Representative images of a vehicle-treated Pten-cre (left) and rapamycin-treated Pten-cre (right) neuron infected with the cre-RFP-expressing lentivirus showing the fluorescence from the RFP fusion protein (red) and the immunoreactivity against phospho-S6 (green). B, Representative images from vehicle-treated (left) and rapamycin-treated (right) Pten-cre glutamatergic neurons showing immunoreactivity for MAP2 (green) and VGLUT1 (red). C, Normalized values (mean ± SEM) for the soma area, VGLUT1 punctae per neuron, and dendritic length of vehicle-treated (red bars) and rapamycin-treated (gray bars) glutamatergic Pten-cre neurons. The values are normalized to the corresponding values from vehicle-treated control neurons, which are represented by the dashed black line and surrounding error bars. D, Representative images from vehicle-treated (left) and rapamycin-treated (right) Pten-cre GABAergic neurons showing immunoreactivity for MAP2 (green) and VGAT (red). E, Normalized values (mean ± SEM) for the soma area, VGAT punctae per neuron, and dendritic length of vehicle-treated (red bars) and rapamycin-treated (gray bars) GABAergic Pten-cre neurons. The values are normalized to the corresponding values from vehicle-treated control neurons, which are represented by the dashed black line and surrounding error bars. All statistical tests are one-way ANOVA with a Student–Newman–Keuls post-test. *p ≤ 0.05; **p ≤ 0.01 versus control.
Shorter-term rapamycin treatment
Next, we tested whether Pten-cre neurons treated with rapamycin for durations <72 h would show a reduction in synaptic output. First, we directly applied 100 nm rapamycin to glutamatergic Pten-cre neurons during voltage-clamp recordings on day 9–12 in vitro. None of the parameters tested, including the RRP size, evoked response amplitudes, or miniature event amplitudes were affected by acute application of the drug (data not shown). Next we applied 100 nm rapamycin at 9–12 d in vitro and waited 12 h before electrophysiological analysis. Although immunostaining showed a decrease in pS6 intensity in neurons treated for 12 h, the treatment was ineffective at reducing the synaptic output of Pten-cre neurons (Fig. 8A,B). The EPSC peak amplitude, RRP size, PPR, and Cm of 12 h rapamycin-treated Pten-cre neurons were all not significantly different from vehicle-treated Pten-cre neurons, and were still significantly greater than control neurons (Table 2). The mEPSC amplitude was slightly reduced by 12 h rapamycin treatment; however, the value fell in the narrow range between the control and Pten-cre neurons, and was not significantly different from either of the other two groups (Table 2). All of these parameters were rescued by 72 h treatment (Table 2).
Figure 8.
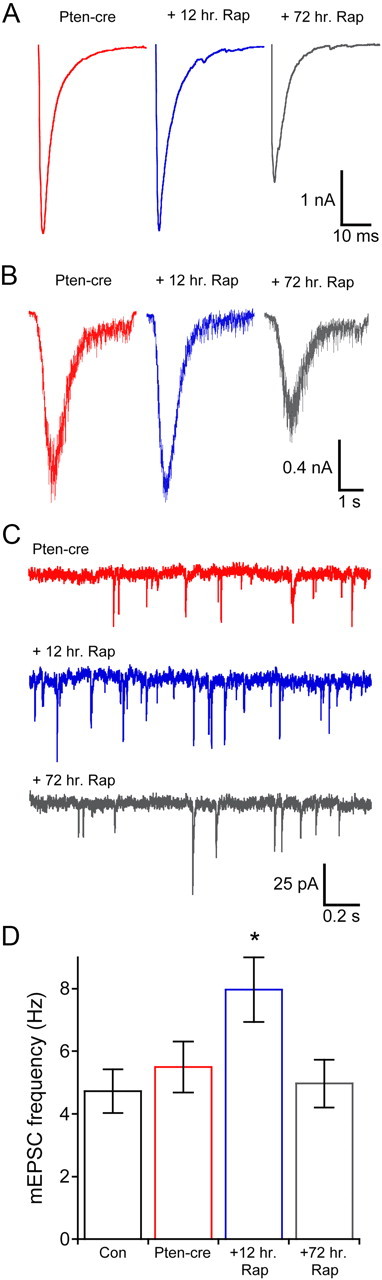
Twelve hour rapamycin treat does not prevent the increase in synaptic transmission, but increases the minifrequency. A, Representative traces showing evoked synaptic responses from vehicle-treated Pten-cre (red traces), 12 h rapamycin-treated Pten-cre (blue traces), and 72 h rapamycin-treated Pten-cre (gray traces) neurons. B, Representative traces showing sucrose application responses from vehicle-treated Pten-cre (red traces), 12 h rapamycin-treated Pten-cre (blue traces), and 72 h rapamycin-treated Pten-cre (gray traces) neurons. C, Representative traces showing miniature EPSC activity from vehicle-treated Pten-cre (red traces), 12 h rapamycin-treated Pten-cre (blue traces), and 72 h rapamycin-treated Pten-cre (gray traces) neurons. D, Miniature event frequencies (mean ± SEM) of vehicle-treated control (black) and Pten-cre (red) neurons, 12 h rapamycin-treated Pten-cre neurons (blue), and 72 h rapamycin-treated Pten-cre neurons (gray). *p ≤ 0.05 versus all other groups (one-way ANOVA with a Student–Newman–Keuls post-test).
Table 2.
Results of 12 and 72 h rapamycin treatment of Pten-cre neurons
| Pten-cre | 12 h rapamycin | 72 h rapamycin | |
|---|---|---|---|
| EPSC amplitude (in nA) | 3.83 ± 0.5; n = 39 | 3.91 ± 0.6; n = 46 | 2.49 ± 0.5**; n = 48 |
| RRP charge (in nC) | 1202 ± 170; n = 36 | 1166 ± 124; n = 42 | 442 ± 60***; n = 48 |
| RRP vesicles | 9397 ± 1662; n = 35 | 9723 ± 1043; n = 40 | 4888 ± 605**; n = 45 |
| mEPSC frequency (in Hz) | 5.50 ± 0.8; n = 36 | 7.97 ± 1.0*; n = 42 | 4.85 ± 0.8; n = 45 |
| mEPSC amplitude (in pA) | 41.2 ± 2.9; n = 36 | 36.1 ± 2.2; n = 42 | 25.7 ± 1.4***; n = 45 |
| Spontaneous release rate (in s−1) | 0.0007 ± 0.0001; n = 35 | 0.0012 ± 0.0002; n = 40 | 0.0011 ± 0.0002; n = 44 |
| Cm (pF) | 38.6 ± 4.6; n = 30 | 34.5 ± 3.4; n = 30 | 26.1 ± 1.8*; n = 34 |
| PPR (20 ms) | 1.29 ± 0.1; n = 30 | 1.26 ± 0.06; n = 26 | 1.11 ± 0.05; n = 28 |
| Pvr (in %) | 3.44 ± 0.5; n = 39 | 3.10 ± 0.2; n = 46 | 5.20 ± 0.8*; n = 48 |
Measurements are shown as mean ± SEM.
*p ≤ 0.05;
**p ≤ 0.01;
***p ≤ 0.001, compared to the other two groups with one-way ANOVA and Student–Newman–Keuls post-test.
The shorter time treatment did, however, have interesting effects on spontaneous release (Fig. 8C,D). It increased the mEPSC frequency ∼1.8 fold above both vehicle-treated and control neurons. Thus, at the 12 h time point there was no change in the number of synaptic vesicles in the RRP, but the rate constant for spontaneous release was already increased, which caused an increase the frequency. This result provides further evidence that mTOR signaling regulates the rate of spontaneous release and an explanation for the apparent lack of an increase in the miniature frequency upon Pten loss (Fig. 2G; Discussion).
Inhibiting mTOR signaling reduces synaptic transmission of wild-type glutamatergic but not wild-type GABAergic neurons
Rapamycin treatment of wild-type animals and slices has been shown to impair both learning and memory and synaptic plasticity (Hoeffer and Klann, 2010). Since rapamycin is a clinically used drug, and treatment in vivo has the potential to affect synaptic transmission, we wanted to test whether inhibiting mTOR signaling with rapamycin affects normal synaptic function in vitro. We therefore treated wild-type glutamatergic and GABAergic neurons with 100 nm rapamycin for 72 h. In glutamatergic neurons, rapamycin significantly reduced the amplitude of the EPSC (Fig. 9A, left bars) and the number of vesicles contained in the RRP (9057 ± 1198 vesicles, n = 44 for vehicle treated vs 6018 ± 912 vesicles, n = 38 for rapamycin treated; p = 0.03) (Fig. 9B,C, left). The mEPSC amplitude was also reduced from 41.7 ± 1.8 pA for vehicle to 34.1 ± 2.1 pA for rapamycin (Fig. 9D,E, left).
Figure 9.
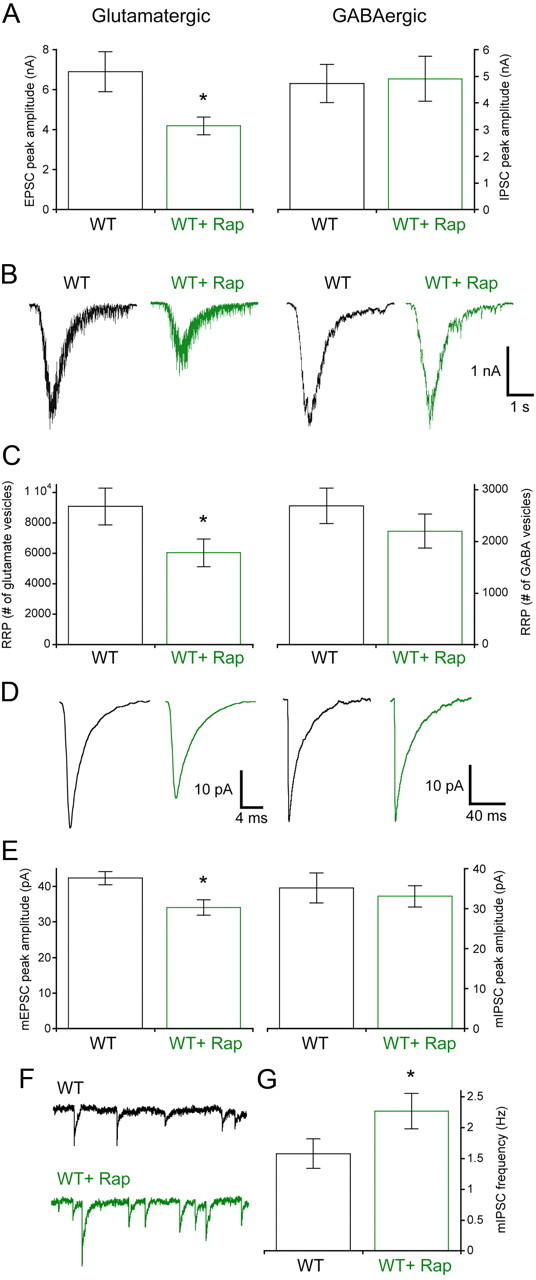
Seventy-two hour rapamycin treatment decreases synaptic transmission in glutamatergic but not GABAergic neurons. A, Peak amplitudes of evoked EPSCs from glutamatergic (left bars) and evoked IPSCs from GABAergic (right bars) neurons. Black bars are vehicle-treated wild-type neurons, and green bars are 72 h 100 nm rapamycin-treated neurons. B, Representative traces showing sucrose application responses from glutamatergic (left traces) and GABAergic (right traces) wild-type neurons treated with vehicle (black traces) or rapamycin (green traces). C, The number of synaptic vesicles (mean ± SEM) contained in the RRP of glutamatergic (left bars) and GABAergic (right bars) neurons treated with vehicle (black) or rapamycin (green). D, Representative traces showing average mEPSCs from glutamatergic (left traces) or average mIPSCs from GABAergic (right traces) wild-type neurons treated with vehicle (black traces) or rapamycin (green traces). E, mEPSC peak amplitudes (left bars) and mIPSC peak amplitudes from (right bars) neurons treated with vehicle (black) or rapamycin (green). F, Representative traces showing miniature IPSC activity from vehicle-treated wild-type (black trace) or 72 h rapamycin-treated wild-type (green trace) GABAergic neurons. G, The frequency of miniature events collected from vehicle-treated and rapamycin-treated wild-type neurons. All values are presented as mean ± SEM. *p ≤ 0.05.
The response of wild-type GABAergic neurons to 72 h rapamycin treatment was notably different from those of the glutamatergic neurons. Although the rapamycin treatment was effective at reducing both pS6 staining and the Cm of GABAergic neurons (21.6 ± 1.7 pF, n = 36 for vehicle treated vs 15.1 ± 0.9 pF, n = 37 for rapamycin; p = 0.003), indicating that it was effective at reducing mTOR signaling, there was no significant change in either the IPSC amplitude (Fig. 9A, right bars) or number of vesicles in the RRP (Fig. 9B,C, right). There was also no reduction in the mIPSC amplitude (Fig. 9D,E, right).
The lack of reduction in the RRP of GABAergic cells in response to rapamycin offered another opportunity to assess the effect of altered mTOR signaling on miniature event frequency in the absence of changes in synaptic vesicle number. In GABAergic neurons, treatment with rapamycin increased the mIPSC rate from 1.57 ± 0.20 Hz to 2.27 ± 0.28 Hz (Fig. 9F,G), further supporting the conclusion that mTOR presynaptically regulates the rate of spontaneous vesicle fusion.
Discussion
In this study, we performed a detailed analysis of synaptic transmission in neurons with altered mTOR signaling. Since misregulation of mTOR signaling is a common cause of neurodevelopmental disorders in humans and mouse models, and rapamycin is a commonly used clinical immunosuppressant and cancer therapeutic, it is important to understand their impact on synaptic transmission. Here we confirm previous studies of synaptic transmission in glutamatergic neurons with hyperactive mTOR signaling by showing that it leads to an increase in excitatory neuron growth and synaptic transmission. We extend these findings by showing that GABAergic neuron growth and synaptic transmission are also regulated by mTOR signaling, and that the synaptic and morphological phenotypes in both cell types are blocked by rapamycin in a time-dependent manner. Finally, we show in wild-type neurons that decreasing mTOR signaling with rapamycin significantly alters normal synaptic transmission, but that these alterations preferentially affect glutamatergic synapses. Since all of the changes occurred in single postmitotic neurons grown on wild-type astrocytes, we conclude they are cell autonomous.
Regulation of synaptic transmission by mTOR
By taking advantage of the autaptic culture system, we show for both neuronal subtypes that mTOR regulates two of the three major parameters that govern the size of evoked synaptic responses. First, our results show changes in the number of synaptic vesicles available for release. This is likely due to the observed increase in synapse formation, and previous studies have linked loss of Pten to increased spine density in glutamatergic neurons (Kwon et al., 2006; Fraser et al., 2008; Luikart et al., 2011; Xiong et al., 2012) Since we did not count synaptic vesicles with electron microscopy, it is also possible that there are also more vesicles per synapse, as reported for Pten loss (Fraser et al., 2008) and the Lis 1 mutant mouse model of epilepsy (Greenwood et al., 2009).
Second, there is an increase in the postsynaptic response to single vesicle fusion. Although we did not conduct experiments to determine whether the increase in miniamplitude was due solely to postsynaptic changes in the number or type of receptors, or whether presynaptic changes in the amount of neurotransmitter released also occur, previous studies have shown changes in mEPSC amplitude when Pten is lost in the postsynaptic neuron (Luikart et al., 2011; Xiong et al., 2012), and that mTOR signaling can regulate surface expression of AMPA receptors through a variety of mechanisms (Wang et al., 2006; Jurado et al., 2010; Moult et al., 2010). However, the fact that we found that both mEPSC amplitude and mIPSC amplitude are regulated by mTOR argues that the presumed change in postsynaptic receptor number does not work exclusively through molecules that are specific to glutamatergic signaling.
Our current-clamp analysis indicated that the AP half-width was lengthened by loss of Pten, which could theoretically increase Ca2+ entry to the presynaptic terminal and increase evoked release. We did not, however, detect a change in Pvr, which would be expected if this were contributing to the increased evoked release.
In contrast to other studies, we did not observe an increase in mEPSC frequency, although we did find an increase in the number of synaptic vesicles in the RRP and number of synapses, which usually leads to an increase in minifrequency. The most likely explanation for this is that the previous studies examined effects of Pten loss in postsynaptic neurons, while in the autaptic culture system the pre- and postsynaptic neurons are one and the same. However, since we experimentally measured both the number of vesicles in the RRP and the rate of spontaneous release, we were able to show that the rate constant for spontaneous fusion was lowered by loss of Pten. Therefore, while Pten loss in a postsynaptic neuron can increase minifrequency by increasing the number of synapses formed, Pten loss affects presynaptic function by inhibiting spontaneous vesicle fusion. Only under conditions where altering mTOR signaling does not lead to a change in the number of available synaptic vesicles, such as rapamycin treatment of wild-type GABAergic neurons (Fig. 9C,F,G), does the presynaptic effect on spontaneous vesicle fusion produce a demonstrable change in minifrequency. This predicts that single synapses formed by axons from neurons with hyperactive mTOR signaling with normal postsynaptic cells would have lower rates of spontaneous release. Since spontaneous vesicle release has been shown to be important for the formation and maintenance of developing synaptic connections, these changes likely have a functional consequence on the development and operations of neuronal networks throughout life.
Loss of Pten in GABAergic neurons
A major finding of this study is that increased mTOR signaling results in nearly identical functional changes in both excitatory and inhibitory neurons. Since mutations or deletions of proteins that negatively regulate mTOR often lead to epilepsy, it is likely that increased synaptic output or connectivity of affected glutamatergic neurons causes network hyperexcitability and seizures in these cases (Wang et al., 2007; Ljungberg et al., 2009). For GABAergic neurons, there is evidence both for and against their contribution to established mTOR-related disease phenotypes (Wang et al., 2007; Fu et al., 2011), although several studies have linked increased inhibitory transmission with both epilepsy and autism (Khalilov et al., 2005; Jones and Baraban, 2007; Tabuchi et al., 2007). GABAergic transmission is thought to regulate important developmental processes such as dendritic maturation, synaptogenesis, and circuit formation (Akerman and Cline, 2007), and can be depolarizing in the developing brain (Mueller et al., 1984; Ben-Ari et al., 1989) and in disease states such as epilepsy (Cohen et al., 2002). It is therefore possible that enhanced GABAergic transmission due to mTOR hyperactivity could cause or facilitate at least some of the neurological deficits currently associated with increased mTOR signaling, or perhaps lead to neurological symptoms that are not currently defined as mTOR related. Independent of the impact on human disease, the finding that mTOR signaling regulates GABAergic transmission is important for our basic understanding of mTOR function in the brain.
The finding that disruption of Pten and hyperactivation of mTOR causes similar increases in synaptic output in two very different classes of neurons implies that the effects of a mutation in a single gene may be very different depending on where in the brain and also when during development the mutation occurs. Mutations in humans that cause mTOR-related diseases are likely often de novo events in progenitor neurons that affect subpopulations of mature neurons. Loss of Pten function in a dentate granule neuron may therefore lead to hyperexcitability of the hippocampal circuit and epilepsy, while loss of function in striatal GABAergic neurons may lead to an imbalance in basal ganglia circuits. GABAergic neurons are incredibly diverse, with subpopulations having distinct roles in circuit function. The effects of altered mTOR on striatal interneurons used in this study may therefore differ from those found in cortex or hippocampus, or at least lead to different effects on the network in which they are present. Thus, our data suggest that even mutations in the same gene may lead to strikingly different disease phenotypes, and that effective treatment in humans may be dependent on the functional consequences of the molecular abnormality in subpopulations of neurons.
Effects of rapamycin on synaptic transmission
Since 72 h rapamycin treatment blocked all of the changes in both neurophysiology and morphology resulting from Pten loss, it is likely that these changes are results of increased mTOR signaling. Twelve hour rapamycin treatment of Pten-cre neurons was ineffective at blocking the increase in the evoked response, the dendrite arbor complexity, and the synapse and synaptic vesicle numbers. This is consistent with the idea that these changes are the result of misregulation of neuron growth, since it would take time to block and/or reverse overgrowth after normalizing translation rates and protein levels with rapamycin. We could not determine whether 12 h rapamycin treatment reduced the mEPSC amplitude, so it is possible that this change occurs on a shorter time scale. We did not test different concentrations of rapamycin, which would likely alter the absolute time scale of the changes we observed.
Rapamycin treatment of wild-type neurons reduced excitatory but not inhibitory synaptic output, even though both phospho-S6 and Cm measurements indicated that mTOR signaling was effectively reduced in both, and rapamycin was shown previously to inhibit axon sprouting of interneurons in response to status epilepticus (Buckmaster and Wen, 2011). It is possible that higher levels of mTOR activity are necessary to support a higher basal level of transcription needed to maintain excitatory synapses than inhibitory synapses. Interestingly, both 12 h rapamycin treatment of Pten-cre neurons and rapamycin treatment of wild-type GABAergic neurons had no effect on synaptic vesicle or synapse number, but increased the miniature event frequency. This provides evidence that mTOR signaling's effects on synaptic transmission are not limited to effects on neuron growth and supports the idea that mTOR regulates synaptic transmission not only through changes in the number of synapses, but also by modifying the function and information coding properties of individual synapses.
Footnotes
This work was supported by an NIH–NINDS Training Grant T32 NS043124 (M.C.W.) and Grants NS018309, NS062992, and P30HD024064. We thank Dr. Ralf Nehring for the cre-RFP lentivirus, and Drs. Mingshan Xue and Christian Rosenmund for critical reading of this manuscript.
References
- Akerman CJ, Cline HT. Refining the roles of GABAergic signaling during neural circuit formation. Trends Neurosci. 2007;30:382–389. doi: 10.1016/j.tins.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, Tsao MS, Shannon P, Bolon B, Ivy GO, Mak TW. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet. 2001;29:396–403. doi: 10.1038/ng782. [DOI] [PubMed] [Google Scholar]
- Bateup HS, Takasaki KT, Saulnier JL, Denefrio CL, Sabatini BL. Loss of Tsc1 in vivo impairs hippocampal mGluR-LTD and increases excitatory synaptic function. J Neurosci. 2011;31:8862–8869. doi: 10.1523/JNEUROSCI.1617-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci U S A. 1991;88:7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol. 1989;416:303–325. doi: 10.1113/jphysiol.1989.sp017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckmaster PS, Wen X. Rapamycin suppresses axon sprouting by somatostatin interneurons in a mouse model of temporal lobe epilepsy. Epilepsia. 2011;52:2057–2064. doi: 10.1111/j.1528-1167.2011.03253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R. On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science. 2002;298:1418–1421. doi: 10.1126/science.1076510. [DOI] [PubMed] [Google Scholar]
- Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61:10–26. doi: 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Castillo J, Katz B. Quantal components of the end-plate potential. J Physiol. 1954;124:560–573. doi: 10.1113/jphysiol.1954.sp005129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser MM, Bayazitov IT, Zakharenko SS, Baker SJ. Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience. 2008;151:476–488. doi: 10.1016/j.neuroscience.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu C, Cawthon B, Clinkscales W, Bruce A, Winzenburger P, Ess KC. GABAergic interneuron development and function is modulated by the tsc1 gene. Cereb Cortex. 2011 doi: 10.1093/cercor/bhr300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood JS, Wang Y, Estrada RC, Ackerman L, Ohara PT, Baraban SC. Seizures, enhanced excitation, and increased vesicle number in Lis1 mutant mice. Ann Neurol. 2009;66:644–653. doi: 10.1002/ana.21775. [DOI] [PubMed] [Google Scholar]
- Habets RL, Borst JG. Post-tetanic potentiation in the rat calyx of Held synapse. J Physiol. 2005;564:173–187. doi: 10.1113/jphysiol.2004.079160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci. 2005;25:11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DL, Baraban SC. Characterization of inhibitory circuits in the malformed hippocampus of Lis1 mutant mice. J Neurophysiol. 2007;98:2737–2746. doi: 10.1152/jn.00938.2007. [DOI] [PubMed] [Google Scholar]
- Jurado S, Benoist M, Lario A, Knafo S, Petrok CN, Esteban JA. PTEN is recruited to the postsynaptic terminal for NMDA receptor-dependent long-term depression. EMBO J. 2010;29:2827–2840. doi: 10.1038/emboj.2010.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalilov I, Le Van Quyen M, Gozlan H, Ben-Ari Y. Epileptogenic actions of GABA and fast oscillations in the developing hippocampus. Neuron. 2005;48:787–796. doi: 10.1016/j.neuron.2005.09.026. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. 2001;29:404–411. doi: 10.1038/ng781. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungberg MC, Sunnen CN, Lugo JN, Anderson AE, D'Arcangelo G. Rapamycin suppresses seizures and neuronal hypertrophy in a mouse model of cortical dysplasia. Dis Model Mech. 2009;2:389–398. doi: 10.1242/dmm.002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- Luikart BW, Schnell E, Washburn EK, Bensen AL, Tovar KR, Westbrook GL. Pten knockdown in vivo increases excitatory drive onto dentate granule cells. J Neurosci. 2011;31:4345–4354. doi: 10.1523/JNEUROSCI.0061-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, Jensen FE, Kwiatkowski DJ. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. 2007;27:5546–5558. doi: 10.1523/JNEUROSCI.5540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moult PR, Cross A, Santos SD, Carvalho AL, Lindsay Y, Connolly CN, Irving AJ, Leslie NR, Harvey J. Leptin regulates AMPA receptor trafficking via PTEN inhibition. J Neurosci. 2010;30:4088–4101. doi: 10.1523/JNEUROSCI.3614-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller AL, Taube JS, Schwartzkroin PA. Development of hyperpolarizing inhibitory postsynaptic potentials and hyperpolarizing response to gamma-aminobutyric acid in rabbit hippocampus studied in vitro. J Neurosci. 1984;4:860–867. doi: 10.1523/JNEUROSCI.04-03-00860.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Schikorski T, Stevens CF, Zhu Y. Inactivity produces increases in neurotransmitter release and synaptic size. Neuron. 2001;32:673–682. doi: 10.1016/s0896-6273(01)00500-1. [DOI] [PubMed] [Google Scholar]
- Orlova KA, Parker WE, Heuer GG, Tsai V, Yoon J, Baybis M, Fenning RS, Strauss K, Crino PB. STRADalpha deficiency results in aberrant mTORC1 signaling during corticogenesis in humans and mice. J Clin Invest. 2010;120:1591–1602. doi: 10.1172/JCI41592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reim K, Mansour M, Varoqueaux F, McMahon HT, Südhof TC, Brose N, Rosenmund C. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell. 2001;104:71–81. doi: 10.1016/s0092-8674(01)00192-1. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Rost BR, Breustedt J, Schoenherr A, Grosse G, Ahnert-Hilger G, Schmitz D. Autaptic cultures of single hippocampal granule cells of mice and rats. Eur J Neurosci. 2010;32:939–947. doi: 10.1111/j.1460-9568.2010.07387.x. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Stevens CF. Neurotransmitter release at central synapses. Neuron. 2003;39:381–388. doi: 10.1016/s0896-6273(03)00643-3. [DOI] [PubMed] [Google Scholar]
- Tabuchi K, Blundell J, Etherton MR, Hammer RE, Liu X, Powell CM, Südhof TC. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science. 2007;318:71–76. doi: 10.1126/science.1146221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci. 2001;21:4249–4258. doi: 10.1523/JNEUROSCI.21-12-04249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Spronsen M, Hoogenraad CC. Synapse pathology in psychiatric and neurologic disease. Curr Neurol Neurosci Rep. 2010;10:207–214. doi: 10.1007/s11910-010-0104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waites CL, Garner CC. Presynaptic function in health and disease. Trends Neurosci. 2011;34:326–337. doi: 10.1016/j.tins.2011.03.004. [DOI] [PubMed] [Google Scholar]
- Wang Y, Barbaro MF, Baraban SC. A role for the mTOR pathway in surface expression of AMPA receptors. Neurosci Lett. 2006;401:35–39. doi: 10.1016/j.neulet.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Wang Y, Greenwood JS, Calcagnotto ME, Kirsch HE, Barbaro NM, Baraban SC. Neocortical hyperexcitability in a human case of tuberous sclerosis complex and mice lacking neuronal expression of TSC1. Ann Neurol. 2007;61:139–152. doi: 10.1002/ana.21058. [DOI] [PubMed] [Google Scholar]
- Xiong Q, Oviedo HV, Trotman LC, Zador AM. PTEN regulation of local and long-range connections in mouse auditory cortex. J Neurosci. 2012;32:1643–1652. doi: 10.1523/JNEUROSCI.4480-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Stradomska A, Chen H, Brose N, Zhang W, Rosenmund C, Reim K. Complexins facilitate neurotransmitter release at excitatory and inhibitory synapses in mammalian central nervous system. Proc Natl Acad Sci U S A. 2008;105:7875–7880. doi: 10.1073/pnas.0803012105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, Sinton C, Powell CM, Parada LF. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knockout mice. J Neurosci. 2009;29:1773–1783. doi: 10.1523/JNEUROSCI.5685-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]