

Abstract
Endometrial cancer is the most frequent gynecologic cancer in women. Long-term outcomes for patients with advanced stage or recurrent disease are poor. Targeted molecular therapy against the vascular endothelial growth factor (VEGF) and its receptors constitute a new therapeutic option for these patients. The goal of our study was to assess the potential effectiveness of inhibition of VEGF/VEGFR signaling in a xenograft model of endometrial cancer using bevacizumab (Avastin, a humanized antibody against VEGFA). We also aimed to identify molecular markers of sensitivity or resistance to this agent. We show that bevacizumab retards tumor growth in athymic mice by inhibiting molecular components of signaling pathways that sustain cell survival and proliferation. We also demonstrate that resistance to bevacizumab may involve up-regulation of anti-apoptotic genes and certain proto-oncogenes. We propose that down-regulation of ARHGAP6 and MMP15 transcripts indicates that tumors are sensitive to bevacizumab whereas inhibition of PKCδ- or S6K-dependent signaling and up-regulation of TNFRS4 or MMP13 and MMP14 mark a developing resistance to bevacizumab therapy. Interestingly, the significant activation of c-Jun oncogene detected in bevacizumab-treated tumors suggests that, in endometrial cancers, the c-Jun-mediated pathway(s) contribute to bevacizumab resistance.
Keywords: endometrial cancer, vascular endothelial growth factor, bevacizumab
Introduction
Endometrial (uterine) cancer is the fourth most common cancer in women and the most frequent gynecologic malignancy with approximately 43000 new cases and 7950 American women lost every year (1). The lifetime risk for developing endometrial cancer is 2.4% and it is on the rise (2). Surprisingly, relative 5-year survival for endometrial cancer has decreased from 88% in 1975 to 84% in 2003 according to the American Cancer Society (3). In comparison, 5-year survival for breast and prostate cancers now exceeds 90%. Thus, finding new therapies for recurrent or advanced endometrial cancer is a priority in cancer research.
Inhibitors of VEGF/VEGFR pathway constitute a novel therapy to treat endometrial cancer. For example, VEGF expression is associated with elevated tumor vascularization, measured by microvessel density in endometrial cancer tissues (4), and is a predictive marker for decreased 5-year survival in patients with advanced endometrial carcinoma (5–7). Current VEGF-targeted therapeutics have shown promise in animal models and clinical trials of endometrial cancer. Using an orthotopic mouse model of endometrial carcinoma, Kamat et al, found that bevacizumab (a monoclonal antibody against VEGFA), reduced tumor volume by 77% compared to control treatment (4). Mechanistically, it is hypothesized that bevacizumab mimics the differentiating effects of progesterone on endometrium and inhibits growth of endometrial cancers through down-regulation of a number of angiogenic and proliferative signaling pathways (8).
In a retrospective study of nine patients with recurrent endometrial carcinoma, partial response was observed in two patients and three individuals demonstrated stable disease with a median survival of 8.7 months (9). A Phase II trial, GOG (Gynecologic Oncology Group)-229E, evaluated bevacizumab single-agent therapy in recurrent or persistent endometrial carcinoma. Early results from this study of 53 patients include one complete and seven partial responses (15.1% response rate) and 35.8% of subjects demonstrated 6-month progression-free survival (10).
While these preclinical and clinical results are encouraging, longer studies in other types of cancer have detected disease progression, which suggests eventual resistance to bevacizumab therapy (11–14). Understanding the pathways that are responsible for resistance is crucial for extending progression-free survival. In this study we aimed to investigate the effects of bevacizumab in a xenograft model of human endometrial cancer at the transcriptional and protein signaling levels and to identify potential markers of sensitivity and resistance to bevacizumab-based therapy. Defining potential biomarkers of bevacizumab sensitivity and resistance has immediate application for clinical trials now underway in patients with endometrial cancer.
Materials and methods
Cells and reagents
Hec50 endometrial cancer cells were provided by Dr Erlio Gurpide (New York University). An aggressive sub-clone, Hec50co, has been previously characterized and validated in a model for advanced endometrial cancer by our laboratory (15,16). Cells were cultured in DMEM (Sigma, St Louis, MO) supplemented with 10% FBS (Gemini Bio Products, Inc., Calabasas, CA) and antibiotic/antimycotic solution containing 100 units/ml penicillin-G, 100 μg/ml streptomycin and 0.25 μg/ml amphotericin B (Gibco Life Technologies, Grand Island, NY).
Mouse xenograft of human endometrial cancer
The experimental procedures for the creation of tumor xenografts and injection of therapeutic agents were reported previously (17). The experimental protocols were approved by the University of New Mexico Health Sciences Center Institutional Animal Care and Use Committee. Briefly, 6–8 week old, Crl: NU/NU-nuBR female athymic mice (NCI, Frederic, MD) were injected subcutaneously with 5×106 cancer cells in 0.1 ml of RPMI cell culture medium using a syringe with a 22G5/8 needle. Bevacizumab reconstituted in saline was given at a dose of 0.2 mg/mouse through intraperitoneal (i.p.) injection twice a week, starting the day of cancer cell injection. The control group was given saline i.p. Tumors were measured weekly with a caliper, and tumor cross-sectional areas (mm2) were calculated using the formula: length (mm) × width (mm) × π/4. The experiment was terminated when a tumor reached 2 cm in any dimension, and animals were then euthanized in a CO2 chamber. The control group had 8 mice and the treatment group had 16 mice. Data were analyzed by one-way ANOVA and Holm-Sidak t-test to compare the tumor sizes between control and treatment groups with a p-value of ≤0.05 considered to be significant. At sacrifice, tumors from control animals as well as those tumors that grew in the presence of bevacizumab were collected and snap-frozen in liquid nitrogen for RNA and protein extraction.
RNA extraction
RNA was extracted using RNeasy spin columns (Qiagen Corp., Valencia, CA). The RNA quality was checked using the Agilent 2100 Bioanalyzer (Agilent Technologies, Foster City, CA) and the concentration determined with the Nanodrop spectrophotometer (Thermo Scientific, Wilmington, DE).
Affymetrix data analysis
Functional replicate Affymetrix microarrays were performed for two independent experiments using the human HG-U133A plus 2.0 chips (Santa Clara, CA), which queries the expression of 54000 transcripts. All procedures for the chip preparation and cDNA production were performed per manufacturer’s instructions (v. 701025 Rev.5). Briefly, total RNA was used to generate double-stranded cDNA with an oligo dT-primer containing the T7 RNA polymerase promoter site and the One-Cycle Target Labeling kit. cDNA was purified via a column using the GeneChip Sample Cleanup Module and biotinylated cRNA was synthesized by in vitro transcription using the GeneChip IVT Labeling kit. Biotin-labeled cRNA was purified (GeneChip Sample Cleanup Module) and the absorbance measured at 260 nm to determine yield (Nanodrop spectrophotometer). Twenty micrograms of the labeled cRNA was fragmented, and its quality was assessed for purified cRNA and fragmented cRNA using the Agilent 2100 Bioanalyzer and the RNA 6000 Nano LabChip kit. The labeled fragmented cRNA was hybridized to Affymetrix GeneChip HG-U133A plus 2.0 arrays for 16 h at 45°C. Array washing and staining was performed on the Affymetrix fluidics (450) station according to the antibody amplification protocol (Fluidics script: EukGE-WS2v5). The GeneChips were scanned using the Affymetrix GeneChip Scanner 3000 (a wide-field, epifluorescent near-confocal microscope with a patented flying objective).
Analysis approaches and data interpretation
Initial data analysis was performed using Affymetrics Microarray Suite v 5.0 software, setting the scaling of all probe sets to a constant value of 500 for each GeneChip. Silicon Genetics’ (now Agilent Technologies) GeneSpring 7.3 (Redwood City, CA) was used to filter data using several criteria. First, only transcripts with a fluorescent signal above the background level were retained for the subsequent 2-fold change filter. Starting with over 54000 transcripts this eliminated all but 5592 transcripts. Next, we used an ANOVA calculation to identify transcripts with a p-value <0.05. Four hundred and fifty-four transcripts meet these stringent criteria. Data (GSE18195) were deposited at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE18195. Affymetrics microarray data were additionally analyzed by the University of Iowa DNA Facility Molecular Biology Computing Service.
Real-time RT-PCR
cDNA was synthesized from 1 μg of total RNA and PCR reactions were carried out in 50 μl reaction mixtures using 50 ng of template per well. Amplification of GADPH was used as an endogenous control to standardize the amount of RNA in each sample. The Assay on Demand™ protocol was carried out as directed in the ABI manual (Applied Biosystems, Foster City, CA). The raw data were presented as the cycle number associated with initial amplification. The data were then normalized to an endogenous control (GAPDH) to allow for variance in RNA template amounts added to the reverse transcription reaction. The data could then be compared to a calibrator and analyzed using the 2−ΔΔCT method (18).
Isolation of tissue lysates
Protein was extracted according to the Kinetworks’ guidelines (Kinexus, Victoria, BC). Tumor tissues were incubated in 0.5% Triton X-100 lysis buffer (0.5% Triton X-100, 10 mM Tris-HCl pH 7.4, 5 mM EDTA, 50 mM NaCl, 50 mM NaF, 20 μg/ml aprotinin, 1 mM PMSF and 2 mM Na3VO4), homogenized and disrupted using a rotor-stator homogenizer for 90 sec. The samples were then sonicated for 10 sec on ice to rupture the cells. Cell lysates were ultra-centrifuged for 30 min at 100000 × g at 4°C. The supernatant was then transferred to fresh tubes, and protein concentrations determined by the Bradford assay (Bio-Rad Laboratories, Hercules, CA).
Kinetworks signal transduction profiling
Phosphoproteomic analysis of lysates from control and bevacizumab-treated tumors was performed in duplicate. Briefly, tissue lysates (400 μg) were mixed with 4X sample buffer (125 mM Tris-HCl, pH 6.8, 4% w/v SDS, 50% v/v glycerol, 0.08% w/v bromophenol blue and 5% β-mercaptoethanol) in a ratio of 1:4 sample to buffer, boiled for 4 min and analyzed using Kinetworks Phospho-Site Screen 1.3, which tracks the abundance of 31 phosphoprotein targets (Kinexus Bioinformatics Corp., Vancouver, Canada). The results were reported as the percent of binding compared with control, and peptide densities that vary (±25% from the controls are considered to be legitimate signaling targets reliably detected using this method.
Western blotting
Tissue lysates were re-suspended in Laemmli’s buffer (Bio-Rad Laboratories) at 1:1 dilution and 100 μg of total protein extract was separated on Criterion™ XT precast 4–12% gradient gels (Bio-Rad Laboratories) at a 150 V for 1 h. The proteins were then transferred to a nitrocellulose membrane (Bio-Rad Laboratories) for 1 h at 100 V. The membrane was blocked for 1 h in 5% non-fat dry milk and incubated with the anti-phospho-c-Jun antibody (Upstate Cell Signaling, Lake Placid, NY) overnight at 4°C. Subsequently, the membranes were incubated with the HRP-conjugated secondary antibody (Bio-Rad Laboratories) at room temperature for 1 h. Signal was visualized using Pierce® ECL Western blotting developing solution (Thermo Scientific, Rockford, IL). Anti-actin rabbit polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA), overnight at 4°C, was used to assess equal loading of samples.
Results
Bevacizumab controls tumor growth in mouse xenograft model of endometrial cancer
Bevacizumab has been shown to be effective in limiting the tumor growth in several types of cancers, although it is unclear whether it exerts the same effects in endometrial cancers. Therefore, we examined the effect of bevacizumab on tumor growth in a xenograft model of endometrial cancer using Hec50co cells, an aggressive endometrial cancer cell line (15,16). Animals were implanted with Hec50co cells subcutaneously, and treatment with bevacizumab began on the day of tumor cell implantation and was repeated twice per week for the duration of the study. The data in Fig. 1 demonstrate that bevacizumab treatment alters the rate and extent of endometrial tumor formation. The bevacizumab-treated animals had a statistically reduced tumor volume (Fig. 1, bevacizumab, open circles; mean cross-sectional area 23.2 mm2) compared to control animals (control, closed circles; mean cross-sectional area 79.5 mm2, p≤0.05), indicating that some sensitivity to bevacizumab treatment occurs. Interestingly, we observed a 19% reduction in tumor incidence in the bevacizumab-treated animals compared to controls: 75% of control animals developed tumors, whereas only 56% of bevacizumab-treated animals had measureable tumors (data not shown). However, the fact that tumors are present at all in the treated group indicates some degree of resistance to bevacizumab.
Figure 1.

Bevacizumab inhibits growth of endometrial cancer in mouse xenograft model. Athymic mice injected with Hec50co endometrial cancer cells were treated with vehicle control (saline, n=8) or bevacizumab (n=16) for 5 weeks. Tumor growth in mice in control (closed circles ●) and bevacizumab (open circles ○) treated groups was estimated weekly for 5 weeks. Differences in tumor size (mm2) were assessed by ANOVA and post-hoc Bonferroni’s test. *Indicates statistically significant differences between groups (p≤0.05) and #indicates that tumor sizes differ significantly between control and experimental groups at week 5 of treatment (p≤0.05).
The results from this experiment suggest that two distinct events mediating sensitivity and resistance are occurring in the bevacizumab-treated tumors: i) bevacizumab exerts an inhibitory effect in human endometrial cancers and this effect may be mediated by the intracellular pathways that confer sensitivity to the drug; and ii) tumor growth, albeit slow, in bevacizumab-treated animals implicates the activation of escape mechanisms that may ultimately lead to the resistance to this agent. Therefore, we next sought to identify the divergent signaling pathways that are responsible for the sensitivity and resistance to bevacizumab treatment.
Bevacizumab regulates expression of putative sensitivity and resistance genes in tumors
Since the endometrial tumors displayed differing responses to bevacizumab therapy, it is imperative to identify putative pathways of sensitivity and resistance that are activated in response to bevacizumab. Therefore, an array strategy was employed to identify genes that changed in expression following exposure to bevacizumab (microarray data, GSE18195, have been deposited at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE18195). Many genes of interest were regulated, including TNFRSF4, MMP13, MMP14, TP53BP1 and COL5A2 that were up-regulated (Table I) and ARHGAP6, PDCD14, MMP15, PRDM16 and COL9A3 that were down-regulated (Table II). The regulation of these genes was confirmed by quantitative real-time RT-PCR (Fig. 2). Transcript levels of TNFRSF4 and MMP13 genes were 8 and 9.5 times higher in bevacizumab-treated animals, respectively, and expression of MMP14, TP53BP1 and COL5A2 genes was ~2–2.5 times higher than in control animals (Fig. 2A). In bevacizumab-treated tumors ARHGAP6, PDCD14, MMP15, PRDM16 and COL9A3 genes showed 20–50% lower expression levels compared to non-treated controls (Fig. 2B). Full names and accession numbers of these genes are given in Tables I and II.
Table I.
Genes up-regulated in response to bevacizumab treatment.
| Gene | Abbreviation | Accession no. | Fold-change
|
Proposed reason for regulation | |
|---|---|---|---|---|---|
| Affymetrix | qRT-PCR | ||||
| Tumor protein p53 binding protein 1 | TP53BP1 | NM_001141980.1 | 2.62 | 2.41 | Sensitivity |
| Tumor necrosis factor receptor superfamily member 4 | TNFRSF4 | NM_003327.2 | 11.80 | 8.40 | Resistance |
| Matrix metallopeptidase 13 (collagenase 3) | MMP13 | NM_002427.2 | 11.30 | 10.25 | Resistance |
| Matrix metallopeptidase 14 | MMP14 | NM_004995.2 | 1.61 | 1.64 | Resistance |
| Collagen, type V, α 2 | COL5A2 | NM_000393.3 | 3.13 | 2.09 | Resistance |
Table II.
Genes down-regulated in response to bevacizumab treatment.
| Gene | Abbreviation | Accession no. | Fold-change
|
Proposed reason for regulation | |
|---|---|---|---|---|---|
| Affymetrix | qRT-PCR | ||||
| Rho GTPase activating protein 6 transcript variant 1 | ARHGAP6 | NM_013427.2 | −1.15 | −0.49 | Sensitivity |
| Matrix metallopeptidase 15 | MMP15 | NM_002428.2 | −6.70 | −0.36 | Sensitivity |
| Collagen, type IX, α 3 | COL9A2 | NM_001853.3 | −1.27 | −0.25 | Sensitivity |
| PR domain containing 16 | PRDM16 | NM_022114.2 | −2.47 | −0.40 | Resistance |
| Programmed cell death 4 | PDCD4 | NM_145341.2 | −1.56 | −0.23 | Resistance |
Figure 2.

Identification of putative biomarkers for bevacizumab resistance and sensitivity. Nude mice were injected with Hec50co endometrial cancer cells and subsequently treated with bevacizumab or saline (controls) as in Fig. 1. Following a 5-week treatment tumors were extracted and RNA content analyzed by quantitative RT-PCR. Expression levels of genes (A) up-regulated or (B) down-regulated by bevacizumab treatment, normalized to an internal control. A value of 0 on the y-axis represents basal (control) levels. Grey bars denote predicted markers of sensitivity to bevacizumab treatment; black bars represent predicted markers of resistance to bevacizumab.
Bevacizumab activates putative signaling pathways of sensitivity and resistance in tumors
The changes at the mRNA level led us to investigate how bevacizumab affects post-translational modifications of proteins, such as phosphorylation, using the Kinetworks phosphoproteomic assay. The Kinetworks multilane immunoblotting is a phosphoproteomic assay that assesses phosphorylation levels of multiple signal transduction molecules. An increase in phosphorylation of intracellular signaling pathways indicates that activation has occurred in response to therapeutic/experimental interventions; therefore, we used this approach to compare activation of tumor signaling pathways between control and bevacizumab-treated animals. Data indicate that an increase as well as a decrease in phosphorylation occurred following bevacizumab treatment. In particular, signaling pathways that respond with increased phosphorylation include proliferative and pro-survival pathway components, such as c-Jun, MEK 1/2, Akt/PKBα, STAT-3 and SMAD 1/5/9 (Fig. 3, Table III). In contrast, p70S6Kα and PKCδ demonstrated reduced phosphorylation levels (Fig. 3, Table III).
Figure 3.
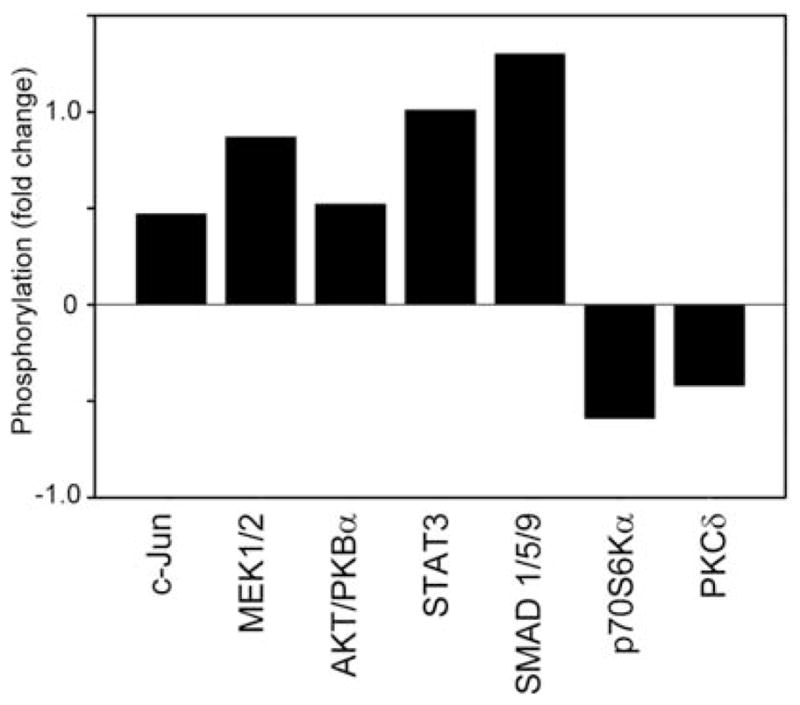
Phosphoproteomic analysis of putative pathways involved bevacizumab sensitivity and resistance. Nude mice were injected with Hec50co human endometrial cancer cells and treated with bevacizumab or vehicle (controls) for 5 weeks. Subsequently, tumors were excised and total cell lysates were isolated. Phosphorylation levels of signaling molecules from two independent experiments were assessed using Kinetworks immunoblotting approach. Results obtained from bevacizumab-treated mice were averaged, normalized to controls and presented as a fold increase/decrease in phosphorylation levels. A value of 0 on the y-axis indicates the phosphorylation levels in vehicle-treated mice.
Table III.
Phosphoproteomic comparison of bevacizumab-treated and control tumors.
| Phosphorylated protein | Phosphorylation site(s) | Accession no.(s) |
|---|---|---|
| c-Jun | S73 | NP_002219.1 |
| MEK1/2 | S218, S222 | NP_002746.1 |
| AKT/PKBα | T308 | NP_005154.2 |
| STAT3 | S727 | NP_644805.1 |
| SMAD 1/5/9 | S463 | NP_00589.1 |
| S465 | NP_005894.3 | |
| S467 | NP_001120689.1 | |
| p70S6Kα | T389 | NP_003152.1 |
| PKCδ | T507 | NP_006245.2 |
The activation of c-Jun was of particular interest
c-Jun is a proto-oncogene of the AP-1 family of proteins that is most frequently associated with tumor progression in endometrial cancers (19). Differential phosphorylation of c-Jun in non-treated vs. treated animals was verified by immunoblotting (Fig. 4). Consistent with the phosphoproteomic data, we detected an increase in phosphorylation of c-Jun in bevacizumab-treated tumors compared to control tumors. Quantitation of immunoblotting signals indicate that compared to the vehicle-treated mice, a 3- to 10-fold increase in phosphorylation levels of c-Jun was detected in experimental animals. These data suggest that elevated c-Jun phosphorylation is one mechanism by which bevacizumab-treated tumors continue to grow. Furthermore, inhibition of components of the c-Jun signaling pathway may represent one approach to circumvent the resistance to bevacizumab.
Figure 4.
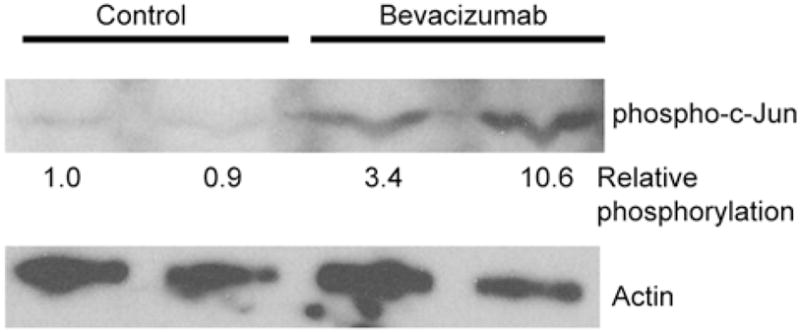
Bevacizumab activates c-Jun. Nude mice were injected with Hec50co endometrial cancer cells and treated with bevacizumab or saline (controls) for 5 weeks. Subsequently, tumors were excised and total cell lysates were isolated. Signaling pathways identified as activated/inhibited by Kinetworks immunoblotting were further tested by immunoblotting lysates from control and bevacizumab-treated tumors. Representative immunoblot shows an increase in phosphorylation levels of c-Jun (upper panel, 45 kDa) in bevacizumab-treated vs. vehicle-treated control group. Actin (lower panel, 40 kDa) was used as a loading control. Densitometric analysis was performed on phospho-c-jun blot, normalized to actin expression, and the relative c-jun phosphorylation (to control, lane 1) is listed below phospho-c-jun blot.
In conclusion, our results demonstrate that, in an animal model of endometrial cancer, bevacizumab induces both the activation and the inhibition of proteins downstream of VEGFR. Further studies would be required to validate the use of these molecules as markers of therapeutic response or resistance to VEGF inhibitors.
Discussion
The interest in anti-angiogenic therapy is due to the proven effectiveness of bevacizumab (the anti-VEGFA antibody) in treating several solid tumors, for example lung, prostate and breast (20–22). It is unclear however whether malignant lesions of endometrium respond to bevacizumab and whether resistance to bevacizumab-based therapy is an issue in endometrial cancers. This study aimed to identify potential molecular indicators of sensitivity and/or resistance to bevacizumab. Specifically, in a xenograft model of endometrial cancer, we used mRNA expression profiling and a phosphoproteomic approach to assess the regulation of intracellular signaling pathways at both the mRNA and protein levels. The potential markers of response would be the pro-growth genes or signaling molecules for which bevacizumab is inhibitory and, alternatively, the pro-growth factors up-regulated/activated by bevacizumab treatment may be used as markers of resistance.
At the level of transcription, bevacizumab inhibits the expression of proliferative genes, such as ARHGAP6 and MMP15. MMP15 is a transmembrane metalloproteinase involved in the breakdown of extracellular matrix and induction of transmigration thus propagating metastasis (23,24). ARHGAP6 activates RhoA GTPases of the ras superfamily of oncogenes and promotes cell migration via inhibition of focal adhesion molecules and actin remodeling (25,26). We hypothesize that these genes may represent molecular markers of sensitivity; down-regulation of each of these transcripts would be an indicator(s) of the inhibition of tumor growth by bevacizumab.
However, there was a significant enhancement of phosphorylation of targets downstream of ras, including c-Jun, MEK 1/2, Akt/PKBα, STAT3 and SMAD1/5/9, suggesting a Rho/ROCK-independent compensatory activation of pro-oncogenic pathways (27–30). Other findings from the gene expression and phosphoprotein immunoblotting assays also suggest augmentation of intracellular signaling that favors survival of cancer cells. Both PDCD4 and PRDM16, which were down-regulated in bevacizumab-treated tumors, have been previously shown to possess tumor suppressor activity (31–34). PDCD4 phosphorylation by p70S6Kα and subsequent degradation by the proteasome serve to regulate protein synthesis and cell growth (35). Combined, up-regulation of PDCD4 and decreased p70S6Kα activity would contribute to indefinite cell proliferation that is characteristic of cancer growth and therefore may be one marker of resistance to bevacizumab.
We also detected an up-regulation of the anti-apoptotic gene TNFRS4 and metalloproteinases MMP13 and MMP14. In normal endometrium, progesterone inhibits MMP13 gene expression (36), thus the up-regulation of MMPs in these studies suggests that the tumors may be insensitive to progesterone. Therefore, elevated MMP13 expression may represent a compensatory effect in response to bevacizumab treatment. MMP14 plays a role in the regulation of angiogenesis, and increased expression may indicate a developing resistance to anti-angiogenic therapy such as bevacizumab (37,38).
PKCδ functions as a pro-apoptotic gene in multiple cell types (39,40). In endometrial cancers, in particular, a loss of PKCδ expression or function is associated with a higher grade of cancer and possibly is an indicator of malignant transformation of endometrial epithelium (41). In accord with previously published results, our study suggests that the loss of PKCδ function, as indicated by the attenuated phosphorylation of Thr507 in the enzyme activation loop, has occurred and this may be a result of the inhibitory effect of bevacizumab on VEGFA-stimulated PKCδ activation (42,43). The signaling pathways that depend on PKCδ for activation include, among others, Akt/mTOR/p70S6K and STAT3. Phosphorylation of Thr389 on p70S6Kα by PKCδ regulates the Akt/mTOR/p70S6K pathway to inhibit ERK1/2 signaling, thereby decreasing cell proliferation (44,45). Hence, combined inhibition of PKC and p70S6Kα may mark the escape mechanism that would contribute to the cancer growth despite the administration of bevacizumab.
The finding that bevacizumab activates the oncogene c-Jun is particularly interesting. c-Jun is overexpressed in poorly differentiated human endometrial cancer cells and is down-regulated by chemotherapeutic agents (19). An increase in phosphorylation of c-Jun in animals that received bevacizumab is another potential indicator of developing resistance to treatment. Further studies are necessary to investigate whether changes in phosphorylation levels of c-Jun can be used as molecular markers of sensitivity/resistance to bevacizumab therapy.
The role of STAT3 Ser727 phosphorylation for transcriptional activity in general and in endometrial cancers in particular, is unclear. Conflicting data report both an increase and a decrease in transcriptional activity of STAT3 upon phosphorylation of Ser727, and in some cancers, PKCδ-dependent Ser727 phosphorylation results in an inhibition of cell proliferation (46,47). Thus, whether augmented phosphorylation of STAT3 Ser727 indicates developing resistance to bevacizumab in endometrial cancers or whether this is a marker of sensitivity requires further elucidation. Similarly, the importance of increased phosphorylation of SMAD1/5/9 is unclear since, depending upon the location of the cancer; both induction and inhibition of tumor growth have been linked to the activation of this pathway (48,49).
In conclusion, these studies confirm that the VEGF pathway is a promising target for new therapies against endometrial cancer. The inhibition of this pathway results in changes in gene expression that enhance apoptosis and reduce cellular proliferation and tumor invasion. This study also suggests that endometrial cancers have the potential to become resistant to bevacizumab-based treatments and identify potential markers of resistance that include the induction of compensatory phosphoproteins and transcripts involved in cell survival. We propose that ARHGAP6 and MMP15 transcripts are putative markers of sensitivity of endometrial cancers to bevacizumab, and down-regulation of these factors correlates with tumor responsiveness to therapy. We also suggest that differential regulation of PDCD4, PRDM16, TNFRS4 and MMP13 and MMP14 transcripts and inhibition of p70S6Kα and PKCδ signaling indicate that tumors have become resistant to bevacizumab. The exact role of activation of c-Jun, MEK1/2, Akt/PKB, STAT3 and SMAD 1/5/9 in endometrial cancers requires further elucidation.
Acknowledgments
This study was supported by NIH grant R01CA99908-1 (K.K.L.), the Cory Beach Family Fund (K.K.L., D.D. and S.D.) and the Oxnard and Anderson Foundations (K.K.L. and S.D.). We also thank our benefactors, Dean and Alice Irvin and Mrs. Shirley Leslie, for supporting this research. We thank the Keck-UNM Genomics Shared Resource supported by the University of New Mexico Cancer Research and Treatment Center, the W.M. Keck foundation and the State of New Mexico, for making this study possible. We wish to express our appreciation to Rebecca Lobb and Marilee Morgan for their experimental assistance and expertise in the performance of the Affymetrix arrays and subsequent real-time RT-PCR.
Abbreviations
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
- VEGFA
vascular endothelial growth factor A
- DMEM
Dulbecco modified Eagle’s medium
- FBS
fetal bovine serum
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer Statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.McCann SE, Freudenheim JL, Marshall JR, Brasure JR, Swanson MK, Graham S. Diet in the epidemiology of endometrial cancer in Western New York (United States) Cancer Causes Control. 2000;11:965–974. doi: 10.1023/a:1026551309873. [DOI] [PubMed] [Google Scholar]
- 3.Cancer Facts and Figures. 2010 http://www.cancer.org/acs/groups/content/@nho/documents/document/acspc-024113.pdf.; Am Cancer Soc. 2010 [Google Scholar]
- 4.Kamat AA, Merritt WM, Coffey D, et al. Clinical and biological significance of vascular endothelial growth factor in endometrial cancer. Clin Cancer Res. 2007;13:7487–7495. doi: 10.1158/1078-0432.CCR-07-1017. [DOI] [PubMed] [Google Scholar]
- 5.McMeekin DS, Sill MW, Benbrook D, et al. A phase II trial of thalidomide in patients with refractory endometrial cancer and correlation with angiogenesis biomarkers: a Gynecologic Oncology Group study. Gynecol Oncol. 2007;105:508–516. doi: 10.1016/j.ygyno.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanseverino F, Santopietro R, Torricelli M, et al. pRb2/p130 and VEGF expression in endometrial carcinoma in relation to angiogenesis and histopathologic tumor grade. Cancer Biol Ther. 2006;5:84–88. doi: 10.4161/cbt.5.1.2345. [DOI] [PubMed] [Google Scholar]
- 7.Hirai M, Nakagawara A, Oosaki T, Hayashi Y, Hirono M, Yoshihara T. Expression of vascular endothelial growth factors (VEGF-A/VEGF-1 and VEGF-C/VEGF-2) in postmenopausal uterine endometrial carcinoma. Gynecol Oncol. 2001;80:181–188. doi: 10.1006/gyno.2000.6056. [DOI] [PubMed] [Google Scholar]
- 8.Cerezo L, Cardenes H, Michael H. Molecular alterations in the pathogenesis of endometrial adenocarcinoma. Therapeutic implications. Clin Transl Oncol. 2006;8:231–241. doi: 10.1007/BF02664933. [DOI] [PubMed] [Google Scholar]
- 9.Wright JD, Powell MA, Rader JS, Mutch DG, Gibb RK. Bevacizumab therapy in patients with recurrent uterine neoplasms. Anticancer Res. 2007;27:3525–3528. [PubMed] [Google Scholar]
- 10.Aghajanian C, Sill MW, Darcy K, et al. A phase II evaluation of bevacizumab in the treatment of recurrent or persistent endometrial cancer: a Gynecologic Oncology Group (GOG) study. J Clin Oncol. 2009;27 doi: 10.1200/JCO.2010.32.6397. abs. 5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ortega J, Vigil CE, Chodkiewicz C. Current progress in targeted therapy for colorectal cancer. Cancer Control. 2010;17:7–15. doi: 10.1177/107327481001700102. [DOI] [PubMed] [Google Scholar]
- 12.Koutras AK, Fountzilas G, Makatsoris T, Peroukides S, Kalofonos HP. Bevacizumab in the treatment of breast cancer. Cancer Treat Rev. 2010;36:75–82. doi: 10.1016/j.ctrv.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 13.Tamaskar I, Pili R. Update on novel agents in renal cell carcinoma. Expert Rev Anticancer Ther. 2009;9:1817–1827. doi: 10.1586/era.09.157. [DOI] [PubMed] [Google Scholar]
- 14.Norden AD, Drappatz J, Wen PY. Antiangiogenic therapies for high-grade glioma. Nat Rev Neurol. 2009;5:610–620. doi: 10.1038/nrneurol.2009.159. [DOI] [PubMed] [Google Scholar]
- 15.Albitar L, Laidler LL, Abdallah R, Leslie KK. Regulation of signaling phosphoproteins by epidermal growth factor and Iressa (ZD1839) in human endometrial cancer cells that model type I and II tumors. Mol Cancer Ther. 2005;4:1891–1899. doi: 10.1158/1535-7163.MCT-05-0274. [DOI] [PubMed] [Google Scholar]
- 16.Albitar L, Pickett G, Morgan M, Davies S, Leslie KK. Models representing type I and type II human endometrial cancers: Ishikawa H and Hec50co cells. Gynecol Oncol. 2007;106:52–64. doi: 10.1016/j.ygyno.2007.02.033. [DOI] [PubMed] [Google Scholar]
- 17.Dai D, Albitar L, Nguyen T, Laidler LL, Singh M, Leslie KK. A therapeutic model for advanced endometrial cancer: systemic progestin in combination with local adenoviral-mediated progesterone receptor expression. Mol Cancer Ther. 2005;4:169–175. [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2[−Delta DeltaC(T)] method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Dai D, Litman ES, Schonteich E, Leslie KK. Progesterone regulation of activating protein-1 transcriptional activity: a possible mechanism of progesterone inhibition of endometrial cancer cell growth. J Steroid Biochem Mol Biol. 2003;87:123–131. doi: 10.1016/j.jsbmb.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 20.Horn L, Sandler A. Epidermal growth factor receptor inhibitors and antiangiogenic agents for the treatment of non-small cell lung cancer. Clin Cancer Res. 2009;15:5040–5048. doi: 10.1158/1078-0432.CCR-09-0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jenab-Wolcott J, Giantonio BJ. Bevacizumab: current indications and future development for management of solid tumors. Expert Opin Biol Ther. 2009;9:507–517. doi: 10.1517/14712590902817817. [DOI] [PubMed] [Google Scholar]
- 22.Aragon-Ching JB, Dahut WL. VEGF inhibitors and prostate cancer therapy. Curr Mol Pharmacol. 2009;2:161–168. doi: 10.2174/1874467210902020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hotary K, Li X-Y, Allen E, Stevens SL, Weiss SJ. A cancer cell metalloprotease triad regulates the basement membrane transmigration program. Genes Dev. 2006;20:2673–2686. doi: 10.1101/gad.1451806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia H, Qi Y, Ng SS, et al. microRNA-146b inhibits glioma cell migration and invasion by targeting MMPs. Brain Res. 2009;1269:158–165. doi: 10.1016/j.brainres.2009.02.037. [DOI] [PubMed] [Google Scholar]
- 25.Prakash SK, Paylor R, Jenna S, et al. Functional analysis of ARHGAP6, a novel GTPase-activating protein for RhoA. Hum Mol Genet. 2000;9:477–488. doi: 10.1093/hmg/9.4.477. [DOI] [PubMed] [Google Scholar]
- 26.Lu Q, Longo FM, Zhou H, Massa SM, Chen YH. Signaling through Rho GTPase pathway as viable drug target. Curr Med Chem. 2009;16:1355–1365. doi: 10.2174/092986709787846569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dogan T, Harms GS, Hekman M, et al. X-linked and cellular IAPs modulate the stability of C-RAF kinase and cell motility. Nat Cell Biol. 2008;10:1447–1455. doi: 10.1038/ncb1804. [DOI] [PubMed] [Google Scholar]
- 28.Voisin L, Julien C, Duhamel S, et al. Activation of MEK1 or MEK2 isoform is sufficient to fully transform intestinal epithelial cells and induce the formation of metastatic tumors. BMC Cancer. 2008;8:337. doi: 10.1186/1471-2407-8-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colomiere M, Ward AC, Riley C, et al. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial-mesenchymal transition in ovarian carcinomas. Br J Cancer. 2008;100:134–144. doi: 10.1038/sj.bjc.6604794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fromigué O, Hamidouche Z, Marie PJ. Blockade of the RhoA-JNK-c-Jun-MMP2 cascade by atorvastatin reduces osteosarcoma cell invasion. J Biol Chem. 2008;283:30549–30556. doi: 10.1074/jbc.M801436200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nieves-Alicea R, Colburn N, Simeone AM, Tari A. Programmed cell death 4 inhibits breast cancer cell invasion by increasing tissue inhibitor of metalloproteinases-2 expression. Breast Cancer Res Treat. 2009;114:203–209. doi: 10.1007/s10549-008-9993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goke R, Barth P, Schmidt A, Samans B, Lankat-Buttgereit B. Programmed cell death protein 4 suppresses CDK1/cdc2 via induction of p21Waf1/Cip1. Am J Physiol Cell Physiol. 2004;287:C1541–C1546. doi: 10.1152/ajpcell.00025.2004. [DOI] [PubMed] [Google Scholar]
- 33.Takahata M, Inoue Y, Tsuda H, et al. SKI and MEL1 cooperate to inhibit transforming growth factor-beta signal in gastric cancer cells. J Biol Chem. 2009;284:3334–3344. doi: 10.1074/jbc.M808989200. [DOI] [PubMed] [Google Scholar]
- 34.Xinh PT, Tri NK, Nagao H, et al. Breakpoints at 1p36.3 in three MDS/AML(M4) patients with t(1;3)(p36;q21) occur in the first intron and in the 5′ region of MEL1. Genes Chromosomes Cancer. 2003;36:313–317. doi: 10.1002/gcc.10176. [DOI] [PubMed] [Google Scholar]
- 35.Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314:467–471. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- 36.Zong W, Meyn LA, Moalli PA. The amount and activity of active matrix metalloproteinase 13 is suppressed by estradiol and progesterone in human pelvic floor fibroblasts. Biol Reprod. 2009;80:367–374. doi: 10.1095/biolreprod.108.072462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stratman AN, Saunders WB, Sacharidou A, et al. Endothelial cell lumen and vascular guidance tunnel formation requires MT1-MMP-dependent proteolysis in 3-dimensional collagen matrices. Blood. 2009;114:237–247. doi: 10.1182/blood-2008-12-196451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Devy L, Huang L, Naa L, et al. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 2009;69:1517–1526. doi: 10.1158/0008-5472.CAN-08-3255. [DOI] [PubMed] [Google Scholar]
- 39.Liu H, Lu ZG, Miki Y, Yoshida K. Protein kinase C delta induces transcription of the TP53 tumor suppressor gene by controlling death-promoting factor Btf in the apoptotic response to DNA damage. Mol Cell Biol. 2007;27:8480–8491. doi: 10.1128/MCB.01126-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu W, Finnis S, Xiang C, et al. Tyrosine 311 is phosphorylated by c-Abl and promotes the apoptotic effect of PKCdelta in glioma cells. Biochem Biophys Res Commun. 2007;352:431–436. doi: 10.1016/j.bbrc.2006.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reno EM, Haughian JM, Dimitrova IK, Jackson TA, Shroyer KR, Bradford AP. Analysis of protein kinase C delta (PKC delta) expression in endometrial tumors. Hum Pathol. 2008;39:21–29. doi: 10.1016/j.humpath.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kitamura K, Mizuno K, Etoh A, et al. The second phase activation of protein kinase C delta at late G1 is required for DNA synthesis in serum-induced cell cycle progression. Genes Cells. 2003;8:311–324. doi: 10.1046/j.1365-2443.2003.00635.x. [DOI] [PubMed] [Google Scholar]
- 43.Gliki G, Wheeler-Jones C, Zachary I. Vascular endothelial growth factor induces protein kinase C (PKC)-dependent Akt/PKB activation and phosphatidylinositol 3′-kinase-mediates PKC delta phosphorylation: role of PKC in angiogenesis. Cell Biol Int. 2002;26:751–759. doi: 10.1016/s1065-6995(02)90926-1. [DOI] [PubMed] [Google Scholar]
- 44.Toschi A, Lee E, Xu L, Garcia A, Gadir N, Foster DA. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol Cell Biol. 2009;29:1411–1420. doi: 10.1128/MCB.00782-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koike K, Fujii T, Nakamura AM, et al. Activation of protein kinase C delta induces growth arrest in NPA thyroid cancer cells through extracellular signal-regulated kinase mitogen-activated protein kinase. Thyroid. 2006;16:333–341. doi: 10.1089/thy.2006.16.333. [DOI] [PubMed] [Google Scholar]
- 46.Shi X, Zhang H, Paddon H, Lee G, Cao X, Pelech S. Phosphorylation of STAT3 serine-727 by cyclin-dependent kinase 1 is critical for nocodazole-induced mitotic arrest. Biochemistry. 2006;45:5857–5867. doi: 10.1021/bi052490j. [DOI] [PubMed] [Google Scholar]
- 47.Lacreusette A, Barbieux I, Nguyen JM, et al. Defective activations of STAT3 Ser727 and PKC isoforms lead to oncostatin M resistance in metastatic melanoma cells. J Path. 2009;217:665–676. doi: 10.1002/path.2490. [DOI] [PubMed] [Google Scholar]
- 48.Le Page C, Puiffe ML, Meunier L, et al. BMP-2 signaling in ovarian cancer and its association with poor prognosis. J Ovarian Res. 2009;2:4. doi: 10.1186/1757-2215-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnsen IK, Kappler R, Auernhammer CJ, Beuschlein F. Bone morphogenetic proteins 2 and 5 are down-regulated in adrenocortical carcinoma and modulate adrenal cell proliferation and steroidogenesis. Cancer Res. 2009;69:5784–5792. doi: 10.1158/0008-5472.CAN-08-4428. [DOI] [PubMed] [Google Scholar]