

Abstract
The p53 tumor suppressor pathway is inactivated in cancer either via direct mutation or via deregulation of upstream regulators or downstream effectors. P53 mutations are rare in uveal melanoma. Here we investigated the role of the p53 inhibitor Hdmx in uveal melanoma. We found Hdmx over-expression in a subset of uveal melanoma cell lines and fresh-frozen tumor samples. Hdmx depletion resulted in cell-line dependent growth inhibition, apparently correlating with differential Hdm2 levels. Surprisingly, p53 knockdown hardly rescued cell cycle arrest and apoptosis induction upon Hdmx knockdown, whereas it effectively prevented growth suppression induced by the potent p53 activator Nutlin-3. In addition, two compounds inhibiting Hdmx function or expression, SAH-p53-8 and XI-011, also elicited a growth inhibitory effect in a partly p53-independent manner. These findings suggest a novel, growth-promoting function of Hdmx that does not rely on its ability to inhibit p53. We provide evidence for a contribution of p27 protein induction to the observed p53-independent G1 arrest in response to Hdmx knockdown. In conclusion, our study establishes the importance of Hdmx as an oncogene in a subset of uveal melanomas and widens the spectrum of its function beyond p53 inhibition.
Keywords: Uveal melanoma, Hdmx, p53, Nutlin-3, p27, SAH-p53-8, XI-011, retinoblastoma
Introduction
After the skin, primary melanoma most commonly affects the eye [1]. Uveal melanoma arises in the uveal tract, which comprises the iris, ciliary body, and the choroid. Current treatments mostly involve plaque radiotherapy (brachytherapy), proton beam irradiation or enucleation [2]. However, these local treatments do not prevent distant metastases. Up to 50% of patients with uveal melanoma develop metastases after the initial diagnosis and treatment, most frequently in the liver. Prognosis is poor when the tumor has metastasized; metastases are only sporadically curable and median survival is about 10 - 18 months [3,4]. Therefore, a better understanding of the molecular mechanisms underlying uveal melanomagenesis is needed to develop more efficient treatment modalities.
The molecular pathogenesis of uveal melanoma is different from that in cutaneous melanoma. For example, mutations of NRAS, BRAF and CDKN2A (the gene encoding p16INK4A and p14ARF) are frequently observed in cutaneous melanoma, but not in uveal melanoma [5-7]. Uveal melanomas have been reported to show frequent loss of chromosome 3, correlating with poor prognosis [8]; over-expression of Cyclin D1 [9,10] and inactivating mutations of BAP1 [11], both associated with metastasis. Furthermore, activating mutations of GNAQ and GNA11 [12,13] and promoter methylation of the tumor suppressors p16INK4A [14] and RassF1A [15] have been reported. Interestingly, mutations of p53 are uncommon in cutaneous melanoma [16] as well as in uveal melanoma [17-19]. DNA damage induces p53 stabilization in uveal melanoma cell lines, although downstream functional defects may be common [20].
Functional inactivation of the p53 tumor suppressor pathway is believed to be involved in virtually all human cancers [21]. Direct gene mutation is found in about 50% of tumors [22,23], whereas those tumors that retain wild type p53 contain other genetic changes that prevent p53’s tumor suppressor function [24]. P53 maintains genomic integrity following a variety of stress signals by orchestrating cellular responses that include cell cycle arrest, DNA repair, senescence and apoptosis [25]. Controlled p53 activation requires tight regulation of the main p53 inhibitors, Hdm2 and Hdmx [26]. Hdm2 ubiquitinates p53 to target it for degradation [27], whereas Hdmx functions mostly by inhibiting p53 activity through interaction with its transcription activation domain [28,29]. Furthermore, Hdmx and Hdm2 dimerize via their RING finger domains [30], which promotes Hdm2’s E3 ligase activity towards p53 [31,32].
About 5-10% of all human tumors show Hdm2 overexpression [33]. In addition, the presence of increased Hdmx mRNA levels in 20% of common tumor types [34] and Hdmx gene amplification and overexpression in a high percentage of retinoblastomas [35] and in a subset gliomas [36] indicate an oncogene function for Hdmx. Aberrant Hdmx expression in a large number of human tumor cell lines correlated with wild-type p53 status [37]. In addition, a few reports suggested p53-independent activities for Hdmx. For example, Hdmx has been implicated to suppress transcriptional activity of E2F1 [38] and Smad proteins [39,40], and to downregulate p21 protein levels [41]. However, p53 remains its major cellular target. Since uveal melanomas usually harbor wild-type p53, a subset of these cancers probably relies on increased levels of Hdm2 or Hdmx. To investigate this, we evaluated the status of the p53 pathway in uveal melanoma, with particular focus on Hdmx. Interestingly, when performing functional analysis of Hdmx in several selected uveal melanoma cell lines, we encountered a growth promoting function of Hdmx that is independent of p53 inhibition. Our findings suggest that a novel p53-independent function of Hdmx is relevant in uveal melanoma and that targeting Hdmx may be beneficial in a subset of these tumors.
Materials and methods
Cell lines, lentiviral transductions, drug treatments
Human uveal melanoma cell lines 92.1 [65], Mel202 and Mel285 (a gift of Dr. Ksander BR, Schepens Eye Research Institute, Boston, USA) were cultured in RPMI + F10 medium (1:1 ratio) with 10% fetal bovine serum (FBS) and antibiotics. Lentiviral shRNA expression constructs have been described before [66] or obtained from the Mission shRNA library (Sigma-Aldrich, St Louis, MO). For lentiviral transductions, cells were seeded at a density of 4.0*105 (92.1 and Mel285) or 6.0*105 (Mel202) cells per 6 cm dish. The next day, cells were transduced using MOI = 1.0 in medium containing 8.0 μg/mL polybrene and were puromycin-selected for stable expression. Nutlin-3 was purchased from Cayman Chemical (Ann Arbor, MI, USA). XI-011 was obtained from Tocris Bioscience (Ellisville, Missouri, USA), and the SAH-p53-8 peptide was a gift from F. Bernal.
Immunoblotting
Cells were lysed in Giordano buffer (50 mM Tris-HCl, pH 7.4, 250 mM NaCl, 0.1% Triton X-100, 5 mM EDTA) with protease- and phosphatase inhibitors. Proteins were separated by SDS-PAGE, blotted onto Polyvinylidene Fluoride Transfer membranes, incubated with the appropriate primary and secondary antibodies, and bands were visualized by chemoluminescence (West Dura, Pierce Biotechnology, Rockford, IL). Antibodies used: p53 (DO-1 and PAb1801), Hdm2 (SMP14) and E2F1 (KH-95) were all obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA); phospho-Ser15-p53, PARP and Survivin from Cell Signaling Technology (Beverly, MA, USA); phospho-Ser46-p53 and p27KIP1 were from Epitomics (CA, USA); Hdmx and USP7 (A300-287A, A300-033A) were from Bethyl Laboratories (Montgomery, TX, USA); PUMA (Nter), β-tubulin and vinculin were from Sigma-Aldrich (St Louis, MO, USA); p21 (CP74) was obtained from Upstate Biotechnology (Lake Placid, NY, USA); Rb (G3-245) was obtained from BD Pharmingen (Franklin Lakes, New Jersey, USA). Anti-Mdm2 4B2 was a gift from Dr. Levine A. Anti-BrdU-FITC was obtained from Roche Biochemicals (Indianapolis, IN, USA).
RNA isolation, qRT-PCR
RNA was isolated using the SV Total RNA isolation kit (Promega, Madison, WI). cDNA was synthesized using 2.0 μg RNA in a total volume of 30 μL reverse transcriptase reaction mixture (Promega). Samples were analyzed in triplicate using SYBR Green mix (Roche Biochemicals, Indianapolis, IN, USA) in a 7900ht Fast Real-Time PCR System (Applied Biosystems, Foster City, CA). For normalization the geometric mean of at least two housekeeping genes was used. Primer sequences are listed in Table 1.
Table 1.
Sequences of DNA primers used for qRT-PCR analyses.
| GENE | Forward primer | Reverse primer |
|---|---|---|
| Hdm2 | 5'-acgcacgccactttttctct-3' | 5'-tccgagctggaatctgtgag-3' |
| PUMA | 5'-gacctcaacgcacagta-3' | 5'-ctaattgggctccatct-3' |
| p21 | 5'-agcagaggaagaccatgtgga-3' | 5'-aatctgtcatgctggtctgcc-3' |
| GADD45α | 5'-gcgacctgcagtttgcaata-3' | 5'-atcccccaccttatccatcct-3' |
| p27 | 5'-caaatgccggttctgtggag-3' | 5'-tccattccatgagtcagcgata-3' |
| CAPNS1 | 5'-atggttttggcattgacacatg-3 | 5'-gcttgcctgtggtgtcgc-3' |
| GAPDH | 5'-tgccatgtagaccccttgaag-3' | 5'-atggtacatgacaaggtgcgg-3' |
| ÁRP | 5'-caccattgaaatcctgagtgatgt-3 | 5'-accagccgaaaggagaag-3' |
| RPS11 | 5'-aagcagccgaccatctttca-3' | 5'-cgggagcttctccttgcc-3' |
| Hdmx | 5'-tgcatgcagcaggtgcg-3' | 5'-cattacttctaggtgtat-3' |
Flow cytometry
Cells were harvested, washed in PBS and fixed in ice-cold 70% EtOH. Prior to FACS analysis, cells were washed in PBS and resuspended in PBS containing 50 μg/mL RNase A and 50 μg/mL propidium iodide (PI). Flow cytometry was performed in the BD LSR II system (BD Biosciences). For Annexin V staining, cells were washed twice in PBS and resuspended in Annexin V-binding buffer containing Fluorescein isothyocyanate (FITC)-labeled Annexin-V (Sigma-Aldrich) and PI. After 10 min RT incubation cells were analyzed by flow cytometry. Positive PI stained cells, indicating necrotic or late apoptotic cells, were excluded from the analysis. PI-negative, Annexin V-positive cells represent early apoptotic cells. For bromodeoxyuridine (BrdU) incorporation, we added BrdU to the culture medium at a final concentration of 20 μM for 2h. Cells were harvested, washed in PBS and fixed in ice-cold 70% EtOH. Subsequently, cells were treated with 50 μg/ml RNase A (30 min 37 °C), washed and resuspended in 5 M HCl / 0.5% Triton (20 min RT). Cells were then neutralized in 1 M Tris/HCl pH 7.5, washed in PBS and incubated with anti-BrdU-FITC antibody in PBS/Tween with 1% BSA (30 min RT). Cells were washed twice in PBS/Tween, resuspended in PBS containing 50 μg/mL PI and analyzed by flow cytometry to detect BrdU and PI staining.
WST-1 proliferation assay
Cells were counted and seeded in triplicate in 96-well plates at a density of 3000 (92.1 and Mel285) or 6000 (Mel202) cells per well, in a total volume of 100 μL culture medium. To determine the survival/cell growth, 10 μL WST-1 (Roche Biochemicals, Indianapolis, IN, USA) was added to the wells and absorbance (450 nm) was measured 2 hrs later in a microplate reader (Victor3 Multilabel Counter 1420-042, Perkin-Elmer, Wellesley, MA, USA).
Results
Levels of Hdmx and Hdm2 are increased in a subset of uveal melanomas
We started to investigate whether indeed high expression of Hdmx or Hdm2 would occur in uveal melanoma as a means to inactivate p53. Therefore, a panel of ten uveal melanoma cell lines was analyzed for basal levels of several key proteins in the p53 pathway (Figure 1A). The levels of p53 itself were found to be more or less constant in all cell lines and they were comparable to the levels seen in the wild-type p53 expressing osteosarcoma cell line U2OS that was used as control, suggesting the absence of stabilizing p53 mutations. The double band pattern observed in some lanes most likely represents the p53 codon 72 polymorphism [42]. Levels of Hdm2 and Hdmx varied greatly between cell lines, with most cell lines showing increased levels of at least one of these proteins.
Figure 1.

High expression of Hdmx in a subset of uveal melanomas. A. Total lysates of ten uveal melanoma cell lines and the osteosarcoma cell line U2OS were analyzed by western blot using the indicated antibodies. B. RT-PCR analysis of the mRNA levels of Hdmx (exon 3 – exon 8) and GAPDH. FL = full length Hdmx; S = Hdmx-S. C. Protein extracts of 23 fresh-frozen uveal melanoma tumor samples were analyzed for Hdm2, Hdmx and Vinculin protein levels by western blot. Band intensities were quantified from two different blots using Image J software. As example, blots of 11 fresh uveal melanoma samples, 3 control cell lines and of normal uveal melanocytes (NUM) is shown. For all samples, the expression levels of Hdm2 and Hdmx were calculated relative to 92.1, lysate of which was loaded on each gel, and corrected for Vinculin levels on each blot. Statistical comparison of each sample with NUM levels was performed using a two-tailed t-test; an asterisk indicates p < 0.05.
In addition to OCM8, especially the cell lines derived from a metastasis (Omm1, Omm2.3 and Omm2.5) show very low levels of Hdmx protein. Recently, we found that especially in later stage tumors, the relative expression of an alternative splice variant of Hdmx, Hdmx-S, is increased, which is accompanied by lower Hdmx protein levels, correlating with lower survival of patients [43]. Therefore, we investigated the levels of Hdmx and Hdmx-S mRNA in the panel of uveal melanoma cell lines. Indeed, the results revealed that all three metastasis-derived cell lines express relatively high levels of Hdmx-S mRNA (Figure 1B). We also analyzed Hdm2 and Hdmx protein levels in lysates of freshly-frozen uveal melanoma tumor tissue and compared these with normal uveal melanocytes (NUM), OCM8, 92.1 and U2OS lysates, representing low and high level Hdm2 and Hdmx controls (Figure 1C). Hdm2 levels were significantly elevated in 5 out of 23 tumor samples (22%) compared to NUM. In 7 samples (30%) we found increased levels of Hdmx; three of these tumor samples overlapped. These findings indicate Hdm2 and/or Hdmx over-expression in a subset of uveal melanomas.
Cell line-dependent growth inhibition upon Hdmx knockdown
To investigate whether high Hdmx expression contributes to the growth of uveal melanoma cell lines, we selected three cell lines from the panel based on their differential expression of Hdmx and Hdm2. The 92.1 cells express high levels of Hdmx and low Hdm2, Mel202 cells express high levels of Hdmx and moderate Hdm2, and Mel285 cells express moderate levels of Hdmx and high Hdm2. We reduced Hdmx expression using shRNA and analyzed cell proliferation/survival. Hdmx knockdown strongly suppressed growth of both 92.1 and Mel202 cells, whereas the growth of Mel285 cells was largely unaffected (Figure 2A, 2B). Also flow cytometry analyses showed that the cell cycle profile of Mel285 was not affected upon Hdmx knockdown (not shown). This difference in sensitivity is most likely the result of differences in Hdm2 levels, which are highest in Mel285 cells. We have previously shown that Mel285 cells are sensitive to Nutlin-3 treatment, accompanied by induction of p53 protein levels and p53 target gene expression, indicating a wild-type p53 expression in Mel285 cells [44].
Figure 2.

Cell line-dependent, partly p53-independent growth inhibition upon Hdmx knockdown. A. 92.1, Mel202 and Mel285 cells were transduced with shCtrl or shHdmx#1 RNAs, and protein extracts were analyzed by western blot using the indicated antibodies. B. Cells were counted and seeded for WST-1 proliferation assay, and cell viability was measured at several time points during five days. C. 92.1 cells were stably transduced using shCtrl or shp53 RNAs. The resulting cell lines were transduced with shCtrl or with three different shHdmx RNAs, and protein extracts were analyzed by western blot using the indicated antibodies. D. Cells were counted and seeded for WST-1 proliferation assay, and cell viability was measured after five days. E. Stable 92.1-shCtrl and 92.1-shp53 cells were transiently transduced with shCtrl or shHdmx#1 RNA and after six days, cell cycle profiles were analyzed by flow cytometry. Bars represent the mean and s.e. of two independent experiments. F,G. Evaluation of Sub-G1 fractions and Annexin V staining, four days after transduction. Bars represent the mean and s.e. of three independent experiments. Statistical analysis was performed using a two-tailed t-test.
P53-independent growth inhibition upon Hdmx knockdown
To investigate whether the growth inhibitory effect of Hdmx knockdown was p53-dependent, we generated stable shp53 and shCtrl cell lines, which were subsequently transduced with shCtrl or three different shHdmx RNAs (Figure 2C). Surprisingly, p53 depletion did not rescue the effects of Hdmx knockdown in a 5-day growth assay, in any of the knockdown constructs (Figure 2D). We further evaluated the biological effects of Hdmx depletion by flow cytometry and found an accumulation of cells in G1 phase concomittant with decreased numbers of S-phase cells, which was largely p53-independent (Figure 2E). The inhibition of cell cycle progression upon Hdmx knockdown was confirmed by analysis of BrdU incorporation (not shown, and Figure 6E). In addition to cell cycle arrest, loss of Hdmx also resulted in an enhanced Sub-G1 fraction (5% > ~ 15%; Figure 2F) and a slight increase in Annexin V staining (3% > 5%; Figure 2G), indicating a minor increase in apoptosis upon Hdmx knockdown, partly p53-independent.
Figure 6.
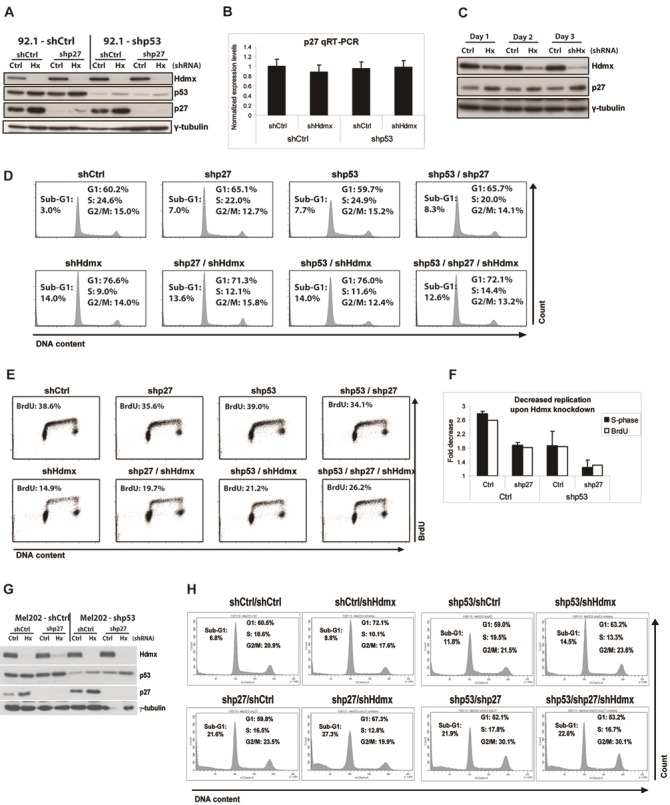
Induction of p27 protein levels upon Hdmx knockdown occurs independently of p53 and contributes to G1 arrest. A. 92.1 cells were stably transduced with shCtrl, shp53, shp27 or with a combination of shp53 and shp27 RNAs. The resulting cell lines were transduced with shCtrl or shHdmx#1 RNAs and after four days protein extracts were analyzed by western blot using the indicated antibodies. B. Stable 92.1/shCtrl and 92.1/shp53 cells were transduced with shCtrl or shHdmx#1 RNAs as indicated and analyzed by qRT-PCR for p27 expression levels, normalized for the geometric mean of CAPNS1, ARP and RPS11. C. 92.1-shp53 cells were transduced with shCtrl or shHdmx#1 RNAs. Protein extracts were isolated at the indicated time-points and analyzed by western blot using the indicated antibodies. D. Cells transduced as described in A were analyzed by flow cytometry. E. Cells transduced as described in A were incubated with 20 μM BrdU for 2 hrs and analyzed by flow cytometry. F. Quantification of D and E. Graphs indicate the fold reductions upon Hdmx knockdown of S-phase cells (mean and s.e. of two independent experiments) and BrdU positive cells. G. Mel202 cells were stably transduced with shCtrl, shp53, shp27 or with a combination of shp53 and shp27 RNAs. The resulting cell lines were transduced with shCtrl or shHdmx#1 RNAs, and four days later analyzed by western blotting. H. Cells transduced as described in G were analyzed by flow cytometry.
The above-described effects of Hdmx depletion in shp53 cells would be easily explained if the knockdown of p53 would be far from complete. In that case, Hdmx knockdown would still lead to p53 reactivation and p53-dependent growth inhibition. Therefore, we tested the efficiency of p53 knockdown by treating the cells with Nutlin-3, a potent activator of p53 [45]. Nutlin-3 binds Hdm2 with high affinity, but also interacts with Hdmx and can reduce the p53-Hdmx interaction, albeit with lower efficiency [35,46]. Nutlin-3 treatment for 24 hrs strongly induced the levels of p53 and of its target genes Hdm2, PUMA and p21 in 92.1/shCtrl and Mel202/shCtrl cells (Figure 3A, 3B and data not shown). Of note, Hdmx knockdown in shCtrl cells also slightly increased the expression of p53 targets, indicating some p53 activation in response to Hdmx depletion in these cells. Importantly, the 92.1/shp53 and Mel202/shp53 cells showed strongly reduced effects of Nutlin-3 on the protein and mRNA levels of the p53 target genes (Figure 3A, 3B). These differences are reflected in the biological response: while Nutlin-3 strongly suppressed cell proliferation and mildly induced apoptosis in shCtrl cells (Figure 3C, 3D, 3E, 3F), these effects were nearly abrogated in shp53 cells, indicating that the p53 knockdown was indeed sufficient to prevent p53 activation by Nutlin-3. In addition, Nutlin-3 enhanced the effect of Hdmx knockdown only in the shCtrl cells. Together, these findings indicate that the growth inhibition upon Hdmx knockdown is at least in part due to a novel function of Hdmx that stretches beyond p53 inhibition.
Figure 3.

Hdmx knock-down sensitizes for Nutlin-3 treatment. Stable 92.1-shCtrl and 92.1-shp53 cells were transiently transduced with shCtrl or shHdmx#1 RNA and 4 days later seeded for various assays, and subsequently mock-treated or treated with 10 μM Nutlin-3 for 24h. A. Protein extracts were analyzed by western blot using the indicated antibodies. B. qRT-PCR analysis of the expression levels of Hdm2, PUMA and p21, normalized for the geometric mean of CAPNS1, GAPDH and ARP. C. Cells were counted and seeded for WST-1 proliferation assay. Cells were mock-treated or treated with 10 μM Nutlin-3, and cell viability was measured at several time points during five days. D. Subsequently, cells were mock-treated or treated with 10 μM Nutlin-3 for 24h and analyzed by flow cytometry. E, F. Stable Mel202-shCtrl and Mel202-shp53 cells were transiently transduced with shCtrl or shHdmx#1 RNA and 5 days later seeded WST1 proliferation assay and flow cytometry analysis, and subsequently mock-treated or treated with 10 μM Nutlin-3 for 24h.
Hdmx inhibitors show partly p53-independent effects in uveal melanoma cells
The results presented above strongly indicate that Hdmx performs proliferative and pro-survival function(s) in addition to its p53 inhibition activity. To address this in another manner, we decided to investigate the effect of two compounds recently reported to particularly target Hdmx, i.e. the stapled peptide SAH-p53-8 [47] and the small molecule XI-011 [48]. We selected the same three cell lines (92.1, Mel202 and Mel285) as used for the Hdmx knockdown experiments, based on their differential expression of Hdmx and Hdm2 (Figure 1A). All three cell lines are growth-inhibited by the SAH-p53-8 peptide; the cell lines that show high/intermediate Hdmx levels (92.1 and Mel202) are more sensitive than the Hdm2-overexpressing cell line Mel285 (Figure 4A). However, the Mel285 cells are also less sensitive to Nutlin-3 treatment. This differential sensitivity could be caused by the differential expression of two p53 target genes, PUMA and p21. While basal levels of p21 are higher in Mel285 compared to 92.1 and Mel202 cells, PUMA levels are lower.
Figure 4.

Inhibition of uveal melanoma cell growth by Hdmx inhibitors SAH-p53-8 and XI-011. Stable 92.1/shCtrl, 92.1/shp53, Mel202/shCtrl, Mel202/shp53, Mel285/shCtrl and Mel285/shp53 cells were counted and seeded for WST-1 assay and for immunoblot analyses. Next day cells were treated with the indicated concentrations of SAH-p53-8, XI-011 or Nutlin-3 for the WST-1 proliferation assay. Relative survival was measured after 72 hrs of treatment (Figure 4A, 4C). For immunoblot analyses cells were treated with 20 μM SAH-p53-8 or 5 μM Nutlin-3 (Figure 4B), or with 0.5 μM XI-011 (Figure 4D) for indicated time periods. Protein extracts were analysed with indicated antibodies; expression of USP7 or Vinculin serves as loading control.
Notably, the effects of SAH-p53-8 are only partly p53 dependent; this might be explained to some extent by the incomplete knockdown of p53 as some p53-activation can still be detected (Figure 4B), although the effects of Nutlin-3 are strongly reduced in the shp53 cells as shown also above. These results are consistent with the previous observation that Hdmx performs p53-independent proliferative functions.
The XI-011 compound was found to activate p53 via downregulation of Hdmx gene transcription [48]. When tested on the same three uveal melanoma cell lines, the cells that expressed the higher levels of Hdmx, were indeed more sensitive than the high Hdm2-expressing cell line Mel285 (Figure 4C). Interestingly, the higher sensitivity of 92.1 and Mel202 cells correlated with induction of phosphorylation of Ser46 of p53 (Figure 4D) and PARP cleavage (not shown), both regarded as markers of an apoptotic response. Accordingly, XI-011 clearly elicited a DNA damage response in 92.1 and Mel202 cells, as illustrated by increased phosphorylation of KAP1, an ATM target protein (not shown). The growth inhibitory effects of XI-011 were also found to be only partly p53-dependent. Again, this might be caused by the p53-independent functions of Hdmx, expression of which is very strongly downregulated upon XI-011 treatment (Figure 4D). However, the observed DNA damage response might also contribute to the p53-independent effects of XI-011.
Inhibition of growth and survival upon Hdmx knockdown is not rescued by knocking down expression of the retinoblastoma gene
To characterize the aforementioned p53-independent function of Hdmx, we investigated the involvement of a few obvious candidates. First, we investigated a putative involvement of the tumor suppressor protein Rb. We hypothesized that, similar to the reported activity of Hdm2 towards Rb [49,50], Hdmx may also function in stimulating cell cycle progression via Rb inhibition. Furthermore, although Hdmx knockdown results in slightly lower total levels of Rb, especially in 92.1/shCtrl cells, the amount of hypo-phosphorylated Rb is increased (Figure 5A). Therefore, we stably reduced Rb expression via shRNA expression (Figure 5A) and investigated the effect of Hdmx knockdown on proliferation and cell cycle progression (Figure 5B, 5C). Reduced Rb levels failed to rescue the growth suppression and the reduction in S-phase cells upon Hdmx knockdown, although the Rb knockdown was not complete.
Figure 5.
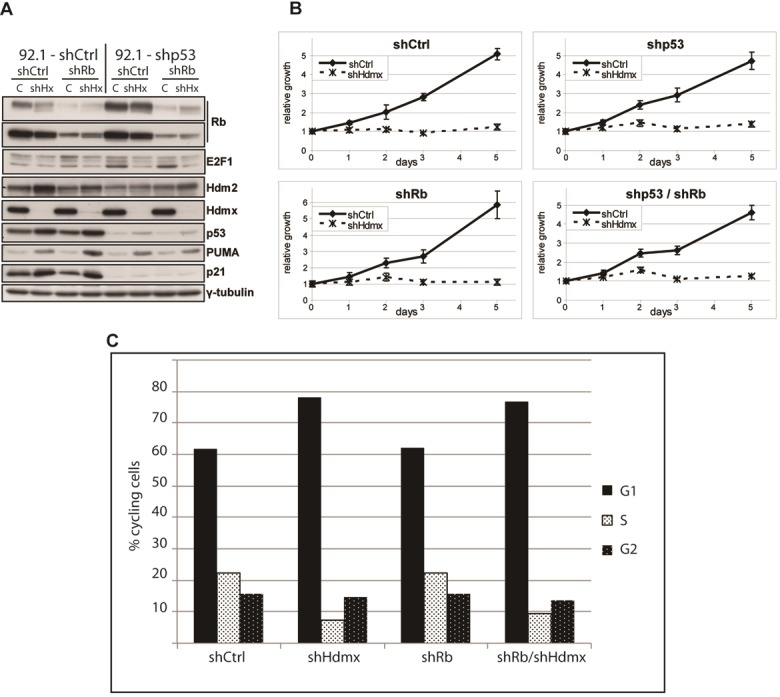
Rb knockdown fails to rescue growth inhibition upon Hdmx knockdown. A. 92.1 cells were stably transduced with shCtrl, shp53, shRb or with a combination of shp53 and shRb RNAs. The four resulting cell lines were transduced with shCtrl or shHdmx#1 RNAs and after four days protein extracts were analyzed by western blot using the indicated antibodies. B. Cells from A were counted and seeded for WST-1 proliferation assay and cell viability was measured at several time points during five days. C. Cells from A were seeded for flow cytometry analysis. Shown is the percentage of the cycling cells in a certain cell cycle phase.
Induction of p27 protein levels upon Hdmx knockdown occurs independently of p53 and contributes to G1 arrest
The G1 arrest that is induced by Hdmx knockdown could be caused by increased levels of Cdk inhibitor(s), as for example, p21 has been reported to interact with Hdmx, which leads to p21 degradation [41]. However, we found strongly reduced mRNA and protein levels of p21 in shp53 cells as compared to shCtrl cells (Figures 3A, 5A and data not shown), and no significant upregulation upon shHdmx in the shp53 cells. Therefore, a functional contribution of p21 to the observed effects of Hdmx knockdown is highly unlikely.
Another Cdk inhibitor possibly involved in the growth-inhibitory effects of Hdmx knockdown is p27. The p27 gene is a transcriptional target of FOXO proteins, and Hdm2 has been reported to inhibit activity of FOXO proteins by inducing the poly-ubiquitination of FOXO1 and FOXO3A [51] and mono-ubiquitination of FOXO4 [52]. Therefore, Hdmx might also function via regulation of FOXO proteins. Indeed, we observed an induction of p27 protein levels after Hdmx knockdown in 92.1 (Figure 6A) and Mel202 cells (Figure 6G), irrespective of p53 levels. However, p27 mRNA was not induced (Figure 6B), and protein levels of FOXO3A and FOXO4 were not affected (not shown), indicating that the increased p27 levels in response to Hdmx knockdown does not occur at the transcriptional level. Importantly, we found p27 protein induction already one day after Hdmx knockdown (Figure 6C), a time frame during which the G1 arrest is not yet maximal (not shown), suggesting that the increased p27 protein level is not a secondary effect of cell cycle arrest [53-55]. To further examine the relevance of p27 induction in the responses to Hdmx knockdown, we reduced p27 expression in 92.1 cells by shRNA (Figure 6A) and analyzed cell cycle progression. Interestingly, the reduction of cells in S-phase (Figure 6D) as well as the amount of BrdU positive cells (Figure 6E) upon Hdmx knockdown was partially rescued by p27 depletion. A summary of these experiments (Figure 6F) illustrates that p27 knockdown partially prevents the G1 arrest induced by Hdmx knockdown, on top of a partial rescue by p53 depletion. Similarly, reducing p27 levels in Mel202 cells partly prevented the S-phase reduction upon Hdmx knockdown (Figures 6G, 6H).
Discussion
A pathological high expression of Hdmx is found in a subset of human cancers, and generally correlated with the presence of wild-type p53 protein [34-37]. Constitutive Hdmx overexpression contributes to the oncogenic transformation of cultured cells, thereby functionally resembling loss of p53 [34,56]. These findings emphasize that high Hdmx expression in cancer appears mainly to serve to block p53 activity. Indeed, in this study, we show high Hdmx expression in a subset of cell lines and fresh-frozen tumor samples from uveal melanoma, which rarely contain p53 mutations. Increased levels of Hdm2 were also observed in some cell lines and tumor samples, although the extent of over-expression was not impressive when compared to normal uveal melanocytes. Interestingly, our experiments in uveal melanoma cell lines also suggest the existence of an additional growth promoting function of Hdmx. Reducing Hdmx levels via shRNA knockdown constructs reduced proliferation and survival of 92.1 and Mel202 cells, whereas the growth of Mel285 cells remained largely unaffected. This indicates that the proliferation in a subset of uveal melanomas depends on the high Hdmx expression. The resistance of Mel285 cells reduces the likelihood of non-specific effects caused by the Hdmx knockdown constructs in 92.1 and Mel202 cells. Unexpectedly, knocking down Hdmx still reduced growth of the 92.1 and Mel202 cells in which p53 levels were strongly reduced by shRNA. The lack of growth inhibition by Nutlin-3 in these shp53-cells confirmed the efficiency of the p53 knockdown, indicating that Hdmx promotes uveal melanoma growth through p53-dependent and -independent pathways.
Similarly, two recently described Hdmx inhibitors, although certainly less specific than knocking down Hdmx, also inhibited the growth of these uveal melanoma cells in a partly p53-independent manner. These results strongly suggest that high Hdmx levels stimulate cell proliferation and survival in a manner stretching beyond p53 inhibition.
At first sight, this finding is a little surprising, particularly with regard to the complete rescue of the embryonic lethality of Mdmx deletion by loss of p53 [57,58], which would argue against the importance of p53-independent effects of Hdmx. On the other hand, the physiological role of basal Hdmx levels during development may not be identical to the pathological effects of Hdmx overexpression during tumorigenesis. In addition, it is becoming increasingly clear that Hdm2 activity, and especially pathologically high levels of Hdm2, are not restricted to p53 regulation. Because of the homology between Hdm2 and Hdmx, our search for the mechanisms underlying p53-independent activities of Hdmx was primarily based on known functions of Hdm2. Enhanced Hdm2 activity has been reported to inhibit Rb function [49,50], which prompted us to investigate whether activation of Rb would be involved in the inhibition of S-phase entry upon Hdmx knockdown. However, Rb knockdown did not rescue the decrease in S-phase cells upon Hdmx knockdown, suggesting that Hdmx does not function via Rb inactivation. Interestingly, our results point to p27 protein induction as one of the factors contributing to the growth-inhibiting effects of Hdmx knockdown. Although p27 may exert some functions that are potentially oncogenic, it is generally considered to be a tumor suppressor [59]. The main role of p27 is to regulate the G0/G1 to S transition by binding and inhibiting cyclin E/CDK2 and cyclin D/CDK4,6 complexes, and thereby reducing phosphorylation of pocket proteins. P27 itself is highly regulated at multiple levels, including transcription, translation, phosphorylation and ubiquitination [59,60]. P27 protein levels are maximal during G0 and early G1, mainly due to differences in cap-independent translation [61] and ubiquitin-dependent proteolysis [62] in different stages of the cell cycle. However, the induction of p27 protein levels in response to Hdmx knockdown probably occurred too quickly to be a secondary event of the G1 arrest. Moreover, p27 knockdown partially prevented the G1 arrest in response to Hdmx knockdown. This indicates that Hdmx somehow limits p27 levels via an unknown mechanism and thereby stimulates cell cycle progression.
A few reports suggest that the inhibitor of apoptosis (IAP) family member Survivin is overexpressed in uveal melanoma [63,64]. We found that, although endogenous levels of Survivin are slightly reduced upon Hdmx knockdown, constitutive Survivin expression did not affect the outcome of Hdmx knockdown experiments (not shown).
In conclusion, Hdmx over-expression is present in a subset of uveal melanomas, most likely to promote tumorigenesis by inhibiting p53, which is rarely mutated in this type of tumors. Interestingly, however, we show that Hdmx also has an important p53-independent role in promoting cell proliferation and survival. It will be important to analyze the relevance of this role of Hdmx in other cell types as well. Our attempts to uncover the molecular basis of a p53-independent function of Hdmx reveal a contribution for p27 in the induction of G1 arrest. Future studies are required to provide more insight into the mechanism by which Hdmx affects p27 protein levels. However, our data strongly suggest the involvement of additional, yet unknown factors, although unraveling these factors thus far proved difficult. In this respect, it may be worthwhile to investigate the involvement of proteins reported to interact with Hdm2, but not tested in this study, since they might interact with Hdmx as well. Alternatively, detailed analysis of Hdmx- containing protein complexes in uveal melanoma by mass spectrometry and subsequent functional characterization of newly found interactors might open new avenues to clarify p53-independent activities of Hdmx. Together, this will improve our understanding of Hdmx over-expressing tumors and ultimately may lead to the development of new therapeutic strategies to target such tumors.
Acknowledgements
We thank Prof. Ksander BR for providing the Mel cell lines and Martijn Rabelink for help with the shRNA viruses. This study was supported by EC FP6 funding (contract 503576 to AG Jochemsen). This publication reflects the authors’ views and not necessarily those of the European Community. The EC is not liable for any use that may be made of the information contained. This research was further supported in part by grants from the Association for International Cancer Research (grant 05-273) and by the Intramural Research Program of the NIH/NCI.
References
- 1.Chang AE, Karnell LH, Menck HR. The National Cancer Data Base report on cutaneous and noncutaneous melanoma: a summary of 84,836 cases from the past decade. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer. 1998;83:1664–1678. doi: 10.1002/(sici)1097-0142(19981015)83:8<1664::aid-cncr23>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 2.Shields CL, Shields JA. Ocular melanoma: relatively rare but requiring respect. Clin Dermatol. 2009;27:122–133. doi: 10.1016/j.clindermatol.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 3.Kivela T, Eskelin S, Kujala E. Metastatic uveal melanoma. Int Ophthalmol Clin. 2006;46:133–149. doi: 10.1097/01.iio.0000195861.71558.13. [DOI] [PubMed] [Google Scholar]
- 4.Augsburger JJ, Correa ZM, Shaikh AH. Effectiveness of treatments for metastatic uveal melanoma. Am J Ophthalmol. 2009;148:119–127. doi: 10.1016/j.ajo.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 5.Cruz F III, Rubin BP, Wilson D, Town A, Schroeder A, Haley A, Bainbridge T, Heinrich MC, Corless CL. Absence of BRAF and NRAS mutations in uveal melanoma. Cancer Res. 2003;63:5761–5766. [PubMed] [Google Scholar]
- 6.Rimoldi D, Salvi S, Lienard D, Lejeune FJ, Speiser D, Zografos L, Cerottini JC. Lack of BRAF mutations in uveal melanoma. Cancer Res. 2003;63:5712–5715. [PubMed] [Google Scholar]
- 7.Goldstein AM, Stacey SN, Olafsson JH, Jonsson GF, Helgason A, Sulem P, Sigurgeirsson B, Benediktsdottir KR, Thorisdottir K, Ragnarsson R, Kjartansson J, Kostic J, Masson G, Kristjansson K, Gulcher JR, Kong A, Thorsteinsdottir U, Rafnar T, Tucker MA, Stefansson K. CDKN2A mutations and melanoma risk in the Icelandic population. J Med Genet. 2008;45:284–289. doi: 10.1136/jmg.2007.055376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prescher G, Bornfeld N, Hirche H, Horsthemke B, Jockel KH, Becher R. Prognostic implications of monosomy 3 in uveal melanoma. Lancet. 1996;347:1222–1225. doi: 10.1016/s0140-6736(96)90736-9. [DOI] [PubMed] [Google Scholar]
- 9.Coupland SE, Bechrakis N, Schuler A, Anagnostopoulos I, Hummel M, Bornfeld N, Stein H. Expression patterns of cyclin D1 and related proteins regulating G1-S phase transition in uveal melanoma and retinoblastoma. Br J Ophthalmol. 1998;82:961–970. doi: 10.1136/bjo.82.8.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coupland SE, Anastassiou G, Stang A, Schilling H, Anagnostopoulos I, Bornfeld N, Stein H. The prognostic value of cyclin D1, p53, and MDM2 protein expression in uveal melanoma. J Pathol. 2000;191:120–126. doi: 10.1002/(SICI)1096-9896(200006)191:2<120::AID-PATH591>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 11.Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, Council ML, Matatall KA, Helms C, Bowcock AM. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–1413. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O'Brien JM, Simpson EM, Barsh GS, Bastian BC. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, Sozen MM, Baimukanova G, Roy R, Heguy A, Dolgalev I, Khanin R, Busam K, Speicher MR, O'Brien J, Bastian BC. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363:2191–2199. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Velden PA, Metzelaar-Blok JA, Bergman W, Monique H, Hurks H, Frants RR, Gruis NA, Jager MJ. Promoter hypermethylation: a common cause of reduced p16(INK4a) expression in uveal melanoma. Cancer Res. 2001;61:5303–5306. [PubMed] [Google Scholar]
- 15.Maat W, Beiboer SH, Jager MJ, Luyten GP, Gruis NA, van der Velden PA. Epigenetic regulation identifies RASEF as a tumor-suppressor gene in uveal melanoma. Invest Ophthalmol Vis Sci. 2008;49:1291–1298. doi: 10.1167/iovs.07-1135. [DOI] [PubMed] [Google Scholar]
- 16.Houben R, Hesbacher S, Schmid CP, Kauczok CS, Flohr U, Haferkamp S, Muller CS, Schrama D, Wischhusen J, Becker JC. High-level expression of wild-type p53 in melanoma cells is frequently associated with inactivity in p53 reporter gene assays. PLoS One. 2011;6:e22096. doi: 10.1371/journal.pone.0022096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brantley MA Jr, Harbour JW. Deregulation of the Rb and p53 pathways in uveal melanoma. Am J Pathol. 2000;157:1795–1801. doi: 10.1016/s0002-9440(10)64817-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chana JS, Wilson GD, Cree IA, Alexander RA, Myatt N, Neale M, Foss AJ, Hungerford JL. c-myc, p53, and Bcl-2 expression and clinical outcome in uveal melanoma. Br J Ophthalmol. 1999;83:110–114. doi: 10.1136/bjo.83.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hussein MR. The relationships between p53 protein expression and the clinicopathological features in the uveal melanomas. Cancer Biol Ther. 2005;4:57–59. doi: 10.4161/cbt.4.1.1480. [DOI] [PubMed] [Google Scholar]
- 20.Sun Y, Tran BN, Worley LA, Delston RB, Harbour JW. Functional analysis of the p53 pathway in response to ionizing radiation in uveal melanoma. Invest Ophthalmol Vis Sci. 2005;46:1561–1564. doi: 10.1167/iovs.04-1362. [DOI] [PubMed] [Google Scholar]
- 21.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 22.Hainaut P, Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res. 2000;77:81–137. doi: 10.1016/s0065-230x(08)60785-x. [DOI] [PubMed] [Google Scholar]
- 23.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 24.Wynford-Thomas D, Blaydes J. The influence of cell context on the selection pressure for p53 mutation in human cancer. Carcinogenesis. 1998;19:29–36. doi: 10.1093/carcin/19.1.29. [DOI] [PubMed] [Google Scholar]
- 25.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 26.Wade M, Wang YV, Wahl GM. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 2010;20:299–309. doi: 10.1016/j.tcb.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haupt Y, Barak Y, Oren M. Cell type-specific inhibition of p53-mediated apoptosis by mdm2. EMBO J. 1996;15:1596–1606. [PMC free article] [PubMed] [Google Scholar]
- 28.Marine JC, Jochemsen AG. Mdmx as an essential regulator of p53 activity. Biochem Biophys Res Commun. 2005;331:750–760. doi: 10.1016/j.bbrc.2005.03.151. [DOI] [PubMed] [Google Scholar]
- 29.Shvarts A, Steegenga WT, Riteco N, van Laar T, Dekker P, Bazuine M, van Ham RC, van der Houven van Oordt W, Hateboer G, van der Eb AJ, Jochemsen AG. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996;15:5349–5357. [PMC free article] [PubMed] [Google Scholar]
- 30.Sharp DA, Kratowicz SA, Sank MJ, George DL. Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J Biol Chem. 1999;274:38189–38196. doi: 10.1074/jbc.274.53.38189. [DOI] [PubMed] [Google Scholar]
- 31.Gu J, Kawai H, Nie L, Kitao H, Wiederschain D, Jochemsen AG, Parant J, Lozano G, Yuan ZM. Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J Biol Chem. 2002;277:19251–19254. doi: 10.1074/jbc.C200150200. [DOI] [PubMed] [Google Scholar]
- 32.Linares LK, Hengstermann A, Ciechanover A, Muller S, Scheffner M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc Natl Acad Sci USA. 2003;100:12009–12014. doi: 10.1073/pnas.2030930100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Momand J, Wu HH, Dasgupta G. MDM2--master regulator of the p53 tumor suppressor protein. Gene. 2000;242:15–29. doi: 10.1016/s0378-1119(99)00487-4. [DOI] [PubMed] [Google Scholar]
- 34.Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, Frenk R, de Graaf P, Francoz S, Gasparini P, Gobbi A, Helin K, Pelicci PG, Jochemsen AG, Marine JC. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol. 2004;24:5835–5843. doi: 10.1128/MCB.24.13.5835-5843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, Teunisse A, Lam S, Ramos Y, Mohan A, Johnson D, Wilson M, Rodriguez-Galindo C, Quarto M, Francoz S, Mendrysa SM, Guy RK, Marine JC, Jochemsen AG, Dyer MA. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–66. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- 36.Riemenschneider MJ, Knobbe CB, Reifenberger G. Refined mapping of 1q32 amplicons in malignant gliomas confirms MDM4 as the main amplification target. Int J Cancer. 2003;104:752–757. doi: 10.1002/ijc.11023. [DOI] [PubMed] [Google Scholar]
- 37.Ramos YF, Stad R, Attema J, Peltenburg LT, van der Eb AJ, Jochemsen AG. Aberrant expression of HDMX proteins in tumor cells correlates with wild-type p53. Cancer Res. 2001;61:1839–1842. [PubMed] [Google Scholar]
- 38.Wunderlich M, Ghosh M, Weghorst K, Berberich SJ. MdmX represses E2F1 transactivation. Cell Cycle. 2004;3:472–478. [PubMed] [Google Scholar]
- 39.Kadakia M, Brown TL, McGorry MM, Berberich SJ. MdmX inhibits Smad transactivation. Oncogene. 2002;21:8776–8785. doi: 10.1038/sj.onc.1205993. [DOI] [PubMed] [Google Scholar]
- 40.Yam CH, Siu WY, Arooz T, Chiu CH, Lau A, Wang XQ, Poon RY. MDM2 and MDMX inhibit the transcriptional activity of ectopically expressed SMAD proteins. Cancer Res. 1999;59:5075–5078. [PubMed] [Google Scholar]
- 41.Jin Y, Zeng SX, Sun XX, Lee H, Blattner C, Xiao Z, Lu H. MDMX promotes proteasomal turnover of p21 at G1 and early S phases independently of, but in cooperation with, MDM2. Mol Cell Biol. 2008;28:1218–1229. doi: 10.1128/MCB.01198-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matlashewski GJ, Tuck S, Pim D, Lamb P, Schneider J, Crawford LV. Primary structure polymorphism at amino acid residue 72 of human p53. Mol Cell Biol. 1987;7:961–963. doi: 10.1128/mcb.7.2.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lenos K, Grawenda AM, Lodder K, Kuijjer ML, Teunisse AF, Repapi E, Grochola LF, Bartel F, Hogendoorn PC, Wuerl P, Taubert H, Cleton-Jansen AM, Bond GL, Jochemsen AG. Alternate splicing of the p53 inhibitor HDMX offers a superior prognostic biomarker than p53 mutation in human cancer. Cancer Res. 2012;72:4074–4084. doi: 10.1158/0008-5472.CAN-12-0215. [DOI] [PubMed] [Google Scholar]
- 44.De Lange J, Ly LV, Lodder K, Verlaan-de Vries M, Teunisse AF, Jager MJ, Jochemsen AG. Synergistic growth inhibition based on small-molecule p53 activation as treatment for intraocular melanoma. Oncogene. 2012;31:1105–1116. doi: 10.1038/onc.2011.309. [DOI] [PubMed] [Google Scholar]
- 45.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 46.Joseph TL, Madhumalar A, Brown CJ, Lane DP, Verma CS. Differential binding of p53 and nutlin to MDM2 and MDMX: computational studies. Cell Cycle. 2010;9:1167–1181. doi: 10.4161/cc.9.6.11067. [DOI] [PubMed] [Google Scholar]
- 47.Bernal F, Wade M, Godes M, Davis TN, Whitehead DG, Kung AL, Wahl GM, Walensky LD. A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Cell. 2010;18:411–422. doi: 10.1016/j.ccr.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang H, Yan C. A Small-Molecule p53 Activator Induces Apoptosis through Inhibiting MDMX Expression in Breast Cancer Cells. Neoplasia. 2011;13:611–619. doi: 10.1593/neo.11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uchida C, Miwa S, Kitagawa K, Hattori T, Isobe T, Otani S, Oda T, Sugimura H, Kamijo T, Ookawa K, Yasuda H, Kitagawa M. Enhanced Mdm2 activity inhibits pRB function via ubiquitin-dependent degradation. EMBO J. 2005;24:160–169. doi: 10.1038/sj.emboj.7600486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sdek P, Ying H, Chang DL, Qiu W, Zheng H, Touitou R, Allday MJ, Xiao ZX. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol Cell. 2005;20:699–708. doi: 10.1016/j.molcel.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 51.Fu W, Ma Q, Chen L, Li P, Zhang M, Ramamoorthy S, Nawaz Z, Shimojima T, Wang H, Yang Y, Shen Z, Zhang Y, Zhang X, Nicosia SV, Zhang Y, Pledger JW, Chen J, Bai W. MDM2 acts downstream of p53 as an E3 ligase to promote FOXO ubiquitination and degradation. J Biol Chem. 2009;284:13987–14000. doi: 10.1074/jbc.M901758200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brenkman AB, de Keizer PL, van den Broek NJ, Jochemsen AG, Burgering BM. Mdm2 induces mono-ubiquitination of FOXO4. PLoS One. 2008;3:e2819. doi: 10.1371/journal.pone.0002819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 54.Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev. 1997;11:1464–1478. doi: 10.1101/gad.11.11.1464. [DOI] [PubMed] [Google Scholar]
- 55.Sutterluty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Muller U, Krek W. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol. 1999;1:207–214. doi: 10.1038/12027. [DOI] [PubMed] [Google Scholar]
- 56.Lenos K, De Lange J, Teunisse AF, Lodder K, Verlaan-de Vries M, Wiercinska E, Van der Burg MJM, Szuhai K, Jochemsen AG. Oncogenic functions of Hdmx in in vitro transformation of primary human fibroblasts and embryonic retinoblasts. Mol Cancer. 2011;10:111. doi: 10.1186/1476-4598-10-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 58.Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 59.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008;8:253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 60.Kedde M, van KM, Zwart W, Oude Vrielink JA, Elkon R, Agami R. A Pumilio-induced RNA structure switch in p27-3' UTR controls miR-221 and miR-222 accessibility. Nat Cell Biol. 2010;12:1014–1020. doi: 10.1038/ncb2105. [DOI] [PubMed] [Google Scholar]
- 61.Miskimins WK, Wang G, Hawkinson M, Miskimins R. Control of cyclin-dependent kinase inhibitor p27 expression by cap-independent translation. Mol Cell Biol. 2001;21:4960–4967. doi: 10.1128/MCB.21.15.4960-4967.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 63.An J, Wan H, Zhou X, Hu DN, Wang L, Hao L, Yan D, Shi F, Zhou Z, Wang J, Hu S, Yu J, Qu J. A comparative transcriptomic analysis of uveal melanoma and normal uveal melanocyte. PLoS One. 2011;6:e16516. doi: 10.1371/journal.pone.0016516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li H, Niederkorn JY, Neelam S, Alizadeh H. Downregulation of survivin expression enhances sensitivity of cultured uveal melanoma cells to cisplatin treatment. Exp Eye Res. 2006;83:176–182. doi: 10.1016/j.exer.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 65.De Waard-Siebinga I, Blom DJ, Griffioen M, Schrier PI, Hoogendoorn E, Beverstock G, Danen EH, Jager MJ. Establishment and characterization of an uveal-melanoma cell line. Int J Cancer. 1995;62:155–161. doi: 10.1002/ijc.2910620208. [DOI] [PubMed] [Google Scholar]
- 66.Lam S, Lodder K, Teunisse AF, Rabelink MJ, Schutte M, Jochemsen AG. Role of Mdm4 in drug sensitivity of breast cancer cells. Oncogene. 2010;29:2415–2426. doi: 10.1038/onc.2009.522. [DOI] [PubMed] [Google Scholar]