

Abstract
Dysregulation of mechanisms that govern the control of epithelial cell polarity, morphology and plasticity are emerging as key processes in tumor progression. In this study we report amplification and overexpression of PAR6B, an essential component in epithelial cell tight junction (TJ) formation and maintenance of apico-basal polarity, in breast cancer cell lines. Analysis of chromosome 20q13.13 in 11 breast cancer cell lines by fluorescence in situ hybridization (FISH) identified a novel small amplicon centered at PARD6B in 5 cell lines, with copy number ranging from 7 to 27. The presence of the PARD6B amplicon correlated with PARD6B transcript and PAR6B protein abundance. Expression of related isoforms PARD6A and PARD6G were detectable at significantly lower levels. PARD6B overexpression correlated with TJ network formation in cultured cell monolayers. SiRNA-mediated inhibition of PAR6B in MCF7 resulted in loss of TJ assembly and membrane localization of atypical PKCζ (aPKC), but did not affect adherens junction formation. SiRNA-mediated inhibition of CDC42 in MCF7 also resulted in loss of TJ networks, confirming the requirement of a complete PAR6-aPKC-CDC42-PAR3 complex to activate and stabilize TJs. Immunohistochemical analysis of PAR6B expression on breast tumor microarrays indicated exquisite epithelial cell-specificity. Few quantitative differences in staining were observed between normal epithelium and adjacent tumor margins. However staining appeared reduced and cytoplasmic in more poorly differentiated tumors. We propose that quantitative imbalances in the components of pathways governing normal epithelial cell polarity arising from gain or loss of function may radically alter epithelial cell architecture and contribute to tumor progression.
Keywords: Breast Cancer, DNA amplification, tight junction, siRNA, polarity, adhesion, PARD6B, PAR6B, CDC42, PKCζ
Introduction
Asymmetric cell division and cell polarization are fundamental traits required for both embryonic development and tissue remodeling. Maintenance of tissue architecture requires preservation of cell polarity, accurate asymmetric cell division and directional cell migration. Disturbance of these features of normal tissue organization is a hallmark of malignancy. In epithelial tissues, tight junctions (TJs), adherens junctions (AJs), gap junctions and desmosomes are all essential complexes which mediate cell-cell interactions required to maintain epithelial cell polarity. TJs, the most apical component of cell-cell junctional complexes create a physical barrier which functions to selectively regulate paracellular solute trafficking through epithelial sheets, prevent lateral migration of lipids and proteins between apical and basolateral membrane domains, thereby maintaining compartmentalization and tissue homeostasis.
The physical interaction of polarized epithelial cells with the basement membrane also ensures correct positioning and acts as a survival factor for epithelial cells. Cells that lose contact with the basement membrane undergo apoptosis or anoikis [1-3] however, in carcinomas, this positional control and homeostasis is disrupted or absent. Dissolution of epithelial TJs, AJs and desmosomes, reorganization of the actin cytoskeleton and loss of apical-basal polarity are events indicative of neoplastic transition from polarized epithelial cells to depolarized mesenchymal cell phenotypes. Reduced expression of several structural components of both AJs and TJs in progressively poorly differentiated breast cancers implies a tumor suppressive role for these junctional complexes. Loss of E-cadherin, the prominent molecule localized at AJs is linked to tumor progression in epithelial cancers including lobular breast cancer [4]. ZO-1 and claudin-7, both components of TJs show decreased staining in several invasive breast cancer cell lines and in poorly differentiated breast carcinomas [5,6] and are becoming increasingly viewed as prognostic biomarkers and predictors of tumor grade. Loss of heterozygosity of ZO-1 in a proportion of breast tumors suggests ZO-1 may play a direct role in malignant progression [6]. A large number of studies have contributed significantly to our understanding of the complex protein networks involved in assembly and stability of TJs. The topological role these complexes play in regulating TJs provides mechanistic insight into how deregulation or loss of stoichiometric co-ordination between key components required for maintaining epithelial cell polarity and plasticity might contribute to tumorigenisis providing a novel avenue for the investigation of tumor biology.
There has been considerable progress in elucidating the biochemical mechanisms underlying these processes in model organisms and in 2D and 3D mammalian cell culture models such as Martin-Darby canine kidney cells (MDCK), NMuNG, EpH4 and EMT-6 mouse mammary tumor cells [7]. This body of work has made it clear that proteins involved in apico-basal polarity and TJ formation are functionally connected in a complex network with several other pathways. Three evolutionarily conserved molecular complexes regulate epithelial cell polarization and TJ polymerization (for review, refer to [8]): 1) integral membrane proteins that constitute TJ strands, including occludins, claudins and Junctional Adhesion Molecule; 2) Peripherally associated cytoplasmic proteins that have the ability to organize the integral membrane TJ components, connecting them to actin filaments and/or other cytoplasmic proteins, completing formation of the TJ molecular complex; and 3) signaling proteins required for junctional assembly, regulation of barrier function and regulation of gene transcription. A series of molecules now recognized as indispensable participants in establishment of apico-basal asymmetry and TJ assembly are PAR6, atypical protein kinase C (aPKC), the RHO guanosine triphosphatase (GTPase) CDC42 and PAR3, a PDZ (PSD95/Dlg/ZO-1)-domain-containing membrane-scaffold adapter protein. This highly conserved quaternary complex functionally regulates asymmetric cell division from worms to mammals, by affecting cytoskeleton re-organization, post-golgi vesicle trafficking and microtubule network polarization [9-13]. In mammalian epithelial cells, interference of PAR3, PAR6 or aPKC have been shown to inhibit TJ but not AJ [11-13]. PAR6 interacts directly with aPKC isoforms ι/λ and ζ forming the core of this complex [14]. Binding of CDC42•GTP to inactive PAR6-aPKC complex induces a conformational change in PAR6, leading to phosphorylation and activation of aPKC, and subsequent binding of this complex to PAR3 (reviewed in [15]). CDC42•GTP is required for maintaining this polarization but alone is not sufficient to establish polarity. Studies in D. melanogaster have shown the par3 homologue bazooka, to be the pivotal component controlling asymmetric cell division [16]. Two additional binding partners for PAR6 have now been identified as necessary for maintenance of cell asymmetry: STARDUST-CRUMBS-PATJ [17,18] and DLG-SCRIB-LGL [19,20]. There are 3 PARD6 gene family members in mammalian cells (homologous to a single pard6 member in Drosophila and C-elegans), PARD6A, PARD6B and PARD6G [11]. Expression of each PAR6 isoform is tissue specific [21], with apparent differences in function, localization and effector interactions, which have not yet been well defined.
PAR6 has recently risen as central to the coordinated maintenance of cell polarity by physically linking several of the multi-component complexes described above, converging CDC42- and aPKC-mediated signal transduction (for review see [22]). Recent studies have identified an important association between PAR6 and TGFβ receptors, regulating interaction of PAR6 with SMURF1, an E3 ubiquitin ligase which targets RHO GTPases for degradation leading to TJ dissolution, TGFβ -induced EMT and metastasis [7,23,24]. Cai et al have demonstrated that G-protein-activated plospholipase C-β (PLCβ) interacts with both PAR6 and PAR3, mediating downstream signal transduction by hydrolysis of phosphatidylinositol-4,5-bisphosphate, releasing intracellular calcium and diacylglycerol, all important second messengers required for cell polarity and asymmetric cell division processes [25]. Signaling networks impinging on the PAR6 complex have been further expanded to signaling through TIAM1 guanine nucleotide exchange factor, which is a regulator of cell motility and invasion [26]. In addition, activation of several oncogenes (and oncogenic signaling mechanisms), including ERBB2, KRAS, RAF, FOS, JUN, VSRC, RHO and RAC have been shown to disrupt apical-basal polarity by altering the localization of apical membrane markers [27,28] and references therein). While the mechanisms by which this occurs are still under investigation, Aranda and colleagues have shown that ERBB2 disrupts apical-basal polarity by associating directly with the PAR6-aPKC core complex [27]. These studies further highlight the need to understand how extracellular effectors signal to the polarity machinery to maintain correct cell architecture in an appropriate biochemical and biophysical context. Moreover, molecular aberrations contributing to loss of stoichiometric equilibrium of molecules governing cell morphology are likely to play a major role in progression of epithelial tumors [28].
Our laboratory and others have extensively investigated genomic DNA copy number alterations as key genetic events in the development and progression of human cancers. The advent of microarray-based Comparative Genome Hybridization (aCGH) has vastly increased the resolution at which we can view and map regions of chromosomal gain and loss in tumors that are likely to harbor oncogenes and tumor suppressor genes [29]. aCGH-based studies in breast cancer [30,31] have provided a comprehensive view of regions of DNA gain at chr20q11-13 a common region of amplification in breast cancer. In this study, a significant peak in amplification was observed at chromosome 20q13.13, a region containing the BCAS4 gene, previously reported to be highly amplified in breast cancer cell lines [32]. The gene immediately centromeric to BCAS4 is PARD6B. In this study, we find amplification and overexpression of PARD6B in a proportion of breast cancer cell lines. Given that PAR6 plays a central role in TJ assembly, maintenance of cell polarity, and is implicated in breast tumor progression [7,28], including overexpression in premalignant neoplasms [33], we have investigated the requirement of PAR6, CDC42 and aPKCζ to promote TJ polymerization breast cancer cells. We also examine in situ PAR6 expression and localization in normal polarized epithelia and epithelial breast tumors. Our observations uncover a previously unreported mechanism for dysregulation of PAR6 expression in breast cancer, and highlight the importance of further investigating the disturbances of the mechanisms that control cell polarity in cancer.
Materials and methods
Cell culture and siRNA electroporation
All cell lines were obtained from the ATCC (Rockville, MD) and maintained at 37°C in 5 % CO2 in ATCC-recommended growth media. Double-stranded siRNAs were purchased from Qiagen (Valencia, CA). 25mg siRNAs were electroporated into 8x106 cells using the 005RG electroporator from MaxCyteTM (Rockville, MD) using proprietary conditions optimized by the manufacturer. siRNA sequences (sense and antisense respectively): GFP [34], gca agc uga ccc uga agu uca u, gaa cuu cag ggu cag cuu gcc g; P6B30, cgu auu gcg ucc uga caa c-dtdt, guu guc agg acg caa uac g-dtdt; P6B42, gua cag gac uau uag cug u-dtdt, aca gcu aau agu ccu gua c-dtdt; CDC42-9, cua ugc agu cac agu uau g-dtdt, cau aac ugu gac ugc aua g-dtdt; CDC42-11, gag gau uau gac aga uua c-dtdt, gua auc ugu cau aau ccu c-dtdt; CDC42-35, ggu agu gcu gua uau ucu a-dtdt, uag aau aua cag cac uuc -dtdt.
Cell adhesion assay
siRNA- or mock-electroporated cells were trypsinized, counted, and seeded in quadruplicate in 96-well plates at a density of 50,000 cells per well for 5, 15, 30, 45 and 60 min. Each time point was terminated by aspirating floating cells, washing adherent cells with PBS, and adding MTT reagent (Molecular probes, Eugene, OR). Solubilized formazan concentration was determined at 570 nM. Assays were repeated on cells electroporated on three separate occasions.
Fluorescence in situ hybridization
BAC clone DNA was purified using the QIAgen midi purification kit, and labeled with Spectrum Orange-d-UTP using the Nick Translation kit from Vysis (Downers Grove, IL). BAC clones were obtained from Invitrogen (Carlsbad, CA).
Quantitative RT-PCR
Total RNA was prepared from cells lysed in TRIzol reagent (Invitrogen) as described [35]. 1 mg total RNA was reverse transcribed using the Random Hexamer protocol supplied with SuperScriptTMII Rnase H- enzyme (Invitrogen). Quantitative RT-PCR analysis was conducted in triplicate using the 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA) using the Qiagen SYBR Green Kit and protocol. Parallel amplification of b-actin (ACTB) cDNA was used to normalize cDNA concentration, and was determined to be linearly co-amplified with respect to all gene specific primers (data available on request). Each experiment was repeated three times, and each cDNA was measured in triplicate in each experiment. All gene-specific primers are exon-spanning: PARD6B Fwd 5’ ggg cac tat gga ggt gaa ga Rev 5’ tcc atg gat gtc tgc ata gc; PARD6A Fwd 5’ct ggacgt gct act tgg cta Rev 5’cag gca gta ggt cca cgt ct; PARD6G Fwd 5’tca gac ctt gcg att cta cg Rev 5’tgc aca tct gca tag cca at; ACTB Fwd 5’gca aag acc tgt acg cca aca Rev 5’cct cgg cca cat tgt gaac.
PAR6B antibody production
A PAR6B-specific peptide (sequence HAPDQKLLEEDGTI, amino acids 356-369) was synthesized and purified by Sigma (St. Louis, MO). Sera from two immunized rabbits were tested for the presence of PAR6B antibodies by western blot from breast cancer cell lines with known high and low PAR6B expression (results not shown). After the final immunization boost, IgG was affinity purified from serum by Sigma. We rigorously tested the purified antibody for PAR6B specificity as follows. A293 cells, which express low endogenous levels of PAR6B were transiently electroporated with a constitutive PARD6B expression construct (pCDNA3-FLAG-PARD6B, detailed in the next section) or empty vector control. After 24h, cell lysates were harvested for SDS-PAGE, and a high level of PAR6B protein was detected by western blot only in extracts where pCDNA3-FLAG-PARD6B had been introduced (not shown). Accuracy of the PAR6B -immunoreactive product was verified by probing the same extracts with an anti-FLAG monoclonal antibody (Sigma), which recognizes the expressed fusion protein.
Construction of pCDNA3-FLAG-PARD6B expression vector
cDNA was synthesized from normal adult kidney RNA (Stratagene, La Jolla, CA) and used to amplify the entire coding region of PARD6B, using the Accuprime PCR kit (Invitrogen) with forward primer 5’-ctttccgagatccccagtc and reverse primer 5’-gcctattccacgtcactggt. Nested Primers were designed with flanking restriction sites and were used to directionally clone the full length PARD6B cDNA into pCDNA3.1 (Invitrogen). A Kozak sequence and N-terminal FLAG epitope were added to this construct using synthetic oligonucleotides. Double-stranded DNA sequence from pCMV-FLAG-PARD6B was obtained to verify the in-frame fusion of the FLAG tag, and wild type PARD6B coding sequence. Expression of the correct size fusion protein (42 kda) was verified by western blot using anti FLAG antibody (Sigma, St. Louis, MO).
Immunoblotting and immunofluorescence cytostaining
Immunoblotting of whole cell lysates was performed as described [35]. Fluorescent immunocytostaining was conducted in chamber slides (Nunc, Rochester, NY). Cells were washed twice in PBS, fixed for 15 min in 4% paraformaldehyde, and incubated for 1 h in blocking buffer (5% normal serum, 1% casein, 0.5% BSA, 0.1% Triton X-100). Cells were incubated with primary antibodies for 1 h at RT in blocking buffer, washed 3 times with PBS with 0.1% tween-20 (PBS-T), then incubated with 2 μg/ml Cy3- or FITC-conjugated IgG secondary antibodies (Jackson Immunoresearch, West Grove, PA) in blocking buffer for 1 h at RT in the dark. After 3 washes in PBS-T, slides were mounted in Vectashield with DAPI (Vector Laboratories, Burlingame, CA) and analyzed using a fluorescence microscope (Zeiss Axiophot). Antibodies for ZO-1, E-cadherin and claudin 7 were from Invitrogen (formerly Zymed Laboratories, San Francisco, CA), and anti-alpha tubulin was purchased from MP Biomedicals.
Immunohistochemistry on tissue micorarrays
Tissue Microarrays (TMA) used in this study were as follows; High Density Breast Cancer Array (HD-BR-1 Lot# 031003, Cytomyx Holdings PLC, UK-Based. Formerly Clinomics Biosciences, Inc, Fredrick, MD), and Breast Carcinoma progression Tissue Microarray (CHTNBrCaProg1 TMA00052, Cooperative Human Tissue Network). Immunohistochemical staining was performed as previously described [36]. Briefly, slides were deparaffinized using a xylene-ethanol series. Antigen retrieval was carried out by steaming slides for 15 min in Antigen Unmasking Solution (Cat No. H-3300 Vector Laboratories, Burlingame, CA) followed by slides incubation with primary antibodies overnight at 4°C. Antigen-antibody reactions were revealed with UltraVision Detection System (Cat. No: TR-015-HD Labvisition Corporation, Fremont, CA). Slides were counter- stained with hematoxylin and fixed. The primary antibodies were as follows; Rabbit anti-PAR6B polyclonal (this study); Mouse anti-ZO-1 (Cat. No. 33-9100) Invitrogen (formerly Zymed Laboratories); Rabbit IgG (Cat. No1-1000, Vector laboratories) was used as a negative control. Based on scoring systems previously described [36,37], all TMAs were reviewed and scored on a five-point scale, as negative=0, no staining or equivocal staining in <5% of cells; weak=1+, definite weak-moderate staining in ≤ 20% of cells; moderate=2+, moderate staining in 21%-50% of cells; strong=3+, moderate staining in the majority of cells; and very strong positive=4+, strong staining in > 50% of cells.
Results
Fine mapping of chromosome 20q13.13 in breast cancer cells identifies a small novel amplicon centered at PARD6B
Recent genomic amplification studies in breast cancer [31,32], prompted us to conduct a more thorough FISH analysis of gene amplification at 20q13.13 – 20q13.2, surrounding the BCAS4 gene in breast cancer cells (Figure 1). We observed a previously unidentified amplification peak at 20q13.13 with a BAC FISH probe containing the PARD6B gene in a number of breast cancer cell lines (Figure 1). The most highly amplified region in our FISH survey is centered at the PARD6B gene. In MCF7, consistent with the FISH data, high-resolution oligonucleotide aCGH defines a small amplicon containing only PARD6B and BCAS4 (data not shown). As previously reported [32,38], BCAS4 is interrupted at the amplicon boundary, leaving PARD6B as the only intact gene in this small region of amplification. One of these cell lines, BT474, also showed multiple copies of chromosome 20 centromeres (not shown). This suggests that amplification of the PARD6B gene may be a mechanism contributing to deregulated expression in breast cancer.
Figure 1.
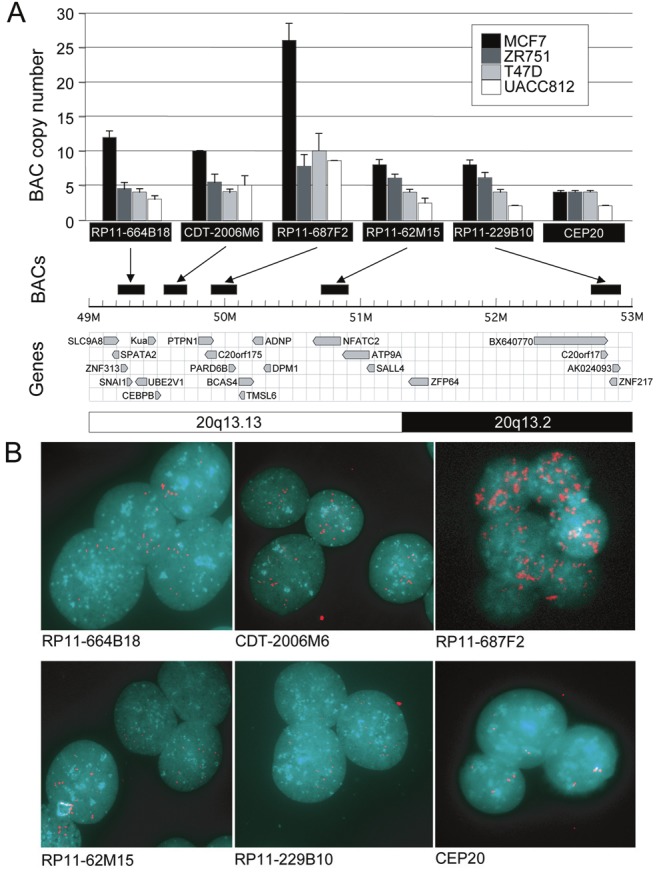
Identification of PARD6B amplification by interphase FISH. A. Five BAC clones spanning a 4 megabase region of chromosome 20q13.12–13.2 used to examine DNA copy number changes in 4 breast cancer cell lines. The average number of BAC signals identified from approximately 20 interphase nuclei is presented. Arrows, relative to known and predicted genes, indicate the genomic position of each BAC. CEP20, control centromere probe. B. Panel of representative MCF7 interphase nuclei probed with each BAC. Each BAC probe was first hybridized to metaphase spreads from normal peripheral blood lymphocytes to confirm signal localization to chromosome 20q.
PAR6B is highly expressed in luminal epithelial breast cancer cells
We first examined the level of mRNA expression of PARD6B in a panel of breast cancer cell lines by quantitative RT-PCR (Figure 2A). We observed a dramatic variation in expression of PARD6B across our panel, and observed levels were consistent with protein abundance as determined by western blot from whole cell lysates. The 4 cell lines which carry additional copies of the PARD6B gene (MCF7, T47D, UACC812 and ZR751; Figure 1) all show very high levels of PARD6B mRNA and PAR6B protein expression. This suggests that PARD6B amplification may be an important mechanism for increased expression in these cell lines. We were interested to note that all four of these cell lines are luminal epithelial-derived. Low levels of PARD6B mRNA were detected in non-tumorigenic MCF10A and MCF12A cell lines (data not shown), highlighting the high abundance of PARD6B in a proportion of breast cancer cell lines. The level of mRNA expression of PARD6A and PARD6G isoforms was also variable across our cell line panel, but was uniformly very low relative to PARD6B (Figure 2B). Immunofluoresence (IF) detection of endogenous PAR6B protein showed a cytoplasmic and membrane localization pattern, clearly detectable in cell lines with high expression level (examples for MCF7 and T47D shown in Figure 2C).
Figure 2.
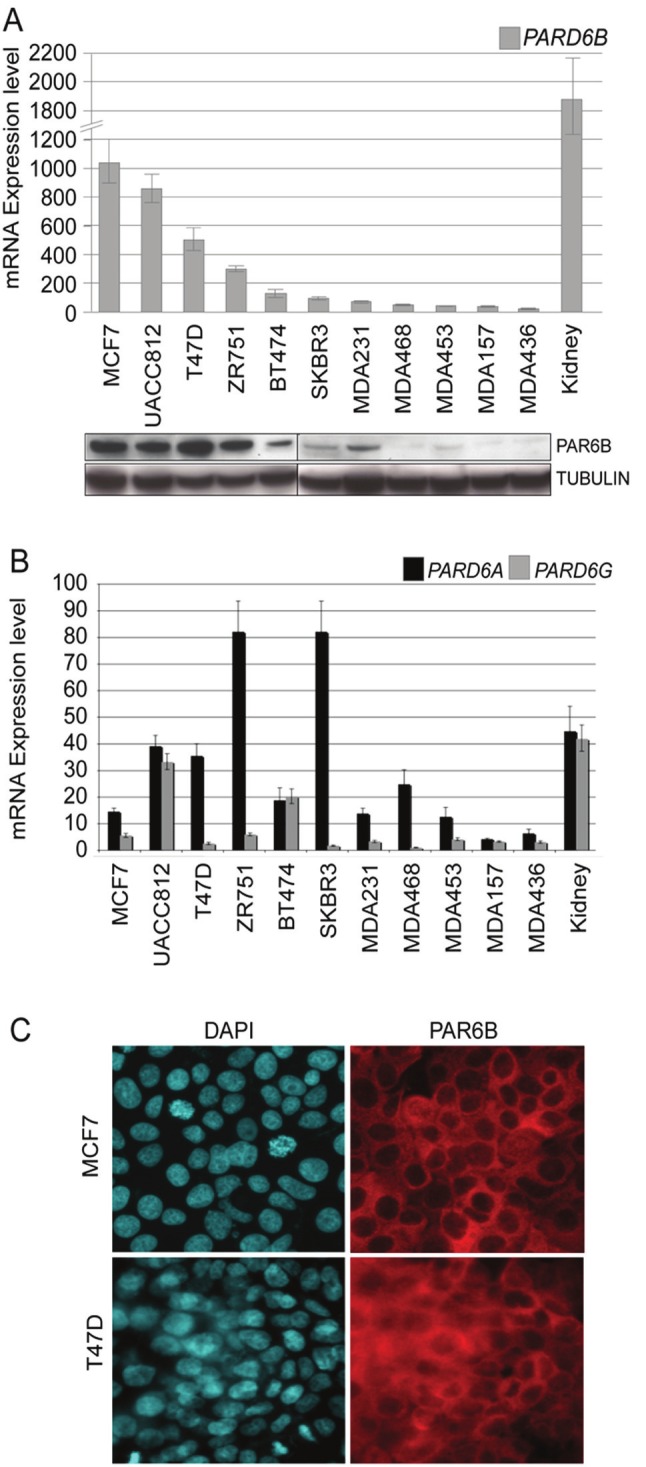
Survey of endogenous PARD6B expression in breast cancer cell lines. A. Quantitative-RT-PCR analysis was performed on 11 breast cancer cell lines for PARD6B. Human adult kidney total RNA was used as a positive control. PAR6B protein level was determined by western blot from breast cancer cell line whole cell lysates. Blots were re-probed with anti -tubulin to control for protein loading. B. Q-RTPCR for PARD6A and PARD6G. The level of expression is comparable with PARD6B in Figure 2A, as data shown in both graphs has been normalized to the level of PARD6G expression. C. IF staining of endogenous PAR6B protein in MCF7 and T47D (polyclonal antibody isolated in this study).
PAR6B expression correlates with the ability of breast cancer cells to form tight junctions
As PAR6 proteins play an essential role in coordinating assembly of several protein complexes required for TJ polymerization during epithelial cell polarization, we conducted a survey of TJ and AJ formation in our panel breast cancer cell lines (Figure 3). After staining all cell lines with TJ-associated proteins ZO-1, occludin-1 and caludin-7, and AJ-associated protein E-cadherin, we observed that cell lines with abundant PARD6B mRNA expression exhibited strong formation of structured TJ networks in confluent cell monolayers. Cell lines consistently staining for ZO-1 showed a similar immunoreactive pattern for occludin-1 and claudin-7 (representative result for MCF7 shown in Figure 3A). IF detection of ZO-1 (for TJ) and E-cadherin (for AJ) is shown for 6 cell lines representative of variable PARD6B mRNA expression shown in Figure 3. We observed strong TJ staining in the luminal-epithelial-like cells, and complete lack of TJ staining in the mesenchymal- or stromal-like cell lines. Structurally complete TJ networks were also observed in the non-tumorigenic MCF12A and MCF10A breast cell lines (not shown), but staining was relatively weak. However this is consistent with the low level of PARD6B expression observed in these cell lines. Interestingly, TJ networks were completely polymerized 24h after seeding MCF7 cells, however all other cell lines required at least 48-72h for strong polymerization to occur (not shown). Together, our data suggests PAR6B protein level may be a rate-limiting step in TJ assembly.
Figure 3.
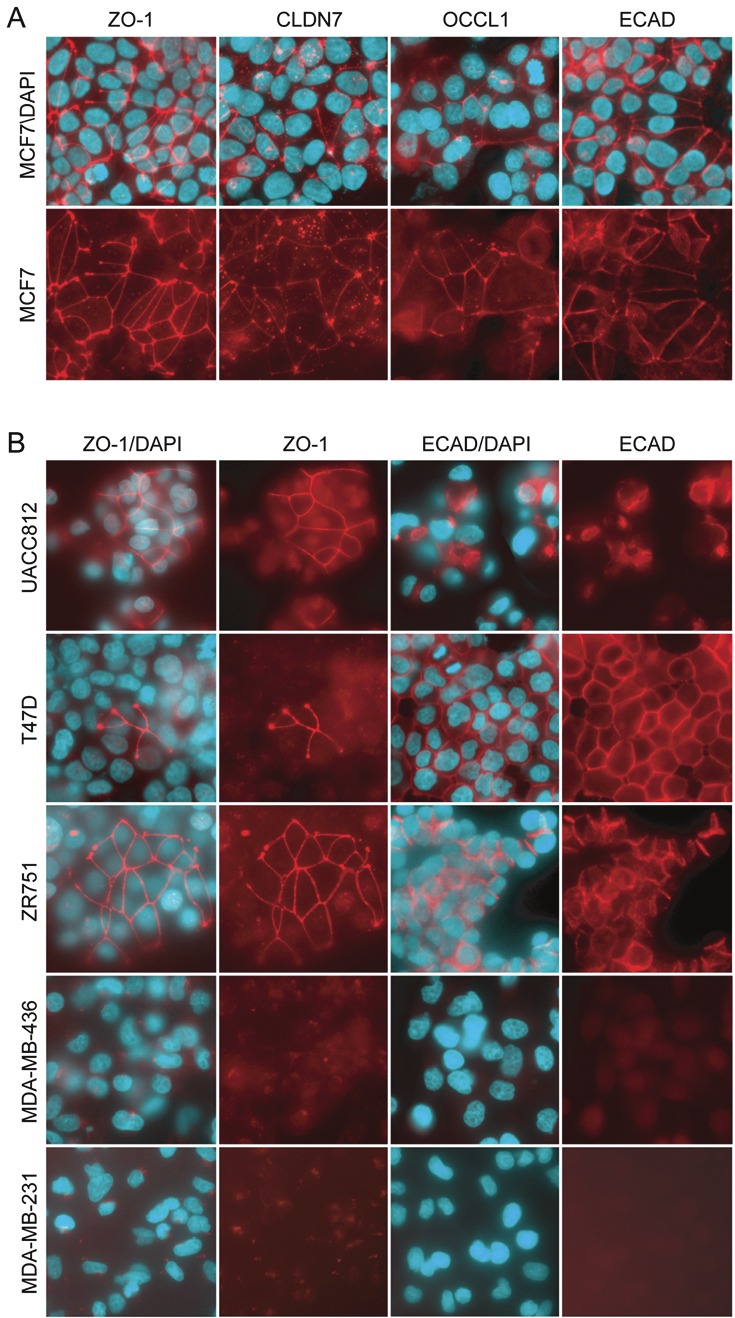
Immunofluorescence staining survey of tight and adherens junction proteins in breast cancer cell lines. A. MCF7 cells stained for TJ proteins zona occludens 1 (ZO-1), claudin 7, occludin 1 (OCCL1) and adherens junction associated protein, E-cadherin (ECAD). Nuclei are DAPI counterstained (blue). B. Four additional breast cancer cell lines are shown from a total of 11 that were stained separately for the presence of ZO-1 and ECAD. A strong correlation was observed between the presence and absence of both cell junctional networks.
To further investigate the dynamics of TJ polymerization in our panel of breast cancer cell lines, we also investigated RNA expression level of PAR3, an apico-lateral membrane-associated scaffold protein that binds PAR6, by RT-PCR in all cell lines. We observed a moderate and relatively uniform level of PAR3 expression across all samples, (data not shown).
From our present understanding of the sequential assembly process for mammalian epithelial cell TJs, both PAR6 and aPKC in association with CDC42, constitute the functional core protein complex required for establishment of TJs and maintenance of apico-basal polarization [11-13]. Our first goal was to confirm whether loss of one or more component(s) from this core complex in breast cancer cells would result in a loss of potential to form a TJ network.
Inhibition of PAR6B expression disrupts tight junction but not adherens junction network formation
To investigate the requirement of PAR6B protein isoform for TJ assembly, small interfering RNA (siRNA) molecules were designed to specifically inhibit PARD6B expression (detailed in the Materials and Methods section). Two siRNAs (P6B30 and P6B42) were designed to different regions of the PARD6B mRNA sequence. Following electroporation of the siRNAs into MCF7 cells, we observed a significant reduction of the level of PARD6B mRNA and protein levels compared to control non-specific siRNA or mock-electroporated cells (Figure 4A and 4B). The rate of endogenous PAR6B protein turnover following cycloheximide treatment of MCF7 was determined previously by western blot to be >90% at 24h (data not shown), indicating incubation of introduced siRNAs for 24h was sufficient to produce a PAR6B-deficient state in MCF7 cells. Using IF to examine the visual appearance of junctional network proteins, siRNA-mediated inhibition of PAR6B resulted in a dramatic disruption in formation of TJ networks in confluent MCF7 monolayers visualized by a very unusual pattern of ZO-1 staining (Figure 4C), with the majority of adjacent cells not displaying any structured network at all. Staining patterns for occludin-1 and claudin-7 were very similar to the ZO-1 result (data not shown). Our IF staining in MCF7 also indicated enhanced levels of ZO-1 in the cytoplasm after PAR6B inhibition, indicating that loss of PAR6B disrupts the appropriate sub-cellular localization of ZO-1 rather than leading to reduced protein levels. The same TJ-disrupted phenotype was observed for both P6B30 and P6B42 siRNAs (results for P6B30 only are shown) suggesting this effect of these siRNAs is PAR6B. Mock and control siRNA-electroporated cells both exhibited visually identical TJ networks to that observed for untreated MCF7 cells. Therefore, our results indicate that PAR6B is essential for polymerization of TJ networks in MCF7 breast cancer cells.
Figure 4.
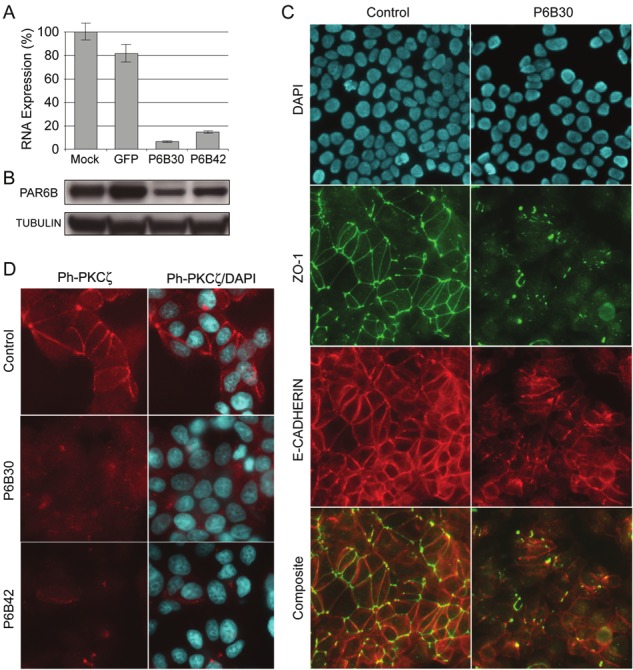
Inhibition of PAR6B in MCF7 cells disrupts tight junction but not adherens junction assembly. A. Quantitative RTPCR analysis of PARD6B expression level in MCF7 cells following siRNA electroporation and 24h incubation. siRNAs were control, and PARD6B-spedcific P6B30 and P6B42. Mock indicates electroporated cells without siRNA. B. Western blot to detect PAR6B protein in MCF7 whole cell lysate following 24h siRNA treatment. Immunoblot was re-probed with anti-tubulin antibody to control for protein loading. C. Dual immunofluoresence stain of MCF7 cells for ZO-1 and E-cadherin following siRNA electroporaiton and 24h incubation. SiRNAs are control or PARD6B-spedcific P6B30. Composite images show the merge of ZO-1 and ECAD stains without DAPI. AJ networks are intact following PAR6B knockdown, whereas TJ networks are lost. D. IF stain for phospho-PKCζ in MCF7 cells following siRNA-mediated knockdown of PAR6B.
To determine whether AJs were affected by PAR6B inhibition, or whether the role of PAR6B was specific to regulation of TJ assembly, dual IF stains were performed for both ZO-1 and E-cadherin in MCF7 cells with and without PAR6B knockdown (Figure 4C). Inhibition of PAR6B resulted in a slight decrease in E-cadherin staining intensity, but did not disrupt AJ network assembly. The composite images (Figure 4C) clearly show the presence of AJs (red) between immediately adjacent epithelial cells without TJs (green). The result for both P6B30 and P6B42 siRNAs was identical. Our observation is consistent with the current understanding that early events in epithelial cell polarity involving formation of nectin and E-cadherin mediated cell-cell contacts precede subsequent recruitment of PAR6-aPKC-CDC42 and PALS1/PATJ complexes to apicolateral cell-cell contact points.
Depletion of PAR6B disrupts membrane localization of activated PKCζ in MCF7 cells
Our results lead us to further hypothesize that loss of PAR6B (and thus disruption of the PAR6-aPKC core complex) would result in the inability of aPKC to become activated (phosphorylated) and recruited to the apicolateral membrane TJ scaffold proteins. We tested this hypothesis by electroporating MCF7 cells with control or PARD6B-specific siRNAs, then immunostaining cells for the presence of phospho-aPKCζ. We observed a complete loss of phospho-aPKCζ staining at the cell membrane after PAR6B knockdown (Figure 4D), which suggests that incorrect TJ assembly abrogates downstream signaling from aPKCζ.
Inhibition of CDC42 expression results in loss of tight junction formation and reduced adhesion in MCF7
Activated CDC42 (CDC42•GTP) is a key component required to tether the PAR6-aPKC core complex to PAR3 and JAM1 at the apico-lateral membrane. We therefore predicted that disruption of CDC42 expression would result in loss of TJ polymerization and establishment of cellular asymmetry by inhibiting completion and localization of the PAR3-CDC42-PAR6-aPKC signaling complex. Similar to our siRNA-mediated inhibition of PAR6B in MCF7, we designed siRNAs to silence CDC42. Two of the three siRNAs designed were able to substantially reduce the level of CDC42 protein in MCF7 cells (Figure 5A) and were used in subsequent experiments. siRNA-mediated inhibition of CDC42 in MCF7 resulted in aborted TJ formation as predicted (Figure 5B) but not AJ formation by E-cadherin IF staining (not shown). No change in PAR6B protein level (by western blot) or subcellular localization (by IF staining) was observed following CDC42 knockdown (not shown). A significant change in cell morphology in 2-D culture was observed following CDC42 knockdown in MCF7. Cells appeared rounded, tending to grow in columnar rather than radial clusters with less evidence of ruffling and focal adhesion. This altered cell morphology correlated with reduced cell adhesion (Figure 5C) and is consistent with the known role of CDC42 in regulation of actin polymerization into filopodia. Taken together, our siRNA knockdown results are consistent with the predicted model that CDC42 and PAR6B each play an essential role in TJ assembly and subsequent signal transduction mechanisms required for normal epithelial cell homeostasis.
Figure 5.
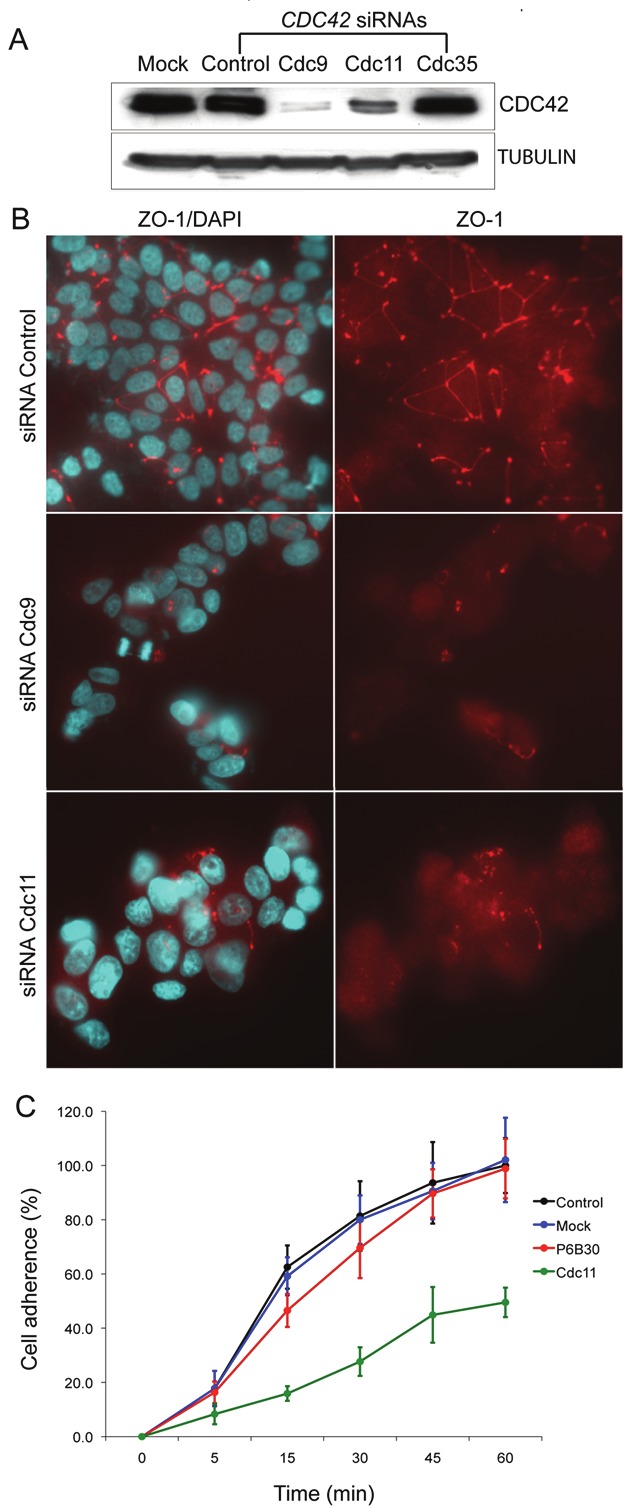
Inhibition of endogenous CDC42 expression in MCF7 cells disrupts tight junction formation. A. Western blot for total CDC42 protein in MCF7 whole cell lysates following electroporation of control or CDC42-specific siRNAs and 24h incubation. siRNA CDC42-35 did not reduce CDC42 levels and was not used further. B. Immunofluoresence stain for ZO-1 in MCF7 cells after siRNA-mediated CDC42 knockdown, indicates the requirement of CDC42 as well as PAR6B for polymerization of TJ network. C. Cell adhesion assay demonstrates significant loss of cell adhesion of siRNA-mediated CDC42 knockdown in MCF7.
PAR6B is present in normal epithelia and in a large proportion of breast tumors
We lastly sought to examine the pattern of PAR6B expression in breast cancer tissue specimens. We first developed and tested a polyclonal antibody specific to the PAR6B isoform (this study). To evaluate antibody specificity, a mammalian expression construct containing full length PARD6B (this study) was cloned and transiently expressed in both A293 and NIH-3T3 cells. Whole cell lysates prepared from PARD6B-transfeced cells showed a strong immunoreactive product at 42 kda by western blot and by IF. No product was observed with cells transfected with empty vector, or with control naive rabbit serum (data not shown). We next conducted an immunohistochemical (IHC) survey of breast tumor specimens from whole sections and from commercially available normal multi-tissue and breast tumor microarrays (TMAs) with our affinity-purified PAR6B antibody. Our results indicate highly specific staining in normal epithelia from multiple organs. Staining was moderate to very strong in all epithelial structures with increased staining often observed at the apical surface in mature glandular epithelial structures (Figure 6A-C) indicative of polarized cell architecture. Our IHC survey evaluated a total of 17 normal samples and 133 samples of invasive ductal carcinoma. All normal specimens stained positively for PAR6B. Tumor staining was 90/133 (68%) 4+, 8/133 (6%) 3+, 16/133 (12%) 2+, 6/133 (4%) 1+, 13/133 (10%) negative indicating that in the majority of cases, expression is maintained in tumors. We did not observe dramatic quantitative differences in staining between normal epithelial structures and adjacent tumor. Expression of PAR6B did appear to be less in very poorly differentiated tumors showing no tubular or glandular features. IHC of the same TMA series with anti ZO-1 antibody yielded a pattern of staining consistent with the PAR6B results (data not shown) suggesting that the presence of PAR6B correlates predominantly with the presence of TJs. Additional studies on a larger number of clinically annotated tumors (and premalignant neoplasms) with more variable histopathology and etiology will be necessary to investigate whether quantitative level of PAR6B expression might correlate with stage, histological grade, biomarker status (hormone receptors, ERBB2, TP53, etc) or outcome.
Figure 6.
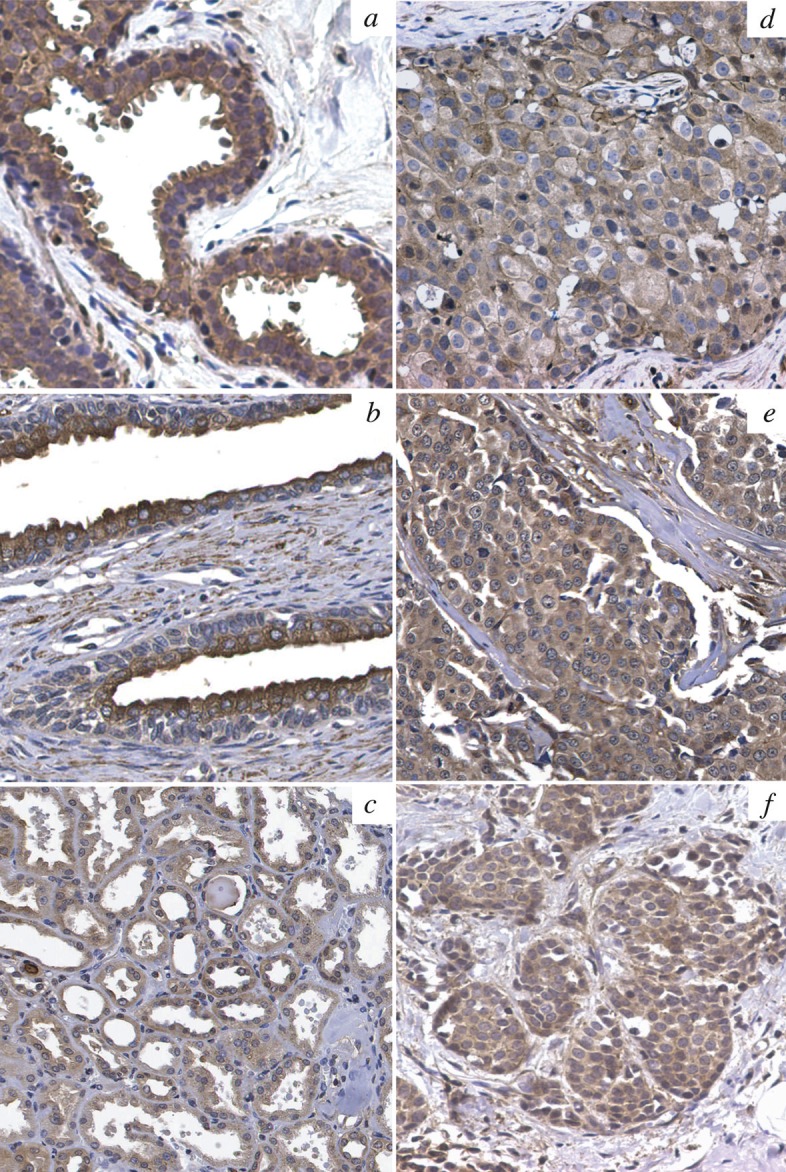
In situ analysis of PAR6B epithelial cell specificity. IHC evaluation with PAR6B isoform-specific polyclonal antibody (this study). A, normal breast, B, normal prostate, C, normal kidney, D, breast ductal carcinoma in situ, E, breast invasive ductal carcinoma, F, breast invasive lobular carcinoma.
Discussion
PAR6B is known to play a key role in mammary epithelial cell biology. Several studies have now been reported which indicate deregulated PAR6B signaling contributes to malignant epithelial cell phenotypes due predominantly to disrupted polymerization and maintenance of TJs [7,27,33]. In this study, we have identified significant copy number amplification of PARD6B (located at chromosome 20q13.13) in several breast cancer cell lines, which correlated strongly with overexpression of PAR6B protein. 20q13.13 has previously been reported as amplified in a significant proportion of breast tumors, however data in this study defines a small amplicon common to multiple breast cancer cell lines centered at the PARD6B gene. We and others hypothesize that abnormalities in the level of PAR6B protein, or quantitative imbalances in the components of pathways governing normal epithelial polarity (through gain or loss of function), may disrupt cell architecture and contribute to breast cancer progression. This study supports amplification of the PARD6B gene as a previously unreported mechanism contributing to PAR6B overexpresson. While we did not observe dramatic quantitative differences in PAR6B staining by IHC in breast tumors relative to adjacent normal epithelial structures, we did observe reduced PAR6B expression in very poorly differentiated tumors showing no tubular or glandular features. This finding is consistent with loss of TJs during epithelial to mesenchymal transition (EMT) and tumor progression.
In this study, we confirm the critical requirement of PAR6B and CDC42 to facilitate TJ assembly. Localization of activated aPKCζ to the plasma membrane is necessary to promote interaction of the core PAR6B complex with PAR3, CDC42 and TJ structural proteins. Our study demonstrates that silencing of PAR6B completely blocks the ability of activated PKCζ to localize to the cell membrane. We further demonstrate the necessity of CDC42 to facilitate TJ assembly in mammary epithelial cells. While depletion of PAR3 has been shown previously to disrupt TJ formation in MDCK cells through spatial regulation of Rac activity [39], this has not yet been demonstrated in breast cancer to our knowledge. Our findings support the prediction that altered expression level of polarity-pathway components by either gain or loss of function, or mislocalization will alter cell physiology, resulting in radical changes in epithelial cell architecture.
A large number of binding partners for PAR6 are now known that play essential roles in maintaining epithelial cell morphology and asymmetric division, and thus, PAR6 overexpression is likely to result in aberrant spatial sequestration of key cell polarity and cell signaling molecules, potentially contributing to perturbation of the many pathways connected to the PAR6-PAR3-aPKC complex. Interestingly, Nolan and colleagues have recently shown that PAR6 is not only a regulator of cell polarity and migration, but also a mediator of cell proliferation [33]. In their study, PAR6 expression was shown to induce epidermal growth factor (EGF)-independent proliferation of normal mammary epithelial cells by promoting activation of mitogen-activated protein kinase (MAPK) signaling. This function was found to be dependant on the ability of PAR6 to interact with aPKC and CDC42 [33]. Given that PAR6 is overexpressed in premalignant breast neoplasms [33] and has been proposed to pay a role in TGFβ-mediated EMT [7], this highlights the significant impact that dysregulation of essential polarity proteins such as PAR6 may have on both carcinogenesis and tumor progression.
The diversity of binding partners for PAR6 and the stoichiometric importance of this molecule in accurate modulation of several key cellular functions, suggest several possibilities that may be disease promoting upon dysregulation. With respect to altered cell polarity, the main effectors in the PAR3-PAR6-aPKC-CDC42-PLCβ complex are aPKC and PLCβ. Transduction of extracellular signals by G-protein coupled receptors, and hydrolysis of PIP2 by activated PLCβ releases second messengers essential for calcium homeostasis and dynamic rearrangement of the actin cytoskeleton via the SRC- and RHO-dependant pathways. The specific targets of activated aPKC affecting cell asymmetry are not yet clear, however our studies show a significant degree of phosphorylated aPKC resides complexed with PAR6 at cell junctions in polarized epithelial cells indicating a specific spatial requirement for this activated molecule in TJ maintenance. Yue and colleagues [40] have shown PLCβ to be a possible phosphorylation target of aPKC. The known spatial proximity of these two enzymes suggests a mechanism for rapid modulation to balance PIP2, IP3, DAG and intracellular calcium levels. Disrupting the interaction of these molecules due to imbalance of polarity molecules such as PAR6 might predict a direct impact cell homeostasis.
While PAR6 is overexpressed in premalignant breast lesions [33], it is not yet clear whether upregulation of PAR6 can itself provide breast cancer cells with a selective growth advantage. Interestingly, we and others have observed PAR6 overexpression in estrogen receptor alpha (ER) positive breast tumors [33]. Moreover, PAR6 is a regulatory target of another gene amplified and overexpressed in MCF7, the nuclear receptor coactivator AIB1 [41], suggesting an important functional link between deregulated hormone signaling and PAR6 overexpression [42]. Regardless of the mechanism driving PAR6 overexpression, previous reports have importantly shown the ability of PAR6 overexpression to sequester PAR3 and PKCζ away from cell boundaries, disrupt ZO-1 localization, but not E-cadherin or b-catenin [11]. This data suggests that overexpression of PAR6 and resultant aberrant sequestration of crucial cell junctional molecules might interfere with TJ formation and lead to disordered tissue architecture, which could contribute to tumor progression. These findings highlight the necessity to further investigate in situ patterns of altered expression and sub-cellular localization of PAR6 and PAR6-binding partners in a progression series (stage, and histological grade) of breast cancer samples to gain further temporal insight into which cell polarity molecules play a role in triggering disease progression.
The potential involvement of PAR6 in protein complexes which regulate EMT also highlights the need for accurate regulation and subcellular localization of this molecule. Recent studies have shown an important association of the PAR6 polarity protein with TGFβRI, a key regulator of EMT [7,23,24]. Other EMT-inducing cell membrane receptors, such as EGFR, IGF1R and c-MET colocalize at AJs, have been shown to physically interact with E-cadherin, and bidirectional regulation between these two classes of molecules is important for EMT [43]. Of particular interest, the association of TIAM1 with PAR6 establishes a connection between the apico-basal polarity system and RAC signaling which has been demonstrated to be essential for the transforming effects of RAS [26]. Moreover, while the biochemistry and structural features of cell-cell junctional molecules have largely been characterized, further investigation of the role of extracellular signals (such as hormones, growth factors and cytokines, and the extracellular matrix) in the context of polarity component disequilibrium is needed to provide insight into possible mechanisms further contributing to neoplastic EMT. It will also be necessary to investigate the role that CDC42 GTPase (activated in association with PAR6) and other small GTPases (such as RHOA, which has an opposing action to CDC42 on the actin cytoskeleton, and RAC, linked to PAR6 through TIAM1) may play in these processes. Emerging literature describing unexpected new functional associations between oncogenes and dysregulated cell polarity genes highlights important new areas of investigation for potential targeted uncoupling of oncogene-mediated mechanisms in breast cancer (for example PAR6-aPKC mediated disruption of ERBB2-induced apoptosis) [27,28].
In summary, our in vitro data strongly supports a role for PAR6B as the primary PAR6 isoform regulating TJ formation in mammary epithelial cells. Our quantitative and qualitative IHC survey of normal tissues and breast tumors with a PAR6B-specific antibody establish strong epithelial specific expression for PAR6B. These results and the known involvement of other components of cell polarity pathways in cancer suggest that it may prove important to comprehensively investigate the role of components in polarity systems with respect to tumor progression. The complexity of the pathways impinged upon by these molecules suggests a number of mechanisms by which alterations in this system could deregulate growth factor signaling, cell adhesion, motility and invasion, as well as cellular organization during mitosis.
Acknowledgements
We thank Drs. Thomas Dennis and Sarah Anzick for their assistance with FISH protocols.
References
- 1.Ruoslahti E, Reed JC. Anchorage dependence, integrins, and apoptosis. Cell. 1994;77:477–478. doi: 10.1016/0092-8674(94)90209-7. [DOI] [PubMed] [Google Scholar]
- 2.Boudreau N, Sympson CJ, Werb Z, Bissell MJ. Suppression of ICE and apoptosis in mammary epithelial cells by extracellular matrix. Science. 1995;267:891–893. doi: 10.1126/science.7531366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slade MJ, Coope RC, Gomm JJ, Coombes RC. The human mammary gland basement membrane is integral to the polarity of luminal epithelial cells. Exp Cell Res. 1999;247:267–278. doi: 10.1006/excr.1998.4340. [DOI] [PubMed] [Google Scholar]
- 4.Birchmeier W. E-cadherin as a tumor (invasion) suppressor gene. Bioessays. 1995;17:97–99. doi: 10.1002/bies.950170203. [DOI] [PubMed] [Google Scholar]
- 5.Hoover KB, Liao SY, Bryant PJ. Loss of the tight junction MAGUK ZO-1 in breast cancer: relationship to glandular differentiation and loss of heterozygosity. Am J Pathol. 1998;153:1767–1773. doi: 10.1016/S0002-9440(10)65691-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kominsky SL, Argani P, Korz D, Evron E, Raman V, Garrett E, Rein A, Sauter G, Kallioniemi OP, Sukumar S. Loss of the tight junction protein claudin-7 correlates with histological grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast. Oncogene. 2003;22:2021–2033. doi: 10.1038/sj.onc.1206199. [DOI] [PubMed] [Google Scholar]
- 7.Viloria-Petit AM, David L, Jia JY, Erdemir T, Bane AL, Pinnaduwage D, Roncari L, Narimatsu L, Bose R, Moffat J, Wong JW, Kerbel RS, O'Malley FP, Andrulis IL, Wrana JL. A role for the TGFbeta-Par6 polarity pathway in breast cancer progression. Proc Natl Acad Sci USA. 2009;106:14028–14033. doi: 10.1073/pnas.0906796106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Itoh M, Bissell MJ. The organization of tight junctions in epithelia: implications for mammary gland biology and breast tumorigenesis. J Mammary Gland Biol Neoplasia. 2003;8:449–462. doi: 10.1023/B:JOMG.0000017431.45314.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kay AJ, Hunter CP. CDC-42 regulates PAR protein localization and function to control cellular and embryonic polarity in C. elegans. Curr Biol. 2001;11:474–481. doi: 10.1016/s0960-9822(01)00141-5. [DOI] [PubMed] [Google Scholar]
- 10.Gotta M, Abraham MC, Ahringer J. CDC-42 controls early cell polarity and spindle orientation in C. elegans. Curr Biol. 2001;11:482–488. doi: 10.1016/s0960-9822(01)00142-7. [DOI] [PubMed] [Google Scholar]
- 11.Joberty G, Petersen C, Gao L, Macara IG. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol. 2000;2:531–539. doi: 10.1038/35019573. [DOI] [PubMed] [Google Scholar]
- 12.Gao L, Joberty G, Macara IG. Assembly of epithelial tight junctions is negatively regulated by Par6. Curr Biol. 2002;12:221–225. doi: 10.1016/s0960-9822(01)00663-7. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki A, Yamanaka T, Hirose T, Manabe N, Mizuno K, Shimizu M, Akimoto K, Izumi Y, Ohnishi T, Ohno S. Atypical protein kinase C is involved in the evolutionarily conserved par protein complex and plays a critical role in establishing epithelia-specific junctional structures. J Cell Biol. 2001;152:1183–1196. doi: 10.1083/jcb.152.6.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohno S. Intercellular junctions and cellular polarity: the PAR-aPKC complex, a conserved core cassette playing fundamental roles in cell polarity. Curr Opin Cell Biol. 2001;13:641–648. doi: 10.1016/s0955-0674(00)00264-7. [DOI] [PubMed] [Google Scholar]
- 15.Henrique D, Schweisguth F. Cell polarity: the ups and downs of the Par6/aPKC complex. Curr Opin Genet Dev. 2003;13:341–350. doi: 10.1016/s0959-437x(03)00077-7. [DOI] [PubMed] [Google Scholar]
- 16.Muller HA, Wieschaus E. Armadillo, bazooka, and stardust are critical for early stages in formation of the zonula adherens and maintenance of the polarized blastoderm epithelium in Drosophila. J Cell Biol. 1996;134:149–163. doi: 10.1083/jcb.134.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hurd TW, Gao L, Roh MH, Macara IG, Margolis B. Direct interaction of two polarity complexes implicated in epithelial tight junction assembly. Nat Cell Biol. 2003;5:137–142. doi: 10.1038/ncb923. [DOI] [PubMed] [Google Scholar]
- 18.Nam SC, Choi KW. Interaction of Par-6 and Crumbs complexes is essential for photoreceptor morphogenesis in Drosophila. Development. 2003;130:4363–4372. doi: 10.1242/dev.00648. [DOI] [PubMed] [Google Scholar]
- 19.Plant PJ, Fawcett JP, Lin DC, Holdorf AD, Binns K, Kulkarni S, Pawson T. A polarity complex of mPar-6 and atypical PKC binds, phosphorylates and regulates mammalian Lgl. Nat Cell Biol. 2003;5:301–308. doi: 10.1038/ncb948. [DOI] [PubMed] [Google Scholar]
- 20.Betschinger J, Mechtler K, Knoblich JA. The Par complex directs asymmetric cell division by phosphorylating the cytoskeletal protein Lgl. Nature. 2003;422:326–330. doi: 10.1038/nature01486. [DOI] [PubMed] [Google Scholar]
- 21.Noda Y, Takeya R, Ohno S, Naito S, Ito T, Sumimoto H. Human homologues of the Caenorhabditis elegans cell polarity protein PAR6 as an adaptor that links the small GTPases Rac and Cdc42 to atypical protein kinase C. Genes Cells. 2001;6:107–119. doi: 10.1046/j.1365-2443.2001.00404.x. [DOI] [PubMed] [Google Scholar]
- 22.Bose R, Wrana JL. Regulation of Par6 by extracellular signals. Curr Opin Cell Biol. 2006;18:206–212. doi: 10.1016/j.ceb.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 23.Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science. 2005;307:1603–1609. doi: 10.1126/science.1105718. [DOI] [PubMed] [Google Scholar]
- 24.Barrios-Rodiles M, Brown KR, Ozdamar B, Bose R, Liu Z, Donovan RS, Shinjo F, Liu Y, Dembowy J, Taylor IW, Luga V, Przulj N, Robinson M, Suzuki H, Hayashizaki Y, Jurisica I, Wrana JL. High-throughput mapping of a dynamic signaling network in mammalian cells. Science. 2005;307:1621–1625. doi: 10.1126/science.1105776. [DOI] [PubMed] [Google Scholar]
- 25.Cai Y, Stafford LJ, Bryan BA, Mitchell D, Liu M. G-protein-activated phospholipase C-beta, new partners for cell polarity proteins Par3 and Par6. Oncogene. 2005;24:4293–4300. doi: 10.1038/sj.onc.1208593. [DOI] [PubMed] [Google Scholar]
- 26.Mertens AE, Pegtel DM, Collard JG. Tiam1 takes PARt in cell polarity. Trends Cell Biol. 2006;16:308–316. doi: 10.1016/j.tcb.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 27.Aranda V, Haire T, Nolan ME, Calarco JP, Rosenberg AZ, Fawcett JP, Pawson T, Muthuswamy SK. Par6-aPKC uncouples ErbB2 induced disruption of polarized epithelial organization from proliferation control. Nat Cell Biol. 2006;8:1235–1245. doi: 10.1038/ncb1485. [DOI] [PubMed] [Google Scholar]
- 28.Aranda V, Nolan ME, Muthuswamy SK. Par complex in cancer: a regulator of normal cell polarity joins the dark side. Oncogene. 2008;27:6878–6887. doi: 10.1038/onc.2008.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y, Dairkee SH, Ljung BM, Gray JW, Albertson DG. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- 30.Volik S, Zhao S, Chin K, Brebner JH, Herndon DR, Tao Q, Kowbel D, Huang G, Lapuk A, Kuo WL, Magrane G, De Jong P, Gray JW, Collins C. End-sequence profiling: sequence-based analysis of aberrant genomes. Proc Natl Acad Sci USA. 2003;100:7696–7701. doi: 10.1073/pnas.1232418100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pollack JR, Sorlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Borresen-Dale AL, Brown PO. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci USA. 2002;99:12963–12968. doi: 10.1073/pnas.162471999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barlund M, Monni O, Weaver JD, Kauraniemi P, Sauter G, Heiskanen M, Kallioniemi OP, Kallioniemi A. Cloning of BCAS3 (17q23) and BCAS4 (20q13) genes that undergo amplification, overexpression, and fusion in breast cancer. Genes Chromosomes Cancer. 2002;35:311–317. doi: 10.1002/gcc.10121. [DOI] [PubMed] [Google Scholar]
- 33.Nolan ME, Aranda V, Lee S, Lakshmi B, Basu S, Allred DC, Muthuswamy SK. The polarity protein Par6 induces cell proliferation and is overexpressed in breast cancer. Cancer Res. 2008;68:8201–8209. doi: 10.1158/0008-5472.CAN-07-6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caplen NJ, Parrish S, Imani F, Fire A, Morgan RA. Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc Natl Acad Sci USA. 2001;98:9742–9747. doi: 10.1073/pnas.171251798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cunliffe HE, Ringner M, Bilke S, Walker RL, Cheung JM, Chen Y, Meltzer PS. The gene expression response of breast cancer to growth regulators: patterns and correlation with tumor expression profiles. Cancer Res. 2003;63:7158–7166. [PubMed] [Google Scholar]
- 36.Baird K, Davis S, Antonescu CR, Harper UL, Walker RL, Chen Y, Glatfelter AA, Duray PH, Meltzer PS. Gene expression profiling of human sarcomas: insights into sarcoma biology. Cancer Res. 2005;65:9226–9235. doi: 10.1158/0008-5472.CAN-05-1699. [DOI] [PubMed] [Google Scholar]
- 37.Gonzalez AL, Roberts RL, Massion PP, Olson SJ, Shyr Y, Shappell SB. 15-Lipoxygenase-2 expression in benign and neoplastic lung: an immunohistochemical study and correlation with tumor grade and proliferation. Hum Pathol. 2004;35:840–849. doi: 10.1016/j.humpath.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 38.Hampton OA, Den Hollander D, Miller CA, Delgado DA, Li J, Coarfa C, Harris RA, Richards S, Scherer SE, Muzny DM, Gibbs RA, Lee AV, Milosavljevic A. A sequence-level map of chromosomal breakpoints in the MCF-7 breast cancer cell line yields insights into the evolution of a cancer genome. Genome Res. 2009;19:167–177. doi: 10.1101/gr.080259.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen X, Macara IG. Par-3 controls tight junction assembly through the Rac exchange factor Tiam1. Nat Cell Biol. 2005;7:262–269. doi: 10.1038/ncb1226. [DOI] [PubMed] [Google Scholar]
- 40.Yue C, Ku CY, Liu M, Simon MI, Sanborn BM. Molecular mechanism of the inhibition of phospholipase C beta 3 by protein kinase C. J Biol Chem. 2000;275:30220–30225. doi: 10.1074/jbc.M004276200. [DOI] [PubMed] [Google Scholar]
- 41.Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, Sauter G, Kallioniemi OP, Trent JM, Meltzer PS. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–968. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- 42.Labhart P, Karmakar S, Salicru EM, Egan BS, Alexiadis V, O'Malley BW, Smith CL. Identification of target genes in breast cancer cells directly regulated by the SRC-3/AIB1 coactivator. Proc Natl Acad Sci USA. 2005;102:1339–1344. doi: 10.1073/pnas.0409578102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. Embo J. 2004;23:1739–1748. doi: 10.1038/sj.emboj.7600136. [DOI] [PMC free article] [PubMed] [Google Scholar]