

Abstract
Accumulated evidence suggests that thyroid hormone receptor β (TRβ) could function as a tumor suppressor, but the detailed mechanisms by which TRβ inhibits tumorigenesis are not fully understood. The present studies explored the mechanisms by which TRβ acted to inhibit thyroid tumor development mediated by simian virus-40 (SV40). In mouse xenograft models, SV40 large T antigen (SV40Tag)-immortalized human thyroid epithelial (HTori) cells rapidly induced tumors, but the tumor development was totally blocked by TRβ stably expressed in HTori cells. Previous studies showed that the SV40Tag oncoprotein binds to and inactivates tumor suppressors p53 and retinoblastoma protein (Rb), thereby inducing tumorigenesis. Here we showed that one of the mechanisms by which TRβ suppressed tumor development was by competing with p53 and Rb for binding to SV40Tag. The interaction of TRβ with SV40Tag led to reactivation of Rb to inhibit cell cycle progression. TRβ- SV40Tag interaction also resulted in reactivating p53 to increase the expression of Pten, thus attenuating PI3K-AKT signaling to decrease cell proliferation and to induce apoptosis. The present study uncovered a novel action of TRβ as a tumor suppressor initiated via interfering with the recruitment of Rb and p53 by SV40Tag oncoprotein through protein-protein interaction, thereby acting to block tumor development.
Keywords: Thyroid hormone receptor, tumor suppressor, tumorigenesis, thyroid hormone, xenograft models
Introduction
Thyroid hormone receptors are ligand-dependent transcription factors that mediate the biological activities of the thyroid hormone (T3) in development, growth, differentiation, and metabolism. Two human TR genes, THRA and THRB, located on different chromosomes, encode thyroid hormone (T3) binding TR isoforms (TRα1, β1, β2, and β3). These TR isoforms share extensive sequence homology in the DNA and ligand-binding domains, but differ in the length and amino acid sequence at the amino terminal A/B domain. These TR isoforms are expressed in a tissue- and development-dependent manner. Besides sharing common functions, TRβ and TRα also mediate isoform-dependent actions [1]. In the past decades, significant progress has been made in understanding the molecular mechanisms by which TR functions to maintain normal physiological T3-mediated homeostasis. However, the roles of TR in human cancers are less well understood.
Early evidence to suggest that mutated TR could be involved in carcinogenesis came from the discovery that TRα1 is the cellular counterpart of the retroviral v-ERBA oncoprotein involved in the neoplastic transformation leading to acute erythroleukemia and sarcomas [2,3]. v-ERBA oncoprotein is a highly mutated chicken TRα1 that does not bind T3 and loses the ability to activate gene transcription. That mutated TRs could be involved in human cancers was supported by the findings that somatic mutations of TRs have been found in human hepatocellular carcinoma [4], renal clear cell carcinoma [5,6], breast cancer [7], pituitary tumor [8,9], and thyroid cancer [10]. Many of these TR mutants have lost T3 binding activity and transcription capacity, and some exhibit dominantly negative activity [5,10].
The proposal that the loss of TR functions could be involved in the development of human cancers gained further support by association studies. Loss in the expression of the THRB gene because of the truncation/deletion of chromosome 3p where the gene is located was reported in many malignancies including lung, melanoma, breast, head and neck, renal cell, uterine cervical, ovarian, testicular and gastrointestinal tumors [11-16]. Moreover, decreased expression due to silencing of the THRB gene by promoter hypermethylation has been found in human cancers including breast, lung, and thyroid carcinoma [17-20]. These association studies raised the possibility that TRs could function as tumor suppressors in human cancers. This possibility has gained additional support from recent studies showing that the loss of normal TRβ functions by mutations leads to spontaneous development of follicular thyroid cancer [21,22]. However, the molecular mechanisms by which TRβ acts to suppress tumor development and progression are largely unknown.
In the present study, we aimed to understand how TRβ could act as a tumor suppressor in the carcinogenesis of human thyroid epithelial cells mediated by Simian virus-40 (designated as HTori cells)[23]. HTori cells were derived from transfection of human primary thyroid follicular epithelial cells with a plasmid containing an origin-defective SV40 genome (SVori-)[23]. Sequences of the SV40 virus have been found in human tumors such as pleural mesotheliomas, ependymomas, choroid plexus tumors, and other brain tumors [24-27]. Particularly, evidence has been presented to show the association of SV40 virus with human papillary thyroid carcinomas [28-30]. Moreover, transgenic mice in which the expression of SV40Tag was driven by the thyroglobulin gene promoter developed thyroid hyperplasia and adenocarcinomas [31]. These observations suggested that SV40 virus could be involved in the development of thyroid cancer. Thus, HTori cells presented an opportunity to elucidate the role of TRβ in thyroid carcinogenesis.
We therefore stably expressed TRβ in HTori cells and assessed how TRβ affected SV40-Tag induced thyroid tumorigenesis. We found that TRβ suppressed tumorigenesis by physical interaction with SV40Tag, leading to inactivation of the oncogenic actions of SV40Tag by blocking its recruitment of the retinoblastoma protein (Rb) and p53 tumor suppressors. The re-actavited Rb and p53 decreased cell proliferation and activated apoptosis, thereby inhibiting tumorigenesis. The present study uncovered a novel action of TRβ as a tumor suppressor via interfering with the recruitment of Rb and p53 by SV40Tag oncoprotein.
Materials and methods
Xenograft mouse model
Athymic mice (AthymicNCr-nu/nu; 4-weeks old) were obtained from the NCI-Frederick Animal facility. HTori cells were a generous gift from Yuri Nikiforov of University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania. Establishment of HTori cells stably expressing either TRβ or the control gene (Neo) was described previously [32]. HTori-Neo or HTori-TRβ cells (5X106 cells) in 200 μl of suspension mixture were injected subcutaneously into the right flank of athymic mice. Tumors were measured weekly and the end point was when the tumors reached 3 cm in diameter.
Western analysis and co-immunoprecipitation assay
Preparation of total cell lysate from HTori-Neo and HTori-TRβ and Western blot analysis were carried out as described previously [33]. Anti-TRβ antibodies used were: C4 (2 μg/ml)[34], C91 (3μg/ml)[35] and J53 (1:300 dilution)[36]. Anti-p53 (1:1000 dilution, sc-6243, Santa Cruz, CA.), anti-Rb (1:1000 dilution, sc-50, Santa Cruz, CA.), anti-cyclin E1 (1:1000 dilution, sc-481, Santa Cruz, CA.), anti-PARP (1:1000 dilution, sc-7150, Santa Cruz, CA), and anti-Caspase3 (1:1000 dilution, sc-2118, Santa Cruz, CA), anti-BAX (1:1000 dilution, sc-7480, Santa Cruz, CA), anti-CDK4 (1:1000 dilution, sc-260, Santa Cruz, CA), anti-CDK6 (1:1000 dilution, sc-177, Santa Cruz, CA), anti-cyclin A (1:1000 dilution, RB007P0, Thermo Scientific, PA), anti-pRb (Ser807/811)( 1:1000 dilution, #9271, Cell Signaling, MA.), anti-AKT (1:1000 dilution, #9272, Cell Signaling, MA.), anti-pAKT (1:1000 dilution, #9271, Cell Signaling, MA.), anti-PTEN (1:1000 dilution, #9552, Cell Signaling, MA.), anti-pBad (Ser136)(1:500 dilution, #9295, Cell Signaling, MA.) and anti-GAPDH (1:1000 dilution, #2118, Cell Signaling, MA.) were used to detect endogenous expression levels of proteins. For the co-immunoprecipitation assay, HTori-Neo and HTori-TRβ cells were grown in 60mm dish in culture medium (RPMI1640 supplemented with 10% FBS) for 24hr. Nuclear extracts (200 μg) prepared as described previously [37] were immunoprecipitated with anti-SV40Tag antibody [PAb416] (2 μg/ml, ab16879, Abcam, MA), followed by Western analysis using anti-TRβ antibodies J53, anti-p53 (1:1000 dilution, sc-6243, Santa Cruz, CA), or anti-Rb (1:1000 dilution, sc-50, Santa Cruz, CA.). Total tumor cell lysates (500 μg) were prepared described previously [38] for co-immunoprecipitation assays similarly. For the negative control, anti-MOPC (2 μg/ml, Sigma Inc, MOPC141) was used. Band intensities were quantified by using ImageJ software (NIH, Bethesda, MD).
Reporter assays
Reporter assays were carried out as described previously [37]. In brief, 0.2 μg/well Pal-luciferase reporter plasmids [39] were transfected with Lipofectamine 2000 (Invitrogen, CA) according to manufacturer’s instructions. luciferase activity was measured using Victor 3 (PerkinElmer Life and Analytical Sciences, Waltham, MA). Luciferase values were normalized to protein concentration.
Cell cycle analysis and propidium iodide staining
HTori-Neo and HTori-TRβ cells were seeded in 150-mm dishes at a density of 3X106 in culture medium. After 24-hour serum starvation, the medium was replaced with 5% FBS for an addition 8 hours. Cells were next washed with PBS, an aliquot of 1X106 cells were centrifuged and re-suspended in 1 mL of ice-cold 70% ethanol. After fixation, cells were incubated with 100 μL of RNAs (100 units) at 37 °C for 20 minutes, followed by staining with propidium iodide solution (50 μg/ml) for 30 minutes. Cell clumps were filtered through 50-micro nylon meshes for cell analysis using the FACSCalibur flow cytometer (BD, MA). To detect late apoptotic cells via a microscope as described previously [40], cells were seeded in 12-well dishes at a density of 8X104 in the culture medium. After 24-hour culture, cells were washed with PBS and stained with 10 μg/ml propidium iodide for 20 minutes at room temperature. Fluorescent images were scanned via a fluorescence microscopy using an X20 objective lens (Olympus).
RNA extraction and real-time polymerase chain reaction (PCR)
HTori-Neo and HTori-TRβ cells were grown in 60-mm dishes at a density of 5X105 in culture medium for 24 hours. Total RNA was prepared using TRIzol (Invitrogen, CA) solution according to the manufacturer’s protocol. Real-time RT-PCR was carried out as described previously [32]. The primers used are as follows: for human PTEN, forward: 5’-ACA CCG CCA AAT TTA ACT GC-3’, reverse: 5’-TAC ACC AGT CCG TCC CTT TC-3’; for human BAX, forward: 5’-TTT GCT TCA GGG TTT CAT CC- 3’, reverse: 5’-CAG TTG AAG TTG CCG TCA GA- 3’.
Mapping the interaction region of TRβ with SV40Tag
The mammalian expression vectors for the truncated forms of TRβ1 were described previously [41]. The expression vector for each truncated mutant of TRβ1 (2 μg) was transfected into HTori-Neo cells using Lipofectamin 2000 according to manufacturer’s protocols. After 24hr incubation, cell lysates were prepared and 500 μg of total proteins were immunoprecipitated with anti-SV40Tag antibody, followed by Western blot analysis using anti-TRβ1 (C91), anti-p53 or anti-Rb antibodies.
Immunohistochemical analysis
Immunohistochemical analysis of the expression of cellular key regulators in tumorigenesis was carried out as described by Lu et al [42]. Anti-phospho-AKT (Ser473) (D9E) XP rabbit mAb in 1:50 dilution (Cat #4060 Cell Signaling); cleaved caspase 3 (Asp175) in 1:300 dilution (Cat# 9661 Cell Signaling); p-Rb (ser807/811; in 1:300 dilution) (Cat.# 9308S Cell Signaling); Ki-67 in 1:300 dilution (Cat# RB-9043-PO Thermo) were used as primary antibodies. The quantification of positive cells was performed on digital images captured under × 200 magnification of each tissue section. Two randomly selected slides from each staining group were analyzed. For each slide, ratios of positive cells to total cells from 10–12 bright fields under × 200 amplification were calculated.
Confocal fluorescence microscopy
Subcellular localization of TRβ and SV40Tag was visualized by confocal fluorescence microscopy. HTori-Neo and HTori-TRβ cells were seeded in a Chambered Coverglass (Thermo scientific, NY, #155380) at a density of 4 X 104 cells/well in culture medium for 24 hrs. All subsequent steps were carried out at room temperature. Cells were washed twice with 1X PBS, fixed with 4% paraformaldehyde for 10 min, and permeabilized with 0.5% Triton X-100 in PBS for 10 min. Nonspecific binding of the antibodies was blocked with 3% BSA before incubation with the anti-TRβ (2 μg/ml, C4) and anti-SV40Tag (2 μg /ml, sc-20800, Santa Cruz, CA) for 1 hr. The cells were subsequently incubated with Alexa Fluor 488 fragment of goat anti-mouse IgG (1 μg/ml, Invitrogen, A11017) or tetrametylrhodamine goat anti-rabbit IgG (1 μg/ml, Invitrogen, T2769) for 1hr. Nuclei were also stained with Hoechst 33342 (5 μg /ml, Sigma, B2261) for 10 min at room temperature. Laser confocal scanning images were captured with an ultraview confocal head on a TV200 inverted microscope (Zeiss, Thornwood, NY).
Statistical analysis
All data are expressed as mean ± the standard error of the mean (SEM). Significant differences between groups were calculated using Student's t-test with the use of GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA). P< 0.05 is considered statistically significant.
Result
Thyroid hormone receptor β blocks SV40Tag-induced tumor development induced in xenograft models
We stably expressed TRβ in HTori cells to examine the impact of TRβ in tumor development. Figure 1A-a shows the expressed protein abundance of TRβ in two representative clone lines, designated as TRβ#1 (lane 2) and TRβ#2 (lane 3), as compared with control cells in which only the Neomycin gene was expressed in HTori cells (designated as HTori-Neo cells; lane 1). That the expressed TRβ was functional was demonstrated by the transcription assay using a reporter system. As shown in Figure 1A-b, no T3-dependent transcriptional activity was detected in the control Neo cells. In contrast, 12- and 13-fold increases of T3-dependent transcriptional activation were observed in TRβ#1 and TRβ#2, respectively.
Figure 1.

TRβ inhibits tumor development in mouse xenograft models. (A-a) Protein abundance of TRβ in two representative clones of HTori-Neo and HTori-TRβ by Western blot analysis using anti-TRβ specific antibody, C4. (A-b) Transcriptional activity of TRβ as determined using Pal-luciferase reporter plasmid harboring the thyroid hormone response element (Pal-TRE) in the presence or absence of T3 (100 nM). Reporter activities were normalized to total protein concentrations. (B) Cloned HTori-Neo or HTori-TRβ cells (#1 and #2) (each 5X106 cells) were injected into the right flank of athymic nude mice. Tumor size was measured every week. Data are the mean ± SEM; n = 7). (C) Tumors derived from HTori-Neo cells (panel a) and the very small growth (“bump’) derived from HTori-TRβ cells (panel b) were fixed, and the slides were prepared and stained by H & E as described in Materials and Methods. Panel a shows dedifferentiated cells with the arrow pointing to abnormally enlarged nuclei that are highly pleomorphic and closely packed. Panel b shows a morphology distinct from that shown in panel a, with the arrow pointing to normal nuclei. These cells show prominent myxoid (resembling mucus, marked as “m”) differentiation. (D) Nuclear Ki-67 staining of tumor cells derived from HTori-Neo cells (panel a) and cells derived from HTori-TRβ (panel b). Arrows point to the positive nuclear staining of Ki-67. The positively Ki-67 stained cells are counted and graphed (panel c). The difference in the number of positively stained cells between tumors cells and cells derived from the HTori-TRβ cell-induced growth is highly significant (p<0.01).
HTori-Neo cells were inoculated subcutaneously into athymic mice. Tumors were visible within 2-3 weeks (Figure 1B, solid line). Remarkably, only very small “bumps” at the sites of inoculation were visible from the inoculated TRβ#1 and TRβ#2 cells after 5 weeks, at which time the size of tumors developed from inoculation of HTori-Neo cells was sufficiently large to meet the end point of the study (indicated by broken line). Histological analysis of the HTori-Neo cells-induced tumors by H & E staining shows malignant morphology with loss of differentiation (panel a, Figure 1C). The arrows point to abnormally enlarged nuclei that are highly pleomorphic and closely packed. Histological analysis of the small growth (a small “bump”) at the sites of injection of TRβ cells shows a morphology distinct from that seen in panel a. The arrows point to normal nuclei (panel b, Figure 1C). These cells show prominent myxoid (resembling mucus) differentiation. These differentiated cells were suspended in a myxoid/mik-soid matrix (m) in the majority of tissue present. That rapid growth of HTori-Neo cells-induced tumors was evident by the strong Ki-67 nuclear staining (Figure 1D-a; marked by arrows). In contrast, fewer cells with Ki-67 nuclear staining were detected (Figure 1D-b). Ki-67 nuclear stained cells were counted and the data are shown in Figure 1D-c. There was a 3-fold increase in Ki-67 stained cells in Neo cells-induced tumors, indicating a marked increase in cell proliferation. Taken together, these results clearly indicated that TRβ could function to inhibit SV40Taginduced tumor cell proliferation in vivo.
Thyroid hormone receptor β physically interacts with SV40Tag
To be certain that SV40Tag also associated with p53 and Rb in tumors derived from HTori-Neo cells, we carried out co-immunoprecipitation assays of tumor extracts. Indeed, in extracts of tumors dissected from athymic mice, with the use of anti-SV40Tag antibodies followed by Western blotting, we detected a specific association of SV40Tag with p53 (lane 3, upper panel, Figure 2A) and with Rb (lane 3, lower panel), but when we used a control antibody (MOPC), no signals were detected (lane 2, upper and lower panels, Figure 2A). These results showed that, consistent with current understanding of the molecular basis of SV40Tag-induced tumorigenesis [43], SV40Tag was associated with p53 and Rb in HTori-Neo cells-induced tumors.
Figure 2.
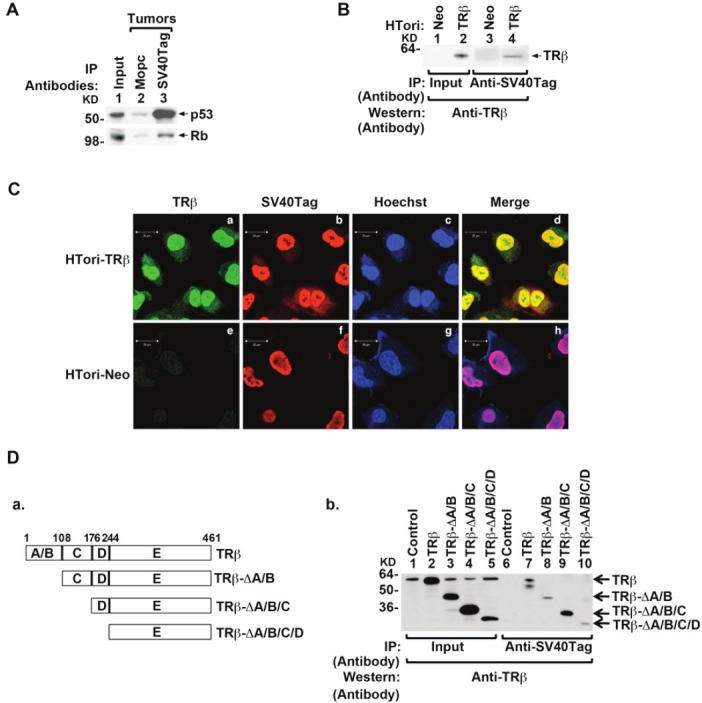
Physical interaction of TRβ with SV40Tag in the nucleus. (A) Association of p53 (upper panel) and Rb (lower panel) with SV40Tag in tumors derived from HTori-Neo cells in athymic mice. Tumor extracts were prepared and immunoprecipitated with anti-SV40Tag antibodies, followed by Western blotting with anti-p53 (upper panel) or anti-Rb (lower panel) as described in Materials and Methods. (B) The physical interaction of TRβ with SV40Tag in HTori-TRβ cells. Nuclear extracts were prepared from HTori-Neo cells (lanes 1 & 3) and from HTori-TRβ cells (lanes 2 & 4). The nuclear extracts were first immunoprecipitated with anti-SV40Tag antibodies, followed by Western blotting with the anti-TRβ antibody J53 (lanes 3 & 4). Lanes 1 and 2 show that respective input amount (3%). (C) TRβ is colocalized with SV40Tag in the nucleus. HTori-Neo and HTori-TRβ cells were plated in chamber slide and cells were cultured for 24 hr before fixation as described in Materials and Methods. Cells were incubated with anti-TRβ1 (C4, 2 μg/ml) (a and e) and anti-SV40Tag antibody (2 μg/ml)(b and f) followed by secondary antibody conjugated with Alexa Fluor 488 (green) or tetramethyrhodamine (red), respectively. Nuclei were stained with Hoechst 33342 (5 μg/ml) as described in Materials and Methods. (D) Identification of ligand binding domain of TRβ as a binding site with SV40Tag. (D-a) Schematic representation of the serial deletion truncated mutants of TRβ. (D-b) Serial deletion truncated mutants of TRβ were transiently transfected into HTori-Neo cells. Total lysates were prepared and immunoprecipitated with anti-SV40Tag antibody, followed by anti-TRβ antibody (C91) as described in Materials and Methods. Lanes 1-5 were the input amount (4%).
To understand how TRβ acted to suppress SV40Tag tumorigenesis, we hypothesized that TRβ could interact with SV40Tag and thereby reactivating the tumor suppressor actions of Rb and p53. Because the tumor growth induced from the injected HTori-TRβ cells was an inadequate size, we could not test this hypothesis by using tumors from athymic mice. We therefore first ascertained this possibility by co-immunoprecipitation assays using HTori-Neo and HTori-TRβ cells (Figure 2B). Co-immunoprecipitation of nuclear extracts (immunoprecipitated first by anti-SV40Tag followed by Western blot with anti-TRβ antibodies) shows that TRβ physically interacted with SV40Tag (lane 4). Because there was no TRβ in HTori-Neo cells (lane 1), and so no signals were detected (lane 3; Figure 2B). The interaction of TRβ with SV40Tag in the nuclear compartment was further demonstrated by confocal microscopy. Figure 2C-a shows that TRβ was predominantly localized in the nucleus with some distribution in the cytoplasm; while SV40Tag was totally in the nucleus (panel b). That the interaction of TRβ with SV40Tag occurred in the nucleus was evident by the merged yellow images (Figure 2C-d). In HTori-Neo cells in which no TRβ, only background green fluorescence was detected (panel e). Panel h shows the merged images of SV40Tag with nuclear Hoechst staining (blue). These confocal fluorescence findings further support that interaction of TRβ with SV40Tag in the nucleus.
The region of TRβ that interacted with SV40Tag was mapped by using serial deletion of functional domains of TRβ. HTori-Neo cells were transfected with the expression plasmids of intact or truncated TRβ (Figure 2D-a). Coimmunoprecipitation assays show that SV40Tag interacted with the intact TRβ (lane 7, Figure 2D-b), TRβ-rA/B (lane 8), TRβ-rA/B/C (lane 9) and TRβ-rA/B/C/D (lane 10). These data indicate that the interaction region was located in amino acids 244 – 461 of TRβ.
The effect of the interaction of TRβ with SV40Tag on the SV40Tag/Rb complex was evident, as shown in lane 4 of Figure 3A-I, in that the association of Rb with SV40Tag was decreased by the presence of TRβ (compare lane 4 with lane 3). Quantitation of the intensities of the bands indicated a reduction of 86% in association of Rb with SV40Tag (Figure 3A-II). Similarly, the association of SV40Tag with p53 was decreased by the presence of TRβ in HTori-TRβ cells (compare lane 4 with lane 3, Figure 3B-I). Quantitation of the intensities of the bands indicated a reduction of 75% in association of p53 with SV40Tag (Figure 3B-II). These results indicate that interaction of TRβ with SV40Tag disrupted the recruitment of these two important tumor suppressors by SV40Tag.
Figure 3.
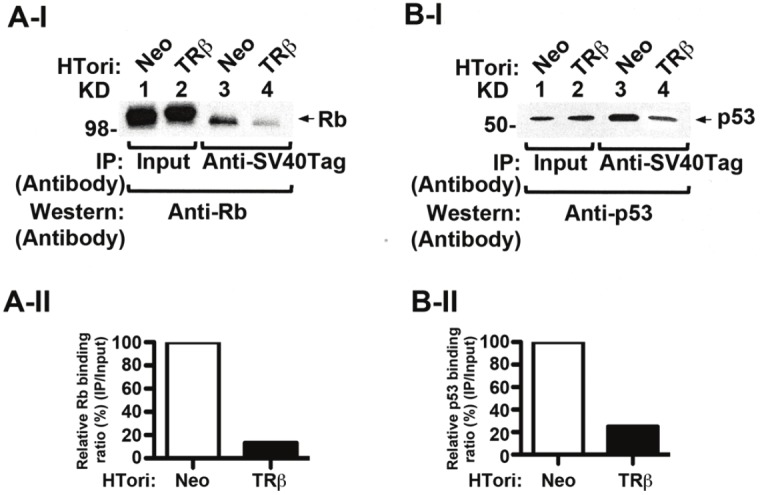
Disruption of SV40Tag-Rb and SV40Tag-p53 complexes by the physical interaction of TRβ with SV40Tag. (AI) Decreased association of SV40Tag with Rb in HTori-TRβ cells. Nuclear extracts were prepared from HTori-Neo cells (lanes 1 & 3) and from HTori-TRβ cells (lanes 2 & 4). The nuclear extracts were first immunoprecipitated with anti-SV40Tag antibodies, followed by Western blotting with the anti-Rb antibody. Lane 4 shows that the interaction of SV40Tag with Rb was lower in HTori-TRβ cells (lane 4) than in HTori-Neo cells (lane 3). Lanes 1 and 2 show respective input amount by direct Western blot analysis. (A-II). Quantification of relative binding intensity of Rb to SV40Tag in HTori-Neo and HTori-TRβ cells. Binding intensities were normalized to that of input. (B-I). Decreased association of SV40Tag with p53 in HTori-TRβ cells. Co-immunoprecipitation was carried out as described in (A), but with the use of anti-p53 antibodies in the Western blot analysis. Lane 4 shows that the interaction of SV40Tag with p53 was lower in HTori-TRβ cells (lane 4) than in HTori-Neo cell (lane3). Lanes 1 and 2 show respective input amount (4%) by direct Western blot analysis. (B-II) Quantification of relative binding intensity of p53 to SV40Tag in HTori-Neo and HTori-TRβ cells. Binding intensities were normalized to the input.
TRβ reactivates Rb functions by impeding cell cycle progression
That the interaction of TRβ with SV40Tag led to the disruption of association of SV40Tag with Rb and p53 prompted us to probe whether the tumor suppressor functions of these two tumor suppressors were reactivated. Rb, in its hypophosphorylated state, is known to induce cell cycle arrest by blocking cells entering from the G1 phase to the S phase [44]. We therefore first evaluated the phosphorylation status of Rb. Indeed, we found a lower abundance of phosphorylated-Rb (S807/811) in HTori-TRβ cells (lane 2, Figure 4A-I-a) than in HTori-Neo cells (lane 1 Figure 4A-I-a). No apparent changes of total Rb were visible between HTori-Neo and HTori-TRβ cells (panel b). Consistent with the decreased phosphorylated Rb, cyclin E1 and cyclin A were markedly decreased in HTori-TRβ cells (lane 2, panels c and d, Figure 4A-I). The protein abundance of Cdk 6 was also decreased in HTori-TRβ cells (lane 2, Figure 4A-I-e). However, no changes were detected for Cdk 4 in HTori-TRβ cells (lane 2, Figure 4A-I-f). The band intensities of each protein were determined and relative expression ratios (HTori-TRβ cells/HTori-Neo cells) were shown in Figure 4A-II.
Figure 4.
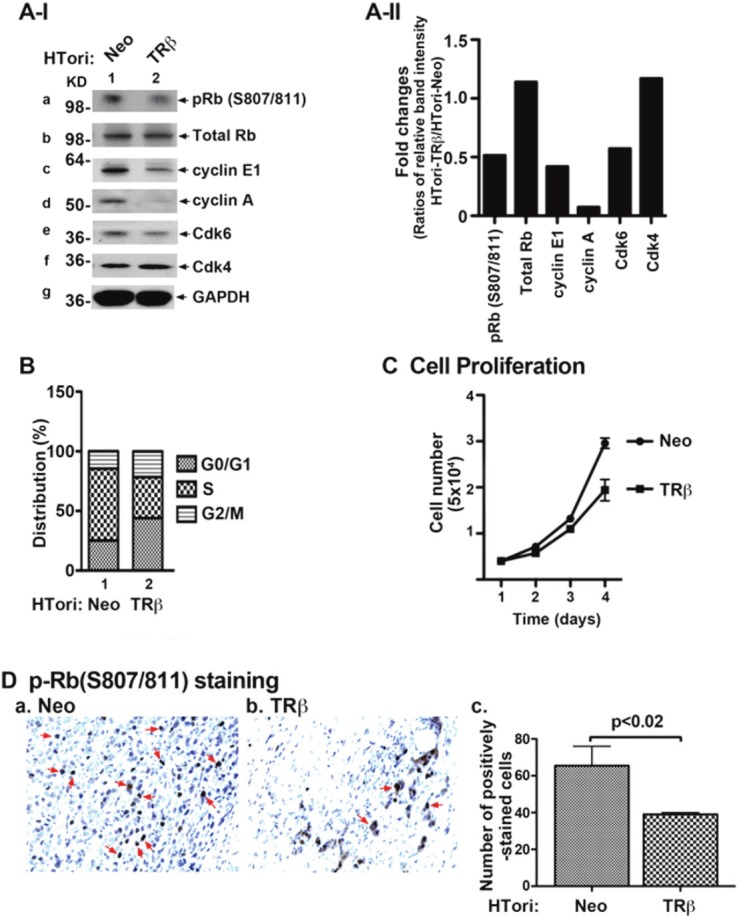
Reactivation of Rb by physical interaction of TRβ with SV40Tag. (A-I) HTori-Neo and HTori-TRβ cells were grown in 60mm dish for 24 hours and the cell extracts were prepared, followed by Western blot analyses (20 μg cell lysates were used) for key regulators of cell cycle progression (as marked), as described in Materials and Methods. The reactivation of Rb was indicated by decreased phosphorylation (panel a), cyclin E1 (panel c), cyclin A (panel d), and Cdk 6 (panel e). (A-II) Fold of changes in the protein abundance of key regulators in HTori-TRβ cells as compared with those in HTori-Neo cells. Each protein level was normalized to GAPDH. (B) Cell cycle distribution was determined in HTori-Neo cells (bars 1) and HTori-TRβ cells (bars 2), as described Materials and Methods. Delayed entries of cells from the G1 to the S phase were observed in cells expressing TRβ. (C) Proliferation of HTori-TRβ cells was less than that of HTori-Neo cells. Data are the mean ± SEM; n=3. (D) pRb (S807/811) staining of tumor cells derived from HTori-Neo cells (panel a) and cells derived from HTori-TRβ (panel b). Arrows point to the positive staining of pRb (S807/811). The positively pRb (S807/811) stained cells were counted and graphed (panel c). The difference in the number of positively stained cells is highly significant (p<0.02).
The functional consequences of changes in the expression levels of key cell cycle regulators shown in Figure 4A-II were assessed by cell cycle analysis. As shown in Figure 4B, a delay in the cell entry from the G0/G1 phase to the S phase was observed (Figure 4B, compare bar 1 to bar 2, 28% to 41%). Consistently, the delay in cell cycle progression led to decreases in proliferation of HTori-TRβ cells (Figure 4C).
That Rb was re-activated by decreased extent of phosphorylation at S807/811 in Neo-cellsinduced tumors was further confirmed by immunohistochemical analysis (Figure 4D). More cells from HTori-Neo cells-induced tumors stained positive for p-Rb (S807/811) (Figure 4D-a) than from HTori-TRβ cells-induced small growths (Figure 4D-b). The p-Rb (S807/811)-stained positive cells were counted and, as shown in Figure 4D-c, 32% fewer HTori-TRβ cells were stained for p-Rb (S807/811) cells than in tumor cells induced by HTori-Neo cells. These results show that in Rb was reactivated when the SV40Tag-Rb complex was disrupted by TRβ in vivo, leading to inhibition of cell cycle progression.
TRβ reactivates p53 functions by inducing apoptosis
One of the important functions of p53 is to initiate apoptosis to eliminate cells with damaged DNA caused by irradiation or other cellular stress. Bax (Bcl2-associated X protein), an apoptosis promoter, is transcriptional targets of p53. We therefore evaluated whether p53, released from complexes with SV40Tag (see Figure 3B), could act to modulate this apoptosis regulator. Figure 5A shows that BAX mRNA expression levels were 2-fold greater in HTori-TRβ cells than in HTori-Neo cells. Consistent with the increase at the mRNA level, BAX protein abundance was also increased (Figure 5B-I-a). We also determined two apoptosis markers-cleaved PARP (poly ADP-ribose polymerase) and cleaved caspase 3. Caspase 3, a member of the caspase family of 13 aspartate-specific cysteine proteases that play a central role in the execution of the apoptotic program [45,46], is primarily responsible for the cleavage of PARP during cell death [47-49]. As shown in Figure 5B-I-b, the cleaved PARP was markedly elevated in HTori-TRβ cells (lower band, lane 2; Figure 5B-I-b) as compared with HTori-Neo cells (lane 1, Figure 5B-I-b). The protein abundance of cleaved caspase 3 was also elevated, indicative of activation of caspase 3 in apoptosis (lane 2, Figure 5B-I-c). Quantitative analysis of relative band intensities was shown in Figure 5B-II, indicating a 4, 6 and 6 fold increases for Bax, cleaved RAPR and cleaved caspase 3 protein abundance. Importantly, consistent with the changes shown in Figure 5A and B, more apoptotic cells were detected in HTori-TRβ cells (Figure 5C-c) than in HTori-Neo cells (Figure 5C-a). The quantification of the apoptotic cells (Figure 5C-e) revealed a 2-fold increase of apoptotic cells in HTori-TRβ cells. The changes in the apoptotic activity due to the disruption of p53/SV40Tag complex by TRβ was further assessed in vivo using the small growth from HTori-TRb in nude mice. Comparison of the number of the cells immuno-stained with anti-cleaved caspase-3 antibodies in HTori-Neo induced tumors (Figure 5D-a) and the small growths from HTori-TRβ show that more apoptotic cells in the latter (Figure 5D-b). Immuno-positively stained cells were counted and the results are shown in Figure 5D-c, indicating clearly that more cells exhibit apoptotic activities in HTori-TRβ cells in vivo.
Figure 5.
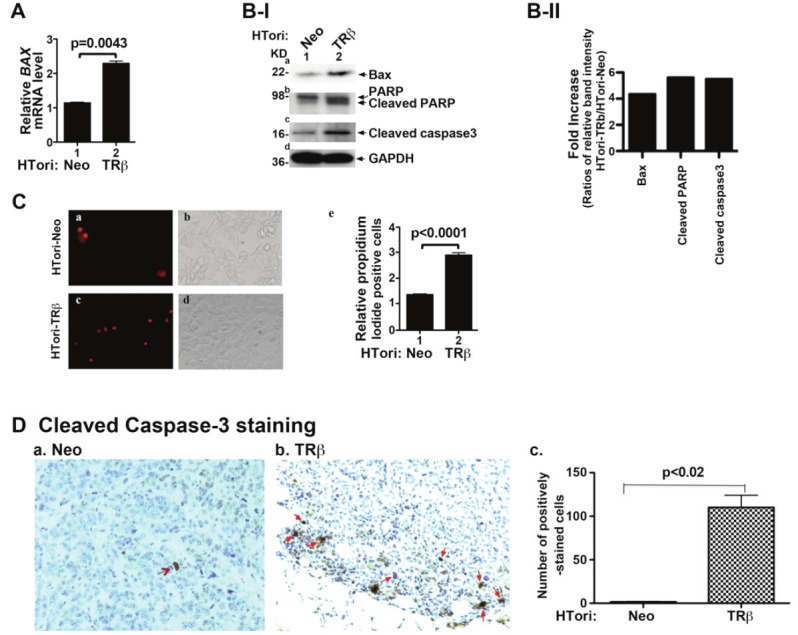
Reactivation of p53 to stimulate apoptosis during tumorigenesis by physical interaction of TRβ with SV40Tag. (A) Activation of the expression of p53 downstream target BAX gene. Total RNA of HTori-Neo and HTori-TRβ was prepared, and mRNA expression was determined, as described in Materials and Methods. The increases in the expression of BAX mRNA are significant with p values as shown. Data are the mean ± SEM (duplicate experiments). (B-I) Increased protein abundance of apoptotic markers in HTori-TRβ cells. Western blot analysis was carried out as described in Materials and Methods. (B-II) Fold of changes of apoptotic markers as shown as ratios of each protein in HTori-TRβ cells to that of HTori-Neo cells after quantification of band intensities shown in B-I. Each protein level was normalized to GAPDH. (C) Increased number of late apoptotic cells in HTori-TRβ cells. Cell survival was determined, as described Materials and Methods. Representative images from three experiments are shown. Late apoptotic cells were counted and the quantitative data are shown in (panel e), indicating significant increases in the number of late apoptotic cells in cells expressing TRβ (HTori-TRβ cells). Data are the mean ± SEM; n=3. (D) Cleaved caspase 3 staining of tumor cells derived from HTori-Neo cells (panel a) and cells derived from HTori-TRβ (panel b). Arrows point to the positive nuclear staining of cleaved caspase 3. The positively cleaved caspase 3 stained cells were counted and graphed (panel c). The difference in the number of positively stained cells between tumors cells and cells derived from the HTori-TRβ cell-induced growth is highly significant (p<0.02).
PTEN (phosphatase and tensin homologue deleted from chromosome 10) is a lipid phosphatase that negatively regulates the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway, critical for cell survival and proliferation. Because the expression of the PTEN gene is transcriptionally regulated by p53 [50], we further examined whether, in HTori-TRβ cells, the expression of the PTEN mRNA was affected by re-activation of p53 due to the interaction of TRβ with SV40Tag. Indeed, Figure 6A shows that the PTEN mRNA was increased ~2-fold. Figure 6B-I further shows that the protein abundance of PTEN was greater in HTori-TRβ cells (lane 2, Figure 6B-I-a) than in HTori-Neo cells (lane 1, Figure 6B-I-a). Importantly, the increased expression of PTEN led to the deactivation of AKT, as evidenced by decreased phosphorylation of AKT (lane 2, Figure 6B-I-b), without changing the total AKT protein abundance (panel c). BAD, when phosphorylated by AKT, suppresses apoptosis and promotes cell survival. In HTori-TRβ cells, the decreased AKT activity led to less phosphorylated BAD, thereby decreasing survival (lane 2, Figure 6B-I-d). Quantification of protein band intensities in Figure 6B-I was determined and fold of changes of key regulators in HTori-TRβ cells were compared with those in HTori-Neo-cell (Figure 6B-II).
Figure 6.

Reactivation of p53 to attenuate AKT signaling by physical interaction of TRβ with SV40Tag. (A). Activation of the expression of the p53 downstream target PTEN gene. Total RNA of HTori-Neo and HTori-TRβ was prepared and mRNA expression was determined, as described in Materials and Methods. Data are the mean ± SEM (duplicate experiments). The increases in the expression of PTEN mRNA are significant with p values as shown. Data are the mean ± SEM (duplicate experiments). (B-I) Decreased activities of AKT signaling in HTori-TRβ cells. Cell extracts of HTori-Neo and HTori-TRβ cells were prepared, followed by Western blot analyses (20 μg cell lysates were used) for PTEN (panel a), pAKT (S473) (panel b), total AKT (panel c), pBAD(S136) (panel d), and the loading control GAPDH (panel e). Increased PTEN expression (panel a) led to attenuation of AKT signaling by decreased phosphorylation of AKT (panel b) and BAD (panel d). (B-II). Fold of changes of protein abundance of key regulators. The band intensities shown in B-I was quantified and compared as ratios of band intensity of each protein in HTori-TRβ to that of HTori-Neo cells. Each protein level was normalized to GAPDH. (C). pAKT (S473) staining of tumor cells derived from HTori-Neo cells (panel a) and cells derived from HTori-TRβ (panel b). Arrows point to the positive nuclear staining of pAKT (S473). The positively pAKT(S473) stained cells were counted and graphed (panel c). The difference in the number of positively stained cells between tumors cells and cells derived from the HTori-TRβ cell-induced growth is highly significant (p<0.001).
To further support that AKT activity was also attenuated in the HTori-Neo-induced tumor cells, we evaluated the pAKT (S473) protein abundance in the tumor cells and the small growths (“bumps”) from the injection sites of HTori-TRβ cells. As shown in Figure 6C-a, strong staining was detected in the majority of HTori-Neo-induced tumor cells, but only weak staining was detected in a few cells from the HTori-TRβ-induced small growths (Figure 6C-b). The quantification of cells positively stained for AKT (S473) protein is shown in Figure 6C-c, indicating that the number of positively AKT (S473) stained cells was 95% lower for HTori-TRβ cells than for HTori-Neo cells. Taken together, these results indicate that reactivation of p53 from the disrupted p53-SV40Tag interaction by the interaction of TRβ with SV40Tag led to attenuation of AKT signaling and to decreased cell survival of tumor cells.
Discussion
Although association studies have long indicated that TRβ could function as a tumor suppressor in human cancers, the molecular mechanism by which TRβ could act to suppress tumorigenesis is still unclear. In this study, we elucidated the mechanisms by which TRβ prevented tumor development induced by SV40Tag. In the mouse xenograft model, SV40Tag-induced tumor development was blocked by the presence of TRβ. Histopathological analyses showed that TRβ inhibited SV40Tag-induced cell proliferation and activated tumor cell apoptosis. Using cell-based studies, we elucidated that the tumor-suppressing effects of TRβ were mediated by disrupting the interaction of SV40Tag with p53 and Rb, thereby leading to reactivation the tumor suppressing activities of these two tumor suppressors. The reactivated Rb released from the SV40Tag oncoprotein by TRβ delayed cells’ entry into the S phase from the G0/G1 phase. The cell proliferation was correspondingly decreased in cells expressing TRβ (HTori-TRβ). The reactivated p53 released from the SV40Tag oncoprotein by TRβ acted to increase the expression of the PTEN gene, thereby attenuating the PI3K-AKT signaling and inducing apoptosis. Thus, the present study uncovered a novel mechanism by which TRβ acted as a tumor suppressor by impeding the deleterious activities of an oncoprotein, SV40Tag, via direct protein-protein interaction.
Mapping studies show that the interaction region with the oncoprotein SV40Tag was located in the ligand-binding domain E (amino acids 244-461) of TRβ. Though the regions of SV40Tag with which TRβ is associated are currently unknown, the regions of SV40Tag that bind to p53 and Rb have been mapped. The binding regions are located in the amino acids 103-107 with LXCXE motif and amino acids 350–627 for Rb and p53, respectively [51]. The findings that the physical association of TRβ with SV40Tag led to the concurrent reduction in the binding of both Rb and p53 with SV40Tag (Figure 3) suggested that binding of TRβ could grossly affect the concurrent complex formation of SV40Tag with Rb and p53. Alternatively, there could be more than one binding site of TRβ on SV40Tag such that the Rb and p53 sites on SV40Tag are individually affected. Nonetheless, regardless of which of the two possibilities holds true, p53 regained its transcription activity when TRβ was bound to SV40Tag, as evidenced by the increases in the expression of one of its target genes, PTEN (Figure 6). The suppressor functions of Rb were also recovered when TRβ was bound to SV40Tag, as evidenced by its reduced phosphorylation that delayed the entry of cells in the G1/S phase transition (Figure 4). However, because the interaction of TRβ with SV40Tag had a significant functional impact on the oncogenic actions of SV40Tag, it would be of interest examine these two possibilities in future studies.
Previous studies have shown that the expression TRβ in hepatocellular and breast cancer cells reduces tumor growth, and inhibits invasiveness, extravasation, and metastasis in mice. In culture cells, TRβ abolishes anchorage-independent growth and migration, blocks responses to epidermal growth factor, insulin-like growth factor-I, and transforming growth factor beta, and regulates expression of genes that play a key role in tumorigenicity and metastatic growth. These inhibitory effects are mediated by suppression of the expression of growth factor receptors and suppressing activation of ERK and PI3K signaling pathways [52], indicating that TRβ functions as a tumor suppressor via transcription regulation. Moreover, TRβ has also been shown as a suppressor of Ras-mediated transcription, proliferation and transformation via cross talk with Ras oncogene at the transcription level [52,53]. The present studies show that TRβ initiated its tumor suppressor functions through direct protein-protein interaction to block the activities of SV40Tag oncoprotein. This novel mode of action has broadened our understanding of molecular mechanisms by which TRβ can function to inhibit proliferation of tumor cells and development of tumor.
Acknowledgments
The present research was supported by the Intramural Research Program at the Center for Cancer Research, National Cancer Institute, NIH.
References
- 1.Forrest D, Vennstrom B. Functions of thyroid hormone receptors in mice. Thyroid. 2000;10:41–52. doi: 10.1089/thy.2000.10.41. [DOI] [PubMed] [Google Scholar]
- 2.Sap J, Munoz A, Damm K, Goldberg Y, Ghysdael J, Leutz A, Beug H, Vennstrom B. The c-erb-A protein is a high-affinity receptor for thyroid hormone. Nature. 1986;324:635–640. doi: 10.1038/324635a0. [DOI] [PubMed] [Google Scholar]
- 3.Thormeyer D, Baniahmad A. The v-erbA oncogene (review) Int J Mol Med. 1999;4:351–358. [PubMed] [Google Scholar]
- 4.Lin KH, Shieh HY, Chen SL, Hsu HC. Expression of mutant thyroid hormone nuclear receptors in human hepatocellular carcinoma cells. Mol Carcinog. 1999;26:53–61. doi: 10.1002/(sici)1098-2744(199909)26:1<53::aid-mc7>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 5.Puzianowska-Kuznicka M, Nauman A, Madej A, Tanski Z, Cheng S, Nauman J. Expression of thyroid hormone receptors is disturbed in human renal clear cell carcinoma. Cancer Lett. 2000;155:145–152. doi: 10.1016/s0304-3835(00)00416-x. [DOI] [PubMed] [Google Scholar]
- 6.Kamiya Y, Puzianowska-Kuznicka M, McPhie P, Nauman J, Cheng SY, Nauman A. Expression of mutant thyroid hormone nuclear receptors is associated with human renal clear cell carcinoma. Carcinogenesis. 2002;23:25–33. doi: 10.1093/carcin/23.1.25. [DOI] [PubMed] [Google Scholar]
- 7.Silva JM, Dominguez G, Gonzalez-Sancho JM, Garcia JM, Silva J, Garcia-Andrade C, Navarro A, Munoz A, Bonilla F. Expression of thyroid hormone receptor/erbA genes is altered in human breast cancer. Oncogene. 2002;21:4307–4316. doi: 10.1038/sj.onc.1205534. [DOI] [PubMed] [Google Scholar]
- 8.Safer JD, Colan SD, Fraser LM, Wondisford FE. A pituitary tumor in a patient with thyroid hormone resistance: a diagnostic dilemma. Thyroid. 2001;11:281–291. doi: 10.1089/105072501750159750. [DOI] [PubMed] [Google Scholar]
- 9.Ando S, Sarlis NJ, Oldfield EH, Yen PM. Somatic mutation of TRbeta can cause a defect in negative regulation of TSH in a TSH-secreting pituitary tumor. J Clin Endocrinol Metab. 2001;86:5572–5576. doi: 10.1210/jcem.86.11.7984. [DOI] [PubMed] [Google Scholar]
- 10.Puzianowska-Kuznicka M, Krystyniak A, Madej A, Cheng SY, Nauman J. Functionally impaired TR mutants are present in thyroid papillary cancer. J Clin Endocrinol Metab. 2002;87:1120–1128. doi: 10.1210/jcem.87.3.8296. [DOI] [PubMed] [Google Scholar]
- 11.Leduc F, Brauch H, Hajj C, Dobrovic A, Kaye F, Gazdar A, Harbour JW, Pettengill OS, Sorenson GD, van den Berg A, et al. Loss of heterozygosity in a gene coding for a thyroid hormone receptor in lung cancers. Am J Hum Genet. 1989;44:282–287. [PMC free article] [PubMed] [Google Scholar]
- 12.Sisley K, Curtis D, Rennie IG, Rees RC. Loss of heterozygosity of the thyroid hormone receptor B in posterior uveal melanoma. Melanoma Res. 1993;3:457–461. doi: 10.1097/00008390-199311000-00009. [DOI] [PubMed] [Google Scholar]
- 13.Chen LC, Matsumura K, Deng G, Kurisu W, Ljung BM, Lerman MI, Waldman FM, Smith HS. Deletion of two separate regions on chromosome 3p in breast cancers. Cancer Res. 1994;54:3021–3024. [PubMed] [Google Scholar]
- 14.Gonzalez-Sancho JM, Garcia V, Bonilla F, Munoz A. Thyroid hormone receptors/THR genes in human cancer. Cancer Lett. 2003;192:121–132. doi: 10.1016/s0304-3835(02)00614-6. [DOI] [PubMed] [Google Scholar]
- 15.Huber-Gieseke T, Pernin A, Huber O, Burger AG, Meier CA. Lack of loss of heterozygosity at the c-erbA beta locus in gastrointestinal tumors. Oncology. 1997;54:214–219. doi: 10.1159/000227691. [DOI] [PubMed] [Google Scholar]
- 16.Ali IU, Lidereau R, Callahan R. Presence of two members of c-erbA receptor gene family (c-erbA beta and c-erbA2) in smallest region of somatic homozygosity on chromosome 3p21-p25 in human breast carcinoma. J Natl Cancer Inst. 1989;81:1815–1820. doi: 10.1093/jnci/81.23.1815. [DOI] [PubMed] [Google Scholar]
- 17.Joseph B, Ji M, Liu D, Hou P, Xing M. Lack of mutations in the thyroid hormone receptor (TR) alpha and beta genes but frequent hypermethylation of the TRbeta gene in differentiated thyroid tumors. J Clin Endocrinol Metab. 2007;92:4766–4770. doi: 10.1210/jc.2007-0812. [DOI] [PubMed] [Google Scholar]
- 18.Ling Y, Xu X, Hao J, Ling X, Du X, Liu X, Zhao X. Aberrant methylation of the THRB gene in tissue and plasma of breast cancer patients. Cancer Genet Cytogenet. 2010;196:140–145. doi: 10.1016/j.cancergencyto.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 19.Iwasaki Y, Sunaga N, Tomizawa Y, Imai H, Iijima H, Yanagitani N, Horiguchi K, Yamada M, Mori M. Epigenetic inactivation of the thyroid hormone receptor beta1 gene at 3p24.2 in lung cancer. Ann Surg Oncol. 2010;17:2222–2228. doi: 10.1245/s10434-010-0956-9. [DOI] [PubMed] [Google Scholar]
- 20.Li Z, Meng ZH, Chandrasekaran R, Kuo WL, Collins CC, Gray JW, Dairkee SH. Biallelic inactivation of the thyroid hormone receptor beta1 gene in early stage breast cancer. Cancer Res. 2002;62:1939–1943. [PubMed] [Google Scholar]
- 21.Suzuki H, Willingham MC, Cheng SY. Mice with a mutation in the thyroid hormone receptor beta gene spontaneously develop thyroid carcinoma: a mouse model of thyroid carcinogenesis. Thyroid. 2002;12:963–969. doi: 10.1089/105072502320908295. [DOI] [PubMed] [Google Scholar]
- 22.Kim WG, Guigon CJ, Fozzatti L, Park JW, Lu C, Willingham MC, Cheng SY. SKI-606, an Src Inhibitor, Reduces Tumor Growth, Invasion, and Distant Metastasis in a Mouse Model of Thyroid Cancer. Clin Cancer Res. 2012;18:1281–1290. doi: 10.1158/1078-0432.CCR-11-2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lemoine NR, Mayall ES, Jones T, Sheer D, McDermid S, Kendall-Taylor P, Wynford-Thomas D. Characterisation of human thyroid epithelial cells immortalised in vitro by simian virus 40 DNA transfection. Br J Cancer. 1989;60:897–903. doi: 10.1038/bjc.1989.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carbone M, Pass HI, Rizzo P, Marinetti M, Di Muzio M, Mew DJ, Levine AS, Procopio A. Simian virus 40-like DNA sequences in human pleural mesothelioma. Oncogene. 1994;9:1781–1790. [PubMed] [Google Scholar]
- 25.Bergsagel DJ, Finegold MJ, Butel JS, Kupsky WJ, Garcea RL. DNA sequences similar to those of simian virus 40 in ependymomas and choroid plexus tumors of childhood. N Engl J Med. 1992;326:988–993. doi: 10.1056/NEJM199204093261504. [DOI] [PubMed] [Google Scholar]
- 26.Lednicky JA, Garcea RL, Bergsagel DJ, Butel JS. Natural simian virus 40 strains are present in human choroid plexus and ependymoma tumors. Virology. 1995;212:710–717. doi: 10.1006/viro.1995.1529. [DOI] [PubMed] [Google Scholar]
- 27.Martini F, Iaccheri L, Lazzarin L, Carinci P, Corallini A, Gerosa M, Iuzzolino P, Barbanti-Brodano G, Tognon M. SV40 early region and large T antigen in human brain tumors, peripheral blood cells, and sperm fluids from healthy individuals. Cancer Res. 1996;56:4820–4825. [PubMed] [Google Scholar]
- 28.Pacini F, Vivaldi A, Santoro M, Fedele M, Fusco A, Romei C, Basolo F, Pinchera A. Simian virus 40-like DNA sequences in human papillary thyroid carcinomas. Oncogene. 1998;16:665–669. doi: 10.1038/sj.onc.1201552. [DOI] [PubMed] [Google Scholar]
- 29.Ozdarendeli A, Camci C, Aygen E, Kirkil C, Toroman ZA, Dogru O, Doymaz MZ. SV40 in human thyroid nodules. J Clin Virol. 2004;30:337–340. doi: 10.1016/j.jcv.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 30.Vivaldi A, Pacini F, Martini F, Iaccheri L, Pezzetti F, Elisei R, Pinchera A, Faviana P, Basolo F, Tognon M. Simian virus 40-like sequences from early and late regions in human thyroid tumors of different histotypes. J Clin Endocrinol Metab. 2003;88:892–899. doi: 10.1210/jc.2002-020436. [DOI] [PubMed] [Google Scholar]
- 31.Ledent C, Dumont J, Vassart G, Parmentier M. Thyroid adenocarcinomas secondary to tissue-specific expression of simian virus-40 large T-antigen in transgenic mice. Endocrinology. 1991;129:1391–1401. doi: 10.1210/endo-129-3-1391. [DOI] [PubMed] [Google Scholar]
- 32.Guigon CJ, Kim DW, Zhu X, Zhao L, Cheng SY. Tumor suppressor action of liganded thyroid hormone receptor beta by direct repression of beta-catenin gene expression. Endocrinology. 2010;151:5528–5536. doi: 10.1210/en.2010-0475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guigon CJ, Zhao L, Lu C, Willingham MC, Cheng SY. Regulation of beta-catenin by a novel nongenomic action of thyroid hormone beta receptor. Mol Cell Biol. 2008;28:4598–4608. doi: 10.1128/MCB.02192-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhat MK, McPhie P, Cheng SY. Interaction of thyroid hormone nuclear receptor with antibody: characterization of the thyroid hormone binding site. Biochem Biophys Res Commun. 1995;210:464–471. doi: 10.1006/bbrc.1995.1683. [DOI] [PubMed] [Google Scholar]
- 35.Fukuda T, Willingham MC, Cheng SY. Anti-peptide antibodies recognize c-erbA and a related protein in human A431 carcinoma cells. Endocrinology. 1988;123:2646–2652. doi: 10.1210/endo-123-6-2646. [DOI] [PubMed] [Google Scholar]
- 36.Lin KH, Willingham MC, Liang CM, Cheng SY. Intracellular distribution of the endogenous and transfected beta form of thyroid hormone nuclear receptor visualized by the use of domain-specific monoclonal antibodies. Endocrinology. 1991;128:2601–2609. doi: 10.1210/endo-128-5-2601. [DOI] [PubMed] [Google Scholar]
- 37.Fozzatti L, Lu C, Kim DW, Cheng SY. Differential recruitment of nuclear coregulators directs the isoform-dependent action of mutant thyroid hormone receptors. Mol Endocrinol. 2011;25:908–921. doi: 10.1210/me.2010-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim CS, Vasko VV, Kato Y, Kruhlak M, Saji M, Cheng SY, Ringel MD. AKT activation promotes metastasis in a mouse model of follicular thyroid carcinoma. Endocrinology. 2005;146:4456–4463. doi: 10.1210/en.2005-0172. [DOI] [PubMed] [Google Scholar]
- 39.Lin HM, Zhao L, Cheng SY. Cyclin D1 Is a Ligand-independent Co-repressor for Thyroid Hormone Receptors. J Biol Chem. 2002;277:28733–28741. doi: 10.1074/jbc.M203380200. [DOI] [PubMed] [Google Scholar]
- 40.Valero JG, Sancey L, Kucharczak J, Guillemin Y, Gimenez D, Prudent J, Gillet G, Salgado J, Coll JL, Aouacheria A. Bax-derived membrane-active peptides act as potent and direct inducers of apoptosis in cancer cells. J Cell Sci. 2011;124:556–564. doi: 10.1242/jcs.076745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dace A, Zhao L, Park KS, Furuno T, Takamura N, Nakanishi M, West BL, Hanover JA, Cheng S. Hormone binding induces rapid proteasome-mediated degradation of thyroid hormone receptors. Proc Natl Acad Sci USA. 2000;97:8985–8990. doi: 10.1073/pnas.160257997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu C, Zhao L, Ying H, Willingham MC, Cheng SY. Growth activation alone is not sufficient to cause metastatic thyroid cancer in a mouse model of follicular thyroid carcinoma. Endocrinology. 2010;151:1929–1939. doi: 10.1210/en.2009-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pipas JM. SV40: Cell transformation and tumorigenesis. Virology. 2009;384:294–303. doi: 10.1016/j.virol.2008.11.024. [DOI] [PubMed] [Google Scholar]
- 44.Qin XQ, Livingston DM, Ewen M, Sellers WR, Arany Z, Kaelin WG Jr. The transcription factor E2F-1 is a downstream target of RB action. Mol Cell Biol. 1995;15:742–755. doi: 10.1128/mcb.15.2.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- 46.Cryns V, Yuan J. Proteases to die for. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- 47.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 48.Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 49.Le Rhun Y, Kirkland JB, Shah GM. Cellular responses to DNA damage in the absence of Poly(ADP-ribose) polymerase. Biochem Biophys Res Commun. 1998;245:1–10. doi: 10.1006/bbrc.1998.8257. [DOI] [PubMed] [Google Scholar]
- 50.Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S, Mak TW. Regulation of PTEN transcription by p53. Mol Cell. 2001;8:317–325. doi: 10.1016/s1097-2765(01)00323-9. [DOI] [PubMed] [Google Scholar]
- 51.Cheng J, DeCaprio JA, Fluck MM, Schaffhausen BS. Cellular transformation by Simian Virus 40 and Murine Polyoma Virus T antigens. Semin Cancer Biol. 2009;19:218–228. doi: 10.1016/j.semcancer.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martinez-Iglesias O, Garcia-Silva S, Tenbaum SP, Regadera J, Larcher F, Paramio JM, Vennstrom B, Aranda A. Thyroid hormone receptor beta1 acts as a potent suppressor of tumor invasiveness and metastasis. Cancer Res. 2009;69:501–509. doi: 10.1158/0008-5472.CAN-08-2198. [DOI] [PubMed] [Google Scholar]
- 53.Garcia-Silva S, Aranda A. The thyroid hormone receptor is a suppressor of ras-mediated transcription, proliferation, and transformation. Mol Cell Biol. 2004;24:7514–7523. doi: 10.1128/MCB.24.17.7514-7523.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]