

Abstract
Membrane depolarization and intracellular Ca2+ transients generated by activation of voltage-gated Na+ and Ca2+ channels are local signals, which initiate physiological processes such as action potential conduction, synaptic transmission, and excitation-contraction coupling. Targeting of effector proteins and regulatory proteins to ion channels is an important mechanism to ensure speed, specificity, and precise regulation of signaling events in response to local stimuli. This article reviews experimental results showing that Na+ and Ca2+ channels form local signaling complexes, in which effector proteins, anchoring proteins, and regulatory proteins interact directly with ion channels. The intracellular domains of these channels serve as signaling platforms, mediating their participation in intracellular signaling processes. These protein-protein interactions are important for regulation of cellular plasticity through modulation of Na+ channel function in brain neurons, for short-term synaptic plasticity through modulation of presynaptic CaV2 channels, and for the fight-or-flight response through regulation of postsynaptic CaV1 channels in skeletal and cardiac muscle. These localized signaling complexes are essential for normal function and regulation of electrical excitability, synaptic transmission, and excitation-contraction coupling.
Introduction
The electrical signals produced by ion channels and the resulting Ca2+ entry that initiates intracellular responses are local signaling events. Modulation of ion channels is a dynamic process that is precisely controlled in space and time [1, 2]. Targeting and localization of signaling enzymes to discrete subcellular compartments or substrates is an important regulatory mechanism ensuring specificity of signaling events in response to local stimuli [3]. This article describes signaling complexes formed by three representative ion channels: brain Na+ channels (NaV1.2) that initiate and conduct action potentials, presynaptic Ca2+ channels (CaV2.1) that conduct P/Q-type Ca2+ currents and initiate synaptic transmission, and muscle Ca2+ channels (CaV1.1 and CaV1.2) that initiate excitation-contraction coupling. In each case, signaling proteins and anchoring proteins that regulate these channels or are effectors in downstream signaling pathways bind to specific sites on their intracellular domains, and these protein-protein interactions are required for normal signal transduction in nerve and muscle cells.
Experimental Approaches for Analysis of Ion Channel Signaling Complexes
Biochemical, proteomic, and functional approaches have been combined in the analysis of ion channel signaling complexes. The biochemical approach usually begins with purification of an ion channel and identification of associated subunits and other interacting proteins. The initial signaling complexes of voltage-gated sodium and calcium channels were defined in this way as described below. Proteomic methods offer a broader view of ion channel signaling complexes by defining all of their interacting proteins. Both yeast two-hybrid screening methods and identification of ion channel associated proteins by mass spectrometry have been successfully employed in analysis of ion channel signaling complexes. The power of mass spectrometry as a method for detection of associated proteins in ion channel signaling complexes is increasing at a rapid pace and promises to provide the most in-depth view of such macromolecular complexes. However, identification of interacting proteins is not sufficient to define a signaling complex. Demonstration of close co-localization in native cells and co-immunoprecipitation from transfected cells helps to solidify the case for significant protein interactions. Moreover, demonstration of a functional outcome of association of ion channel signaling complexes in transfected cells, native cells, and native tissues is an essential element in defining their physiological significance. Co-expression and functional analysis by electrophysiology is the most common approach to demonstrate functional interactions, but this approach suffers from possible artifacts of over-expression and use of heterologous cells with their own signal transduction pathways. Peptide inhibitors of protein interactions can be powerful tools to demonstrate the significance of ion channel signaling complexes in native cells. Finally, mouse genetics offers the opportunity to analyze the functional significance of ion channel signaling complexes in vivo by disrupting specific protein interactions with mutations. Information from all of these diverse approaches has been integrated in the studies of the three ion channel signaling complexes used as examples here.
A Signaling Complex of Brain Na+ Channels Mediates Cellular Plasticity
Neuromodulation of electrical excitability is a fundamental mechanism in many aspects of learning, memory, and physiological regulation. Voltage-gated Na+ channels are responsible for the initiation and propagation of action potentials [4]. Their regulation by neurotransmitters and second messengers provides an important form of cellular plasticity, which controls the excitability of central neurons in response to the sum of their synaptic inputs, sets the threshold for excitability, and modulates the frequency and form of action potential generation [2].
Na+ channel proteins in mammalian brain consist of an α subunit of approximately 260 kDa in association with auxiliary subunits of 33 to 36 kDa--β1-β4 [1, 5, 6]. The α subunits consist of four homologous domains (I – IV), which each contains six transmembrane segments (S1 – S6) and a membrane-reentrant loop between S5 and S6 (Fig. 1A). The S4 segments in each domain serve as the voltage sensors, the S5 and S6 segments and the reentrant loop between them form the lining of the pore, and the short intracellular loop between domains III and IV forms the inactivation gate. Expression of α subunits alone is sufficient for formation of functional Na+ channels, but β subunits must be co-expressed to give the correct kinetics and voltage dependence of Na+ channel activation and inactivation.
Figure 1. A Na+ channel signaling complex with PKA.
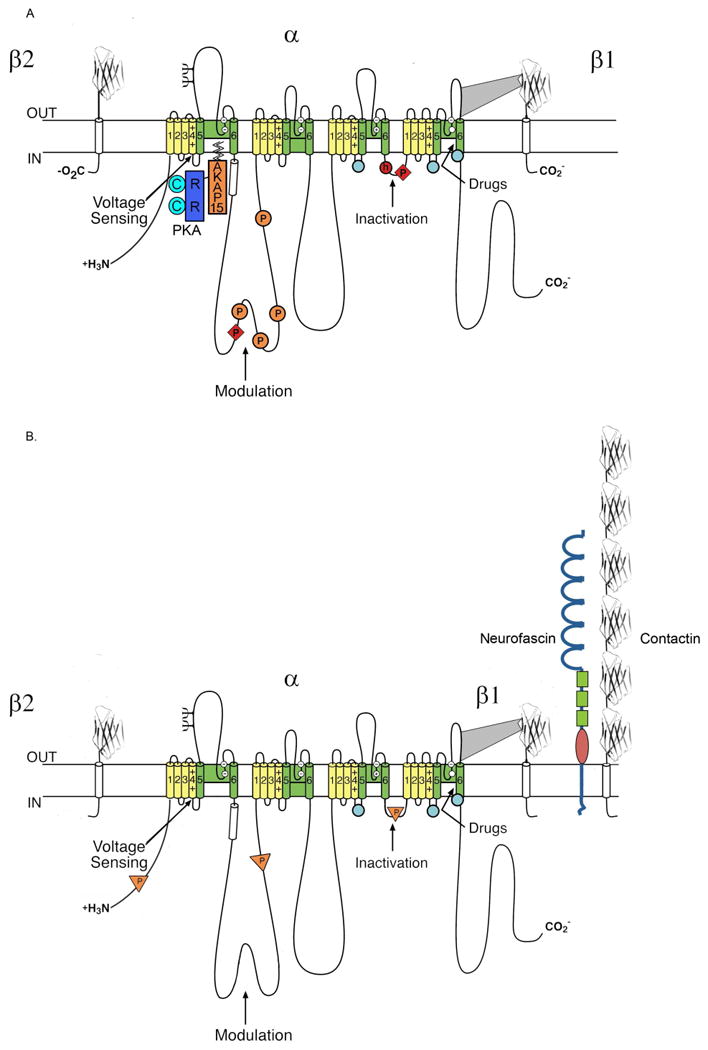
A. The brain NaV1.2 channel is illustrated as a transmembrane folding diagram with α, β1, and β2 subunits and a PKA signaling complex. Transmembrane alpha helical segments are illustrated as cylinders. The gating charges of the S4 segments are denoted by +. The amino acid residues that form the selectivity filter are denoted by circles with -, +, or blank inside. Phosphorylation sites are indicated by P. The inactivation particle in the inactivation gate is indicated by h in a circle. Open circles indicate the regions that form the inactivation gate receptor. The extracellular domains of the β subunits are shown as immunoglobulin-like domains. The interaction of PKA and AKAP15 with an amphipathic alpha helix in beginning of the intracellular loop connecting domains I and II is shown. B. A similar diagram of the NaV1.2 channel with the tyrosine phosphorylation signaling complex.
Biochemical studies of purified brain Na+ channels showed surprisingly that the α subunit is an exceptionally good substrate for phosphorylation by PKA [7] and is rapidly phosphorylated on multiple sites following activation of PKA in synaptosomes by cAMP derivatives [8]. Measurements of neurotoxin-activated Na+ influx in synaptosomes showed that cAMP-dependent phosphorylation is correlated with a reduction of 26% in neurotoxin-activated Na+ conductance [9]. Na+ channels are rapidly phosphorylated by PKA in intact brain neurons in cell culture [10] on up to five sites [11, 12], including serine residues 573, 610, 623, and 687 (Fig. 1A [8]). Activation of PKA modulates the function of the NaV1.2 channel expressed in Xenopus oocytes [13] and mammalian cells [14, 15]. Peak Na+ current is reduced 25 to 40% without change in the kinetics or voltage dependence of activation or inactivation. PKA-mediated reduction in peak Na+ current at typical resting membrane potentials of -70 mV to -80 mV is primarily mediated by phosphorylation at serine 573 in LI-II [15, 16]. PKA-dependent modulation of NaV1.2 channels is voltage-dependent in the range of −110 to −70 mV and is synergistic with phosphorylation by protein kinase C [17, 18]. Systematic mutagenesis of the three additional PKA phosphorylation sites in LI-II revealed the requirement for PKA phosphorylation at serine 687 for voltage-dependent modulation. A double mutation at positions 573 and 687 (SS573/687AA) eliminated modulation at both the permissive and non-permissive membrane potentials [19]. Thus, maximal modulation of NaV1.2 channels by PKA requires phosphorylation at serine residues 573, 576 and 687 and concurrent depolarization. This type of convergent regulation provides a novel mechanism by which information from multiple signaling pathways may be integrated depending upon the state of the cell and its prior history of electrical activity.
Similar modulation of Na+ channels by direct activation of PKA was observed in primary cultures of embryonic rat brain neurons [14]. Na+ channel activity was decreased 25 to 40% by activation of PKA, as shown by a decrease in the ensemble average current in excised membrane patches following application of the catalytic subunit of PKA and ATP. The decrease in peak current resulted from an increase in null sweeps during which no Na+ channel opened after depolarization. Activation of D1-like dopamine receptors, which couple to the activation of adenylyl cyclase, decreases Na+ current via activation of PKA in acutely isolated hippocampal pyramidal neurons without altering the voltage-dependence or kinetics of activation or fast inactivation [15]. The biophysical mechanism of modulation by dopamine or serotonin acting via the PKA pathway is enhancement of the intrinsic slow inactivation process of Na+ channels, which is known to modulate the threshold and firing pattern of brain neurons [20, 21]. This modulation is blocked by mutations of conserved asparagine residues in the S6 segments that selectively impair slow inactivation [21].
Specificity and speed of signaling of kinase signaling are often achieved targeting of protein kinases to specific substrates via protein-protein interactions [3]. PKA is localized by protein–protein interactions between the regulatory (R) subunit dimer and A-kinase anchoring proteins (AKAPs) [22, 23], a family of functionally related proteins defined by their ability to bind R subunits of PKA and target PKA to specific compartments or substrates. AKAPs contain an amphipathic helix that inserts into a hydrophobic pocket in the R subunit dimer [24, 25]. Consistent with this theme, modulation of the Na+ channel by D1-like dopamine receptors in hippocampal neurons requires targeted localization of PKA near the channel via an A-Kinase Anchoring Protein (AKAP15) (Fig. 1A) [26, 27], which we characterized initially in studies of Ca2+ channel modulation (see below). This novel 81-amino acid AKAP has an amino terminal lipid anchor for membrane targeting, an amphipathic helix for PKA binding, and a site for interaction with Na+ channels. PKA, AKAP-15, and the Na+ channel therefore form a signaling complex that controls channel activity. The site of binding of AKAP15 on the Na+ channel is located at the beginning of the large intracellular loop connecting domains I and II (LI-II), which also contains the sites of PKA phosphorylation and regulation (Fig. 1; [19, 27]). This signaling complex places the bound PKA precisely at its target sites.
NaV1.2 channels are also modulated by a tyrosine kinase signaling complex (Fig. 1B; [28, 29]). Brain-derived neurotrophic factor (BDNF) activates its receptor TrkB, which in turn phosphorylates and activates Fyn kinase specifically bound to an SH3 binding motif in LI-II of NaV1.2 channels. Activated Fyn tyrosine residues in LI-II and LIII-IV (Fig. 1B), which reduces sodium channel activity by accelerating inactivation and shifting its voltage dependence to more negative membrane potentials. These effects are reversed by receptor tyrosine kinase β (RPTPβ), which is also bound to NaV1.2 channels through interactions with both α and β subunits (Fig. 1B; [30]). Additional members of this tyrosine kinase signaling complex include the cell adhesion molecules neurofascin [31] and contactin [32-34], the extracellular matrix protein tenascin [35-37], and the intracellular scaffolding protein ankyrin [38]. These interactions are important for localization of sodium channels in the cell surface membrane and for cell-cell interactions between neurons.
A Presynaptic Ca2+ Channel Signaling Complex Mediates Short-term Synaptic Plasticity
Presynaptic CaV2 channels conduct P/Q-, N-, and R-type Ca2+ currents, which initiate synaptic transmission. The efficiency of neurotransmitter release depends on the third or fourth power of entering Ca2+. This steep dependence of neurotransmission on Ca2+ entry makes the presynaptic Ca2+ channel an unusually sensitive target of regulation. CaV2.1 channels conducting P/Q-type Ca2+ currents are the predominant pathway for Ca2+ entry initiating fast release of classical neurotransmitters like glutamate, acetylcholine, and GABA in the brain. They are controlled by protein interactions with their intracellular domains, which serve as a platform for Ca2+-dependent signal transduction.
Interaction with SNARE proteins increases the efficiency of transmitter release
Ca2+ entry through voltage-gated Ca2+ channels initiates exocytosis by triggering the fusion of secretory vesicle membranes with the plasma membrane through actions on the SNARE protein complex of syntaxin, SNAP-25, and VAMP/synaptobrevin (reviewed in [39, 40]). The function of the SNARE complex is regulated by the synaptic vesicle Ca2+-binding protein synaptotagmin. Presynaptic CaV2.1 and CaV2.2 channels interact directly with the SNARE proteins through a specific synaptic protein interaction (synprint) site in the large intracellular loop connecting domains II and III [41, 42]. This interaction is regulated by Ca2+ and protein phosphorylation [43-45]. Synaptotagmin also binds to the synprint site of CaV2 channels [46-48]. Injection of peptide inhibitors of these interactions into presynaptic neurons inhibits synaptic transmission at synapses of sympathetic neurons and at the neuromuscular junction, consistent with the conclusion that binding to SNARE proteins is required to position docked synaptic vesicles near Ca2+ channels for effective fast exocytosis [49, 50]. These results define a second functional activity of the presynaptic Ca2+ channel—targeting docked synaptic vesicles to a source of Ca2+ for effective transmitter release.
Feedback regulation by SNARE proteins
SNARE proteins also have retrograde regulatory effects on Ca2+ channel function. Co-expression of syntaxin or SNAP-25 with CaV2.1 or CaV2.2 channels reduces the level of channel expression and inhibits Ca2+ channel activity by shifting the voltage dependence of steady-state inactivation toward more negative membrane potentials [51, 52]. Co-expression of synaptotagmin to mimic the effect of docking a synaptic vesicle nearby relieves the inhibition of channel [48, 53, 54]. This regulatory mechanism would focus Ca2+ entry on Ca2+ channels that have a nearby docked synaptic vesicle and therefore are poised for effective exocytosis. These opposing effects of interactions with SNARE proteins to enhance docking of synaptic vesicles and to regulate CaV2 channel activity confer biphasic regulation of synaptic transmission on neuromuscular synapses in vivo [55].
G protein modulation
N-type and P/Q-type Ca2+ currents are regulated through G protein-coupled pathways [56-58]. One major pathway is voltage-dependent and membrane-delimited, the inhibition of Ca2+ channel activity is caused by a positive shift in voltage dependence and a slowing of activation [59], and these effects are relieved by strong depolarization resulting in facilitation of Ca2+ currents [59, 60]. Synaptic transmission is inhibited by neurotransmitters through this mechanism, and voltage-dependent facilitation of synaptic transmission is observed when depolarization relieves G protein inhibition [61]. Gβγ subunits are responsible for this form of modulation of Ca2+ channels [62, 63]. Possible sites of G protein βγ subunit interaction with Ca2+ channels have been investigated extensively, and multiple points of interaction have been identified including the intracellular loop between domains I and II (LI-II) [64-67] and the N-terminal and C-terminal domains [68-73]. The inhibition of N-type and P/Q-type Ca2+ currents in dissociated neurons through binding of Gβγ subunits can be reversed by several neurotransmitters acting through protein kinase C [74, 75]. This mechanism of reversal involves phosphorylation of sites in LI-II, just downstream of a principal site of interaction of βγ subunits with the Ca2+ channel [65](Figure 2). Thus, this intracellular loop of the Ca2+ channel integrates G protein, protein kinase C, and voltage signals in regulating Ca2+ channel activity.
Figure 2. A presynaptic Ca2+ channel signaling complex.
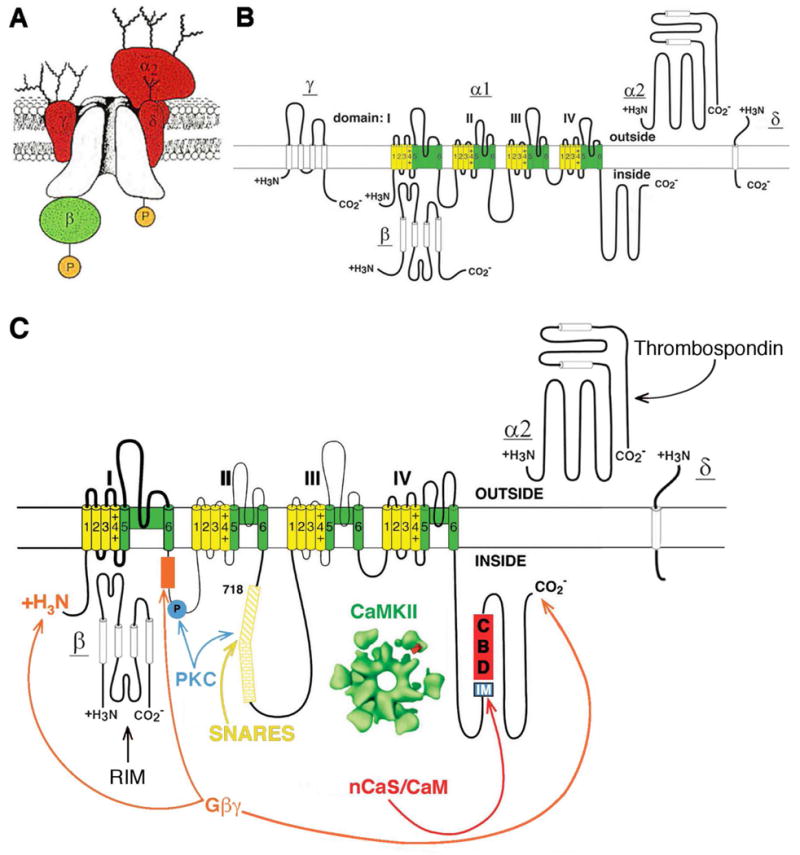
A. The subunits of calcium channels. B. Transmembrane folding diagrams of the calcium channel subunits. C. The presynaptic Ca2+ channel signaling complex. Sites of interaction of SNARE proteins (the synprint site), Gβγ subunits, protein kinase C (PKC), calmodulin and neuronal Ca2+ binding proteins (CBD), and Ca2+/calmodulin-dependent protein kinase II are illustrated.
Ca2+-dependent regulation of Cav2.1 channels
Ca2+-dependent facilitation and inactivation of presynaptic Ca2+ channels was observed in patch clamp recordings of presynaptic nerve terminals in the rat pituitary [76] and the calyx of Held synapse in the rat brainstem [77]. During tetanic stimulation, Cav2.1 currents undergo an initial Ca2+-dependent facilitation followed by progressive inactivation [78, 79]. Simultaneous recording of excitatory postsynaptic responses showed that Ca2+ -dependent facilitation and inactivation of Cav2.1 channels lead to synaptic enhancement and depression, respectively [77-79].
Ca2+-dependent facilitation and inactivation are also observed for cloned and expressed Cav2.1 channels expressed in mammalian cells [80, 81], suggesting an important role for direct, Ca2+-dependent regulation of Cav2.1 channels in activity-dependent synaptic plasticity. The C-terminal domain of CaV2.1 channels contains a bipartite calmodulin-binding site, consisting of a modified IQ-like domain beginning with the sequence IM and a novel calmodulin binding domain (CBD) [80, 82]. CaM binding to the CBD is Ca2+-dependent and both Ca2+-dependent facilitation and inactivation are blocked by co-expression of a CaM inhibitor peptide [80]. The slow intracellular Ca2+ buffer EGTA (10 mM) inhibits Ca2+-dependent inactivation but not Ca2+ -dependent facilitation, suggesting that facilitation has greater Ca2+ sensitivity due to faster binding or higher affinity binding of local Ca2+ [81]. The IQ-like motif is required for facilitation, while the CBD is required for rapid Ca2+-dependent inactivation [82]. The two lobes of calmodulin are differentially involved in these two processes. Mutations of the two EF-hands in the C-terminal lobe prevent facilitation, whereas mutation of the EF-hands in the N-terminal lobe prevents inactivation [82-84]. FRET studies indicate that apo-calmodulin can bind to Cav2.1 channels in intact cells and binding is enhanced by Ca2+ binding to calmodulin [84]. Altogether, these results support a model in which the two lobes of CaM interact differentially with the modified IQ domain and the CBD in order to effect bi-directional regulation, with the C-terminal lobe primarily controlling facilitation through interactions with the IQ-like domain and the N-terminal lobe primarily controlling inactivation through interactions with the CBD.
Differential modulation by CaM and neuronal Ca2+-sensor proteins
CaM is the most well-characterized member of a superfamily of Ca2+-sensor (CaS) proteins, whose members typically have neuron-specific localization and amino acid substitutions that prevent Ca2+ binding to one or more of their four EF-hand motifs [85]. CaS proteins confer Ca2+-dependent and Ca2+-independent regulation of CaV2.1 channels that differs significantly from modulation by CaM. CaBP1 binds to the CBD of α12.1 in a Ca2+ -independent manner [86]. This Ca2+-independent binding of CaBP1 causes strong enhancement of the rate of inactivation, positive shift in the voltage-dependence of activation, and loss of Ca2+-dependent facilitation of Cav2.1 channels [86]. These effects of CaBP1 are absent in channels lacking the CBD, consistent with the hypothesis that CaBP1 displaces CaM from this site [86]. The differences in modulation of Cav2.1 channels by CaBP1 and CaM may result from the structural differences between the two Ca2+ binding proteins, which include an inactivated EF-hand Ca2+ binding site, an extended N-terminal domain with a lipid anchor, and an extended central helix connecting the two lobes of CaBP1 [85]. Myristoylation of CaBP1 is required for its distinctive regulatory properties [87]. Moreover, the distinctive features of regulation by CaBP1 can be transferred in a chimera containing the N-terminal domain and the second, inactive EF-hand motif (Few et al., Soc. Neurosci. Abst, 2008). These results begin to elucidate the molecular logic for differential regulation of CaV2.1 channels by CaS proteins. Since it co-immunoprecipitates and co-localizes with Cav2.1 channels in the brain [86], CaBP1 may contribute to the diversity of regulation of these channels in the nervous system.
Visinin-like protein 2 (VILIP-2) is a neuronal CaS protein that is distantly related to CaBP-1 [85]. Consistent with these substantial structural differences, VILIP-2 has opposite effects on CaV2.1 channels from CaBP-1 [88]. Co-expression of VILIP-2 causes slowed inactivation and enhanced facilitation, but its binding and effects are Ca2+-independent like CaBP-1. Differential expression of CaBP1, VILIP-2, and other CaS proteins at synapses would lead to differential modulation of Ca2+ currents and therefore of synaptic transmission in response to trains of action potentials.
Ca2+/CaM-dependent protein kinase II in the presynaptic Ca2+ channel signaling complex
Ca2+/CaM-dependent protein kinase II (CaMKII) is one of the most important Ca2+ receptors in neurons. In order for it to receive the Ca2+ signal in presynaptic terminals, it must be located in close proximity to the presynaptic Ca2+ channel. Endogenous CaMKII co-immunoprecipitates with CaV2.1 channels from transfected cells [89]. Moreover, inhibition of endogenous CaMKII with the specific organic CaMKII inhibitor KN-93 or the endogenous brain CaMKII inhibitor protein CaMKIIN accelerates inactivation and shifts the voltage dependence of inactivation to more negative membrane potentials, resulting in a substantial reduction in Ca2+ channel activity. These results imply that CaMKII activity slows inactivation and positively shifts its voltage dependence, substantially increasing Ca2+ channel activity. Surprisingly, these effects are caused by binding of CaMKII to a site in the C-terminal domain of CaV2.1 channels and do not require kinase catalytic activity [89]. These results indicate that CaMKII binds to the C-terminal domain of presynaptic Ca2+ channels and enhances their activity by slowing inactivation and positively shifting the voltage dependence of inactivation. CaMKII bound to CaV2.1 channels is well-positioned to respond to entering Ca2+ and phosphorylate neighboring proteins, including the CaV2.1 channel itself.
Short-term synaptic plasticity mediated by regulation of CaV2.1 channels by CaS proteins
The strong regulation of presynaptic Ca2+ channels by CaS proteins suggests that this process may play a role in short-term synaptic facilitation and rapid synaptic depression. To test this idea, wild-type CaV2.1 channels and mutants in the IQ-like domain and CBD were expressed by injection of cDNA in presynaptic sympathetic ganglion neurons in culture, endogenous N-type Ca2+ currents were blocked by ω-contoxin GVIA, and synaptic transmission mediated by the transfected Ca2+ channels was measured with sharp microelectrodes [90]. Transfected CaV2.1 channels mediated synaptic transmission with typical short-term facilitation during the first several action potentials followed by rapid depression [90]. Mutation of the IQ-like domain of the transfected channels substantially reduced facilitation, whereas mutation of the CBD substantially slowed depression [90]. These results show that modulation of CaV2.1 channels by binding of CaM to the IQ-like domain and the CBD contributes substantially to short-term facilitation and rapid depression of synaptic transmission in this synaptic preparation [91].
Short-term synaptic plasticity differs substantially among different types of synapses, but CaM is universally expressed. Therefore, it is possible that neuron-specific expression of CaS proteins overrides modulation by CaM and exerts differential regulation of synaptic plasticity. As expected from their effects on transfected CaV2.1 channels in non-neuronal cells, over-expression of CaBP1 in sympathetic neurons inhibited synaptic facilitation and enhanced synaptic depression, whereas over-expression of VILIP-2 enhanced facilitation and slowed depression (Leal, Mochida, Scheuer, and Catterall, Soc. Neurosci. Abst, 2010). Therefore, these two CaS proteins can mediate biphasic regulation of synaptic transmission by CaV2.1 channels, controlling both the form and direction of short-term synaptic plasticity in synapses in which they are expressed. Similarly, the closely related CaS protein Neuronal Calcium Sensor-1 (NCS-1, called frequenin in Drosophila) enhances synaptic transmission at the Calyx of Held and in cultured hippocampal neurons by increasing presynaptic Ca2+ currents [92, 93]. Differential expression of CaS proteins may be a widespread mechanism of modulation of short-term synaptic plasticity in different classes of synapses.
Proteomic analysis of the presynaptic Ca2+ channel signaling complex
Although the results of protein interaction and functional studies reviewed above indicate that presynaptic Ca2+ channels form an extensive signaling complex, they reveal only the tip of the iceberg of this huge protein structure. Detailed analyses of the components of the presynaptic Ca2+ channel complex using proteomic methods have revealed many additional proteins, including cytoskeletal proteins, extracellular matrix proteins, adaptors and trafficking proteins, G protein-coupled receptors, and additional signaling proteins [94, 95]. The most extensive analysis, using mouse Ca2+ channel knockouts as negative controls, reveals more than 100 specifically associated proteins [94]. Thus, the presynaptic Ca2+ channel proteome is a large universe indeed.
Muscle CaV1 Channel Signaling Complexes Regulate Excitation-Contraction Coupling in the Fight-or-Flight Response
Skeletal muscle CaV1 channels
Skeletal muscle Cav1.1 channels have been the primary experimental model for biochemical studies of Ca2+ channels [96-98]. Both the pore-forming α1 subunit and the auxiliary β subunit are phosphorylated by PKA [98-100]. The α1 subunit is truncated by proteolytic processing of the C-terminal domain [101, 102], and the primary sites of PKA phosphorylation are located in the distal C-terminus beyond the point of proteolytic cleavage [103, 104] Voltage-dependent potentiation of Cav1.1 channels on the 50-msec time scale requires both PKA phosphorylation [105] and PKA anchoring via an AKAP [106, 107], suggesting close association of PKA and Ca2+ channels. A novel, plasma-membrane-targeted AKAP (AKAP15) is associated with Cav1.1 channels and may mediate their regulation by PKA [108, 109]. This AKAP is also known as AKAP18 [110]. AKAP15 binds to the C-terminal domain of Cav1.1 channels via a novel modified leucine-zipper in the C-terminal domain near the primary sites of PKA phosphorylation [111]. Block of this interaction by competing peptides prevents PKA regulation of Ca2+ currents in intact skeletal myoblasts, indicating that direct interaction with the Cav1.1 channel is required for effective regulation in situ in skeletal muscle cells [111].
Cardiac Ca2+ channels
Release of catecholamines from the adrenal medulla and the autonomic nervous system stimulates β-adrenergic receptors, adenylyl cyclase, cAMP, and PKA to induce chronotropic, inotropic, and lusitropic regulation of heart rate, contractility, and relaxation, respectively. In cardiac myocytes, Ca2+ influx through Cav1.2 channels contributes to the plateau phase of the cardiac action potential and is responsible for initiating excitation-contraction coupling. Cav1.2 channels are modulated by the β-adrenergic receptor/cAMP signaling pathway [112, 113]. As for skeletal muscle Cav1.1 channels, both the pore-forming α1 and auxiliary β subunits of Cav1.2 channels are substrates for phosphorylation by PKA [114-117]. PKA phosphorylates the α1 subunit prominently on serine 1928 in the distal C-terminal domain [115, 118]. Activation of β-adrenergic receptors increases L-type Ca2+ currents through PKA-mediated phosphorylation of the Cav1.2 channel protein and/or associated proteins [119-122]. As for skeletal muscle Cav1.1 channels, PKA regulation of Cav1.2 channels in cardiac myocytes is rapid and requires the anchoring of PKA through direct association with an AKAP [123, 124]. These results suggest that a Ca2+ channel signaling complex containing an AKAP and PKA is formed in cardiac muscle, as in skeletal muscle.
AKAP-15 has been identified as the anchoring protein that targets PKA to Cav1.2 channels in cardiac muscle via a conserved leucine-zipper motif in the distal C-terminus of the pore-forming α1 subunit ([124]; Fig. 3). Mutation of this motif prevents PKA anchoring, and disruption of this interaction with competing peptides prevents β-adrenergic and PKA-dependent regulation of L-type Ca2+ currents in ventricular myocytes [124]. Thus, PKA anchored directly to the Cav1.2 channel by AKAP15 via a leucine-zipper interaction is required for regulation of Ca2+ channels in cardiac myocytes by the autonomic nervous system.
Figure 3. The cardiac Ca2+ channel signaling complex.
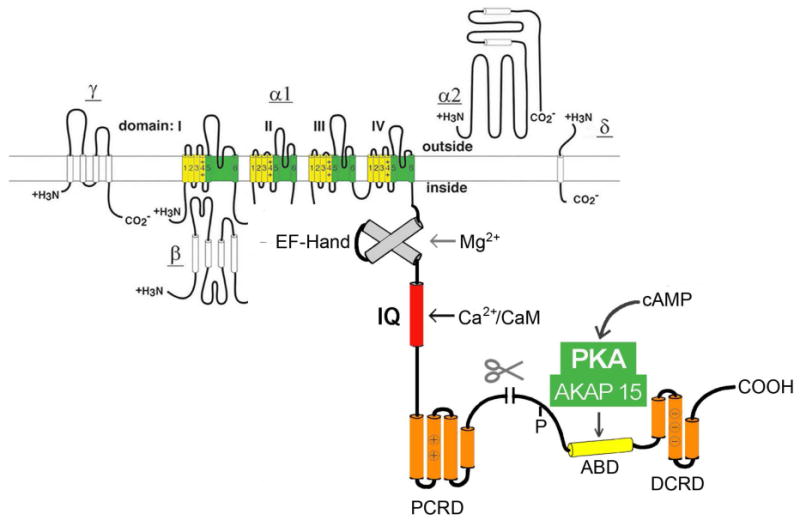
The CaV1.2 channel is illustrated as a transmembrane folding diagram. Proteolytic processing of the distal C-terminal domain is indicated. ABD, AKAP15 binding domain; DCRD, distal C-terminal regulatory domain; PCRD, proximal C-terminal regulatory domain; scissors, site of proteolytic processing.
An autoinhibitory signaling complex of CaV1.1 and CaV1.2 channels
The distal C-terminal domains of skeletal muscle and cardiac Ca2+ channels are proteolytically processed in vivo [101, 102, 115, 125] at a specific site, Ala1664 in CaV1.1 channels, which is conserved at position 1700 in CaV1.2 channels [125]. Nevertheless, the most prominent PKA phosphorylation sites of both proteins are located beyond the site of proteolytic truncation [103, 104, 115], and interaction of AKAP15 and PKA with the distal C-terminal domain through a leucine zipper motif is required for regulation of cardiac Ca2+ channels in intact myocytes [124]. These results imply that the distal C-terminal domain remains associated with the proteolytically processed cardiac CaV1.2 channel [126, 127]. The distal C-terminal domain of both CaV1.1 and CaV1.2 channels bind to the proximal C-terminal domain in a specific complex [125, 128]. Moreover, formation of this complex dramatically inhibits cardiac Ca2+ channel function [128]. Deletion of the distal C-terminal near the site of proteolytic processing increases Ca2+ channel activity [128, 129]; however, noncovalent association of the cleaved distal C-terminal reduces channel activity more than several fold, to a level much below that of Ca2+ channels with an intact C-terminus [128]. These effects are caused primarily by reduction of the coupling of voltage sensing to channel opening [128]. Thus, proteolytic processing produces an autoinhibited Ca2+ channel complex containing noncovalently bound distal C-terminus with AKAP15 and PKA associated through a modified leucine zipper interaction. This autoinhibited complex is primary substrate for regulation of cardiac Ca2+ channels by the β-adrenergic receptor/PKA pathway in vivo [128].
Reconstitution of regulation of CaV1.2 channels in the fight-or-flight response
The definitive test of our understanding of Ca2+ channel regulation in the fight-or-flight response is reconstitution of that regulation in a non-muscle cell from well-defined molecular components. Reconstitution of regulation of CaV1.2 channels in nonmuscle cells has been difficult to achieve, perhaps because their α1 subunits are not proteolytically processed in heterologous cells [130]. Formation of a stoichiometric complex of truncated CaV1.2 channels, their distal C-terminal domain, and AKAP15 by controlled expression of each component as a separate protein successfully restores physiologically normal regulation of CaV1.2 channels by the PKA signaling pathway [131]. Parallel experiments with full-length α1 subunits do not yield substantial channel regulation. These results provide direct evidence that the autoinhibited signaling complex of truncated Ca2+ channel α1 subunits, their noncovalently associated distal C-terminal domain, and AKAP15 is the physiological PKA substrate for regulation of Ca2+ channels in the fight-or flight response in vivo.
The Effector Checkpoint Hypothesis for Ca2+ Channel Regulation
Three well-defined examples of Ca2+ channel regulation suggest the effector-checkpoint hypothesis for Ca2+ channel regulation [89]. Skeletal muscle CaV1.1 channels in transverse tubules interact directly with the ryanodine-sensitive Ca2+ release channels in the sarcoplasmic reticulum, which serve as their effectors in excitation-contraction coupling [132]. Deletion of the gene for the ryanodine-sensitive Ca2+ release channel dramatically reduces the activity of the CaV1.1 channels [133]. Thus, effector interaction enhances Ca2+ channel activity in this case. SNARE proteins that are involved in docking and exocytosis of neurotransmitter vesicles are the effectors of the Ca2+ signal conducted by presynaptic Ca2+ channels. Moreover, the SNARE proteins regulate CaV2.1 and CaV2.2 channels in a biphasic manner [134-137]. The plasma membrane SNARE proteins syntaxin and SNAP-25 inhibit Ca2+ channel activity by shifting the voltage dependence of inactivation to more negative membrane potentials, but formation of a complete SNARE complex with synaptobrevin and synaptotagmin relieves the inhibition and enhances Ca2+ channel activation. Evidently, formation of a complete SNARE complex, which serves as the effector of the Ca2+ signal in synaptic transmission, increases both CaV2.1 and CaV2.2 channel activity. Finally, an effector of the Ca2+ signal, CaMKII, up-regulates the activity of presynaptic CaV2.1 channels when bound. This regulation increases the activity of those CaV2.1 channels whose Ca2+ signal would be utilized physiologically by CaMKII. In all three of these examples, the regulation is organized to enhance Ca2+ entry through channels that have appropriate effectors bound in place. Thus, this mechanism serves as an ‘effector-checkpoint’ to ensure the functional fitness of individual Ca2+ channel molecules for signal transduction to bound effector proteins like ryanodine receptors, SNARE proteins, and CaMKII before they are allowed to open with high efficiency. This regulatory process serves to focus Ca2+ entry on those Ca2+ channels that are poised to use the Ca2+ signal most effectively and thereby limits unproductive Ca2+ entry that might be deleterious.
Perspective
The idea that ion channels are multi-protein complexes stems from early biochemical studies showing that they are multi-subunit proteins and now is amplified by the extensive evidence for signaling complexes that are nucleated by ion channels. The work reviewed here shows that voltage-gated Na+ and Ca2+ channels form specific signaling complexes that are essential for their physiological functions and for their regulation. Electrical signals generated by Na+ and Ca2+ channels are inherently local. Similarly, the Ca2+ transients generated by activation of Ca2+ channels by electrical signals are also local, forming Ca2+ microdomains of molecular dimensions. Evidently, these local signaling events require locally bound effector proteins and regulatory proteins for effective signal transduction. Understanding receptor signaling to and through ion channels will require determination of the structure of ion channel signaling complexes and the functional significance of each member of these complexes.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Catterall WA. Structure and regulation of voltage-gated calcium channels. Annu Rev Cell Dev Bio. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 2.Cantrell AR, Catterall WA. Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat Rev Neurosci. 2001;2:397–407. doi: 10.1038/35077553. [DOI] [PubMed] [Google Scholar]
- 3.Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- 4.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qu Y, Curtis R, Lawson D, Gilbride K, Ge P, DiStefano PS, Silos-Santiago I, Catterall WA, Scheuer T. Differential modulation of sodium channel gating and persistent sodium currents by the beta1, beta2, and beta3 subunits. Mol Cell Neurosci. 2001;18:570–580. doi: 10.1006/mcne.2001.1039. [DOI] [PubMed] [Google Scholar]
- 6.Yu FH, Westenbroek RE, Silos-Santiago I, McCormick KA, Lawson D, Ge P, Ferriera H, Lilly J, DiStefano PS, Catterall WA, et al. Sodium channel beta4, a new disulfide-linked auxiliary subunit with similarity to beta2. J Neurosci. 2003;23:7577–7585. doi: 10.1523/JNEUROSCI.23-20-07577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costa MR, Casnellie JE, Catterall WA. Selective phosphorylation of the alpha subunit of the sodium channel by cAMP-dependent protein kinase. J Biol Chem. 1982;257:7918–7921. [PubMed] [Google Scholar]
- 8.Murphy BJ, Rossie S, De Jongh KS, Catterall WA. Identification of the sites of selective phosphorylation and dephosphorylation of the rat brain Na+ channel α subunit by cAMP-dependent protein kinase and phosphoprotein phosphatases. J Biol Chem. 1993;268:27355–27362. [PubMed] [Google Scholar]
- 9.Costa MR, Catterall WA. Cyclic AMP-dependent phosphorylation of the alpha subunit of the sodium channel in synaptic nerve ending particles. J Biol Chem. 1984;259:8210–8218. [PubMed] [Google Scholar]
- 10.Rossie S, Catterall WA. Cyclic-AMP-dependent phosphorylation of voltage-sensitive sodium channels in primary cultures of rat brain neurons. J Biol Chem. 1987;262:12735–12744. [PubMed] [Google Scholar]
- 11.Rossie S, Catterall WA. Phosphorylation of the alpha subunit of rat brain sodium channels by cAMP-dependent protein kinase at a new site containing Ser686 and Ser687. J Biol Chem. 1989;264:14220–14224. [PubMed] [Google Scholar]
- 12.Rossie S, Gordon D, Catterall WA. Identification of an intracellular domain of the sodium channel having multiple cAMP-dependent phosphorylation sites. J Biol Chem. 1987;262:17530–17535. [PubMed] [Google Scholar]
- 13.Smith RD, Goldin AL. Phosphorylation of brain sodium channels in the I-II linker modulates channel function in Xenopus oocytes. J Neurosci. 1996;16:1965–1974. doi: 10.1523/JNEUROSCI.16-06-01965.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li M, West JW, Lai Y, Scheuer T, Catterall WA. Functional modulation of brain sodium channels by cAMP-dependent phosphyrlation. Neuron. 1992;8:1151–1159. doi: 10.1016/0896-6273(92)90135-z. [DOI] [PubMed] [Google Scholar]
- 15.Cantrell AR, Scheuer T, Catterall WA. Dopaminergic modulation of sodium current in hippocampal neurons via cAMP-dependent phosphorylation of specific sites in the sodium channel α subunit. J Neurosci. 1997;17:7330–7338. doi: 10.1523/JNEUROSCI.17-19-07330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith RD, Goldin AL. Phosphorylation at a single site in the rat brain sodium channel is necessary and sufficient for current reduction by protein kinase A. J Neurosci. 1997;17:6086–6093. doi: 10.1523/JNEUROSCI.17-16-06086.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li M, West JW, Numann R, Murphy BJ, Scheuer T, Catterall WA. Convergent regulation of sodium channels by protein kinase C and cAMP-dependent protein kinase. Science. 1993;261:1439–1442. doi: 10.1126/science.8396273. [DOI] [PubMed] [Google Scholar]
- 18.Cantrell AR, Scheuer T, Catterall WA. Voltage-dependent neuromodulation of Na+ channels by D1-like dopamine receptors in rat hippocampal neurons. J Neurosci. 1999;19:5301–5310. doi: 10.1523/JNEUROSCI.19-13-05301.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cantrell AR, Tibbs VC, Yu FH, Murphy BJ, Sharp EM, Qu Y, Catterall WA, Scheuer T. Molecular mechanism of convergent regulation of brain Na+ channels by protein kinase C and protein kinase A anchored to AKAP-15. Mol Cell Neurosci. 2002;21:63–80. doi: 10.1006/mcne.2002.1162. [DOI] [PubMed] [Google Scholar]
- 20.Carr DB, Day M, Cantrell AR, Held J, Scheuer T, Catterall WA, Surmeier DJ. Transmitter modulation of slow, activity-dependent alterations in sodium channel availability endows neurons with a novel form of cellular plasticity. Neuron. 2003;39:793–806. doi: 10.1016/s0896-6273(03)00531-2. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Yu FH, Surmeier DJ, Scheuer T, Catterall WA. Neuromodulation of Na+ channel slow inactivation via cAMP-dependent protein kinase and protein kinase C. Neuron. 2006;49:409–420. doi: 10.1016/j.neuron.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 22.Scott JD, McCartney S. Localization of A-kinase through anchoring proteins. Mol Endocrinol. 1994;8:5–11. doi: 10.1210/mend.8.1.8152430. [DOI] [PubMed] [Google Scholar]
- 23.Rubin CS. A kinase anchor proteins and the intracellular targeting of signals carried by cyclic AMP. Biochim Biophys Acta. 1994;1224:467–479. [PubMed] [Google Scholar]
- 24.Newlon MG, Roy M, Morikis D, Carr DW, Westphal R, Scott JD, Jennings PA. A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. EMBO J. 2001;20:1651–1662. doi: 10.1093/emboj/20.7.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newlon MG, Roy M, Hausken ZE, Scott JD, Jennings PA. The A-kinase anchoring domain of type II alpha cAMP-dependent protein kinase is highly helical. J Biol Chem. 1997;272:23637–23644. doi: 10.1074/jbc.272.38.23637. [DOI] [PubMed] [Google Scholar]
- 26.Cantrell AR, Tibbs VC, Westenbroek RE, Scheuer T, Catterall WA. Dopaminergic modulation of voltage-gated Na+ current in rat hippocampal neurons requires anchoring of cAMP-dependent protein kinase. J Neurosci. 1999;19:RC21. doi: 10.1523/JNEUROSCI.19-17-j0003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Few WP, Scheuer T, Catterall WA. Dopamine modulation of neuronal Na (+) channels requires binding of A kinase-anchoring protein 15 and PKA by a modified leucine zipper motif. Proc Natl Acad Sci U S A. 2007;104:5187–5192. doi: 10.1073/pnas.0611619104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahn M, Beacham D, Westenbroek RE, Scheuer T, Catterall WA. Regulation of NaV1.2 channels by brain-derived neurotrophic factor, TrkB, and associated Fyn kinase. J Neurosci. 2007;27:11533–11542. doi: 10.1523/JNEUROSCI.5005-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beacham D, Ahn M, Catterall WA, Scheuer T. Sites and molecular mechanisms of modulation of NaV1.2 channels by Fyn tyrosine kinase. J Neurosci. 2007;27:11543–11551. doi: 10.1523/JNEUROSCI.1743-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ratcliffe CF, Qu Y, McCormick KA, Tibbs VC, Dixon JE, Scheuer T, Catterall WA. A sodium channel signaling complex: modulation by associated receptor protein tyrosine phosphatase beta. Nat Neurosci. 2000;3:437–444. doi: 10.1038/74805. [DOI] [PubMed] [Google Scholar]
- 31.Ratcliffe CF, Westenbroek RE, Curtis R, Catterall WA. Sodium channel beta1 and beta3 subunits associate with neurofascin through their extracellular immunoglobulin-like domain. J Cell Biol. 2001;154:427–434. doi: 10.1083/jcb.200102086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brackenbury WJ, Calhoun JD, Chen C, Miyazaki H, Nukina N, Oyama F, Ranscht B, Isom LL. Functional reciprocity between Na+ channel Nav1.6 and beta1 subunits in the coordinated regulation of excitability and neurite outgrowth. Proc Natl Acad Sci U S A. 2010;107:2283–2288. doi: 10.1073/pnas.0909434107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brackenbury WJ, Davis TH, Chen C, Slat EA, Detrow MJ, Dickendesher TL, Ranscht B, Isom LL. Voltage-gated Na+ channel beta1 subunit-mediated neurite outgrowth requires Fyn kinase and contributes to postnatal CNS development in vivo. J Neurosci. 2008;28:3246–3256. doi: 10.1523/JNEUROSCI.5446-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kazarinova-Noyes K, Malhotra JD, McEwen DP, Mattei LN, Berglund EO, Ranscht B, Levinson SR, Schachner M, Shrager P, Isom LL, et al. Contactin associates with Na+ channels and increases their functional expression. J Neurosci. 2001;21:7517–7525. doi: 10.1523/JNEUROSCI.21-19-07517.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srinivasan J, Schachner M, Catterall WA. Interaction of voltage-gated sodium channels with the extracellular matrix molecules tenascin-C and tenascin-R. Proc Natl Acad Sci U S A. 1998;95:15753–15757. doi: 10.1073/pnas.95.26.15753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malhotra JD, Kazen-Gillespie K, Hortsch M, Isom LL. Sodium channel beta subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell-cell contact. J Biol Chem. 2000;275:11383–11388. doi: 10.1074/jbc.275.15.11383. [DOI] [PubMed] [Google Scholar]
- 37.Xiao ZC, Ragsdale DS, Malhotra JD, Mattei LN, Braun PE, Schachner M, Isom LL. Tenascin-R is a functional modulator of sodium channel beta subunits. J Biol Chem. 1999;274:26511–26517. doi: 10.1074/jbc.274.37.26511. [DOI] [PubMed] [Google Scholar]
- 38.Zhou D, Lambert S, Malen PL, Carpenter S, Boland LM, Bennett V. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol. 1998;143:1295–1304. doi: 10.1083/jcb.143.5.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bajjalieh SM, Scheller RH. The biochemistry of neurotransmitter secretion. J Biol Chem. 1995;270:1971–1974. doi: 10.1074/jbc.270.5.1971. [DOI] [PubMed] [Google Scholar]
- 40.Sudhof TC. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- 41.Sheng ZH, Rettig J, Takahashi M, Catterall WA. Identification of a syntaxin-binding site on N-type calcium channels. Neuron. 1994;13:1303–1313. doi: 10.1016/0896-6273(94)90417-0. [DOI] [PubMed] [Google Scholar]
- 42.Rettig J, Sheng ZH, Kim DK, Hodson CD, Snutch TP, Catterall WA. Isoform-specific interaction of the α1A subunits of brain Ca2+ channels with the presynaptic proteins syntaxin and SNAP-25. Proc Natl Acad Sci U S A. 1996;93:7363–7368. doi: 10.1073/pnas.93.14.7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheng ZH, Rettig J, Cook T, Catterall WA. Calcium-dependent interaction of N-type calcium channels with the synaptic core-complex. Nature. 1996;379:451–454. doi: 10.1038/379451a0. [DOI] [PubMed] [Google Scholar]
- 44.Yokoyama CT, Sheng ZH, Catterall WA. Phosphorylation of the synaptic protein interaction site on N-type calcium channels inhibits interactions with SNARE proteins. J Neurosci. 1997;17:6929–6938. doi: 10.1523/JNEUROSCI.17-18-06929.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yokoyama CT, Myers SJ, Fu J, Mockus SM, Scheuer T, Catterall WA. Mechanism of SNARE protein binding and regulation of CaV2 channels by phosphorylation of the synaptic protein interaction site. Mol Cell Neurosci. 2005;28:1–17. doi: 10.1016/j.mcn.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 46.Charvin N, Lévêque C, Walker D, Berton F, Raymond C, Kataoka M, Shoji-Kasai Y, Takahashi M, De Waard M, Seagar MJ. Direct interaction of the calcium sensor protein synaptotagmin I with a cytoplasmic domain of the α1A subunit of the P/Q-type calcium channel. EMBO J. 1997;16:4591–4596. doi: 10.1093/emboj/16.15.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sheng ZH, Yokoyama C, Catterall WA. Interaction of the synprint site of N-type Ca2+ channels with the C2B domain of synaptotagmin I. Proc Natl Acad Sci U S A. 1997;94:5405–5410. doi: 10.1073/pnas.94.10.5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wiser O, Tobi D, Trus M, Atlas D. Synaptotagmin restores kinetic properties of a syntaxin-associated N-type voltage sensitive calcium channel. FEBS Lett. 1997;404:203–207. doi: 10.1016/s0014-5793(97)00130-0. [DOI] [PubMed] [Google Scholar]
- 49.Rettig J, Heinemann C, Ashery U, Sheng ZH, Yokoyama CT, Catterall WA, Neher E. Alteration of Ca2+ dependence of neurotransmitter release by disruption of Ca2+ channel/syntaxin interaction. J Neurosci. 1997;17:6647–6656. doi: 10.1523/JNEUROSCI.17-17-06647.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mochida S, Sheng ZH, Baker C, Kobayashi H, Catterall WA. Inhibition of neurotransmission by peptides containing the synaptic protein interaction site of N-type Ca2+ channels. Neuron. 1996;17:781–788. doi: 10.1016/s0896-6273(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 51.Bezprozvanny I, Scheller RH, Tsien RW. Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature. 1995;378:623–626. doi: 10.1038/378623a0. [DOI] [PubMed] [Google Scholar]
- 52.Wiser O, Bennett MK, Atlas D. Functional interaction of syntaxin and SNAP-25 with voltage-sensitive L- and N-type Ca2+ channels. EMBO J. 1996;15:4100–4110. [PMC free article] [PubMed] [Google Scholar]
- 53.Tobi D, Wiser O, Trus M, Atlas D. N-type voltage-sensitive calcium channel interacts with syntaxin, synaptotagmin and SNAP-25 in a multiprotein complex. Receptors and Channels. 1999;6:89–98. [PubMed] [Google Scholar]
- 54.Zhong H, Yokoyama C, Scheuer T, Catterall WA. Reciprocal regulation of P/Q-type Ca2+ channels by SNAP-25, syntaxin and synaptotagmin. Nat Neurosci. 1999;2:939–941. doi: 10.1038/14721. [DOI] [PubMed] [Google Scholar]
- 55.Keith RK, Poage RE, Yokoyama CT, Catterall WA, Meriney SD. Bidirectional modulation of transmitter release by calcium channel/syntaxin interactions in vivo. J Neurosci. 2007;27:265–269. doi: 10.1523/JNEUROSCI.4213-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 57.Ikeda SR, Dunlap K. Voltage-dependent modulation of N-type calcium channels: role of G protein subunits. Adv Second Messenger Phosphoprotein Res. 1999;33:131–151. doi: 10.1016/s1040-7952(99)80008-1. [DOI] [PubMed] [Google Scholar]
- 58.Jones LP, Patil PG, Snutch TP, Yue DT. G-protein modulation of N-type calcium channel gating current in human embryonic kidney cells (HEK 293) J Physiol (Lond) 1997;498:601–610. doi: 10.1113/jphysiol.1997.sp021886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- 60.Marchetti C, Carbone E, Lux HD. Effects of dopamine and noradrenaline on Ca channels of cultured sensory and sympathetic neurons of chick. Pflugers Arch. 1986;406:104–111. doi: 10.1007/BF00586670. [DOI] [PubMed] [Google Scholar]
- 61.Brody DL, Yue DT. Relief of G-protein inhibition of calcium channels and short-term synaptic facilitation in cultured hippocampal neurons. J Neurosci. 2000;20:889–898. doi: 10.1523/JNEUROSCI.20-03-00889.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G protein βγ subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- 63.Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- 64.De Waard M, Liu HY, Walker D, Scott VES, Gurnett CA, Campbell KP. Direct binding of G-protein βγ complex to voltage-dependent calcium channels. Nature. 1997;385:446–450. doi: 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- 65.Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- 66.Herlitze S, Hockerman GH, Scheuer T, Catterall WA. Molecular determinants of inactivation and G protein modulation in the intracelular loop connecting domains I and II of the calcium channel α1A subunit. Proc Natl Acad Sci U S A. 1997;94:1512–1516. doi: 10.1073/pnas.94.4.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Garcia DE, Li B, Garcia-Ferreiro RE, Hernández-Ochoa EO, Yan K, Gautam N, Catterall WA, Mackie K, Hille B. G-protein β-subunit specificity in the fast membrane-delimited inhibition of Ca2+ channels. J Neurosci. 1998;18:9163–9170. doi: 10.1523/JNEUROSCI.18-22-09163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang JF, Ellinor PT, Aldrich RW, Tsien RW. Multiple structural elements in voltage-dependent Ca2+ channels support their inhibition by G proteins. Neuron. 1996;17:991–1003. doi: 10.1016/s0896-6273(00)80229-9. [DOI] [PubMed] [Google Scholar]
- 69.Qin N, Platano D, Olcese R, Stefani E, Birnbaumer L. Direct interaction of Gβγ with a C-terminal Gβγ-binding domain of the Ca2+ channel α1 subunit is responsible for channel inhibition by G protein-coupled receptors. Proc Natl Acad Sci U S A. 1997;94:8866–8871. doi: 10.1073/pnas.94.16.8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Page KM, Stephens GJ, Berrow NS, Dolphin AC. The intracellular loop between domains I and II of the B-type calcium channel confers aspects of G-protein sensitivity to the E-type calcium channel. J Neurosci. 1997;17:1330–1338. doi: 10.1523/JNEUROSCI.17-04-01330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Page KM, Cantí C, Stephens GJ, Berrow NS, Dolphin AC. Identification of the amino terminus of neuronal Ca2+ channel α1 subunits α1B and α1E as an essential determinant of G-protein modulation. J Neurosci. 1998;18:4815–4824. doi: 10.1523/JNEUROSCI.18-13-04815.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Canti C, Page KM, Stephens GJ, Dolphin AC. Identification of residues in the N terminus of alpha 1B critical for inhibition of the voltage-dependent calcium channel by Gβγ. J Neurosci. 1999;19:6855–6864. doi: 10.1523/JNEUROSCI.19-16-06855.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li B, Zhong H, Scheuer T, Catterall WA. Functional role of a C-terminal Gbetagamma-binding domain of Ca(v)2.2 channels. Mol Pharmacol. 2004;66:761–769. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- 74.Swartz KJ, Merritt A, Bean BP, Lovinger DM. Protein kinase C modulates glutamate receptor inhibition of Ca2+ channels and synaptic transmission. Nature. 1993;361:165–168. doi: 10.1038/361165a0. [DOI] [PubMed] [Google Scholar]
- 75.Swartz KJ. Modulation of Ca2+ channels by protein kinase C in rat central and peripheral neurons: Disruption of G protein-mediated inhibition. Neuron. 1993;11:305–320. doi: 10.1016/0896-6273(93)90186-u. [DOI] [PubMed] [Google Scholar]
- 76.Branchaw JL, Banks MI, Jackson MB. Ca2+- and voltage-dependent inactivation of Ca2+ channels in nerve terminals of the neurohypophysis. J Neurosci. 1997;17:5772–5781. doi: 10.1523/JNEUROSCI.17-15-05772.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/s0896-6273(00)81017-x. [DOI] [PubMed] [Google Scholar]
- 78.Borst JG, Sakmann B. Facilitation of presynaptic calcium currents in the rat brainstem. J Physiol. 1998;513:149–155. doi: 10.1111/j.1469-7793.1998.149by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cuttle MF, Tsujimoto T, Forsythe ID, Takahashi T. Facilitation of the presynaptic calcium current at an auditory synapse in rat brainstem. J Physiol. 1998;512:723–729. doi: 10.1111/j.1469-7793.1998.723bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, Catterall WA. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- 81.Lee A, Scheuer T, Catterall WA. Ca2+-Calmodulin dependent inactivation and facilitation of P/Q-type Ca2+ channels. Biophys J. 2000;78:265A. doi: 10.1523/JNEUROSCI.20-18-06830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee A, Zhou H, Scheuer T, Catterall WA. Molecular determinants of Ca2+/calmodulin-dependent regulation of CaV2.1 channels. Proc Natl Acad Sci U S A. 2003;100:16059–16064. doi: 10.1073/pnas.2237000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- 84.Erickson MG, Alseikhan BA, Peterson BZ, Yue DT. Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron. 2001;31:973–985. doi: 10.1016/s0896-6273(01)00438-x. [DOI] [PubMed] [Google Scholar]
- 85.Haeseleer F, Palczewski K. Calmodulin and Ca2+-binding proteins (CaBPs): variations on a theme. Adv Exp Med Biol. 2002;514:303–317. doi: 10.1007/978-1-4615-0121-3_18. [DOI] [PubMed] [Google Scholar]
- 86.Lee A, Westenbroek RE, Haeseleer F, Palczewski K, Scheuer T, Catterall WA. Differential modulation of CaV2.1 channels by calmodulin and Ca2+-binding protein 1. Nat Neurosci. 2002;5:210–217. doi: 10.1038/nn805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Few AP, Lautermilch NJ, Westenbroek RE, Scheuer T, Catterall WA. Differential regulation of CaV2.1 channels by calcium-binding protein 1 and visinin-like protein-2 requires N-terminal myristoylation. J Neurosci. 2005;25:7071–7080. doi: 10.1523/JNEUROSCI.0452-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lautermilch NJ, Few AP, Scheuer T, Catterall WA. Modulation of CaV2.1 channels by the neuronal calcium-binding protein visinin-like protein-2. J Neurosci. 2005;25:7062–7070. doi: 10.1523/JNEUROSCI.0447-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jiang X, Lautermilch NJ, Watari H, Westenbroek RE, Scheuer T, Catterall WA. Modulation of CaV2.1 channels by Ca2+/calmodulin-dependent protein kinase II bound to the C-terminal domain. Proc Natl Acad Sci U S A. 2008;105:341–346. doi: 10.1073/pnas.0710213105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mochida S, Few AP, Scheuer T, Catterall WA. Regulation of presynaptic CaV2.1 channels by Ca2+ sensor proteins mediates short-term synaptic plasticity. Neuron. 2008;57:210–216. doi: 10.1016/j.neuron.2007.11.036. [DOI] [PubMed] [Google Scholar]
- 91.Catterall WA, Few AP. Calcium channel regulation and presynaptic plasticity. Neuron. 2008;59:882–901. doi: 10.1016/j.neuron.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 92.Sippy T, Cruz-Martin A, Jeromin A, Schweizer FE. Acute changes in short-term plasticity at synapses with elevated levels of neuronal calcium sensor-1. Nat Neurosci. 2003;6:1031–1038. doi: 10.1038/nn1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tsujimoto T, Jeromin A, Saitoh N, Roder JC, Takahashi T. Neuronal calcium sensor 1 and activity-dependent facilitation of P/Q-type calcium currents at presynaptic nerve terminals. Science. 2002;295:2276–2279. doi: 10.1126/science.1068278. [DOI] [PubMed] [Google Scholar]
- 94.Muller CS, Haupt A, Bildl W, Schindler J, Knaus HG, Meissner M, Rammner B, Striessnig J, Flockerzi V, Fakler B, et al. Feature Article: Quantitative proteomics of the CaV2 channel nano-environments in the mammalian brain. Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.1005940107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Khanna R, Zougman A, Stanley EF. A proteomic screen for presynaptic terminal N-type calcium channel (CaV2.2) binding partners. J Biochem Mol Biol. 2007;40:302–314. doi: 10.5483/bmbrep.2007.40.3.302. [DOI] [PubMed] [Google Scholar]
- 96.Curtis BM, Catterall WA. Purification of the calcium antagonist receptor of the voltage-sensitive calcium channel from skeletal muscle transverse tubules. Biochem. 1984;23:2113–2118. doi: 10.1021/bi00305a001. [DOI] [PubMed] [Google Scholar]
- 97.Flockerzi V, Oeken HJ, Hofmann F. Purification of a functional receptor for calcium channel blockers from rabbit skeletal muscle microsomes. Eur J Biochem. 1986;161:217–224. doi: 10.1111/j.1432-1033.1986.tb10145.x. [DOI] [PubMed] [Google Scholar]
- 98.Takahashi M, Seagar MJ, Jones JF, Reber BF, Catterall WA. Subunit structure of dihydropyridine-sensitive calcium channels from skeletal muscle. Proc Natl Acad Sci U S A. 1987;84:5478–5482. doi: 10.1073/pnas.84.15.5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Curtis BM, Catterall WA. Phosphorylation of the calcium antagonist receptor of the voltage-sensitive calcium channel by cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1985;82:2528–2532. doi: 10.1073/pnas.82.8.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Flockerzi V, Jaimovich P, Ruth P, Hofmann F. Phosphorylation of purified bovine cardiac sarcolemma and potassium-stimulated calcium uptake. Eur J Biochem. 1983;135:132–142. doi: 10.1111/j.1432-1033.1983.tb07628.x. [DOI] [PubMed] [Google Scholar]
- 101.De Jongh KS, Merrick DK, Catterall WA. Subunits of purified calcium channels: a 212-kDa form of α1 and partial amino acid sequence of a phosphorylation site of an independent β subunit. Proc Natl Acad Sci U S A. 1989;86:8585–8589. doi: 10.1073/pnas.86.21.8585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.De Jongh KS, Warner C, Colvin AA, Catterall WA. Characterization of the two size forms of the α1 subunit of skeletal muscle L-type calcium channels. Proc Natl Acad Sci U S A. 1991;88:10778–10782. doi: 10.1073/pnas.88.23.10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rotman EI, De Jongh KS, Florio V, Lai Y, Catterall WA. Specific phosphorylation of a COOH-terminal site on the full-length form of the α1 subunit of the skeletal muscle calcium channel by cAMP-dependent protein kinase. J Biol Chem. 1992;267:16100–16105. [PubMed] [Google Scholar]
- 104.Rotman EI, Murphy BJ, Catterall WA. Sites of selective cAMP-dependent phosphorylation of the L-type calcium channel α1 subunit from intact rabbit skeletal muscle myotubes. J Biol Chem. 1995;270:16371–16377. doi: 10.1074/jbc.270.27.16371. [DOI] [PubMed] [Google Scholar]
- 105.Sculptoreanu A, Scheuer T, Catterall WA. Voltage-dependent potentiation of L-type Ca2+ channels due to phosphorylation by cAMP-dependent protein kinase. Nature. 1993;364:240–243. doi: 10.1038/364240a0. [DOI] [PubMed] [Google Scholar]
- 106.Johnson BD, Brousal JP, Peterson BZ, Gallombardo PA, Hockerman GH, Lai Y, Scheuer T, Catterall WA. Modulation of the cloned skeletal muscle L-type Ca2+ channel by anchored cAMP-dependent protein kinase. J Neurosci. 1997;17:1243–1255. doi: 10.1523/JNEUROSCI.17-04-01243.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Johnson BD, Scheuer T, Catterall WA. Voltage-dependent potentiation of L-type Ca2+ channels in skeletal muscle cells requires anchored cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1994;91:11492–11496. doi: 10.1073/pnas.91.24.11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gray PC, Tibbs VC, Catterall WA, Murphy BJ. Identification of a 15-kDa cAMP-dependent protein kinase-anchoring protein associated with skeletal muscle L-type calcium channels. J Biol Chem. 1997;272:6297–6302. doi: 10.1074/jbc.272.10.6297. [DOI] [PubMed] [Google Scholar]
- 109.Gray PC, Johnson BD, Westenbroek RE, Hays LG, Yates JR, 3rd, Scheuer T, Catterall WA, Murphy BJ. Primary structure and function of an A kinase anchoring protein associated with calcium channels. Neuron. 1998;20:1017–1026. doi: 10.1016/s0896-6273(00)80482-1. [DOI] [PubMed] [Google Scholar]
- 110.Fraser IDC, Tavalin SJ, Lester LB, Langeberg LK, Westphal AM, Dean RA, Marrion NV, Scott JD. A novel lipid-anchored A-kinase anchoring protein facilitates cAMP-responsive membrane events. EMBO J. 1998;17:2261–2272. doi: 10.1093/emboj/17.8.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hulme JT, Ahn M, Hauschka SD, Scheuer T, Catterall WA. A novel leucine zipper targets AKAP15 and cyclic AMP-dependent protein kinase to the C terminus of the skeletal muscle Ca2+ channel and modulates its function. J Biol Chem. 2002;277:4079–4087. doi: 10.1074/jbc.M109814200. [DOI] [PubMed] [Google Scholar]
- 112.Reuter H. Calcium channel modulation by neurotransmitters, enzymes and drugs. Nature. 1983;301:569–574. doi: 10.1038/301569a0. [DOI] [PubMed] [Google Scholar]
- 113.Tsien RW, Bean BP, Hess P, Lansman JB, Nilius B, Nowycky MC. Mechanisms of calcium channel modulation by beta-adrenergic agents and dihydropyridine calcium agonists. J Mol Cell Cardiol. 1986;18:691–710. doi: 10.1016/s0022-2828(86)80941-5. [DOI] [PubMed] [Google Scholar]
- 114.Hell JW, Yokoyama CT, Wong ST, Warner C, Snutch TP, Catterall WA. Differential phosphorylation of two size forms of the neuronal class C L-type calcium channel α1 subunit. J Biol Chem. 1993;268:19451–19457. [PubMed] [Google Scholar]
- 115.De Jongh KS, Murphy BJ, Colvin AA, Hell JW, Takahashi M, Catterall WA. Specific phosphorylation of a site in the full-length form of the α1 subunit of the cardiac L-type calcium channel by cAMP-dependent protein kinase. Biochemistry. 1996;35:10392–10402. doi: 10.1021/bi953023c. [DOI] [PubMed] [Google Scholar]
- 116.Puri TS, Gerhardstein BL, Zhao XL, Ladner MB, Hosey MM. Differential effects of subunit interactions on protein kinase A- and C-mediated phosphorylation of L-type calcium channels. Biochemistry. 1997;36:9605–9615. doi: 10.1021/bi970500d. [DOI] [PubMed] [Google Scholar]
- 117.Haase H, Bartel S, Karczewski P, Morano I, Krause EG. In-vivo phosphorylation of the cardiac L-type calcium channel beta-subunit in response to catecholamines. Mol Cell Biochem. 1996;163-164:99–106. doi: 10.1007/BF00408645. [DOI] [PubMed] [Google Scholar]
- 118.Mitterdorfer J, Froschmayr M, Grabner M, Moebius FF, Glossmann H, Striessnig J. Identification of PKA phosphorylation sites in the carboxyl terminus of L-type calcium channel α1 subunits. Biochemistry. 1996;35:9400–9406. doi: 10.1021/bi960683o. [DOI] [PubMed] [Google Scholar]
- 119.Reuter H, Scholz H. The regulation of calcium conductance of cardiac muscle by adrenaline. J Physiol. 1977;264:49–62. doi: 10.1113/jphysiol.1977.sp011657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tsien RW. Adrenaline-like effects of intracellular iontophoresis of cyclic AMP in cardiac Purkinje fibres. Nature New Biol. 1973;245:120–122. doi: 10.1038/newbio245120a0. [DOI] [PubMed] [Google Scholar]
- 121.Osterrieder W, Brum G, Hescheler J, Trautwein W, Flockerzi V, Hofmann F. Injection of subunits of cyclic AMP-dependent protein kinase into cardiac myocytes modulates Ca2+ current. Nature. 1982;298:576–578. doi: 10.1038/298576a0. [DOI] [PubMed] [Google Scholar]
- 122.McDonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol Rev. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- 123.Gao T, Yatani A, DellAcqua ML, Sako H, Green SA, Dascal N, Scott JD, Hosey MM. cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron. 1997;19:185–196. doi: 10.1016/s0896-6273(00)80358-x. [DOI] [PubMed] [Google Scholar]
- 124.Hulme JT, Lin TW, Westenbroek RE, Scheuer T, Catterall WA. Beta-adrenergic regulation requires direct anchoring of PKA to cardiac CaV1.2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proc Natl Acad Sci U S A. 2003;100:13093–13098. doi: 10.1073/pnas.2135335100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hulme JT, Konoki K, Lin TW, Gritsenko MA, Camp DG, 2nd, Bigelow DJ, Catterall WA. Sites of proteolytic processing and noncovalent association of the distal C-terminal domain of CaV1.1 channels in skeletal muscle. Proc Natl Acad Sci U S A. 2005;102:5274–5279. doi: 10.1073/pnas.0409885102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gao T, Cuadra AE, Ma H, Bunemann M, Gerhardstein BL, Cheng T, Eick RT, Hosey MM. C-terminal fragments of the α1C (Cav1.2) subunit associate with and regulate L-type calcium channels containing C-terminal-truncated α1C subunits. J Biol Chem. 2001;276:21089–21097. doi: 10.1074/jbc.M008000200. [DOI] [PubMed] [Google Scholar]
- 127.Gerhardstein BL, Gao T, Bunemann M, Puri TS, Adair A, Ma H, Hosey MM. Proteolytic processing of the C terminus of the alpha-1C)subunit of L-type calcium channels and the role of a proline-rich domain in membrane tethering of proteolytic fragments. J Biol Chem. 2000;275:8556–8563. doi: 10.1074/jbc.275.12.8556. [DOI] [PubMed] [Google Scholar]
- 128.Hulme JT, Yarov-Yarovoy V, Lin TW, Scheuer T, Catterall WA. Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C-terminal domain. J Physiol. 2006;576:87–102. doi: 10.1113/jphysiol.2006.111799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wei X, Neely A, Lacerda AE, Olcese R, Stefani E, Perez-Reyes E, Birnbaumer L. Modification of Ca2+ channel activity by deletions at the carboxyl terminus of the cardiac α1 subunit. J Biol Chem. 1994;269:1635–1640. [PubMed] [Google Scholar]
- 130.Hulme JT, Yarov-Yarovoy V, Lin TW, Scheuer T, Catterall WA. Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C-terminal domain. J Physiol. 2006;576:87–102. doi: 10.1113/jphysiol.2006.111799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fuller MD, Emrick MA, Sadilek M, Scheuer T, Catterall WA. Molecular mechanism of calcium channel regulation in the fight-or-flight response. Science Signaling. 2010 doi: 10.1126/scisignal.2001152. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Numa S, Tanabe T, Takeshima H, Mikami A, Niidome T, Nishimura S, Adams BA, Beam KG. Molecular insights into excitation-contraction coupling. Cold Spring Harb Symp Quant Biol. 1990;55:1–7. doi: 10.1101/sqb.1990.055.01.003. [DOI] [PubMed] [Google Scholar]
- 133.Nakai J, Dirksen RT, Nguyen HT, Pessah IN, Beam KG, Allen PD. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 1996;380:72–75. doi: 10.1038/380072a0. [DOI] [PubMed] [Google Scholar]
- 134.Zhong H, Yokoyama CT, Scheuer T, Catterall WA. Reciprocal regulation of P/Q-type Ca2+ channels by SNAP-25, syntaxin and synaptotagmin. Nat Neurosci. 1999;2:939–941. doi: 10.1038/14721. [DOI] [PubMed] [Google Scholar]
- 135.Bezprozvanny I, Scheller RH, Tsien RW. Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature. 1995;378:623–626. doi: 10.1038/378623a0. [DOI] [PubMed] [Google Scholar]
- 136.Wiser O, Tobi D, Trus M, Atlas D. Synaptotagmin restores kinetic properties of a syntaxin-associated N-type voltage sensitive calcium channel. FEBS Lett. 1997;404:203–207. doi: 10.1016/s0014-5793(97)00130-0. [DOI] [PubMed] [Google Scholar]
- 137.Sutton KG, McRory JE, Guthrie H, Murphy TH, Snutch TP. P/Q-type calcium channels mediate the activity-dependent feedback of syntaxin-1A. Nature. 1999;401:800–804. doi: 10.1038/44586. [DOI] [PubMed] [Google Scholar]