

Abstract
Apolipoprotein E4 (apoE4) plays a major role in the pathogenesis of Alzheimer's disease. Brain amyloid-β (Aβ) accumulation depends on age and apoE isoforms (apoE4 > apoE3) both in humans and in transgenic mouse models. Brain apoE levels are also isoform dependent, but in the opposite direction (apoE4 < apoE3). Thus, one prevailing hypothesis is to increase brain apoE expression to reduce Aβ levels. To test this hypothesis, we generated mutant human amyloid precursor protein transgenic mice expressing one or two copies of the human APOE3 or APOE4 gene that was knocked in and flanked by LoxP sites. We report that reducing apoE3 or apoE4 expression by 50% in 6-month-old mice results in efficient Aβ clearance and does not increase Aβ accumulation. However, 12-month-old mice with one copy of the human APOE gene had significantly reduced Aβ levels and plaque loads compared with mice with two copies, regardless of which human apoE isoform was expressed, suggesting a gene dose-dependent effect of apoE on Aβ accumulation in aged mice. Additionally, 12-month-old mice expressing one or two copies of the human APOE4 gene had significantly higher levels of Aβ accumulation and plaque loads than age-matched mice expressing one or two copies of the human APOE3 gene, suggesting an isoform-dependent effect of apoE on Aβ accumulation in aged mice. Moreover, Cre-mediated APOE4 gene excision in hippocampal astrocytes significantly reduced insoluble Aβ in adult mice. Thus, reducing, rather than increasing, apoE expression is an attractive approach to lowering brain Aβ levels.
Introduction
Apolipoprotein E4 (apoE4) is a major genetic risk factor for Alzheimer's disease (AD) (Corder et al., 1993; Saunders et al., 1993; Strittmatter et al., 1993a). AD is pathologically characterized by the presence of amyloid plaques and neurofibrillary tangles, both of which contain apoE (Namba et al., 1991; Wisniewski and Frangione, 1992). Amyloid plaques consist primarily of amyloid-β (Aβ) peptides that are produced from the sequential cleavage of human amyloid precursor protein (hAPP) (Hardy and Selkoe, 2002). Early studies suggested that apoE4 functions as a pathological chaperone to accelerate Aβ fibrillization and deposition (Wisniewski and Frangione, 1992; Strittmatter et al., 1993b; Sanan et al., 1994; Castano et al., 1995). Studies in both humans and transgenic mice showed that brain Aβ levels and amyloid plaque formation are apoE isoform dependent (apoE4 > apoE3 > apoE2) (Bales et al., 2009; Reiman et al., 2009; Castellano et al., 2011). However, apoE isoforms also differentially facilitate the proteolytic degradation of Aβ by microglia and astrocytes, with apoE3 being more effective (Koistinaho et al., 2004; Jiang et al., 2008; Zhao et al., 2009).
Analysis of human plasma samples revealed that apoE4 levels were lower and further decreased in AD patients versus controls (Gupta et al., 2011). Analysis of human brain samples yielded controversial results, with some studies showing decreased apoE mRNA or protein levels and others showing no change or increased apoE mRNA or protein levels in AD versus control brains (Bertrand et al., 1995; Harr et al., 1996; Pirttilä et al., 1996; Lambert et al., 1997; Beffert et al., 1999; Growdon et al., 1999; Bray et al., 2004; Jones et al., 2011). Several studies have shown that brain apoE levels in human apoE knock-in mice vary according to the isoform expressed (apoE4 < apoE3) (Ramaswamy et al., 2005; Riddell et al., 2008; Sullivan et al., 2011). Further studies that measured apoE in hAPP transgenic mouse CSF and brain lysates again found reduced levels of apoE4 versus apoE3 (Bales et al., 2009). These findings suggest a possible mechanism for apoE4's detrimental effects, whereby apoE4, being inherently unstable, becomes targeted for degradation more often, which results in insufficient levels of apoE to facilitate Aβ clearance and leads to Aβ accumulation (Bales et al., 2009). These studies thus suggest a therapeutic strategy whereby elevating apoE expression in the brain stimulates Aβ clearance and thus lowers Aβ levels (Dodart et al., 2005; Riddell et al., 2008; Bales et al., 2009; Sullivan et al., 2011).
To directly test this hypothesis, we generated mutant hAPP transgenic mice expressing one or two copies of the APOE3 or APOE4 gene that was knocked into the endogenous mouse locus. Hemizygous mice expressing one copy of the human APOE gene (and one APOE knock-out allele) or homozygous mice with two copies of the human APOE gene were analyzed at 6 and 12 months for levels of total Aβ, Aβ deposition, and fibrillar amyloid-plaque formation in the hippocampus. Strikingly, we found that reducing human apoE levels attenuates age-dependent Aβ accumulation in mutant hAPP transgenic mice.
Materials and Methods
Mice.
The floxed apoE knock-in (APOE-fKI) mice were generated by homologous recombination of a gene-targeting vector containing exons 2 through 4 of the human APOE gene. LoxP sites were added before exon 2 and after exon 4. The apoE3- and apoE4-targeting vectors were electroporated into embryonic stem cells (ESCs) from mouse strain 129/Svjae, and positive clones were selected and screened as described previously (Xu et al., 2006). One of each of the positive apoE3 and apoE4 ESC clones was microinjected into C57BL/6 blastocysts in the Gladstone Blastocyst Core, yielding >10 chimeric mice. Four to six male chimeras (>80% brown fur) were crossed with C57BL/6 females to generate heterozygous human apoE3 or apoE4 knock-in mice. Multiple additional backcrosses using marker-assisted selection resulted in mice that are >99% C57BL/6. ApoE3/3 and apoE4/4 mice were crossbred with hAPP transgenic mice (line J20 with the Swedish/Indiana mutations) on a mouse apoE knock-out background to generate E3+/−/hAPP and E4+/−/hAPP mice. E3+/−/hAPP mice were then crossbred with E3+/− mice, and E4+/−/hAPP mice were crossbred with E4+/− mice to generate mice with the following genotypes: E3+/−/hAPP, E3+/+/hAPP, E4+/−/hAPP, E4+/+/hAPP, and E−/−/hAPP. Mice at 6–8 or 12 months of age were used for most studies. All experimental groups contained 6–13 mice with equal proportions of males and females. For tissue collection, mice were anesthetized with avertin and transcardially perfused with 0.9% saline. Brains were rapidly harvested, right hemibrains were drop-fixed in 4% paraformaldehyde, and left hemibrains were snap frozen on dry ice.
Western blot analyses.
Snap-frozen tissue was allowed to thaw slowly on ice, dissected to isolate the hippocampus and cortex, and weighed to determine the amount of ice-cold lysis buffer used (10× w/v). For mice not expressing hAPP, the hippocampus and cortex were separately homogenized in a high-detergent buffer (50 mm Tris, 150 mm NaCl, 2% NP-40, 0.5% sodium deoxycholate, 4% SDS, and protease and phosphatase inhibitor cocktails) and centrifuged at 30,000 rpm for 30 min. The supernatant was collected, and protein concentrations were measured with a bicinchoninic acid (BCA) protein assay kit (Thermo Scientific). Snap-frozen hemibrains of APOE-fKI mice that coexpressed the hAPP transgene were dissected to isolate the hippocampus, weighed, resuspended in a low-detergent buffer (50 mm Tris, 150 mm NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and protease and phosphatase inhibitor cocktails) (10× w/v), and homogenized with a Kontes hand-held homogenizer. Fifty microliters was removed and centrifuged at 30,000 rpm for 30 min. The supernatant, which represented the soluble fraction, was collected, and the pellet was rehomogenized in high-detergent buffer and centrifuged at 30,000 rpm for 30 min, which represented the insoluble fraction. Total protein concentrations in these separate fractions were assessed by BCA assay and analyzed by Western blot for relative apoE, APP, and tubulin levels. Fifteen micrograms of total protein were loaded onto 12% Bis-Tris Novex SDS-PAGE gels (Invitrogen). Proteins were transferred onto a nitrocellulose membrane and blocked using LI-COR Biosciences blocking buffer for 2 h before overnight incubation with polyclonal anti-apoE (1:5000), monoclonal anti-APP (1:2000, clone 22C11), and monoclonal anti-tubulin (1:40,000) antibodies. Secondary antibodies coupled to an 800 nm fluorophore were used to detect and quantify the intensity of the resulting bands representing apoE, APP, and tubulin (Odyssey Imager, LI-COR Biosciences).
Aβ ELISA.
The remaining hippocampal homogenates solubilized in low-detergent buffer were centrifuged at 14,000 rpm for 20 min. The pellet was rehomogenized in 5 m guanidine-HCl (GuHCl) and 7.5 m GuHCl was added to the supernatant at a 1:2 (v/v) ratio to obtain a final concentration of 5 m GuHCl. Both the soluble and insoluble fractions were mixed on a nutator for 4 h at room temperature and stored at −20°C before Aβ analyses. Ninety-six-well immunoassay plates were coated with clone m266 or 21F12 anti-Aβ antibodies to capture total Aβ (Aβ1-x) or Aβ42 from the samples, respectively. Samples were diluted from 5 to 0.5 m GuHCl with a casein blocking buffer to final concentrations within range of a simultaneously generated standard curve for each assay. Detection was performed with biotinylated anti-Aβ clone 3D6, HRP-avidin, and TMB as the substrate.
Immunohistochemistry, image collection, and quantitative analysis.
Sections were collected (30 μm) from paraformaldehyde-fixed right hemibrains on a sliding microtome fitted with a freezing stage as described previously (Andrews-Zwilling et al., 2010; Bien-Ly et al., 2011). Briefly, every 10th section for each mouse was immunostained using anti-Aβ 3D6 (1:2000), biotinylated donkey-anti-mouse IgG, HRP-avidin, and diaminobenzidine as the substrate. Stained sections were mounted on Superfrost slides using 1% gelatin, allowed to air dry, and coverslipped. Three sections per mouse were imaged with a 10× objective, and the hippocampal area was outlined and quantified for the percentage area stained using ImageJ software as described previously (Bien-Ly et al., 2011). Before quantification, slides were coded for blind analysis. For immunofluorescent staining, polyclonal anti-apoE (1:4000) and anti-Aβ (1:2000) were incubated overnight and detected with Alexa Fluor 488 donkey anti-goat and Alexa Fluor 594 donkey anti-mouse secondary antibodies (1:2000). Imaging was performed as described previously (Andrews-Zwilling et al., 2010; Bien-Ly et al., 2011).
Thioflavin S staining and quantitative analysis.
One subseries containing every 10th section (30 μm) from each hAPP transgenic mouse was rinsed free of cryoprotectant media with PBS, mounted onto Superfrost slides, and allowed to air dry overnight. The slides were then rinsed in PBS twice, incubated in 0.25% potassium permanganate for 5–10 min, washed three times in PBS, incubated in 2% K2O5S2 and 1% oxalic acid for 5 min, washed three times in PBS, and stained with 0.015% thioflavin S in 50% ethanol for 10 min. Afterward, the slides were differentiated in 50% ethanol, rinsed with water and PBS, and then coverslipped. Analyses were performed on three sections starting from bregma −1.6 on fluorescent images collected with a 10× objective. The hippocampal area was outlined and quantified for the percentage of the area occupied by thioflavin S-positive plaques using ImageJ software. Data were collected by an investigator blinded to the genotypes of the mice and reflect the averages of the percentage area from the three sections.
Stereotaxic brain injection of Ad-Cre virus.
Anesthetized apoE4+/+/hAPP mice aged 6–7 months were given a single unilateral stereotaxic injection of adenovirus expressing a vector containing Cre-recombinase and an EGFP-reporter cassette (Ad-Cre) into the left hippocampus or saline into the right hippocampus using the following coordinates (x = ±1.5, y = 2.1, z = 2.1; distances are from bregma). Injections were infused at 200 nl/min for a total of 2 μl. After 1 month, the brains were collected and analyzed for Aβ levels by the sandwich ELISA or fixed for fluorescent immunostaining as described.
Statistical analysis.
Values are expressed as mean ± SEM. The statistical significance of differences between means was assessed by Student's t test or one-way ANOVA followed by Bonferroni post hoc tests. A p value <0.05 was considered statistically significant.
Results
Generation of floxed human apoE3 and apoE4 knock-in mice
We generated new lines of mice expressing human apoE3 and apoE4 in which a knocked-in human APOE3 or APOE4 gene was flanked with a pair of LoxP sites, allowing for excision of the APOE gene in the presence of Cre-recombinase (Fig. 1A). The “floxed” knock-in mouse lines, herein referred to as APOE3/3-fKI and APOE4/4-fKI (or E3+/+ and E4+/+, respectively), were characterized for apoE expression by immunohistochemistry and Western blot at 6 months of age. Confocal imaging of brain sections immunostained for apoE and glial fibrillary acidic protein (GFAP) revealed that apoE was primarily expressed in astrocytes (Fig. 1B,C), identical to a commercially available traditional apoE knock-in mouse line (apoE-KI) (Fig. 1,E,F). Western blot analysis of hippocampal lysates showed significantly lower levels of apoE4 than apoE3 in both the floxed (two-tailed t test; t(6) = 2.449, p < 0.05) and traditional knock-in mice (two-tailed t test; t(4) = 3.189, p < 0.05) (Fig. 1D,G). We found a similar decrease in cortical lysates, as reported previously (Ramaswamy et al., 2005; Riddell et al., 2008).
Figure 1.
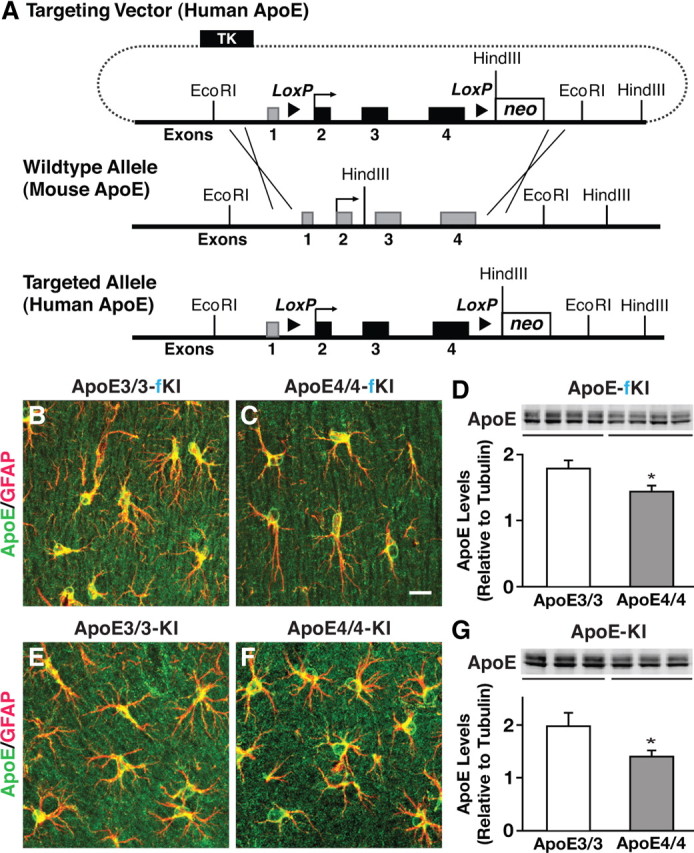
Generation and characterization of floxed human apoE knock-in mice. A, Targeting vector containing human apoE exons 2–4 (represented by black rectangles) flanked by loxP sites with endogenous mouse exon 1 and mouse upstream and downstream elements used for homologous recombination (mouse exons represented by gray rectangles). B, C, E, F, Representative immunofluorescence images for apoE (green) and GFAP (red) in the hippocampal subfield highlighting apoE-producing astrocytes in APOE-fKI mice (B, C) and APOE-KI mice (E, F). D, G, Western blot and densitometric quantification of apoE protein levels in the hippocampus of homozygous APOE-fKI mice (D) and homozygous APOE-KI mice (G). N = 3–4 mice per genotype. Values are mean ± SEM. *p < 0.05 by two-tailed, unpaired t test. Scale bar, 10 μm.
Reducing apoE levels by half does not increase Aβ accumulation in young mice
J20 hAPP mice bred onto a murine apoE knock-out (EKO) background were crossed to APOE-fKI mice to generate the F0 generation of human apoE/hAPP mice hemizygous for apoE3 or apoE4 and with one EKO allele, as well as mice homozygous for the EKO allele (abbreviated E3+/−/hAPP, E4+/−/hAPP, and E−/−/hAPP, respectively). The first generation of hemizygous mice was subsequently crossed to generate homozygous apoE3+/+/hAPP (abbreviated E3+/+/hAPP) and apoE4+/+/hAPP (abbreviated E4+/+/hAPP) mice. In Aβ ELISAs, we found that levels of Aβ1-x in mice expressing one or two copies of APOE3 or APOE4 were similarly low at 6 months of age in low-detergent-soluble and insoluble fractions, as well as the sum of both fractions (Fig. 2A–C). Analysis of Aβ42 levels revealed a similar trend (Fig. 2D–F). Interestingly, E−/−/hAPP mice had significantly higher levels of Aβ1-x and Aβ42 (5-10-fold) in both the soluble (ANOVA with Bonferroni's post hoc test: Aβ1-x, p < 0.001, E−/−/hAPP vs all other genotypes; Aβ42, p < 0.001, E−/−/hAPP vs all other genotypes) and insoluble (ANOVA with Bonferroni's post hoc test: Aβ1-x, p < 0.01, E−/−/hAPP vs all other genotypes; Aβ42, p < 0.001, E−/−/hAPP vs all other genotypes) fractions, suggesting that expression of human apoE facilitates Aβ clearance. Anti-Aβ immunostaining of the mouse brain sections revealed that the percentage area of Aβ deposition was minimal and not significantly different, regardless of whether the mice expressed one or two copies of either APOE3 or APOE4 (Fig. 2G,H,J,K). However, E−/−/hAPP mice had extensive and diffuse Aβ deposition in the hippocampal subfield, resulting in a greater Aβ load than the other genotypes (ANOVA with Bonferroni's post hoc test: p < 0.05, E−/−/hAPP vs all other genotypes), confirming that expression of human apoE facilitates Aβ clearance (Fig. 2L). These findings suggest that in young adult mice reducing apoE levels by half does not increase Aβ accumulation, regardless of the apoE isoform.
Figure 2.

Reducing apoE levels by half does not increase Aβ accumulation in young mice. A–C, Levels of Aβ1-x were measured by sandwich ELISA in combined soluble and insoluble hippocampal lysates (A), low-detergent-soluble lysates (B), and insoluble lysates (C) for E3+/−/hAPP, E3+/+/hAPP, E4+/−/hAPP, E4+/+/hAPP, and E−/−/hAPP mice at 6–8 months of age. D–F, Levels of Aβ42 in combined soluble and insoluble lysates (D), low-detergent-soluble lysates (E), and insoluble lysates (F). G–K, Representative sections from an Aβ immunostain of the hippocampal subfield of E3+/−/hAPP (G), E3+/+/hAPP (H), E4+/−/hAPP (J), E4+/+/hAPP (K), and E−/−/hAPP (I) mice. L, Quantification by densitometry of the percentage area covered by Aβ deposition. N = 7–12 mice per genotype, three sections per mouse. Values are mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 versus all other groups by one-way ANOVA with Bonferroni's post hoc test. Scale bar, 250 μm.
To determine whether differences in apoE levels alter hAPP expression, we analyzed the hippocampal extracts from the 6-month-old mice by Western blot for apoE and hAPP protein levels (Fig. 3A,B). As expected, hemizygous mice with one copy of the APOE3 or APOE4 gene (E3+/− and E4+/−) expressed half as much apoE as mice with two copies (E3+/+ and E4+/+) (Fig. 3C–E). In addition, homozygous apoE4 mice had significantly lower apoE levels than homozygous apoE3 mice (two-tailed t test; total apoE, t(6) = 2.78, p < 0.05; soluble apoE, t(6) = 8.492, p < 0.001) (Fig. 3C,D). This decreased level in E4+/+ mice was found in the soluble fraction, while the insoluble lysate fraction was similar for E3+/+ and E4+/+ mice (Fig. 3E). The ratio of apoE levels from the insoluble to soluble lysate fractions revealed a significant isoform-specific (apoE4 > apoE3) but not gene dose-specific increase in ratio (two-tailed t test; E3+/− vs E4+/−, t(4) = 2.942, p < 0.05; E3+/+ vs E4+/+, t(6) = 2.51, p < 0.05) (Fig. 3F), suggesting that the apoE4 isoform may accumulate more in the insoluble fraction. Analysis of the relative levels of hAPP did not reveal an apoE gene dose-dependent or isoform-dependent difference between the groups (Fig. 3G), and no significant hAPP was isolated or resolved from the insoluble fraction (Fig. 3B). Thus, differences in apoE expression levels did not affect the levels of hAPP expression.
Figure 3.

Human apoE expression levels are reduced in young hemizygous apoE mice without altering APP expression levels. A, B, Western blotting was performed on low-detergent-soluble (A) and insoluble (B) hippocampal lysates and probed for apoE, tubulin, and human full-length APP protein levels in E3+/−/hAPP, E3+/+/hAPP, E4+/−/hAPP, E4+/+/hAPP, and E−/−/hAPP mice at 6–8 months of age. C–F, Quantification of Western blots by densitometry for total apoE levels (C), low-detergent-soluble apoE (D), insoluble apoE (E), and ratios of soluble to insoluble pools of apoE (F). G, H, Western blot quantification of total full-length APP levels (G) and total tubulin levels as loading controls (H). Values are mean ± SEM. N = 3–4 mice per genotype. *p < 0.05, ***p < 0.001, by two-tailed, unpaired t test.
Halving the levels of apoE significantly attenuates Aβ accumulation in aged mice
To determine whether apoE gene dose and isoform affect Aβ levels in aged mice, we analyzed soluble and insoluble hippocampal lysates from mice at 12 months of age. We observed that apoE3 hemizygous mice (E3+/−/hAPP) had significantly lower levels of both Aβ1-x and Aβ42 than homozygous mice (E3+/+/hAPP), within the soluble fractions and the combined insoluble and soluble fractions (two-tailed t test; soluble Aβ1-x, t(20) = 2.723, p < 0.05; total Aβ1-x, t(20) = 2.502, p < 0.05; soluble Aβ42, t(20) = 2.843, p < 0.05; total Aβ42, t(20) = 2.526, p < 0.05) (Fig. 4A,B,D,E). In E3+/−/hAPP and E4+/−/hAPP mice, the levels of Aβ1-x in the insoluble fractions were significantly lower than their homozygous counterparts (two-tailed t test; E3+/− vs E3+/+, t(20) = 2.252, p < 0.05; E4+/− vs E4+/+, t(15) = 3.409, p < 0.01) (Fig. 4C). A similar decrease was observed for Aβ42 in E4+/−/hAPP mice versus E4+/+/hAPP mice (two-tailed t test; t(15) = 4.636, p < 0.001) (Fig. 4F). Thus, there was an apparent gene dose effect on Aβ accumulation in aged mice, whereby increases in apoE resulted in higher soluble as well as insoluble Aβ levels. Interestingly, this effect is independent of isoform, as the genetic reduction of either apoE3 or apoE4 attenuates Aβ accumulation.
Figure 4.

Halving the levels of apoE significantly attenuates Aβ accumulation in aged mice. A–F, Levels of Aβ1-x (A–C) and Aβ42 (D–F) were measured by sandwich ELISA in both soluble and insoluble lysates (A, D), low-detergent-soluble lysates (B, E), and insoluble lysates (C, F) in the hippocampus of E3+/−/hAPP, E3+/+/hAPP, E4+/−/hAPP, E4+/+/hAPP, and E−/−/hAPP mice at 12 months of age. Values are mean ± SEM. N = 6–13 mice per genotype. *p < 0.05, **p < 0.01, and ***p < 0.001, by two-tailed, unpaired t test.
To ensure that the effects of apoE levels on Aβ accumulation were not due to altered expression of hAPP in the hippocampus, we probed the soluble and insoluble lysates by Western blot for apoE and hAPP (Fig. 5). Levels of soluble hAPP were not altered by apoE gene dose or isoform (Fig. 5G). In addition, the hemizygous apoE3 and apoE4 mice had half the apoE protein levels as their homozygous littermates in both the soluble and insoluble fractions, as expected (Fig. 5A–F). There were no significant differences in apoE levels between the isoforms.
Figure 5.

Human apoE expression levels are reduced in aged hemizygous apoE mice without altering APP expression levels. A, B, Western blotting was performed on low-detergent-soluble (A) and insoluble (B) hippocampal lysates, and probed for apoE, tubulin, and human full-length APP protein levels in E3+/−/hAPP, E3+/+/hAPP, E4+/−/hAPP, E4+/+/hAPP, and E−/−/hAPP mice at 12 months of age. C–F, Quantification of Western blots by densitometry for total apoE levels (C), low-detergent-soluble apoE (D), insoluble apoE (E), and ratios of soluble to insoluble pools of apoE (F). G, H, Western blot quantification of total full-length APP levels (G) and total tubulin levels as loading controls (H). Values are mean ± SEM. N = 3–4 mice per genotype.
Halving the levels of apoE significantly attenuates Aβ deposition in aged mice
To visualize Aβ accumulation in the 12-month-old aged mice, we immunostained for Aβ and quantified the percentage area of Aβ deposition in the hippocampus. E3+/−/hAPP mice displayed lower levels of Aβ deposition than E3+/+/hAPP, E4+/−/hAPP, and E4+/+/hAPP mice (Fig. 6A–D). Quantification of the area stained for Aβ revealed that the E3+/−/hAPP mice had a significantly lower Aβ load in the hippocampus than the other groups (ANOVA with Bonferroni's post hoc test; p < 0.01, E3+/−/hAPP vs all other genotypes) (Fig. 6F). The aged E−/−/hAPP mice had a similar Aβ load but exhibited a more diffuse and widespread pattern of deposition than E3+/+/hAPP, E4+/−/hAPP, and E4+/+/hAPP mice (Fig. 6E,F). To investigate whether the Aβ deposition was truly amyloid in nature, neighboring sections were stained with the fluorescent dye thioflavin S for fibrillar Aβ plaques. We analyzed the percentage area occupied by thioflavin S-positive amyloid plaques and found that apoE homozygous mice had significantly more fibrillar Aβ plaques compared with apoE hemizygous mice, and this effect was even more pronounced for apoE4 than apoE3 (two-tailed t test; E3+/− vs E3+/+, t(20) = 2.178, p < 0.05; E4+/− vs E4+/+, t(13) = 2.932, p < 0.05) (Fig. 6G–L). Conversely, although the E−/−/hAPP mice had high levels of Aβ and extensive Aβ deposition detected by immunohistochemistry, there was minimal fibrillar Aβ as detected by thioflavin S (Fig. 6K,L), which corroborates previous reports (Holtzman et al., 1999; Fagan et al., 2002; Bien-Ly et al., 2011).
Figure 6.
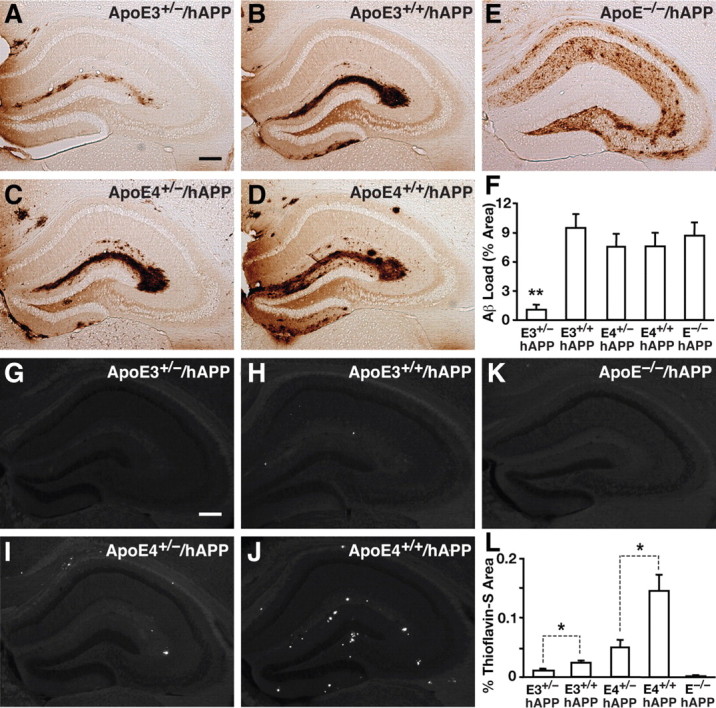
Halving the levels of apoE significantly attenuates Aβ deposition in aged mice. A–E, Representative sections from 12-month-old aged E3+/−/hAPP (A), E3+/+/hAPP (B), E4+/−/hAPP (C), E4+/+/hAPP (D), and E−/−/APP (E) mice immunostained for Aβ. F, Quantification of Aβ immunostain by densitometry for the percentage area of Aβ deposition in the five genotypes of mice. G–K, Representative sections from 12-month-old E3+/−/hAPP (G), E3+/+/hAPP (H), E4+/−/hAPP (I), E4+/+/hAPP (J), and E−/−/hAPP (K) mice stained with thioflavin S dye to fluorescently label fibrillar/amyloid plaques. L, Quantification of percentage area positive for thioflavin S by densitometry. Values are mean ± SEM. N = 6–13 mice per genotype, three sections per mouse. *p < 0.05, by two-tailed, unpaired t test; **p < 0.01 versus all other groups by one-way ANOVA with Bonferroni's post hoc test. Scale bars, 250 μm.
To determine whether the Aβ deposits in the aged mice contained apoE, immunofluorescent staining was performed on brain sections from all five groups of mice. We observed codistribution of apoE and Aβ within plaques from all groups except the E−/−/hAPP mice (Fig. 7A–J). Quantification of the number of Aβ plaques containing apoE revealed a clear gene dose- and isoform-dependent pattern (two-tailed t test; E3+/− vs E3+/+, t(5) = 3.183, p < 0.01; E4+/− vs E4+/+, t(6) = 2.725, p < 0.05) (Fig. 7K). Importantly, apoE3 and apoE4 homozygous mice had a greater number of Aβ plaques containing apoE than their hemizygous littermates (Fig. 7K). These data suggest that in aged mice, apoE promotes Aβ accumulation rather than clearance, and that the apoE4 isoform has a greater propensity to codeposit with Aβ and cause its accumulation.
Figure 7.
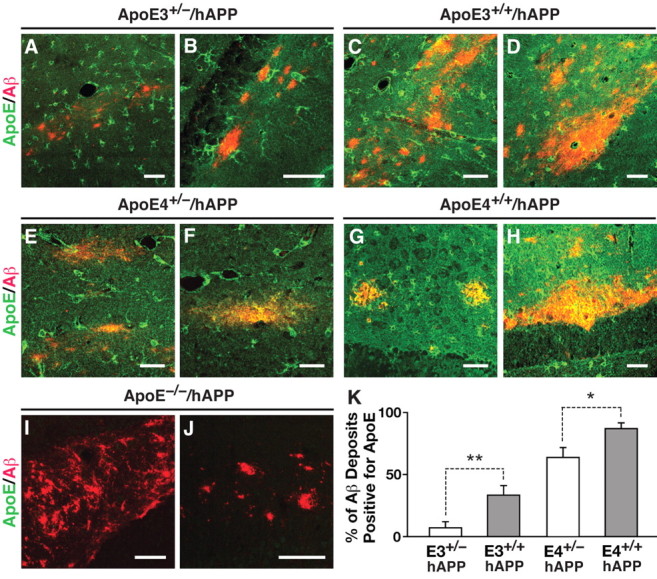
Gene dose- and isoform-dependent codistribution of apoE with Aβ plaques in aged mice. A–J, Representative fluorescent immunostaining for apoE (green) and Aβ (red) in various subregions of the hippocampus in E3+/−/hAPP (A, B), E3+/+/hAPP (C, D), E4+/−/hAPP (E, F), E4+/+/hAPP (G, H), and E−/−/hAPP (I, J) mice at 12 months of age. K, Quantification of Aβ deposits and codistribution with apoE. Mice with two copies of apoE3 or apoE4 have a greater percentage of plaques containing apoE. Values are mean ± SEM. N = 3–4 mice per genotype, two sections per mouse. *p < 0.05, **p < 0.01, by two-tailed, unpaired t test. Scale bar, 25 μm.
Cre-mediated apoE gene excision in hippocampal astrocytes decreases Aβ levels in adult mice
To determine whether acutely decreasing apoE levels in adult mice also lowers Aβ levels, we performed unilateral stereotaxic injections of an adenoviral vector carrying Cre-recombinase and an EGFP reporter (Ad-Cre) into one side of the hippocampus of E4+/+/hAPP mice at 6–7 months of age. The contralateral hippocampus was injected with saline as an internal control. Analysis of the EGFP reporter expression by confocal microscopy revealed that the Ad-Cre-injected hippocampus had EGFP-positive cells concentrated in the dentate gyrus (Fig. 8A). As expected, the saline-injected side of the hippocampus did not have EGFP-positive cells (Fig. 8B). Immunostaining sections for GFP (green), GFAP (blue), and apoE (red) revealed that all EGFP-positive cells were also positive for GFAP (Fig. 8C,E), indicating that the virus selectively targeted astrocytes. Furthermore, all EGFP-positive astrocytes were negative for apoE (Fig. 8D,F), confirming effective excision of the human APOE gene. Interestingly, the area surrounding the EGFP-positive astrocytes lacked apoE immunostaining (Fig. 8D,E), which suggests that the secreted pool of apoE from nontransduced astrocytes does not diffuse into the transduced areas.
Figure 8.
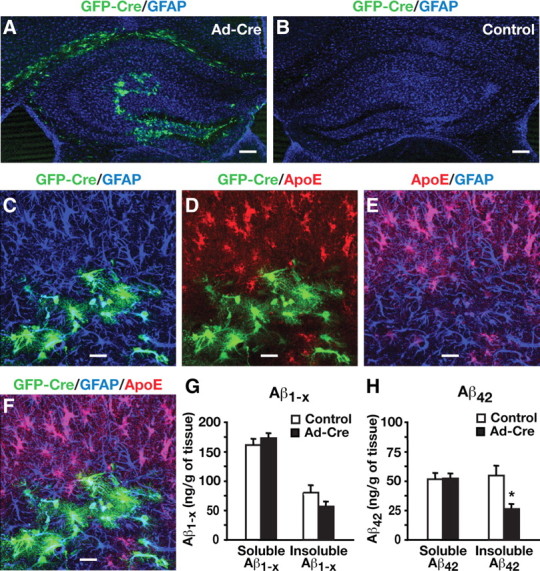
Cre-mediated APOE gene excision in hippocampal astrocytes decreases Aβ levels in young adult mice. A, B, Representative confocal images of brain sections from E4+/+/hAPP mice at 6–7 months of age stained for GFAP (blue) from the hippocampus injected with adenovirus expressing Cre (Ad-Cre) and GFP (green) (A) or saline (B). Brains were collected 1 month after the Ad-Cre virus or saline injection. C–F, Representative confocal images of brain sections from E4+/+/hAPP mice stained for GFP (green; C, D, F), GFAP (blue; C, E, F), and apoE (red; D–F) from the hippocampus injected with Ad-Cre and expressing GFP (green). G, H, Aβ levels in Ad-Cre-injected and saline-injected hippocampi were quantified 1 month after injection. Levels of Aβ1-x (G) in both soluble and insoluble hippocampal lysates were similar in control compared with Ad-Cre-injected hemibrains, whereas levels of Aβ42 (H) showed a specific difference in the insoluble lysates. Values are mean ± SEM. N = 8 mice. *p < 0.05 by two-tailed, paired t test. Scale bars: A, B, 250 μm; C–F, 20 μm.
Strikingly, insoluble Aβ42 decreased by ∼50% in Ad-Cre-injected hippocampi compared with saline-injected controls (two-tailed paired t test; t(7) = 3.43, p < 0.05), although soluble Aβ42 was not altered significantly (Fig. 8H). Insoluble Aβ1-x also showed a trend toward lower levels (∼25%) in Ad-Cre-injected hippocampi, although this difference did not reach statistical significance (Fig. 8G). Thus, Cre-mediated APOE gene excision in hippocampal astrocytes decreases Aβ levels in adult mice.
Discussion
This study shows that genetic modulation of apoE levels significantly alters Aβ accumulation with age. We found that young mice with one copy of either the APOE3 or APOE4 gene had Aβ levels and deposition similar to mice with two copies of the apoE gene, suggesting that one copy of APOE is sufficient to maintain effective Aβ clearance at this age. However, significantly more soluble and insoluble Aβ accumulated in aged mice expressing two copies of APOE than one copy, and these increases were even more pronounced for apoE4- than apoE3-expressing mice. Aβ deposition, thioflavin S-positive plaque formation, and apoE accumulation within plaques were also APOE gene dose dependent and isoform dependent in aged mice. Furthermore, Cre-mediated APOE gene excision in hippocampal astrocytes decreases Aβ levels in adult mice. Thus, reducing apoE expression levels, regardless of apoE isoform or age at treatment, attenuates Aβ accumulation in the brain. Our previous studies suggested that lowering apoE4 levels has beneficial effects independent of Aβ (Huang, 2006, 2010; Mahley et al., 2006). The current study reveals a positive effect of reducing apoE levels directly on Aβ accumulation and plaque formation.
Several studies reported isoform-specific differences in apoE levels in humans and in mouse models. Some human studies demonstrated that AD patients had lower brain apoE levels than controls, although other studies showed no change or increased apoE levels in AD brains (Bertrand et al., 1995; Harr et al., 1996; Pirttilä et al., 1996; Lambert et al., 1997; Beffert et al., 1999; Growdon et al., 1999; Bray et al., 2004; Jones et al., 2011). The differing results are likely due to heterogeneity in patient populations, with subjects at different stages and duration of AD (Kim et al., 2009). A recent study reported that AD patients had lower plasma levels of total apoE and apoE4 than controls, which is associated with higher brain Aβ loads (Gupta et al., 2011). In mouse models where human apoE was expressed under the control of an endogenous murine promoter, apoE4 levels were decreased in plasma, CSF, hippocampus, and/or cortical lysates, compared with apoE3 mice, with no apparent differences in mRNA levels (Ramaswamy et al., 2005; Riddell et al., 2008; Bales et al., 2009; Sullivan et al., 2011). Based on these differences in protein levels, the APOE4 allele was deemed hypomorphic due to relatively lower availability and thus reduced functionality. Further studies in mutant hAPP transgenic mice expressing two copies of the human APOE3 or APOE4 gene demonstrated that lower levels of apoE4 in brains were associated with an increase in Aβ accumulation (Bales et al., 2009). From these studies, the hypothesis was put forward that increasing apoE expression facilitates Aβ clearance (Riddell et al., 2008; Bales et al., 2009; Sullivan et al., 2011). Our current study directly tested this hypothesis and found that decreasing apoE expression reduces Aβ accumulation. It is thus conceivable that increases in apoE expression would enhance Aβ accumulation. In support of this possibility, a recent study showed that brain apoE levels were higher in AD cases than controls, especially in those with apoE4/4, and that more apoE4 than apoE3 interacted with Aβ in amyloid plaques (Jones et al., 2011). Thus, strategies aimed at reducing apoE levels, even for apoE3, may be more beneficial for maintaining lower Aβ levels throughout the lifetime of an individual.
Recently, liver X receptor agonists were shown to elevate murine apoE expression levels in brains of mutant hAPP transgenic mice, which is associated with reduced Aβ levels (Donkin et al., 2010; Fitz et al., 2010). While intriguing, further studies are needed to determine whether these treatments decrease Aβ levels by increasing apoE expression or by altering other lipid metabolism-related processes. In addition, these studies did not test the effect of increasing human apoE3 or apoE4 specifically, and they have not been conducted in aged mice. The effect of murine apoE on Aβ clearly differs from human apoE, particularly with respect to Aβ deposition, which occurs more aggressively in hAPP transgenic mice expressing murine apoE compared with human apoE isoforms (Holtzman et al., 1999; Fagan et al., 2002; Bien-Ly et al., 2011). Additionally, the direct effect of apoE4 on Aβ accumulation may outweigh any beneficial effect of human apoE4 on Aβ clearance, particularly in aged brains, which would not be evident in models testing the effect of increasing murine apoE.
While this article was in preparation, a study reported that haploinsufficiency of the human APOE gene, regardless of isoform, reduces amyloid deposition in mutant hAPP and mutant PS1 double-transgenic mice at a young age (Kim et al., 2011). Our study extends this finding by focusing on the effects of apoE levels on Aβ accumulation in both young and aged mice, and by using an hAPP transgenic mouse model in which Aβ accumulation and plaque formation is less aggressive and more age dependent than mutant hAPP/PS1 double-transgenic mice. In the presence of human apoE expression, the mutant hAPP mice used in the current study do not develop significant brain Aβ deposition at a young age (6 months), providing a model to study the effect of lowering apoE levels on Aβ clearance. However, these mutant hAPP mice do develop Aβ deposition at an older age (12 months), providing a model to study the effect of lowering apoE levels on Aβ deposition. We showed that in young mice, halving the expression levels of human apoE, regardless of isoform, preserves efficient Aβ clearance, and in aged mice expressing one-half levels of apoE, Aβ accumulation is significantly attenuated. Thus, our study suggests that higher levels of apoE expression, rather than being beneficial, likely stimulate Aβ accumulation and deposition during aging. We also took advantage of our floxed human apoE knock-in mice and used adenovirus-mediated Cre expression to show that excision of the human APOE gene in hippocampal astrocytes decreases Aβ levels in adult mice. Thus, our study shows the feasibility of therapeutically lowering apoE levels to substantially reduce Aβ accumulation.
Footnotes
This work was supported by the J. David Gladstone Institutes and National Institutes of Health Grants P01 AG022074, P30 NS065780, and C06RR18928. We thank John Carroll and Chris Goodfellow for graphics; Gary Howard and Anna Lisa Lucido for editorial assistance; and Linda Turney for manuscript preparation.
References
- Andrews-Zwilling Y, Bien-Ly N, Xu Q, Li G, Bernardo A, Yoon SY, Zwilling D, Yan TX, Chen L, Huang Y. Apolipoprotein E4 causes age- and tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J Neurosci. 2010;30:13707–13717. doi: 10.1523/JNEUROSCI.4040-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Liu F, Wu S, Lin S, Koger D, DeLong C, Hansen JC, Sullivan PM, Paul SM. Human APOE isoform-dependent effects on brain-amyloid levels in PDAPP transgenic mice. J Neurosci. 2009;29:6771–6779. doi: 10.1523/JNEUROSCI.0887-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beffert U, Cohn JS, Petit-Turcotte C, Tremblay M, Aumont N, Ramassamy C, Davignon J, Poirier J. Apolipoprotein E and beta-amyloid levels in the hippocampus and frontal cortex of Alzheimer's disease subjects are disease-related and apolipoprotein E genotype dependent. Brain Res. 1999;843:87–94. doi: 10.1016/s0006-8993(99)01894-6. [DOI] [PubMed] [Google Scholar]
- Bertrand P, Poirier J, Oda T, Finch CE, Pasinetti GM. Association of apolipoprotein E genotype with brain levels of apolipoprotein E and apolipoprotein J (clusterin) in Alzheimer's disease. Brain Res Mol Brain Res. 1995;33:174–178. doi: 10.1016/0169-328x(95)00097-c. [DOI] [PubMed] [Google Scholar]
- Bien-Ly N, Andrews-Zwilling Y, Xu Q, Bernardo A, Wang C, Huang Y. C-terminal-truncated apolipoprotein (apo) E4 inefficiently clears amyloid-β (Aβ) and acts in concert with Aβ to elicit neuronal and behavioral deficits in mice. Proc Natl Acad Sci U S A. 2011;108:4236–4241. doi: 10.1073/pnas.1018381108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NJ, Jehu L, Moskvina V, Buxbaum JD, Dracheva S, Haroutunian V, Williams J, Buckland PR, Owen MJ, O'Donovan MC. Allelic expression of APOE in human brain: effects of epsilon status and promoter haplotypes. Hum Mol Genet. 2004;13:2885–2892. doi: 10.1093/hmg/ddh299. [DOI] [PubMed] [Google Scholar]
- Castano EM, Prelli F, Wisniewski T, Golabek A, Kumar RA, Soto C, Frangione B. Fibrillogenesis in Alzheimer's disease of amyloid β peptides and apolipoprotein E. Biochem J. 1995;306:599–604. doi: 10.1042/bj3060599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Marr RA, Koistinaho M, Gregersen BM, Malkani S, Verma IM, Paul SM. Gene delivery of human apolipoprotein E alters brain Aβ burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2005;102:1211–1216. doi: 10.1073/pnas.0409072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donkin JJ, Stukas S, Hirsch-Reinshagen V, Namjoshi D, Wilkinson A, May S, Chan J, Fan J, Collins J, Wellington CL. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem. 2010;285:34144–34154. doi: 10.1074/jbc.M110.108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM. Human and murine apoE markedly alters Aβ metabolism before and after plaque formation in a mouse model of Alzheimer's disease. Neurobiol Dis. 2002;9:305–318. doi: 10.1006/nbdi.2002.0483. [DOI] [PubMed] [Google Scholar]
- Fitz NF, Cronican A, Pham T, Fogg A, Fauq AH, Chapman R, Lefterov I, Koldamova R. Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice. J Neurosci. 2010;30:6862–6872. doi: 10.1523/JNEUROSCI.1051-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Growdon WB, Cheung BS, Hyman BT, Rebeck GW. Lack of allelic imbalance in APOE epsilon3/4 brain mRNA expression in Alzheimer's disease. Neurosci Lett. 1999;272:83–86. doi: 10.1016/s0304-3940(99)00557-1. [DOI] [PubMed] [Google Scholar]
- Gupta VB, Laws SM, Villemagne VL, Ames D, Bush AI, Ellis KA, Lui JK, Masters C, Rowe CC, Szoeke C, Taddei K, Martins RN. Plasma apolipoprotein E and Alzheimer disease risk: the AIBL study of aging. Neurology. 2011;76:1091–1098. doi: 10.1212/WNL.0b013e318211c352. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Harr SD, Uint L, Hollister R, Hyman BT, Mendez AJ. Brain expression of apolipoprotein E, J, and A-I in Alzheimer's disease. J Neurochem. 1996;66:2429–2435. doi: 10.1046/j.1471-4159.1996.66062429.x. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM, Chang LK, Sun Y, Paul SM. Expression of human apolipoprotein E reduces amyloid-β deposition in a mouse model of Alzheimer's disease. J Clin Invest. 1999;103:R15–R21. doi: 10.1172/JCI6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y. Molecular and cellular mechanisms of apolipoprotein E4 neurotoxicity and potential therapeutic strategies. Curr Opin Drug Discov Devel. 2006;9:627–641. [PubMed] [Google Scholar]
- Huang Y. Aβ-independent roles of apolipoprotein E4 in the pathogenesis of Alzheimer's disease. Trends Mol Med. 2010;16:287–294. doi: 10.1016/j.molmed.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Aβ. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PB, Adams KW, Rozkalne A, Spires-Jones TL, Hshieh TT, Hashimoto T, von Armin CA, Mielke M, Bacskai BJ, Hyman BT. Apolipoprotein E: isoform sepecific difference in tertiary structure and interaction with amyloid-β in human Alzheimer brain. PloS One. 2011;6:e14586. doi: 10.1371/journal.pone.0014586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, Yoon H, Basak JM, Finn MB, Holtzman DM. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-β amyloidosis. J Neurosci. 2011;31:18007–18012. doi: 10.1523/JNEUROSCI.3773-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat Med. 2004;10:719–726. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Pérez-Tur J, Dupire MJ, Galasko D, Mann D, Amouyel P, Hardy J, Delacourte A, Chartier-Harlin MC. Distortion of allelic expression of apolipoprotein E in Alzheimer's disease. Hum Mol Genet. 1997;6:2151–2154. doi: 10.1093/hmg/6.12.2151. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt–Jakob disease. Brain Res. 1991;541:163–166. doi: 10.1016/0006-8993(91)91092-f. [DOI] [PubMed] [Google Scholar]
- Pirttilä T, Soininen H, Heinonen O, Lehtimäki T, Bogdanovic N, Paljärvi L, Kosunen O, Winblad B, Riekkinen P, Sr, Wisniewski HM, Mehta PD. Apolipoprotein E (apoE) levels in brains from Alzheimer's disease patients and controls. Brain Res. 1996;722:71–77. doi: 10.1016/0006-8993(96)00183-7. [DOI] [PubMed] [Google Scholar]
- Ramaswamy G, Xu Q, Huang Y, Weisgraber KH. Effect of domain interaction on apolipoprotein E levels in mouse brain. J Neurosci. 2005;25:10658–10663. doi: 10.1523/JNEUROSCI.1922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, Ayutyanont N, Keppler J, Reeder SA, Langbaum JB, Alexander GE, Klunk WE, Mathis CA, Price JC, Aizenstein HJ, DeKosky ST, Caselli RJ. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106:6820–6825. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell DR, Zhou H, Atchison K, Warwick HK, Atkinson PJ, Jefferson J, Xu L, Aschmies S, Kirksey Y, Hu Y, Wagner E, Parratt A, Xu J, Li Z, Zaleska MM, Jacobsen JS, Pangalos MN, Reinhart PH. Impact of apolipoprotein E (apoE) polymorphism on brain apoE levels. J Neurosci. 2008;28:11445–11453. doi: 10.1523/JNEUROSCI.1972-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanan DA, Weisgraber KH, Russell SJ, Mahley RW, Huang D, Saunders A, Schmechel D, Wisniewski T, Frangione B, Roses AD. Apolipoprotein E associates with β amyloid peptide of Alzheimer's Disease to form novel monofibrils. J Clin Invest. 1994;94:860–869. doi: 10.1172/JCI117407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ. Association of apolipoprotein E allele ε4 with late-onset familial and sporadic Alzheimer's disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993a;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD. Binding of human apolipoprotein E to synthetic amyloid β peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993b;90:8098–8102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PM, Han B, Liu F, Mace BE, Ervin JF, Wu S, Koger D, Paul S, Bales KR. Reduced levels of human apoE4 protein in an animal model of cognitive impairment. Neurobiol Aging. 2011;32:791–801. doi: 10.1016/j.neurobiolaging.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Wisniewski T, Frangione B. Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett. 1992;135:235–238. doi: 10.1016/0304-3940(92)90444-c. [DOI] [PubMed] [Google Scholar]
- Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y. Profile and regulation of apolipoprotein E (apoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the apoE locus. J Neurosci. 2006;26:4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Lin S, Bales KR, Gelfanova V, Koger D, Delong C, Hale J, Liu F, Hunter JM, Paul SM. Macrophage-mediated degradation of β-amyloid via an apolipoprotein E isoform-dependent mechanism. J Neurosci. 2009;29:3603–3612. doi: 10.1523/JNEUROSCI.5302-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]