

Macrophage migration inhibitory factor undergoes a localized conformational shift in response to covalent modification by phenethyl isothiocyanate, a natural compound with anti-inflammatory and anticancer properties. The inhibitor sits within a deep hydrophobic pocket and defines a potential target for the development of improved inhibitors.
Keywords: macrophage migration inhibitory factor, phenethyl isothiocyanate, tautomerases, isomerases, cytokines
Abstract
Macrophage migration inhibitory factor is irreversibly inhibited via covalent modification by phenethyl isothiocyanate, a naturally occurring compound with anti-inflammatory and anticancer properties. The structure of the modified protein obtained from X-ray diffraction data to 1.64 Å resolution is presented. The inhibitor sits within a deep hydrophobic pocket between subunits of the homotrimer and is highly ordered. The secondary structure of macrophage migratory inhibitory factor is unchanged by this modification, but there are significant rearrangements, including of the side-chain position of Tyr37 and the main chain of residues 31–34. These changes may explain the decreased binding of the modified protein to the receptor CD74. Together with the pocket, the areas of conformational change define specific targets for the design of more selective and potent inhibitors as potential therapeutics.
1. Introduction
Macrophage migration inhibitory factor (MIF) is a pleiotropic cytokine that plays a role in host microbial defence through promotion and maintenance of the inflammatory response (Calandra & Roger, 2003 ▶). However, in other disease states, including septic shock, inflammatory arthritis and colitis, MIF activity is maladaptive and contributes to the underlying mechanisms of disease. Consequently, inhibition of MIF activity in experimental models of these diseases is beneficial (Calandra et al., 2000 ▶; Leech et al., 2000 ▶; de Jong et al., 2001 ▶). MIF also plays a critical role in diseases in which there is an inflammatory component, including atherosclerosis, diabetes and cancer (Zernecke et al., 2008 ▶; Cvetkovic et al., 2005 ▶; Bifulco et al., 2008 ▶), and efforts are being made to develop small-molecule inhibitors for clinical use (Al-Abed & VanPatten, 2011 ▶).
The biological activity of MIF is triggered by binding to its extracellular receptor CD74 (Leng et al., 2003 ▶) and intracellular signalling proteins (Kleemann et al., 2000 ▶; Jung et al., 2001 ▶). The structure of MIF revealed homology to two bacterial isomerases (Suzuki et al., 1996 ▶) and subsequent studies confirmed isomerase/tautomerase activity (Rosengren et al., 1996 ▶). MIF exists as a homotrimer, and tautomerase activity is dependent on the N-terminal proline residue placed at the bottom of a hydrophobic pocket formed between adjacent subunits (Stamps et al., 1998 ▶). The reactivity of the N-terminal proline is facilitated by neighbouring hydrophobic residues that dramatically lower the pK a of the secondary amine (Swope et al., 1998 ▶). Although no physiologically relevant substrates have been discovered, the tautomerase active site is in the region of the protein that is responsible for CD74 binding. Compounds that bind in the active site and inhibit tautomerase activity are known to interfere in the interaction of MIF with CD74, and these inhibitors display activity in disease models (Al-Abed & VanPatten, 2011 ▶).
A class of compounds called isothiocyanates have been reported to be potent irreversible MIF inhibitors (Brown et al., 2009 ▶; Cross et al., 2009 ▶; Ouertatani-Sakouhi et al., 2009 ▶). Isothiocyanates are plant-derived chemicals that act as noxious deterrents and signalling molecules (Halkier & Gershenzon, 2006 ▶; Holst & Williamson, 2004 ▶; Hopkins et al., 2009 ▶). They have an electrophilic functional group that allows them to react with a range of thiols and amines (Brown & Hampton, 2011 ▶), but there appears to be a degree of selectivity for MIF, as demonstrated by the lowering of MIF in the plasma and urine of individuals ingesting dietary isothiocyanates (Brown & Hampton, 2011 ▶; Healy et al., 2011 ▶).
Mass-spectrometric and mutagenesis studies have revealed that isothiocyanates covalently modify the N-terminal proline of MIF (Brown et al., 2009 ▶). Molecular modelling predicted that isothiocyanate modification would cause conformational change in MIF (Brown et al., 2009 ▶), thereby explaining the observation that isothiocyanates impair CD74 binding (Ouertatani-Sakouhi et al., 2009 ▶). In this study, we present the 1.64 Å resolution structure of MIF with the irreversibly bound inhibitor phenethyl isothiocyanate (PEITC) as the thiourea adduct. Comparisons with noncovalent inhibitors of tautomerase activity revealed conformational changes upon inhibitor binding, confirming previous modelling studies.
2. Materials and methods
2.1. Expression, purification and complex formation
Recombinant human MIF was expressed in Escherichia coli BL21 (DE3) cells and purified by anion-exchange chromatography as described by Bernhagen et al. (1994 ▶). MIF (1 ml at 8 µM) was incubated for 20 min at 291 K with a tenfold excess of PEITC (50 µl at 1.6 mM in dimethyl sulfoxide). The sample was then transferred to 18 MΩ water using an NAP-10 desalting column. A 1 µl aliquot of protein prepared in this way was mixed with 2 µl 10 mg ml−1 α-cyano-4-hydroxycinnamic acid dissolved in 65%(v/v) aqueous acetonitrile containing 0.1%(v/v) trifluoroacetic acid and 10 mM ammonium dihydrogen phosphate and assessed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry to confirm modification of the amino-terminus and to detect any adventitious modifications.
2.2. Crystallization
Crystals were prepared by hanging-drop vapour diffusion in drops composed of a 1:1 mixture of ∼10 mg ml−1 MIF in water and well solution [1.9 M ammonium sulfate, 100 mM Tris pH 8.0, 200 mM NaCl, 4%(v/v) 2-propanol]. The resulting crystals were transferred to the following cryoprotectant for a few minutes before cooling in liquid nitrogen: 2.1 M ammonium sulfate, 100 mM Tris pH 8.0, 200 mM NaCl, 4%(v/v) 2-propanol, 11%(v/v) glycerol.
2.3. Data collection and structure determination
Single-crystal diffraction data were collected using Cu Kα radiation on the University of Otago’s Rigaku MicroMax-007 HF X-ray generator with Osmic VariMax mirrors and an R-AXIS IV++ image-plate detector. Data were collected to 1.53 Å resolution and were indexed in space group P212121. The structure was solved by molecular replacement (using PDB entry 3l5v; McLean et al., 2010 ▶) and refined to an R cryst of 0.189 and an R free of 0.228. Modelling and refinement of the structure was carried out using Coot (Emsley et al., 2010 ▶) and REFMAC (Murshudov et al., 2011 ▶) from the CCP4 suite (Winn et al., 2011 ▶).
3. Results and discussion
3.1. PEITC reaction mechanism
Modification of MIF with PEITC resulted in substantial suppression of the mass-spectrometric peak corresponding to unmodified MIF (12 345 Da) and the appearance of a substantial peak corresponding to PEITC–MIF (12 508 Da, as observed previously; Brown et al., 2009 ▶). No significant signal was detected corresponding to MIF bound to two PEITC moieties (predicted mass 12 671 Da), suggesting that reaction with the ∊-amino group of lysine residues or other secondary sites is much slower than reaction at the amino-terminus or, in the case of the three cysteine residues in MIF, may not produce a stable adduct. Fig. 1 ▶ shows the reaction mechanism in detail. A number of hydrophobic and aliphatic residues create the hydrophobic environment which contributes to the lowered pK a of the N-terminal proline (Swope et al., 1998 ▶).
Figure 1.

Mechanism of inhibition. The N-terminal proline (with a lowered pK a) nucleophilically attacks the central C atom of the isothiocyanate moiety, forming a new covalent bond. This produces a stable thiourea adduct as the inhibited form of the enzyme.
3.2. PEITC binding mode
Crystals grew within 3 d as octahedra of up to 0.5 mm in the largest dimension from solutions containing between 1.8 and 2.1 M ammonium sulfate. Trials under identical conditions except for the replacement of NaCl by 100 mM citrate yielded similar crystals. Trials using the same protein concentration at pH 7 or 7.5 with or without 100 mM citrate and using polyethylene glycol as precipitant [22–32%(w/v) PEG 8000 or 34–44%(w/v) PEG 400] did not yield crystals. Crystals diffracted to beyond 1.5 Å resolution when cooled to 93 K in either mother liquor or cryoprotectant, which was required to prevent ice formation. Data-collection parameters and quality are summarized in Table 1 ▶.
Table 1. Data-collection and reduction statistics.
Values in parentheses are for the highest shell.
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 67.73, b = 68.25, c = 88.71 |
| Unique reflections | 51308 |
| Resolution range (Å) | 31.64–1.53 (1.58–1.53) |
| Multiplicity | 5.310 (2.13) |
| Completeness (%) | 81.4 (14.0) |
| 〈I/σ(I)〉 | 14.00 (2.200) |
| R merge † | 0.057 (0.389) |
R
merge = 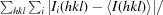
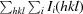 .
.
Initial maps derived from diffraction data from the PEITC–MIF crystal and phases from molecular replacement using native human MIF (PDB entry 3l5v; McLean et al., 2010 ▶) included unambiguous density for the PEITC adduct (Fig. 2 ▶). Refinement was carried out without the use of noncrystallographic symmetry. The final model has been deposited as PDB entry 4f2k (Table 2 ▶).
Figure 2.
Initial OMIT map. Maps (2F o − F c in blue, positive F o − F c in green, negative F o − F c in red) were drawn with phases from the initial molecular-replacement solution using PDB entry 3l5v, which did not contain the PEITC adduct. The final model is shown as sticks (PEITC is shown with C atoms in white, N atoms in blue and S atoms in yellow; other C atoms are shown in green and O atoms in red).
Table 2. Refinement statistics.
Values in parentheses are for the highest shell.
| Resolution range (Å) | 31.64–1.53 (1.57–1.53) |
| Reflections used | 51077 (482) |
| Completeness (%) | 81.1 (11.0) |
| R cryst † | 0.190 (0.50) |
| R free ‡ | 0.228 (0.59) |
| No. of atoms in model | |
| Protein atoms | 2601 |
| PEITC atoms | 33 |
| Other heteroatoms | 36 |
| Solvent atoms | 343 |
| R.m.s.d. from ideal bond lengths (Å) | 0.029 |
| R.m.s.d. from ideal bond angles (°) | 2.193 |
| Average B factor (Å2) | |
| Overall | 18.51 |
| Protein atoms | 17.11 |
| PEITC atoms | 16.17 |
| Solvent atoms | 29.25 |
| Ramachandran plot, residues in | |
| Preferred regions | 313 |
| Allowed regions | 4 |
| Disallowed regions | 2 |
R
cryst = 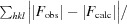
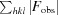 computed over a working set composed of 95% of the data.
computed over a working set composed of 95% of the data.
R
free =
computed over a test set composed of 5% of the data.
As expected, the PEITC is bound to the amino-terminal proline of the polypetide chain. This proline residue is shifted ∼1.6 Å in response to PEITC binding. The PEITC was modelled with full occupancy, which is supported by difference maps (Fig. 2 ▶). The phenyl moiety of PEITC is well ordered; the B factors refined to below the average for the remainder of the protein chain despite being at a terminus (Table 2 ▶). It is bound in a deep pocket (Fig. 3 ▶ b). One side of this binding pocket is substantially aromatic (composed of Tyr96′ and Phe114), while the other is predominantly aliphatic (Met3, Ile65 and Val107). The phenyl moiety of PEITC is sandwiched between Met3 and Val107. An edge-to-face interaction between the phenyl moiety of PEITC and Tyr96′ (from the adjacent chain) provides another critical contact. All noncovalent contacts between the PEITC moiety and the protein appear to be hydrophobic interactions. Specific binding within this pocket prior to reaction may partially explain the specificity of PEITC for the reaction at the amino-terminus of MIF.
Figure 3.

Structural comparison of PEITC–MIF with MIF. The structure of PEITC–MIF (shades of blue; PDB entry 4f2k) is shown superimposed on that of unmodified MIF (yellow; PDB entry 3l5v; McLean et al., 2010 ▶). Separate chains of PEITC–MIF are coloured in different shades of blue; PEITC is shown as sticks with C atoms in white, N atoms in blue and S atoms in yellow. (a) displays the entire trimer, looking approximately down the threefold axis. Two chains are shown as surfaces, with one shown as a cartoon with the PEITC adduct and Pro2 as sticks. The red arrow indicates loop 31–34. (b) shows a view into the binding pocket. The trimer is depicted by a grey surface; residues lining the binding pocket are identified and shown as sticks with non-C atoms coloured as for PEITC.
PEITC–MIF is very similar to underivatized MIF in the disposition of structural elements. However, adjacent to the binding pocket, the first turn of the first helix and the preceding loop (residues 31–34) are displaced approximately 1.6 Å away from the PEITC-binding site and the β-sheet core (red arrow in Fig. 2 ▶ a).
3.3. Functional consequences
Three separate features of the PEITC–MIF structure can explain the inhibition of tautomerase activity: blockage of the catalytic amino-terminus of MIF, competition with substrate binding in the hydrophobic pocket and distortion of the protein surface at residues 32–37. These last two also have the potential to impair CD74 binding by PEITC–MIF.
Residue Tyr37 sits just outside the binding pocket and shows the largest deviation in side-chain position observed in a comparison of PEITC–MIF with MIF (PDB entry 3l5v). Intriguingly, Tyr37 has been identified as a binding target in a fragment-based screen (McLean et al., 2010 ▶). Our data show that this is one of the few areas that undergoes substantial change. Whilst only little variation would be acceptable deep within the pocket, targeting the solvent-accessible cavity offers greater opportunity for the introduction of further interactions with the protein. Together, these features offer novel targets for in silico screening of potential drugs.
Supplementary Material
PDB reference: macrophage migration inhibitory factor, complex with phenethyl isothiocyanate, 4f2k
Acknowledgments
This work was funded by a University of Otago Research Grant. We are grateful to the University of Otago Centre for Protein Research for mass-spectroscopic analysis.
References
- Al-Abed, Y. & VanPatten, S. (2011). Future Med. Chem. 3, 45–63. [DOI] [PubMed]
- Bernhagen, J., Mitchell, R. A., Calandra, T., Voelter, W., Cerami, A. & Bucala, R. (1994). Biochemistry, 33, 14144–14155. [DOI] [PubMed]
- Bifulco, C., McDaniel, K., Leng, L. & Bucala, R. (2008). Curr. Pharm. Des. 14, 3790–3801. [DOI] [PubMed]
- Brown, K. K., Blaikie, F. H., Smith, R. A., Tyndall, J. D., Lue, H., Bernhagen, J., Winterbourn, C. C. & Hampton, M. B. (2009). J. Biol. Chem. 284, 32425–32433. [DOI] [PMC free article] [PubMed]
- Brown, K. K. & Hampton, M. B. (2011). Biochim. Biophys. Acta, 1810, 888–894. [DOI] [PubMed]
- Calandra, T., Echtenacher, B., Roy, D. L., Pugin, J., Metz, C. N., Hültner, L., Heumann, D., Männel, D., Bucala, R. & Glauser, M. P. (2000). Nature Med. 6, 164–170. [DOI] [PubMed]
- Calandra, T. & Roger, T. (2003). Nature Rev. Immunol. 3, 791–800. [DOI] [PMC free article] [PubMed]
- Cross, J. V., Rady, J. M., Foss, F. W., Lyons, C. E., Macdonald, T. L. & Templeton, D. J. (2009). Biochem. J. 423, 315–321. [DOI] [PMC free article] [PubMed]
- Cvetkovic, I., Al-Abed, Y., Miljkovic, D., Maksimovic-Ivanic, D., Roth, J., Bacher, M., Lan, H. Y., Nicoletti, F. & Stosic-Grujicic, S. (2005). Endocrinology, 146, 2942–2951. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Halkier, B. A. & Gershenzon, J. (2006). Annu. Rev. Plant Biol. 57, 303–333. [DOI] [PubMed]
- Healy, Z. R., Liu, H., Holtzclaw, W. D. & Talalay, P. (2011). Cancer Epidemiol. Biomarkers Prev. 20, 1516–1523. [DOI] [PMC free article] [PubMed]
- Holst, B. & Williamson, G. (2004). Nat. Prod. Rep. 21, 425–447. [DOI] [PubMed]
- Hopkins, R. J., van Dam, N. M. & van Loon, J. J. (2009). Annu. Rev. Entomol. 54, 57–83. [DOI] [PubMed]
- de Jong, Y. P. et al. (2001). Nature Immunol. 2, 1061–1066. [DOI] [PubMed]
- Jung, H., Kim, T., Chae, H. Z., Kim, K.-T. & Ha, H. (2001). J. Biol. Chem. 276, 15504–15510. [DOI] [PubMed]
- Kleemann, R., Hausser, A., Geiger, G., Mischke, R., Burger-Kentischer, A., Flieger, O., Johannes, F. J., Roger, T., Calandra, T., Kapurniotu, A., Grell, M., Finkelmeier, D., Brunner, H. & Bernhagen, J. (2000). Nature (London), 408, 211–216. [DOI] [PubMed]
- Leech, M., Metz, C., Bucala, R. & Morand, E. F. (2000). Arthritis Rheum. 43, 827–833. [DOI] [PubMed]
- Leng, L., Metz, C. N., Fang, Y., Xu, J., Donnelly, S., Baugh, J., Delohery, T., Chen, Y., Mitchell, R. A. & Bucala, R. (2003). J. Exp. Med. 197, 1467–1476. [DOI] [PMC free article] [PubMed]
- McLean, L. R., Zhang, Y., Li, H., Choi, Y.-M., Han, Z., Vaz, R. J. & Li, Y. (2010). Bioorg. Med. Chem. Lett. 20, 1821–1824. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Ouertatani-Sakouhi, H., El-Turk, F., Fauvet, B., Roger, T., Le Roy, D., Karpinar, D. P., Leng, L., Bucala, R., Zweckstetter, M., Calandra, T. & Lashuel, H. A. (2009). Biochemistry, 48, 9858–9870. [DOI] [PMC free article] [PubMed]
- Rosengren, E., Bucala, R., Aman, P., Jacobsson, L., Odh, G., Metz, C. N. & Rorsman, H. (1996). Mol. Med. 2, 143–149. [PMC free article] [PubMed]
- Stamps, S. L., Fitzgerald, M. C. & Whitman, C. P. (1998). Biochemistry, 37, 10195–10202. [DOI] [PubMed]
- Suzuki, M., Sugimoto, H., Nakagawa, A., Tanaka, I., Nishihira, J. & Sakai, M. (1996). Nature Struct. Biol. 3, 259–266. [DOI] [PubMed]
- Swope, M., Sun, H.-W., Blake, P. R. & Lolis, E. (1998). EMBO J. 17, 3534–3541. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Zernecke, A., Bernhagen, J. & Weber, C. (2008). Circulation, 117, 1594–1602. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: macrophage migration inhibitory factor, complex with phenethyl isothiocyanate, 4f2k