

Abstract
Endosomal sorting complexes required for transport (ESCRT) regulate diverse processes ranging from receptor sorting at endosomes to distinct steps in cell division and budding of some enveloped viruses. Common to all processes is the membrane recruitment of ESCRT-III that leads to membrane fission. Here, we show that CC2D1A is a novel regulator of ESCRT-III CHMP4B function. We demonstrate that CHMP4B interacts directly with CC2D1A and CC2D1B with nanomolar affinity by forming a 1:1 complex. Deletion mapping revealed a minimal CC2D1A-CHMP4B binding construct, which includes a short linear sequence within the third DM14 domain of CC2D1A. The CC2D1A binding site on CHMP4B was mapped to the N-terminal helical hairpin. Based on a crystal structure of the CHMP4B helical hairpin two surface patches were identified that interfere with CC2D1A interaction as determined by surface plasmon resonance. Introducing these mutations into a C-terminal truncation of CHMP4B that exerts a potent dominant negative effect on HIV-1 budding, revealed that one of the mutants lost this effect completely. This suggests that the identified CC2D1A binding surface might be required for CHMP4B polymerization, which is consistent with the finding that CC2D1A binding to CHMP4B prevents CHMP4B polymerization in vitro. Thus, CC2D1A might act as a negative regulator of CHMP4B function.
Keywords: CC2D1A, FREUD-1, Aki-1, LgD, ESCRT, CHMP4, HIV-1, budding
Introduction
The coiled-coil and C2 domain containing proteins A and B (CC2D1A, CC2D1B) are evolutionary conserved proteins that contain four drosophila melanogaster 14 domains (DM14) of unknown function followed by a C2 domain. CC2D1A and B are also known as Akt kinase interacting protein 1(Aki-1), Five prime REpressor Under Dual repression binding protein 1 (Freud-1 and 2) and Tank-binding kinase 1 (TBK-1) associated protein in endolysosomes (TAPE). The emerging picture for CC2D1 function suggests that it acts as a scaffold protein that touches and/or connects several distinct cellular functions. CC2D1A was initially reported to function as activator of NF-κB 1 and as transcriptional repressor of the serotonin-1A receptor gene 2; 3; its deregulation was associated with major depressive disorder 4. Furthermore, a C-terminal deletion in the CC2D1A gene has been linked to nonsyndromic mental retardation 5.
Besides transcriptional regulation, CC2D1A (TBK-1associated protein in endolysosomes, Aki-1) has been implicated in different signaling pathways including the TBK-1, nuclear factor κB and extracellular-signal-regulated kinase pathways that regulate immunity, inflammation and cell survival 6; 7 and the PDK1/Akt (phosphatidylinositol-2-OH/3-phosphoinosite-dependent protein kinase 1-Akt) pathway in epidermal growth factor signaling 8 associated with cell survival and cell cycle progression.
A direct role in cell division is suggested by the CC2D1A (Aki-1) association with centrosomes and regulation of centriole cohesion 9. Its function during mitosis is further controlled by phosphorylation 10. Consistent with a role in cell division CC2D1A (Aki1) depletion caused the formation of multipolar spindles 9; 11. Interestingly, a similar phenotype was observed upon depletion of some endosomal sorting complexes required for transport (ESCRT) components including ESCRT-III CHMP4B 12.
ESCRT complexes 0, −I, −II, −III and the VPS4 complex catalyze multivesicular body biogenesis leading to plasma membrane receptor down-regulation 13; 14; 15, and some ESCRTs are recruited during cytokinesis and enveloped virus budding 16; 17; 18. The function of ESCRT-III and VPS4 is common to all three processes and required for membrane fission 14; 15; 19. A direct link between ESCRT function and CC2D1A was proposed by the yeast two-hybrid interaction of CC2D1A and CHMP4 20. Further evidence of an ESCRT-CC2D1A, CC2D1B connection originates from studies with the drosophila homologue Lethal giant discs (Lgd) of CC2D1A which is implicated in Notch receptor signaling. In lgd mutant cells, Notch and other transmembrane proteins accumulate in enlarged endosomal compartments positive for Rab5, Rab 7 and the ESCRT-0 factor Hrs. This indicated that Lgd functions in endosomal trafficking downstream of Hrs 21; 22; 23.
In order to test the connection of CC2D1A and ESCRT-III we set out to analyze the structural basis of the CC2D1A interaction with ESCRT-III CHMP4. We show that only one DM14 domain is required for the interaction with C-terminally truncated CHMP4B and identify the interaction surface by site-directed mutagenesis. CC2D1A co-purification with truncated forms of CHMP4B identified the requirement of the helical hairpin of CHMP4B for interaction. Isothermal titration calorimetry (ITC) of CHMP4B deletion constructs and CC2D1A mapped the binding site to the N-terminal end of the CHMP4B hairpin, whose structure was determined to 1.8Å resolution. Mutagenesis of the CHMP4B hairpin fragment confirmed this region as CC2D1A interaction site as tested by surface plasmon resonance (SPR) analyses. Furthermore, we provide evidence that the CC2D1A interaction surface is important for CHMP4B polymerization and required to exert a dominant negative effect on HIV-1 budding. Because the CC2D1A-CHMP4B interaction inhibited CHMP4 polymerization in vitro, our data suggest further that CC2D1A might thus act as a regulator of CHMP4 polymerization.
Results
CC2D1A recruits CHMP4B in vitro
A yeast two-hybrid screen identified CC2D1A and CC2D1B as potential binding partners of CHMP4A, CHMPB and CHMP4C 20. In order to confirm the yeast two hybrid results we set out to determine the direct interaction of CC2D1A or B and CHMP4B. Although attempts to express full length CC2D1A or CC2D1B in E. coli failed, a C-terminal deletion of CC2D1B, which lacks the C2 domain, CC2D1B(1-601) (Figures 1A and B) could be expressed and co-purified with MBP-CHMP4BΔC 24. Both proteins eluted in the same peak from a size-exclusion chromatography (SEC) column (data not shown). However, removal of MBP from CHMP4BΔC by tobacco etch virus (TEV) protease cleavage induced aggregation of the complex. The same construct corresponding to CC2D1A was poorly expressed and not soluble. For enhancement of the solubility of the complex a smaller CHMP4B fragment containing residues 7-110 was identified by limited proteolysis with trypsin and MBP-CHMP4B(7-110) was cloned and expressed. MBP-CHMP4B(7-110) formed a complex with CC2D1B(1-601), which stayed monodisperse after removal of MBP. SEC in combination with multi angle laser light scattering (MALLS) analysis indicated a 1:1 binding stoichiometry based on the ~ 80-kDa molecular weight of the complex (Figure 1C), which is close to the calculated molecular mass of 76.4 kDa for a 1:1 complex (the calculated molecular mass of CC2D1B(1-601) is 64.6 kDa and of CHMP4B(7-110) is 12 kDa). The 1:1 stoichiometry was further confirmed by isothermal titration calorimetry (ITC), which revealed a Kd of 382 nM for the CC2D1B(1-601)-MBP-CHMP4B(7-110) interaction (Figure S1 and Table 1).
Figure 1.

CC2D1B interacts with CHMP4B in vitro.
(A) Schematic drawing of the domain organization of CC2D1 isoforms. Both isoforms contain 4 DM14 domains followed by a C2 domain.
(B) Schematic drawing of the CC2D1A and B constructs tested for CHMP4B interaction by SEC and ITC. Complex formation is indicated with a +.
(C) SEC (S-200 column) in combination with RI (refractive index) and MALLS (Multi Angle Laser Light Scattering) analyses of CC2D1B(1-601) in complex with CHMP4B(7-105) reveals a 1:1 complex. The inset shows the complex eluted from the SEC column (upper band CC2D1B(1-601) and lower band, CHMP4B(7-105).
(D) MALLS (S-75 column) analysis of the CC2D1A(346-455)-CHMP4B(23-97) complex shows a molecular mass of 18.2 kDa. The calculated molecular mass of the complex is 20.9 kDa.
(E) Pull down of CC2D1A(346-455) wild type and mutant (mut; CC2D1A_mut) by MBP-CHMP4B(7-105), shows that the CC2D1A mutations abrogate CHMP4B binding. Lanes 1 and 2 input of CC2D1A(346-455) wild-type and mutant, respectively; lane 3 pull down of CC2D1A346-455) wild-type and lane 4 of the mutant; lanes 5 to 7 shows the controls, MBP-CHMP4B(7-105) alone, CC2D1A(346-455) and mutant alone. Molecular mass markers are indicated.
Table 1.
Isothermal titration calorimetry of the CC2D1 interaction with CHMP4. The standard error associated with the values for N, ΔH and Kd is derived from the nonlinear least-squares fitting of the curves shown in Figure S1.
| Cell (μM) | Syringe (μM) | N | ΔH (kcal/mol) | KD (nMol) | ||
|---|---|---|---|---|---|---|
| CC2D1B(1-601) | 10 | MBP-CHMP4B(7-110) | 85 | 1.02 ± 0.007 | −13710 ± 123.3 | 382 ± 24 |
| MBP-CHMP4B(7-110) | 10 | CC2D1A(309-494) | 253 | 0.84 ± 0.006 | −7381 ± 67.58 | 351 ± 22 |
| MBP-CHMP4B(7-110) | 9 | CC2D1A(346-494) | 145 | 1.03 ± 0.009 | −8164 ± 91.52 | 719 ± 41 |
| MBP-CHMP4B(7-110) | 9 | CC2D1A(346-455) | 117 | 0.90 ± 0.007 | −8193 ± 84.49 | 680 ± 34 |
| MBP-CHMP4B(23-97) | 14 | CC2D1A(346-455) | 200 | 0.90 ± 0.019 | −14600 ± 166.9 | 5495±118 |
In order to further define the CHMP4B interaction site on CC2D1A, we designed several deletion constructs of CC2D1A (Figure 1B) and tested the interaction with CHMP4B(7-110) by SEC (data not shown), ITC or MALLS (Figures S1 and S2). This demonstrated that the third DM14 domain and the region connecting to the fourth DM14 domain are required for interaction. ITC and MALLS confirmed the 1:1 stoichiometry for CHMP4B binding to the minimal binding construct CC2D1A(346-455) (Figures 1D and S1). However, ITC also indicated small differences regarding the individual Kd values. CC2D1A(309-494) produced a Kd of 351 nM, which is comparable to CHMP4B(7-110) binding to CC2D1B(1-601) (Kd=382 nM). Shortening the N-terminus further as in CC2D1A(346-494) increased the Kd 2-fold to 719 nM. In contrast, the C-terminal truncation of CC2D1A(346-494) to CC2D1A(346-455) produced a similar Kd as observed for CC2D1A(346-494) (Table 1). Further truncations such as CC2D1A(291-416), which is short of the region C-terminal of the third DM14 domain and CC2D1A(416-455), which lacks the third DM14 domain did not produce complexes as analyzed by SEC and ITC (data not shown). We conclude from these experiments that amino acids 346 to 455, which include the third DM14 domain, contain the main binding site for C-terminally truncated CHMP4B.
In order to map the CHMP4 binding site on CC2D1A, we compared sequences from different species of CC2D1A and CC2D1B comprising the minimal CHMP4 binding region (residues 346-455) as determined by SEC and ITC. Because both CC2D1A and CC2D1B bind to CHMP4B, we designed a mutant of CC2D1A(346-455) based on sequence conservation. CC2D1A_mut has 7 conserved residues changed to Ala (Figure S3). The mutant protein was soluble and the interaction was tested with MBP-CHMP4B(7-105). While wild-type CC2D1A(346-455) was able to pull-down CHMP4B(7-105) as expected from the ITC measurements, CC2D1A_mut did not interact with CC2D1A in this assay (Figure 2E). We conclude from these experiments that the linear sequence within CC2D1A residues 374-393 (Figure S3) is important, implicating the third DM14 domain in CHMP4B binding.
Figure 2.

Crystal structure of the helical hairpin of CHMP4B.
(A) Stereo image of the electron density map calculated based on the SAD phases without density modification.
(B) Ribbon diagram of the CHMP4B helical hairpin containing residues 23-97. Note that the crystallized construct contained four extra residues at the N-terminus, which are in a helical conformation.
(C) Stereo images of CHMP4B (blue) and CHMP3 (salmon) (Protein Data bank (PDB) ID 3FRT) based on superpositioning of the Cα atoms.
(D) Stereo images of CHMP4B (blue) and IST1 (cyan) (PDB ID 3FRR) based on superpositioning of the Cα atoms.
(E) Superpositioning of the Cα atoms of CHMP4B (blue) and Vps20 (gray) (CHMP6) (PDB ID 3FTU; the CHMP4B residues affecting CC2D1A interaction are labeled in red and the Vps20 residues involved in ESCRT-II Vps25 interaction are shown in yellow.
We tested next whether truncations of both helices of the CHMP4B helical hairpin to residues 23 to 97 affects CC2D1A binding. CHMP4B(23-97) still interacted with CC2D1A(346-455) as determined by SEC (data not shown). However, ITC measurements indicated an ~ 10-fold higher Kd of 5.5 μM (Figure S1 and Table 1). This suggests that the N- and C-terminal ends of the CHMP4B hairpin are important either for CC2D1A interaction or for the stability of the helical hairpin.
Crystal structure of CHMP4B(23-97)
We tested all complexes formed between CC2D1A or CC2D11B and CHMP4B in crystallization trials, but failed to obtain crystals of the complex. However, in one of the crystallization trials performed with the complex of CC2D1A(346-455) and CHMP4B(23-97), CHMP4B(23-97) crystallized on its own. The structure was solved from a selenomethione-substituted crystal using the single-wavelength anomalous dispersion method and diffraction data to 1.8 Å resolution (Table 2), which resulted in a readily interpretable electron density map (Figure 2A). The asymmetric unit contained four molecules forming identical ~64 Å long helical hairpins composed of α1 residues 23-58 and α2 residues 61-97 (Figure 2B). Cα superpositioning of the main-chain atoms of CHMP4B with the main chain of CHMP3 results in an r.m.s.d of 2.68 Å (Figure 2C) and with the main chain of IST1 in an r.m.s.d of 3.32 Å (figure 3D). Likewise part of α helix 1 from yeast Vps20 (CHMP6) fits α1 from CHMP4 with an r.m.s.d. of 0.68 Å (Figure 2D). This corroborates the structural similarity of CHMP proteins despite their low sequence conservation (Figure S4).
Table 2.
Data collection and refinement statistics
| CHMP4 (SeMet) | |
|---|---|
| Data Collection | |
| Space group | P212121 |
| Cell dimensions a, b, c (Å) | 37.59, 71.43, 123.68 |
| Wavelength (Å) | 0.9795 |
| Resolution | 41.23 -1.80 / (1.90- 1.80) |
| Rmerge | 0.074 (0.414) |
| I / σI | 16.3 (4.4) |
| Completeness (%) | 99.7 (100.0) |
| Multiplicity | 7.0 (7.1) |
| Anomalous completeness | 99.6 (100.0) |
| Anomalous multiplicity | 3.6 (3.6) |
| Total observations | 220642 (32311) |
| Unique reflections | 31659 (4520) |
| Wilson B-factor (Å2) | 23.3 |
| Refinement | |
| Resolution (Å) | 1.80 |
| Rwork (number of reflections) | 0.227 (30007) |
| Rfree (number of reflections) | 0.285 (1588) |
| Number of atoms | |
| Protein | 2693 |
| Water | 137 |
| B-factors (Å2) | |
| Protein | 16.78 |
| Water | 27.6 |
| r.m.s d. | |
| Bond lengths (Å) | 0.018 |
| Bond angles (°) | 0.884 |
Values in parentheses are for highest resolution shell. SeMet, selenomethionine.
Figure 3.

CC2D1A binds to the N-terminal end of the CHMP4B helical hairpin.
(A) The molecular surface of CHMP4 is shown in two orientations and charged residues are indicated. The two patches affecting CC2D1A interaction are circled in green; CHMP4Bmut1 carries the R28A, R30A, D31R and E33R mutations and CHMP4Bmut2 has E90R, E94R and E97R mutated.
(B) Co-purification of CC2D1A(346-455) and wild-type or mutant MBP-CHMP4B(7-105); lane 1, wild-type MBP-CHMP4B(7-105); lane 2 MBP-CHMP4B(7-105)mut1; lane 3, MBP-CHMP4B(7-105)mut2; lane 4, MBP-CHMP4B(7-105)mut1.2.
Mapping of the CC2D1A binding site on CHMP4B
We focused our mutational analyses to map the CC2D1A binding site to the N- and C-terminal ends of the helical hairpin for the following reasons. First, deletion of the N- and C-terminal ends of CHMP4B(7-110) that produced CHMP4B(23-97), induced a 10-fold reduction in affinity for CC2D1A. Second, all CHMP4B-CC2D1A complexes were disrupted by high salt treatment indicating a potential role for charged interactions. Third, charged residues for mutagenesis were further chosen based on sequence conservation, taking into account that only CHMP4 isoforms interact with CC2D1A or CC2D1B (Figure S4). Fifth, mapping of the charged residues on the helical hairpin structure revealed one side of the molecule that contains a cluster of acidic and basic residues at the N- and C-terminal ends of the hairpin, while the opposite side is largely uncharged (Figure 3A). Based on these observations, we designed three sets of mutants of CHMP4B(7-105), one containing amino acid changes within α1, CHMP4Bmut1 (R28A, R30A, D31R, E33R), one with amino acid changes within α2, CHMP4Bmut2 (E90R, E94R, E97R) and the double mutant CHMP4Bmut1.2. Co-purification of CC2D1A(346-455) and wild-type or mutant (mut1, mut2, mut1.2) MBP-CHMP4B(7-105) demonstrated that both wild-type and mutant MBP-CHMP4B(7-105)mut1 pulled down CC2D1A(346-455). In contrast, MBP-CHMP4B(7-105)mut2 and MBP-CHMP4B(7-105)mut1.2 no longer interacted with CC2D1A(346-455) (Figure 3B). This interaction pattern was confirmed by SPR. Wild-type CHMP4B(7-105) produced a KD of 320 nM (Table 3 and Figure S5) similar to the affinity measured by ITC (Table 1). In contrast, CHMP4B(7-105)mut1 showed only a ~4.5-fold reduced KD, while CHMP4B(7-105)mut2 and CHMP4B(7-105)mut1.2 did no longer interact with CC2D1A (Table 3 and Figure S5). We conclude that a charged surface patch on α helix 2 (E90, E94 and E97) is important for binding and that neighboring charged residues within helix 1 (R28, R30, D31, E33) contribute to CC2D1A interaction.
Table 3.
SPR analyses of the CHMP4B and CC2D1A interaction. The standard errors (SE) are derived from four measurements.
| Ligand | Analyte | ka (103 M−1 s−1) | kd (10−3 s−1) | KD (nMol) | Chi2 |
|---|---|---|---|---|---|
| CC2D1A(346-455) | MBP-CHMP4B(7-105) | 25 ± 2 | 7.9 ± 0.28 | 320 | 0.244 |
| CC2D1A(346-455) | MBP-CHMP4B(7-105)muti | 1.2 ± 0.13 | 1.7 ± 0.12 | 1400 | 1.16 |
| CC2D1A(346-455) | MBP-CHMP4B(7-105)mut2 | - | - | - | - |
| CC2D1A(346-455) | MBP-CHMP4B(7-105)muti.2 | - | - | - | - |
CC2D1A binding to CHMP4B prevents CHMP4B polymerization in vitro
We have previously shown that C-terminally truncated CHMP4B, MBP-CHMP4BΔC-ALIX can form polymers in vitro 24. For testing whether CC2D1A interferes with CHMP4B polymerization, MBP-CHMP4BΔC-ALIX alone and MBP-CHMP4BΔC-ALIX in complex with CC2D1A(346-455) were cleaved with TEV protease and analyzed by sucrose gradient density centrifugation. This showed that CC2D1A alone is found in the upper fractions of the gradient (Figure 4A) while MBP-CHMP4BΔC-Alix alone forms monomers and high-molecular-weight polymers present in the lower fraction (Figure 4B). When MBP-CHMP4BΔC-Alix is cleaved with TEV protease the monomers shift into the polymer fraction (Figure 4C). However, if MBP-CHMP4BΔC-Alix is incubated with CC2D1A(346-455) prior to TEV protease cleavage, approximately half of monomeric CHMP4BΔC-Alix stays monomeric and floats together with CC2D1A(346-455) in the upper fraction of the gradient, while the other half of CHMP4BΔC-Alix polymerizes and is found in the bottom of the gradient (Figure 4D). When the same experiment is performed with CC2D1A_mut that no longer interacts with CHMP4B (Figure 1E), monomeric CHMP4BΔC-Alix is no longer rescued by CC2D1A and all of cleaved CHMP4BΔC-Alix is found in the bottom fraction of the gradient (Figure 4E). Because CC2D1A is never found in the bottom fraction together with CHMP4BΔC-Alix polymers, it indicates that CC2D1A does not interact with CHMP4B polymers. We confirmed this by incubating CC2D1A(309-494), which migrates in the upper fraction of a sucrose gradient (Figure 4F) with CHMP4BΔC-Alix polymers. Again CC2D1A is not found in the bottom fraction, indicating that it does not interact with CHMP4B polymers (Figure 4G). These results indicate that the CC2D1A binding site is not accessible on CHMP4 filaments and that CC2D1A prevents CHMP4B polymerization in vitro.
Figure 4.

CC2D1A prevents CHMP4B polymerization. Sucrose gradient analyses of MBP-CHMP4BΔC_Alix, CHMP4BΔC-Alix and CC2D1A.
(A) CC2D1A(346-455); (B) MBP-CHMP4BΔC_Alix; (C) MBP-CHMP4BΔC_Alix after TEV protease cleavage; (D) MBP-CHMP4BΔC_Alix was incubated with CC2D1A(346-455) and then subjected to TEV protease cleavage; (E) MBP-CHMP4BΔC_Alix was incubated with CC2D1A_mut and then subjected to TEV protease cleavage.
(F) CC2D1A(309-494) and (G) CHMP4BΔC_Alix polymers were incubated with CC2D1A(309-494).
The CHMP4B mutations affect HIV-1 budding
In order to test whether the mutation of the CC2D1A binding site on CHMP4B affects HIV-1 budding, we introduced the the CHMP4B mutations (mut1, mut2, mut1.2) into CHMP4B(1-153)-flag, which was shown to exert a dominant negative effect on HIV-1 budding when expressed in HIV-1 producing 293T cells 25. As expected wild-type CHMP4B(1-153)-flag inhibited HIV-1 release efficiently in comparison to the vector control (Figure 5A). Similarly, expression of CHMP4Bmut1 (mut1; R28A, R30A, D31R, E33R) is still dominant negative albeit slightly reduced (Figure 5A) while CHMP4Bmut2 (mutation 2, E90R, E94R, E97R) has lost its dominant negative effect completely (Figure 5A). As expected the double mutant CHMP4Bmut1.2, behaved like CHMP4Bmut2 and is no longer dominant negative (Figure 5A). Consistent with the release pattern, wild type CHMP4B(1-153), and CHMP4Bmut1revealed the intracellular accumulation of the Gag cleavage intermediates CAp2 and p41 (Figure 5B, left panel, lanes 1 and 2), which are characteristic for late assembly defects 26; 27. These results indicate that the acidic Mut2 patch on helix 2 is required for the dominant negative effect of CHMP4, while the surface patch of Mut1 seems to be less important.
Figure 5.

Mutations in CHMP4B annul the dominant negative effect of C-terminally truncated CHMP4B.
(A) (Left panel) Expression of wild type CHMP4B1-153FLAG exerts a strong dominant negative effect on HIV-1 budding (lane 2) as compared to the vector control (lane 1). Expression of CHMP4B1-153mut1 shows that it is still dominant negative (lane 3), while CHMP4B1-153mut2 lost the dominant negative effect (lane 4). (Right panel) lane 1 vector only control; lane 2, expression of wild type CHMP4B1-153FLAG exerts a strong dominant negative effect and lane 3, the double mutant CHMP4B1-153mut1.2 is no longer dominant negative.
(B) Western blot revealing the intracellular Gag processing corresponding to the panels shown in (A). (Lower panel), Western blot showing the expression levels of the CHMP4B constructs.
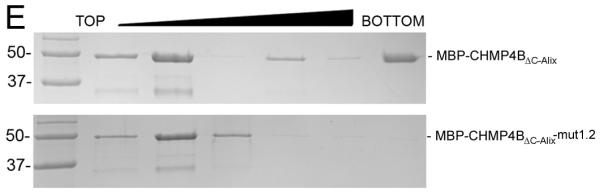
We next compared the cellular localization of CHMP4B(1-153)-flag and its mutated forms in order to test whether localization correlates with the propensity to block HIV-1 budding. Confocal microscopy imaging revealed that approximately 80% of CHMP4B(1-153)-flag, 64% of CHMP4Bmut1 and 79 % of CHMP4Bmut2 localize along the plasma membrane (Figures 6A-C and S7). In contrast CHMP4Bmut1.2 reveals dramatically reduced plasma membrane (19%) versus 81% cytosolic localization (Figures 6D and S6). This indicates that the mut2 mutation still allows plasma membrane localization, while the double mutant (mut1.2) surface has largely lost its ability to be targeted to the plasma membrane. Because mutants mut2 and mut1.2 act no longer dominant negative during HIV-1 budding (Figure 5), we speculate that this might be due to a defect in polymerization rather than membrane association. To test the effect of the mut1.2 surface patch on polymerization, we mutated the surface of MBP-CHMP4BΔC-Alix, which forms polymers in vitro 24. Sucrose gradient analyses of wild-type MBP-CHMP4BΔC-Alix and the mutated form (mut1.2) demonstrates that the wild-type forms monomers detected in the top fraction of the gradient and polymers detected in the bottom fraction of the gradient as expected 24 (Figure 6E). In contrast the mutated form of MBP-CHMP4BΔC-Alix is monomeric and only found in the upper fraction of the gradient (Figure 6E). We thus conclude from the immune localization data and the in vitro polymerization of CHMP4B that the surface patch of mut1.2 plays a role in CHMP4B polymerization.
Figure 6.
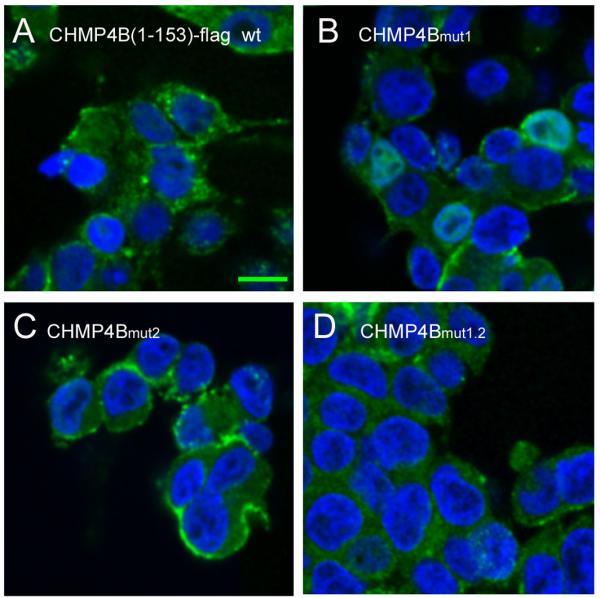
The mut1.2 surface patch is important for plasma membrane localization and polymerization of CHMP4B in vitro.
Confocal microscopy of CHMP4B(1-153)-flag localization in HEK293 cells.
(A) CHMP4B(1-153)-flag wild type, (B) CHMP4B(1-153)-flag carrying the mut1, (C) the mut2 and (D) the mut1.2 surface patch mutations. Individual confocal Z-sections are shown. Nuclei have been stained with 4′6-diamidino-2-phenylindole. The scale bar represents 10 μm.
(E) Sucrose gradient analyses of recombinant wild-type MBP-CHMP4BΔC-ALIX (top panel) and MBP-CHMP4BΔC-ALIX carrying the mut1.2 surface patch mutations (bottom panel). Molecular mass markers are indicated.
Discussion
CC2D1A and CC2D1B have been linked to a wide variety of cellular processes including transcriptional control 2; 3, signaling pathways 1; 6; 7; 8 centriole regulation 9, cell division 9; 11 and endosomal sorting 21; 22; 23. Here, we show that CC2D1A and B serve as adaptor proteins for ESCRT-III CHMP4B as previously indicated by yeast-two hybrid analyses 28. A direct interaction was confirmed in vitro employing recombinant CC2D1A and CC2D1B and CHMP4B. CHMP4 interaction requires the third DM14 repeat to form a 1:1 complex with nanomolar affinity. Although CC2D1A has four DM14 repeats, only one interacts efficiently with C-terminally truncated CHMP4B in vitro. We identified residues within the third DM14 domain that are essential for interaction. Notably sequence alignment of all four DM14 domains from human CC2D1A indicates that the seven residues mutated in the third DM14 domain are not strictly conserved. DM14-1 has five residues, DM14-2 has two residues and DM14-4 has four residues out of the seven mutated residues conserved (Figure S7). This thus suggests that the other domains might provide adaptor function in different pathways.
Structural analysis of a CHMP4B fragment shows that the conformation of the helical hairpin of CHMP4B is very similar to the corresponding structural fragments of CHMP3 and IST1 29; 30; 31. Structure-based mutagenesis analyses demonstrate further that the CC2D1A interaction site on CHMP4B is confined to a charged patch at one end of the helical hairpin formed mostly by α-helix 2 and some contribution from helix 1. Thus, CC2D1A and CC2D1B are the first CHMP4B ligands that bind to the conserved N-terminal core. Other CHMP4 ligands such as Alix and VPS4 bind to peptide motifs present at the extreme C-terminus 32; 33. CHMP proteins exist in a cytosolic closed conformation and membrane targeting is thought to activate them 25; 34; 35, leading to polymer formation in vivo and in vitro 24; 30; 36; 37; 38; 39; 40. Modeling CHMP4B onto IST1 shows that the CC2D1A interaction surface is accessible (Figure 2D). Noteworthy yeast ESCRT-III Vps20 (CHMP6) employs a neighboring surface to interact with ESCRT-II Vps25 41 (Figure 2E).
CHMP interaction with cellular ligands via their C-termini 33; 42; 43; 44; 45 does not affect CHMP polymerization. On the other hand, CC2D1A interaction with the helical hairpin of CHMP4B prevents CHMP4B polymerization in vitro. Our results indicate further that the charged surface patch at the base of the hairpin is required for CHMP4B polymerization in vitro and in vivo. One simple explanation is that CC2D1A binding sterically hinders the formation of CHMP4B polymers in vitro. In the context of ESCRT-III assembly, CC2D1A (or B) could then be considered as a negative regulator of CHMP4 function.
However, we cannot exclude that CC2D1A or B associates with one end of CHMP4 polymers, mimicking the Vps25-Vps20 (CHMP6) interaction that leads to Snf7 (CHMP4) polymerization 46; 47; 48. In the context of ESCRT assembly, CC2D1A (or CC2D1B) could then be considered as a positive regulator of CHMP4 function. Although CC2D1A or CC2D1B has not yet been directly implicated in endosomal sorting, the drosophila homologue Lgd was found at endosomes to regulate Notch receptor trafficking 21; 22; 23. Interestingly, lgd mutants are involved in the ligand-independent activation of the receptor Notch, a process that is disturbed in ESCRT knockdowns 49. One model was proposed where the persistence of internalized Notch at the membrane of the endosomes, prior to internalization into intraluminal vesicles, was responsible for an aberrant activation. Therefore, it is tempting to speculate that a lack of recruitment and/or activation of CHMP4 in the lgd mutant would impair proper internalization of Notch into ILVs, and thus be responsible for the observed lgd phenotype.
Among other processes, CHMP4 interaction might be required during CC2D1A’s function at the centrosome and during cell division. Notably depletion of CC2D1A or B from cells leads to multipolar spindles 9; 11 and the same phenotype is observed upon CHMP4B depletion 12. However, the exact role of ESCRTs during cell division steps other than membrane abscission 37; 50; 51; 52; 53 has yet to be defined.
As HIV-1 budding relies on ESCRTs, 18; 54; 55; 56, HIV-1 budding assays have been used to uncover new aspects of ESCRT function 16; 18; 54; 55; 57. Therefore, we used HIV-1 to test whether the CC2D1A binding site was important for the CHMP4B function in vivo. CHMP4 and CHMP2 isoforms are the essential ESCRT-III players in HIV-1 release 58 and recruited late during assembly 59. Thus, not surprisingly dominant negative CHMP4B is a potent inhibitor of HIV-1 budding 60; 61; 62. The helical hairpin of CHMP4B is required to exert a dominant negative effect 25 and we show that the charged surface patch on helix 2 plays an essential role in this process. Notably, the helix 2 patch in CHMP4B (E90, E94, E97) is distinct from the recently reported patch EVLK104-107 58 which is involved in CHMP2A binding. Moreover, CHMP4A residues 1-116 overexpression leads to the formation of polymers in COS7 cells 36, which implies that such a fragment represents a minimal region for polymerization. Because there is no evidence that CC2D1A or CC2D1B is recruited during HIV-1 budding, we speculate that the helix 2 mutation that abrogates the dominant negative effect as well as CC2D1A binding, affects mainly CHMP4B polymerization during HIV-1 budding, rather than plasma membrane localization. It thus remains to be determined how CC2D1A or CC2D1B influences or controls CHMP4B polymerization during other ESCRT-catalyzed cellular processes. In summary, we characterized CC2D1A in vitro as a novel adaptor molecule for ESCRT-III CHMP4, which will help to elucidate the function of the complex during diverse cellular processes in vivo.
Material and methods
Expression constructs
cDNA encoding CHMP4B(7-105), CHMP4B(7-110) and CHMP4B(23-97) were cloned into expression vector pBADM41 and CHMP4B mutants 1 and 2 (mut 1, R28A, R30A, D31R; mut2, E90R, E94R and E97R) were cloned into PETM41. CC2D1B_1-601 was cloned into expression vector pProEx-Htb (Invitrogen) and all other truncated CC2D1A constructs were cloned into the expression vector PET28-his_TEV (kindly provided from N. Tarbouriech). The CC2D1A(346-455) mutant (K374A, R376A, R380A, K383A, D387A, R390A, K393A) was cloned in pProEx-Htb (Invitrogen), and cDNAs carrying the CC2D1A mutations were synthesized by Genscript Inc.
Protein purification
MBP-CHMP4BΔC-Alix was produced as described previously 24. The monomeric protein was separated from polymeric CHMP4B by gel filtration on a Superdex 200 column (GE Healthcare) in a buffer containing 20 mM Tris pH 7.4 100 mM NaCl. MBP-CHMP4B(7-105) constructs (mutant and wild type) were transformed into BL21 cells, and grown at 37°C to an OD of 0.6, and protein expression was induced with 1mM IPTG. Cultures were grown for 1h, centrifuged, and the pellet was resuspended in lysis buffer A (50 mM Tris pH 7.4, 150 mM NaCl, EDTA-free protease inhibitors) before sonication. After a second step of centrifugation, the supernatant was filtered and loaded onto an amylose column. The resin was washed with lysis buffer containing 1M NaCl and 1M KCl, and eluted in 50 mM Tris pH 7.4, 350 mM NaCl, 10 mM Maltose. Further purification was achieved on a Superdex 200 column (GE Healthcare) in buffer B, 25 mM Tris pH 7.4 100 mM NaCl.
CC2D1A constructs were expressed in BL21, induced with 1 mM IPTG and grown for 3 hours at 37°C. The pellet was resuspended in buffer A, sonicated and the supernatant was filtered and loaded onto a Ni-NTA (Qiagen) resin. The column was washed with buffer A supplemented with 1M NaCl and subsequently with buffer C, 50 mM Tris pH 7.4, 150mM NaCl, 50 mM imidazole. The protein was eluted in 50 mM Tris pH 7.4, 150 mM NaCl, 250 mM imidazole. The 6x-histidine tag was removed with TEV protease at a ratio of 2 μg for 1 mg of protein overnight at 4°C. The processed protein was dialyzed against 50 mM Tris pH 7.4, 150 mM NaCl, 20 mM imidazole and loaded on a Ni-NTA resin, and the flow-through was collected. CC2D1A was further purified on a Superdex 75 column (GE Healthcare) in buffer B.
Complex formation of CC2D1A/B with CHMMP4B
CHMP4B(7-105), CHMP4B(23-97) or CHMP4B(7-110) were transformed into BL21 cells and grown from 1-l flasks at 37°C until an OD of 0.6 was reached, and induced with 0.2% (w/v) arabinose. Cultures were grown for 1h, centrifuged, and the pellet was resuspended in lysis buffer D (50mM Tris pH 7.4, 150 mM NaCl, 3 mM ß-mercaptoethanol, EDTA-free protease inhibitors) before sonication. After centrifugation, the supernatant was filtered and loaded on a Ni-NTA resin, washed with buffer E (buffer D supplemented with 1M NaCl- and 1M KCl) and buffer D plus 50 mM imidazole. The purified CC2D1A or CC2D1B proteins were loaded onto the Ni-resin containing MBP-CHMP4B. Unbound CC2D1A/B protein was washed off from the column in buffer D and the complex was eluted with buffer D plus 250 mM imidazole. MBP was removed from CHMP4B by TEV protease cleavage at a ratio of 2 μg for 1 mg of protein overnight at 4°C. The complex was dialyzed against 50 mM Tris pH 7.4, 150 mM NaCl, 3 mM ß-ME and loaded on a Ni-NTA resin connected to an Amylose column. The CC2D1A/B-CHMP4B complex was in the flow-through and further purified on a Superdex 75 (GE Healthcare) column in buffer C. Further purification of the CC2D1A/B and CHMP4B complexes on an S sepharose column in buffer E (25 mM NaCitrate, pH5.5 and 50 mM NaCl) and elution in buffer E supplemented with 1 M NaCl dissociated the complex and both CC2D1A/B and CHMP4B proteins eluted in separate peaks.
Co-purification experiments
Constructs corresponding to wild-type and mutant (mut1, mut2, mut1.2) MBP-CHMP4B(7-105) were transformed into BL21 cells and grown from 1-l flasks at 37°C until an OD of 0.6 was reached, and induced with 0.2% (w/v) arabinose (for the wild-type construct), or 1mM IPTG for the mutants. Cultures were grown for 2h, and centrifuged, and the pellet resuspended in lysis buffer (20 mM Tris pH 7.4, 100 mM NaCl, 1 mM PMSF) before sonication. After centrifugation, the supernatant was filtered and loaded on an Amylose column. The resin was washed with 5 column volumes of washing buffer (20 mM Tris pH 7.4, 1M NaCl) and then equilibrated with the running buffer (20 mM Tris pH 7.4, 100 mM NaCl). CC2D1A(346-455) was purified separately on a Ni-NTA column as described above. 3 mg of His-tagged CC2D1A(346-455) was incubated with wild-type and mutant MBP-CHMP4B(7-105) bound to the amylose resin for 30 minutes and washed with 5 column volumes of running buffer. MBP-CHMP4B(7-105) wild type and mutants were eluted with a buffer containing 20 mM Tris pH 7.4, 100 mM NaCl, 20 mM maltose and the presence of CC2D1A(346-455) was confirmed by SDS-PAGE.
MBP pulldown experiments
30 μL amylose resin (New England Biolabs) was incubated with an excess of monomeric MBP-CHMP4B 7-105 for 30 minutes at room temperature, except for controls. Unbound MBP-CHMP4B was removed with three washing steps with buffer (25 mM Tris pH 7.4, 100 mM NaCl). The matrices were incubated with similar amounts of CC2D1A 346-455 wild-type and mutant, in 5-fold molar excess for 1 h at room temperature. Unbound CC2D1A was removed by five washing steps in buffer. Bound proteins on matrix were resuspended in SDS-loading buffer; a third of each sample was analyzed by SDS-PAGE and bands were detected by Coomassie Blue Staining.
Sedimentation experiments
Monomeric MBP-CHMP4BΔC-Alix (1.5 μM) was concentrated to a final concentration of 15 μM in the presence of a 5-molar excess of CC2D1A wild-type or mutant; subsequently, MBP was removed by TEV protease cleavage overnight at 4°C at a ratio of 1:10 (w/w) to induce polymerization. The protein(s) were then separated on sucrose gradients in HBS buffer (0.01 M Hepes, pH 7.4, 0.15 M NaCl), by overlaying sucrose solutions of 60% (65 μL), 40% 30%, 20% and 5% (85 μl each). Centrifugation was performed in a Beckman SW55 rotor at 40,000 rpm for 6h at 4°C. Fractions from the gradients were analyzed on a 15% SDS-PAGE, and bands were detected with Coomassie Blue staining.
Isothermal titration calorimetry
Calorimetric measurements were carried out at 25°C using a VP-ITC instrument with a cell volume of 1.4569 ml (MicroCal, LLC). CC2D1B(1-601), CC2D1A-constructs and CHMP4B(7- 107) or CHMP4B(23-97) were exchanged in the same batch of buffer (50 mM Citrate, pH 6, 100 mM NaCl, 1 mM tris(2-carboxyethyl)phosphine) by extensive dialysis. The same batch of buffer was used for further dilutions of the proteins. The protein concentrations used in the cell or in the syringe are indicated in table 2. The solution in the cell was stirred at 286 rpm to ensure rapid mixing. The interaction isotherms were analyzed using Origin software package supplied by MircoCal. An interaction model assuming n independent and equivalent binding sites was applied, and the stoichiometry n, change in enthalpy ΔH, and binding constant Kd were iteratively fitted.
SEC-MALLS
Size exclusion chromatography (SEC) combined with detection by multi-angle laser light scattering (MALLS) and refractometry - SEC was performed with a Superdex S75 or S200 column as indicated (GE Healthcare) equilibrated in a buffer containing 25 mM Tris (pH 7.5), 100 mM NaCl, 3 mM ß-ME. Separations were performed at 20°C with a flow rate of 0.5 ml/min. 50 μl of the complexes as indicated were injected at a concentration of 4 mg/ml. MALLS detection and data analysis were performed as described previously 63.
Surface Plasmon resonance
BIAcore measurements were performed with the Biacore X instrument (BIAcore. Inc.) at 25°C in running buffer (10 mM Tris pH 7.4, 150 mM NaCl, 3.4 mM EDTA, 0.005% Surfactant P20). A CM5 chip was coated with CC2D1A(346-455) to a target of ~ 100 Response units (RU). The analytes MBP-CHMP4B(7-105) wild type and mutants (in running buffer) were passed over the chip surface at concentrations ranging respectively from 0.47 μM to 0.062 μM and 1.4 μM to 0.525 μM for 5 min at a flow rate of 10 μl/min and dissociation was recorded for 10 minutes. The chip was regenerated with 10 μl of 1 M NaCl at 50 μl/min. Binding kinetics were evaluated using the BiaEvaluation software package (BIAcore, Inc.) using a Langmuir model 1:1 with no mass transfer, but corrected for a drifting baseline when necessary.
Crystallization and structure solution of CHMP4(23-97)
CHMP4B(23-97) crystals were obtained by the vapor diffusion method in hanging drops mixing equal volumes of complex and reservoir solution (0.1 M Bis-Tris pH 5.5, 0.2 M KCL, 19% PEG 3350 (w/v)). The crystal was cryo-cooled at 100 K in reservoir buffer containing 25% (v/v) glycerol. The selenomethionine-substituted CHMP4B(23-97) was crystallized under the same conditions. A complete data set was collected at the European Synchrotron Radiation Facility (Grenoble, France) beam line ID14-4. Data were processed and scaled with MOSFLM 64 and SCALA 65; 66. The crystals belong to space group P212121 with unit cell dimensions of a=37.59 Å, b=71.44 Å, c=123.64 Å and contain four molecules per asymmetric unit. The structure was solved by the single anomalous dispersion method employing the data set collected at the peak wavelength using the SAS protocol of Auto-Rickshaw 67. FA values were calculated using the program SHELXC 68. All four heavy-atoms positions were localized using the program SHELXD 69 and the correct hand for the substructure was determined using the programs ABS 70 and SHELXE 71. Initial phases were calculated after density modification using the program SHELXE 71. The initial phases were improved using density modification and phase extension to 1.80 Å resolution using the program RESOLVE 72. An initial model was build with ARP/WARP 73 and completed by several cycles of manual model building with Coot 74 and refinement with REFMAC 75 using data to 1.8 Å resolution to an R-factor of 0.22 and Rfree of 0.28 (Table 1). The final model contains chain A residues 19 to 97, chain B residues 19 to 97, chain C residues 19 to 96 and chain D residues 21 to 97 (residues 19, 20, 21 and 22 derive from the expression vector). Of the residues, 99.6 % are within the most favored and allowed regions of a Ramachandran plot 66. Molecular graphics figures were generated with PyMOL (W. Delano; http://www.pymol.org) and sequence alignments with the ESPript 76. Coordinates and structure factures have been deposited in the Protein Data Bank with accession ID 4abm.
Mammalian expression and immunlocalization
CHMP4B(1-153)-flag and mutated forms carrying the mut1, mut2 and mut1.2 surface patch mutations were expressed in HEK293 cells. Fifteen hours after transfection cells were fixed with 4% paraformaldehyde and stained with a polyclonal anti-flag AB (Sigma) followed by an anti-rabbit Alexa-594-labeled (Invitrogen) antibody. After antibody incubation, slides were washed with phosphate-buffered-saline, mounted in Mowiol and analyzed by confocal microscopy. Microscopy was performed using the TCS SP2 AOBS confocal laser scanning microscope (Leica Microsystems, Germany), with HCX Plan-Apochromat 63× 1.4 oil immersion objective; excitation and emission were set at 405 and 420-470 nm for 4′,6-diamidino-2-phenylindole staining and at 488 and 500-550 nm for Alexa 488 antibody staining. Signals in different colour channels were acquired sequentially. Brightness and contrast of raw images were optimized for presentation purpose.
HIV budding
293T cells (1.2 × 106) were seeded into T25 flasks and transfected 24 h later using a calcium phosphate precipitation technique. The cultures were transfected with 1 μg HXBH10, which encodes HIV-1, together with CHMP4B1-153FLAG wild type or mutants CHMP4Bmut1, CHMP4Bmut2 and CHMP4Bmut1.2 (100 ng each). The total amount of transfected DNA was brought to 8 μg with carrier DNA (pTZ18U). Twenty-four hours post-transfection, the cells were lysed in RIPA buffer (140 mM NaCl, 8 mM Na2HPO4, 2 mM NaH2PO4, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.05% SDS), and the culture supernatants were clarified by low-speed centrifugation and passaged through 0.45-μm filters. Virions released into the medium were pelleted through 20% sucrose cushions and analyzed by SDS-PAGE and Western blotting with anti-HIV CA antibody 183-H12-5C 77. Proteins in the cell lysates were detected by Western blotting with anti-HIV CA or anti-FLAG antibody M2 (Sigma).
Supplementary Material
Acknowledgments
This work was supported by the Agence Nationale de la Recherche (ANR-08-BLAN-0271-01) and the Deutsche Forschungsgemeinschaft (DFG, SPP1175) (W.W.), the National Institute of Allergy and Infectious Diseases (R37AI029873 to H.G.), a postdoctoral fellowship from the European Molecular Biology Organization (B.H.) and a PhD fellowship from the Région Rhône-Alpes (Cluster 10 infectiologie) (N.M.). We thank Dr. T. Klein (Heinrich Heine Universität, Düsseldorf) for the lgd cDNA clone. We acknowledge the Partnership for Structural Biology (PSB; http://www.psb-grenoble.eu) for access to the common platforms including the crystallization facility (J. Marquez), the biophysical platform (M. Jamin) and the European Synchrotron Radiation Facilityfor beam time and assistance during data collection. The HIV-1 p24 monoclonal antibody (183-H12-5C) (provided by Drs. Bruce Chesebro and Kathy Wehrly) was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Abbreviations
- ESCRT
endosomal sorting complex required for transport
- CC2D1A and B
coiled coil and C2 domain containing proteins A and B
- DM14
drosophila melanogaster 14 domains
- Aki-1
Akt kinase interacting protein 1
- Freud-1
Five prime REpressor Under Dual repression binding protein 1
- TBK1
Tank-binding kinase 1
- TAPE
TBK-1 associated protein in endolysosomes
- lgd
Lethal giant discs
- SEC
size exclusion chromatography
- MALLS
multi-angle laser light scattering
- ITC
isothermal titration calorimetry
- SPR
surface plasmon resonance
References
- 1.Matsuda A, Suzuki Y, Honda G, Muramatsu S, Matsuzaki O, Nagano Y, Doi T, Shimotohno K, Harada T, Nishida E, Hayashi H, Sugano S. Largescale identification and characterization of human genes that activate NF-kappaB and MAPK signaling pathways. Oncogene. 2003;22:3307–3318. doi: 10.1038/sj.onc.1206406. [DOI] [PubMed] [Google Scholar]
- 2.Ou XM, Lemonde S, Jafar-Nejad H, Bown CD, Goto A, Rogaeva A, Albert PR. Freud-1: A neuronal calcium-regulated repressor of the 5-HT1A receptor gene. J Neurosci. 2003;23:7415–25. doi: 10.1523/JNEUROSCI.23-19-07415.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rogaeva A, Albert PR. The mental retardation gene CC2D1A/Freud-1 encodes a long isoform that binds conserved DNA elements to repress gene transcription. Eur J Neurosci. 2007;26:965–74. doi: 10.1111/j.1460-9568.2007.05727.x. [DOI] [PubMed] [Google Scholar]
- 4.Szewczyk B, Albert PR, Rogaeva A, Fitzgibbon H, May WL, Rajkowska G, Miguel-Hidalgo JJ, Stockmeier CA, Woolverton WL, Kyle PB, Wang Z, Austin MC. Decreased expression of Freud-1/CC2D1A, a transcriptional repressor of the 5-HT(1A) receptor, in the prefrontal cortex of subjects with major depression. Int J Neuropsychopharmacol. 2011;13:1089–101. doi: 10.1017/S1461145710000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rogaeva A, Galaraga K, Albert PR. The Freud-1/CC2D1A family: transcriptional regulators implicated in mental retardation. J Neurosci Res. 2007;85:2833–8. doi: 10.1002/jnr.21277. [DOI] [PubMed] [Google Scholar]
- 6.Chang CH, Lai LC, Cheng HC, Chen KR, Syue YZ, Lu HC, Lin WY, Chen SH, Huang HS, Shiau AL, Lei HY, Qin J, Ling P. TBK1-associated protein in endolysosomes (TAPE) is an innate immune regulator modulating the TLR3 and TLR4 signaling pathways. J Biol Chem. 2011;286:7043–51. doi: 10.1074/jbc.M110.164632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao M, Li XD, Chen Z. CC2D1A, a DM14 and C2 domain protein, activates NF-kappaB through the canonical pathway. J Biol Chem. 2010;285:24372–80. doi: 10.1074/jbc.M109.100057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakamura A, Naito M, Tsuruo T, Fujita N. Freud-1/Aki1, a novel PDK1-interacting protein, functions as a scaffold to activate the PDK1/Akt pathway in epidermal growth factor signaling. Mol Cell Biol. 2008;28:5996–6009. doi: 10.1128/MCB.00114-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamura A, Arai H, Fujita N. Centrosomal Aki1 and cohesin function in separase-regulated centriole disengagement. J Cell Biol. 2009;187:607–14. doi: 10.1083/jcb.200906019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakamura A, Naito M, Arai H, Fujita N. Mitotic phosphorylation of Aki1 at Ser208 by cyclin B1-Cdk1 complex. Biochem Biophys Res Commun. 2010;393:872–6. doi: 10.1016/j.bbrc.2010.02.103. [DOI] [PubMed] [Google Scholar]
- 11.Neumann B, Walter T, Heriche JK, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U, Cetin C, Sieckmann F, Pau G, Kabbe R, Wunsche A, Satagopam V, Schmitz MH, Chapuis C, Gerlich DW, Schneider R, Eils R, Huber W, Peters JM, Hyman AA, Durbin R, Pepperkok R, Ellenberg J. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature. 2010;464:721–7. doi: 10.1038/nature08869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morita E, Colf LA, Karren MA, Sandrin V, Rodesch CK, Sundquist WI. Human ESCRT-III and VPS4 proteins are required for centrosome and spindle maintenance. Proc Nat Acad Sci. U.S.A. 2010;107:12889–12894. doi: 10.1073/pnas.1005938107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hurley J. ESCRT complexes and the biogenesis of multivesicular bodies. Curr Opin Cell Biol. 2008;20(1):4–11. doi: 10.1016/j.ceb.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peel S, Macheboeuf P, Martinelli N, Weissenhorn W. Divergent pathways lead to ESCRT-III catalyzed membrane fission. Trends Biochem Sci. 2011;36:199–210. doi: 10.1016/j.tibs.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 15.Henne WM, Buchkovich NJ, Emr SD. The ESCRT pathway. Dev Cell. 2011;21:77–91. doi: 10.1016/j.devcel.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 16.Morita E, Sundquist WI. Retrovirus budding. Annu Rev Cell Dev Biol. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- 17.McDonald B, Martin-Serrano J. No strings attached: the ESCRT machinery in viral budding and cytokinesis. J Cell Sci. 2009;122:2167–77. doi: 10.1242/jcs.028308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin-Serrano J, Neil SJ. Host factors involved in retroviral budding and release. Nat Rev Microbiol. 2011;9:519–31. doi: 10.1038/nrmicro2596. [DOI] [PubMed] [Google Scholar]
- 19.Hurley JH, Hanson PI. Membrane budding and scission by the ESCRT machinery: it’s all in the neck. Nat Rev Mol Cell Biol. 2010;11:556–66. doi: 10.1038/nrm2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsang HT, Connell JW, Brown SE, Thompson A, Reid E, Sanderson CM. A systematic analysis of human CHMP protein interactions: Additional MIT domain-containing proteins bind to multiple components of the human ESCRT III complex. Genomics. 2006;88(3):333–46. doi: 10.1016/j.ygeno.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 21.Jaekel R, Klein T. The Drosophila Notch Inhibitor and Tumor Suppressor Gene lethal (2) giant discs Encodes a Conserved Regulator of Endosomal Trafficking. Developmental Cell. 2006;11:655–669. doi: 10.1016/j.devcel.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 22.Gallagher CM, Knoblich JA. The Conserved C2 Domain Protein Lethal (2) Giant Discs Regulates Protein Trafficking in Drosophila. Dev Cell. 2006;11:641–653. doi: 10.1016/j.devcel.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 23.Childress JL, Acar M, Tao C, Halder G. Lethal giant discs, a novel C2-domain protein, restricts notch activation during endocytosis. Curr Biol. 2006;16:2228–33. doi: 10.1016/j.cub.2006.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pires R, Hartlieb B, Signor L, Schoehn G, Lata S, Roessle M, Moriscot C, Popov S, Hinz A, Jamin M, Boyer V, Sadoul R, Forest E, Svergun DI, Gottlinger HG, Weissenhorn W. A crescent-shaped ALIX dimer targets ESCRT-III CHMP4 filaments. Structure. 2009;17:843–56. doi: 10.1016/j.str.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zamborlini A, Usami Y, Radoshitzky SR, Popova E, Palu G, Gottlinger H. Release of autoinhibition converts ESCRT-III components into potent inhibitors of HIV-1 budding. Proc Natl Acad Sci U S A. 2006;103:19140–5. doi: 10.1073/pnas.0603788103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gottlinger H, Dorfman T, Sodroski J, Haseltine W. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc Natl Acad Sci USA. 1991;88:3195–3199. doi: 10.1073/pnas.88.8.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garrus J, von Schwedler U, Pornillos O, Morham S, Zavitz K, Wang H, Wettstein D, Stray K, Cote M, Rich R. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107:55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 28.Tsang HT, Edwards TL, Wang X, Connell JW, Davies RJ, Durrington HJ, O’Kane CJ, Luzio JP, Reid E. The hereditary spastic paraplegia proteins NIPA1, spastin and spartin are inhibitors of mammalian BMP signalling. Hum Mol Genet. 2009;18:3805–21. doi: 10.1093/hmg/ddp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muziol T, Pineda-Molina E, Ravelli RB, Zamborlini A, Usami Y, Gottlinger H, Weissenhorn W. Structural basis for budding by the ESCRT-III factor CHMP3. Dev Cell. 2006;10:821–30. doi: 10.1016/j.devcel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 30.Bajorek M, Schubert HL, McCullough J, Langelier C, Eckert DM, Stubblefield WM, Uter NT, Myszka DG, Hill CP, Sundquist WI. Structural basis for ESCRT-III protein autoinhibition. Nat Struct Mol Biol. 2009;16:754–62. doi: 10.1038/nsmb.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao J, Chen XW, Davies BA, Saltiel AR, Katzmann DJ, Xu Z. Structural basis of Ist1 function and Ist1-Did2 interaction in the multivesicular body pathway and cytokinesis. Mol Biol Cell. 2009;20:3514–24. doi: 10.1091/mbc.E09-05-0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCullough J, Fisher RD, Whitby FG, Sundquist WI, Hill CP. ALIX-CHMP4 interactions in the human ESCRT pathway. Proc Nat Acad Sci USA. 2008;105:7687–7691. doi: 10.1073/pnas.0801567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kieffer C, Skalicky JJ, Morita E, De Domenico I, Ward DM, Kaplan J, Sundquist WI. Two distinct modes of ESCRT-III recognition are required for VPS4 functions in lysosomal protein targeting and HIV-1 budding. Dev Cell. 2008;15:62–73. doi: 10.1016/j.devcel.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 34.Shim S, Kimpler LA, Hanson PI. Structure/Function Analysis of Four Core ESCRT-III Proteins Reveals Common Regulatory Role for Extreme C-Terminal Domain. Traffic. 2007;8:1068–1079. doi: 10.1111/j.1600-0854.2007.00584.x. [DOI] [PubMed] [Google Scholar]
- 35.Lata S, Roessle M, Solomons J, Jamin M, Gottlinger HG, Svergun DI, Weissenhorn W. Structural basis for autoinhibition of ESCRT-III CHMP3. J Mol Biol. 2008;378:818–27. doi: 10.1016/j.jmb.2008.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanson PI, Roth R, Lin Y, Heuser JE. Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J. Cell Biol. 2008;180(2):389–402. doi: 10.1083/jcb.200707031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guizetti J, Schermelleh L, Mantler J, Maar S, Poser I, Leonhardt H, Muller-Reichert T, Gerlich DW. Cortical Constriction During Abscission Involves Helices of ESCRT-III-Dependent Filaments. Science. 2011;331(6024):1616–20. doi: 10.1126/science.1201847. [DOI] [PubMed] [Google Scholar]
- 38.Ghazi-Tabatabai S, Saksena S, Short JM, Pobbati AV, Veprintsev DB, Crowther RA, Emr SD, Egelman EH, Williams RL. Structure and disassembly of filaments formed by the ESCRT-III subunit Vps24. Structure. 2008;16:1345–56. doi: 10.1016/j.str.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 39.Lata S, Schoehn G, Jain A, Pires R, Piehler J, Gottlinger HG, Weissenhorn W. Helical structures of ESCRT-III are disassembled by VPS4. Science. 2008;321:1354–7. doi: 10.1126/science.1161070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bodon G, Chassefeyre R, Pernet-Gallay K, Martinelli N, Effantin G, Lutje Hulsik D, Belly A, Goldberg Y, Chatellard-Causse C, Blot B, Schoehn G, Weissenhorn W, Sadoul R. Charged Multivesicular Body Protein 2B (CHMP2B) of the Endosomal Sorting Complex Required for Transport-III (ESCRT-III) Polymerizes into Helical Structures Deforming the Plasma Membrane. Journal of Biological Chemistry. 2011;286:40276–40286. doi: 10.1074/jbc.M111.283671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Im YJ, Wollert T, Boura E, Hurley JH. Structure and function of the ESCRT-II-III interface in multivesicular body biogenesis. Dev Cell. 2009;17:234–43. doi: 10.1016/j.devcel.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Obita T, Saksena S, Ghazi-Tabatabai S, Gill DJ, Perisic O, Emr SD, Williams RL. Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature. 2007;449:735–739. doi: 10.1038/nature06171. [DOI] [PubMed] [Google Scholar]
- 43.Samson RY, Obita T, Freund SM, Williams RL, Bell SD. A role for the ESCRT system in cell division in archaea. Science. 2008;322:1710–3. doi: 10.1126/science.1165322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stuchell-Brereton MD, Skalicky JJ, Kieffer C, Karren MA, Ghaffarian S, Sundquist WI. ESCRT-III recognition by VPS4 ATPases. Nature. 2007;449:740–4. doi: 10.1038/nature06172. [DOI] [PubMed] [Google Scholar]
- 45.Solomons J, Sabin C, Poudevigne E, Usami Y, Lutje Hulsik D, Macheboeuf P, Hartlieb B, Göttlinger H, Weissenhorn W. Structural basis for ESCRT-III CHMP3 recruitment of AMSH. Structure. 2011;19:1149–59. doi: 10.1016/j.str.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teis D, Saksena S, Emr SD. Ordered Assembly of the ESCRT-III Complex on Endosomes Is Required to Sequester Cargo during MVB Formation. Dev Cell. 2008;15(4):578–89. doi: 10.1016/j.devcel.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 47.Saksena S, Wahlman J, Teis D, Johnson AE, Emr SD. Functional reconstitution of ESCRT-III assembly and disassembly. Cell. 2009;136:97–109. doi: 10.1016/j.cell.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fyfe I, Schuh AL, Edwardson JM, Audhya A. Association of ESCRT-II with VPS20 generates a curvature sensitive protein complex capable of nucleating filaments of ESCRT-III. J Biol Chem. 2011;286:34262–70. doi: 10.1074/jbc.M111.266411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vaccari T, Rusten TE, Menut L, Nezis IP, Brech A, Stenmark H, Bilder D. Comparative analysis of ESCRT-I, ESCRT-II and ESCRT-III function in Drosophila by efficient isolation of ESCRT mutants. J Cell Sci. 2009;122:2413–23. doi: 10.1242/jcs.046391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carlton JG, Martin-Serrano J. Parallels between cytokinesis and retroviral budding: a role for the ESCRT machinery. Science. 2007;316:1908–12. doi: 10.1126/science.1143422. [DOI] [PubMed] [Google Scholar]
- 51.Morita E, Sandrin V, Chung HY, Morham SG, Gygi SP, Rodesch CK, Sundquist WI. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007;26:4215–27. doi: 10.1038/sj.emboj.7601850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee HH, Elia N, Ghirlando R, Lippincott-Schwartz J, Hurley JH. Midbody targeting of the ESCRT machinery by a noncanonical coiled coil in CEP55. Science. 2008;322:576–80. doi: 10.1126/science.1162042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elia N, Sougrat R, Spurlin TA, Hurley JH, Lippincott-Schwartz J. Dynamics of endosomal sorting complex required for transport (ESCRT) machinery during cytokinesis and its role in abscission. Proc Natl Acad Sci U S A. 2011;108:4846–51. doi: 10.1073/pnas.1102714108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bieniasz PD. The cell biology of HIV-1 virion genesis. Cell Host Microbe. 2009;5:550–8. doi: 10.1016/j.chom.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weiss ER, Göttlinger H. The Role of Cellular Factors in Promoting HIV Budding. J Mol Biol. 2011;410(4):525–33. doi: 10.1016/j.jmb.2011.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dordor A, Poudevigne E, Gottlinger H, Weissenhorn W. Essential and supporting host cell factors for HIV-1 budding. Future Microbiol. 2011;6:1159–70. doi: 10.2217/fmb.11.100. [DOI] [PubMed] [Google Scholar]
- 57.Usami Y, Popov S, Popova E, Inoue M, Weissenhorn W, Gottlinger HG. The ESCRT pathway and HIV-1 budding. Biochem Soc Trans. 2009;37:181–4. doi: 10.1042/BST0370181. [DOI] [PubMed] [Google Scholar]
- 58.Morita E, Sandrin V, McCullough J, Katsuyama A, Baci Hamilton I, Sundquist WI. ESCRT-III Protein Requirements for HIV-1 Budding. Cell Host Microbe. 2011;9:235–42. doi: 10.1016/j.chom.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jouvenet N, Zhadina M, Bieniasz PD, Simon SM. Dynamics of ESCRT protein recruitment during retroviral assembly. Nat Cell Biol. 2011;13:394–401. doi: 10.1038/ncb2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Strack B, Calistri A, Popova E, Gottlinger H. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell. 2003;114:689–699. doi: 10.1016/s0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- 61.von Schwedler UK, Stuchell M, Muller B, Ward DM, Chung HY, Morita E, Wang HE, Davis T, He GP, Cimbora DM, Scott A, Krausslich HG, Kaplan J, Morham SG, Sundquist WI. The protein network of HIV budding. Cell. 2003;114:701–713. doi: 10.1016/s0092-8674(03)00714-1. [DOI] [PubMed] [Google Scholar]
- 62.Carlton JG, Agromayor M, Martin-Serrano J. Differential requirements for Alix and ESCRT-III in cytokinesis and HIV-1 release. Proc Natl Acad Sci U S A. 2008;105:10541–6. doi: 10.1073/pnas.0802008105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gerard FCA, Ribeiro E. d. A., Albertini A. l. A. V., Gutsche I, Zaccai G, Ruigrok RWH, Jamin M. Unphosphorylated Rhabdoviridae Phosphoproteins Form Elongated Dimers in Solution. Biochemistry. 2007;46:10328–10338. doi: 10.1021/bi7007799. [DOI] [PubMed] [Google Scholar]
- 64.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Jnt CCP4/ESF-EACMB. Newslett. Protein Crystallogr. 1992;26 [Google Scholar]
- 65.Evans P. Scaling and assessment of data quality. Acta Crystallogr D: Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 66.CCP4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D: Biol. Crystallogr. 1994;50:157–163. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 67.Panjikar S, Parthasarathy V, Lamzin VS, Weiss MS, Tucker PA. Auto-Rickshaw: an automated crystal structure determination platform as an efficient tool for the validation of an X-ray diffraction experiment. Acta Crystallogr D: Biol Crystallogr. 2005;61:449–57. doi: 10.1107/S0907444905001307. [DOI] [PubMed] [Google Scholar]
- 68.Sheldrick GM, Hauptman HA, Weeks CM, Miller R, Uson I. International Tables for Crystallography, Volume F: Crystallography of biological macromolecules, chapter 16.1: Ab initio phasing. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2001. pp. 247–255. [Google Scholar]
- 69.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D: Biol Crystallogr. 2002;58:1772–9. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 70.Hao Q. ABS: a program to determine absolute configuration and evaluate anomalous scatterer substructure. J. Appl. Cryst. 2004;37:498–499. [Google Scholar]
- 71.Sheldrick GM. Macromolecular phasing with SHELXE. Zeitschrift für Kristallographie. 2002;217:644–650. [Google Scholar]
- 72.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D: Biol Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 74.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 75.Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 76.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–8. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 77.Chesebro B, Wehrly K, Nishio J, S P. Macrophage-tropic human immunodeficiency virus isolates from different patients exhibit unusual V3 envelope sequence homogeneity in comparison with T-cell-tropic isolates: definition of critical amino acids involved in cell tropism. J Virol. 1992;66:6547–54. doi: 10.1128/jvi.66.11.6547-6554.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.