

Abstract
Mesencephalic dopamine neurons are central to many aspects of human cognition, motivational, and motor behavior, and they are uniquely vulnerable to degenerative neurologic disorders such as Parkinson's disease. There is growing evidence that in the mature brain these neurons not only remain responsive to neurotrophic support, but are dependent on it for viability and function. Little is known of the cellular signaling pathways that mediate this support, although some evidence suggests that protein kinase Akt/PKB may play such a role. Another candidate for such a role is serum- and glucocorticoid-inducible kinase (SGK), a member of the AGC kinase family that is closely related to Akt. We have herein examined the responsiveness of adult mouse dopamine neurons in vivo to overexpression of wild-type and a constitutively active form of SGK by use of viral vector transfer in normal mice and both before and after 6-OHDA lesion. We find that SGK induces a broad spectrum of neurotrophic effects on these neurons, including induction of neuronal hypertrophy, protection from both neuron death and neurotoxin-induced retrograde axonal degeneration, and axon regeneration. Given the diverse and robust effects of SGK on these neurons, and its abundant expression in them, we suggest that SGK, like closely related Akt, may play a role in their responsiveness to neurotrophic factors and in adult maintenance. It therefore offers a novel target for therapeutic development.
Introduction
The AGC family of kinases includes members that share close sequence relationships in their catalytic domain to cAMP-dependent protein kinase (PKA), cGMP-dependent protein kinase (PKG) and protein kinase C (Hanks and Hunter, 1995; Pearce et al., 2010). One of the most intensively investigated members of this family is Akt (also known as protein kinase B), which has diverse effects on cell survival, growth, and metabolism (Manning and Cantley, 2007). Additionally, it has effects that are unique to neurons, including regulation of axon growth (Namikawa et al., 2000; Markus et al., 2002; Park et al., 2008; Kim et al., 2011a), synaptic strength (Wang et al., 2003), and dendritic growth (Kwon et al., 2006). Given these diverse effects of Akt on neurons, it is not surprising that it has been implicated in many psychiatric and neurologic disorders including autism (Hoeffer and Klann, 2010), schizophrenia (Kvajo et al., 2010), Alzheimer's disease (Baki et al., 2008), and Parkinson's disease (PD)(Greene et al., 2011). In relation to PD, Akt has become of particular interest based on evidence that it may play an important role in the maintenance of mesencephalic dopaminergic neurons, a population that is predominantly affected. Studies have suggested that diminished Akt signaling may play a role in selective neurotoxin injury to these neurons (for review, see Greene et al., 2011). Furthermore, a possible role has been suggested for both sporadic and inherited forms of the disease (Kim et al., 2005; Yang et al., 2005; Malagelada et al., 2008; Timmons et al., 2009; Aleyasin et al., 2010).
This evidence for a critical role for Akt in maintaining dopamine neurons raises the question of whether other members of the AGC kinase family may provide alternate kinase signaling for this essential function. Among other members of the AGC family, serum- and glucocorticoid-inducible kinase (SGK) (Webster et al., 1993; Lang et al., 2006) shares many regulatory elements and functional roles with Akt (Alessi et al., 2009). Both Akt and SGK1 are phosphorylated and activated by another AGC kinase, PDK1, at threonine residues in the activation loop of the catalytic domain (Mora et al., 2004) and by mTORC2 at serine residues in a C-terminal hydrophobic motif (Alessi et al., 2009). SGK mRNA is more highly expressed in the ventral mesencephalon than that of Akt (Iwata et al., 2004). Intriguingly, in several expression analysis studies following brain injury, SGK was highly upregulated, particularly in studies that induced selective degeneration of the dopaminergic system (Imaizumi et al., 1994; Iwata et al., 2004; Stichel et al., 2005). Stichel et al. (2005) proposed that SGK mediates a survival response following MPTP (Schoenebeck et al., 2005), a neurotoxin selective for dopamine neurons.
There has not been a previous direct evaluation of the effects of increased SGK expression on mesencephalic dopamine neurons in vivo. We have therefore undertaken such studies by use of adeno-associated viral (AAV) vector transfer, with both wild-type and a constitutively active form of SGK, in normal mice, and in mice both before and after lesion with 6-hydroxydopamine (6-OHDA).
Materials and Methods
Generation of recombinant AAV.
All vectors used for these studies were AAV1 serotype. The cDNA clone for wild-type human SGK1 was obtained from OriGene Technologies (Catalog No. SC116597; Accession No. NM_005627.2). hSGK1 DNA was amplified and modified to incorporate a FLAG-encoding sequence at the 3′-end by expanded long template PCR (Roche). A constitutively active mutant, in which the serine in the hydrophobic domain at position 422 is modified to a phospho-mimetic aspartate [hSGK1(S422D)], has been previously characterized in in vitro studies (Lang and Cohen, 2001; Sakoda et al., 2003). Kobayashi et al. (1999) demonstrated that this mutation increases the specific activity of SGK by 10-fold. This mutation was introduced by PCR-based site-directed mutagenesis (Phusion Kit; New England Biolabs), and a 3′-end FLAG-encoding sequence was incorporated. These modified forms of hSGK1 were then cloned into an AAV packaging construct that uses the chicken β-actin promoter, and contains a 3′ WPRE (pBL) (Kim et al., 2011a). All nucleotide sequences in the AAV packaging constructs were confirmed before AAV production. AAVs were produced by the University of North Carolina Vector Core. The genomic titer of AAV hSGK1(WT) was 1 × 1012 viral genomes/ml, and that of AAV hSGK1(S422D) was also 1 × 1012 viral genomes/ml. Enhanced green fluorescent protein (GFP), used as a control, was subcloned into the same viral backbone, and viral stocks were produced at a titer of 2.0 × 1011 viral genomes/ml.
Experimental animals.
Adult (8 week) male C57BL/6 mice weighing ∼25 g were obtained from Charles River Laboratories. Tyrosine hydroxylase (TH)-GFP transgenic mice, which express GFP driven by the TH promoter (Sawamoto et al., 2001), were generously made available by Drs. K. Kobayashi and H. Okano and maintained on a C57BL/6 background. All injection procedures, described below, were approved by the Columbia University Animal Care and Use Committee.
6-OHDA lesion.
Mice were first pretreated with desipramine to block 6-OHDA uptake by noradrenergic terminals, thereby limiting the lesion to striatal dopaminergic terminals. They were then anesthetized with ketamine/xylazine solution, and placed in a stereotaxic frame. A solution of 6-OHDA (5.0 μg/μl in 0.9% NaCl/0.02% ascorbate) was injected by microliter syringe at a rate of 0.5 μl/min by pump for a total dose of 15.0 μg/3 μl. The injection was performed into the left striatum at coordinates: anteroposterior (AP): +0.9 mm; mediolateral (ML): +2.2 mm; and dorsoventral (DV): −2.5 mm, relative to bregma. After a wait of 2 min, the needle was withdrawn slowly.
Intranigral AAV injection.
Adult (8 week) male C57BL/6 mice were anesthetized with ketamine/xylazine solution and placed in a stereotaxic frame (Kopf Instruments) with a mouse adapter. The tip of 5.0 μl syringe needle (26S) was inserted to stereotaxic coordinates: AP: −3.5 mm; ML: +1.1 mm; and DV: −3.7 mm, relative to bregma. Viral vector suspension in a volume of 2.0 μl was injected at 0.1 μl/min over 20 min. This slow infusion rate is used to minimize reflux of the injection up along the cannula tract.
Immunohistochemistry.
Immunohistochemical procedures were optimized for each antigen and for the cell structures of interest. Immunoperoxidase staining for TH of neuron cell bodies was performed as described previously (Kholodilov et al., 2004). Mice were perfused intracardially with 0.9% NaCl followed by 4.0% paraformaldehyde (PFA) in 0.1 m phosphate buffer (PB), pH 7.1. The brain was carefully removed and blocked into midbrain and forebrain regions. The region containing the midbrain was postfixed for 1 week, cryoprotected in 20% sucrose overnight, and then rapidly frozen by immersion in isopentane on dry ice. A complete set of serial sections was then cut through the substantia nigra (SN) at 30 μm. Beginning with a random section between 1 and 4, every fourth section was processed, in keeping with the fractionator method of sampling (see below). Sections were processed free floating. The primary antibody was rabbit anti-TH (Calbiochem) at 1:750. Sections were then treated with biotinylated protein A and avidin-biotinylated horseradish peroxidase complexes (ABC; Vector Laboratories). Following immunoperoxidase staining, sections were thionin counterstained. The forebrain region containing the dopaminergic axonal projections to the striatum was postfixed for 48 h, then frozen without cryoprotection, and processed as described previously (Kholodilov et al., 2004).
For anti-FLAG immunostaining, mice were perfused with 4% PFA, and the brains were postfixed for 24 h. Following cryoprotection overnight, brains were frozen and sectioned through the SN at 30 μm. Sections were initially treated with Mouse-on-Mouse Blocking Reagent (Vector Laboratories) and then processed free floating with a mouse monoclonal anti-FLAG antibody (Sigma) at 1:1000. Sections were then incubated with biotinylated anti-mouse IgG (Vector Laboratories), followed by ABC.
Immunostaining for NeuN was performed with a mouse monoclonal antibody (Millipore Bioscience Research Reagents) at 1:100.
Immunostaining for SGK was performed with a rabbit polyclonal antibody (Cell Signaling Technology #3272) at 1:100.
Double immunofluorescence staining.
For TH and FLAG double labeling, mice were perfused and their brains postfixed for 24 h. Following cryoprotection overnight, brains were frozen and sectioned through the SN at 30 μm. Sections were blocked with Mouse-on-Mouse Blocking Reagent (Vector Laboratories) for 1 h at 4°C and then incubated with the primary antibodies, mouse monoclonal anti-FLAG antibody (Sigma) at 1:1000 and rabbit polyclonal anti-TH (Calbiochem) at 1:1000 in PBS containing Mouse-on-Mouse Diluent Working Solution (Vector Laboratories), and normal goat serum for 24 h at 4°C. Control sections were prepared either in the absence of primary antibody or with anti-FLAG or anti-TH alone. Sections were then incubated in a mixture of biotinylated anti-mouse IgG reagent and goat anti-rabbit Texas Red (Vector Laboratories) for 15 min at room temperature, followed by incubation in fluorescein avidin DCS for 1 h at a dilution of 1:100. Following PBS wash, the sections were mounted and then coverslipped with Dako anti-fade medium. For TH and SGK double labeling, a similar protocol was performed, using a rabbit polyclonal antibody anti-SGK (Cell Signaling Technology) and a mouse monoclonal anti-TH (Millipore Bioscience Research Reagents).
Nonradioactive in situ hybridization for SGK1 mRNA.
A probe for in situ hybridization was designed based on the published mouse SGK sequence (Accession No. AF139638). Oligonucleotide primers for probe isolation were synthesized by Gene Link corresponding to sequence as follows: forward TCGGGACGATGACCGTCAAA; reverse GAGCACTCAGAGGAAGGAAT. The PCR product derived from SN mRNA was subcloned in pGEM-T vector (Promega) and used for making sense or antisense riboprobe. Brains were rapidly removed from PND2 and adult mice, and rapidly frozen in OCT (Tissue-Tek) on dry ice. Sections (14 μm) were thaw-mounted on glass slides (Superfrost Plus; Fisher Scientific). For hybridization, sections were warmed to 37°C and then fixed by immersion in 4% PFA in 0.1 m PBS. Sections were then treated with a prehybridization solution as previously described (Burke et al., 1994) for 2 h at room temperature. Sections were then covered with hybridization solution and incubated overnight at 68°C. Hybridization solution contained either antisense or sense SGK1 probe labeled with digoxigenin (DIG)-UTP, prepared as per the manufacturer's instructions (Roche Diagnostics). The size and integrity of labeled probe were confirmed by gel electrophoresis. After washes in 0.2× SSC at 68°C, sections were incubated with an anti-DIG antibody (Roche) at 1:5000 overnight at 4°C. After additional washes, sections were then incubated with a developing solution containing BCIP/NBT (Promega) overnight at room temperature in the dark. Sections were washed and coverslipped with Dako aqueous mounting medium.
Northern analysis of SGK1 and SGK3 in ventral mesencephalon and striatum.
For Northern analysis of SGK1 expression, the same probe as used for nonradioactive in situ hybridization was used. For SGK3, (Accession No. NM_133220) oligonucleotide primers for probe isolation were as follows: forward AGACATTAAATGCAGAGGGA; reverse ATGTCAAAGTGTTCACAAAAAT. Northern analysis was performed as previously described (El-Khodor et al., 2001). Briefly, RNA was isolated from microdissected SN and striatum using the Qiagen RNAeasy Mini kit. RNA was electrophoresed in 1.4% agarose-formaldehyde gel and transferred onto an Immobilon (+) membrane (Millipore). The hybridization was performed overnight at 68 C° in Ultrahyb buffer from Ambion. The membrane was then exposed to PhosphorImager cassettes, scanned, and analyzed by Image Quant software (Molecular Dynamics).
Quantification of dopaminergic axons in the medial forebrain bundle.
Quantification of axons was performed on TH-GFP transgenic mice, which express GFP driven by TH promoter (Sawamoto et al., 2001), as previously described (Cheng et al., 2011). Mice were perfused intracardially with 0.9% NaCl followed by 4.0% PFA in 0.1 m PB, pH 7.1. Following postfixation, the brains were sectioned horizontally at 50 μm with a vibratome. A section containing the posterior third ventricular recess and the A13 dopamine cell group was selected for analysis. Confocal microscopy (Leica TCS SP5 AOBS MP System) was used to acquire images through the entire medial-to-lateral extent of the medial forebrain bundle (MFB). Proceeding from a point midway between the anterior A13 cells and the posterior third ventricle recess, images were acquired with a 20× objective with a zoom factor of 8 applied. The MFB was analyzed over seven contiguous fields (97 × 97 μm). Each field was scanned in the z-axis with 20 0.1 μm thickness optical planes from dorsal to ventral, for a total vertical distance of 2.0 μm in the center of the section. These 20 optical planes were then merged to obtain a single maximal projection of the sampled volume. To count the number of axons passing in the rostrocaudal dimension through each sample volume, two horizontal sampling lines were drawn on the image at a separation distance of 10 μm in the center of the maximal projection. Every intact axon crossing both lines was counted as positive.
Determination of SN neuron numbers by stereologic analysis.
Stereologic analysis was performed under blinded conditions on coded slides. For each animal, the SN on both sides of the brain was analyzed. For each section the entire SN was identified as the region of interest. Using StereoInvestigator software (MicroBright Field), a fractionator probe was established for each section. The number of TH-positive neurons in each counting frame was then determined by focusing down through the section, using 100× objective under oil, as required by the optical disector method. Our criterion for counting an individual TH-positive neuron was the presence of its nucleus either within the counting frame, or touching the right or top frame lines (green), but not touching the left or bottom lines (red). The total number of TH-positive neurons for each side of the SN was then determined by the StereoInvestigator program. The number of NeuN-positive neuronal nuclei was determined in a similar fashion, except that the region of interest was confined to the substantia nigra pars compacta (SNpc).
Determination of SN neuron size.
The cell size of TH neurons in the SN was also determined by use of the StereoInvestigator program. For each brain, five representative sections (one caudal, two middle, and two rostral) were chosen, and five neurons were selected at five random sites from each on both the Control (uninjected) and Experimental (AAV-injected) sides of the brain, for a total of 25 neurons on each side. This procedure was performed on two randomly selected mice from each experimental condition for a total of 50 neurons on each brain side for each experimental condition. Each TH-positive neuron was outlined and measured under a 100× oil-immersion objective.
Determination of striatal TH-positive immunostaining.
The optical density of striatal TH immunostaining was determined with an Imaging Research Analytical Imaging Station as described previously (Burke et al., 1990).
Statistical methods.
The difference between two groups was analyzed by the t test. Multiple comparisons among groups were performed by one-way ANOVA and Tukey's post hoc analysis. All statistical analyses were performed using Sigma Stat software (Systat Software).
Results
SGK1 and SGK3 are expressed in neurons of the SN of adult mouse
Three mammalian isoforms of SGK have been identified and termed SGK1, SGK2, and SGK3 (Kobayashi et al., 1999). Whereas SGK1 and SGK3 mRNA have similar tissue distributions, and are abundant in the brain, SGK2 has a more restricted distribution and is less in the brain (Kobayashi et al., 1999). We therefore investigated the expression of SGK1 and SGK3 in SN. We found that mRNA for both isoforms is expressed (Fig. 1A). At a subregional and cellular level, examined by in situ hybridization, SGK1 mRNA was observed in the SNpc, exclusively in neurons (Fig. 1A). Protein expression, identified by immunostaining with an antibody that recognizes the C terminal of all isoforms, was identified widely in the ventral mesencephalon, including in the SNpc (Fig. 1B). At a cellular level double-label immunofluorescence identified SGK protein expression in the majority of SNpc dopaminergic neurons, identified by staining for TH, predominantly within the nucleus (Fig. 1B).
Figure 1.

Expression of SGK in mouse ventral mesencephalon. A, Northern analysis reveals a distinct band for SGK1 mRNA in mouse ventral mesencephalon (VM). A band is also observed in the striatum (ST). SGK3 mRNA expression is also identified in the ventral mesencephalon, but not in the striatum. At a cellular level (right) SGK1 mRNA expression is identified by in situ hybridization in the SNpc, exclusively in neurons. The neurons bounded by the rectangle are shown at higher magnification in the inset. An adjacent section (right) hybridized with a sense probe [SGK1(S)] reveals an absence of signal. B, Immunoperoxidase staining for SGK protein reveals widespread expression in the ventral mesencephalon, including in the SNpc, (outlined by the dotted line). Double immunofluorescence staining TH (green) in SNpc dopaminergic neurons and SGK protein (red) reveals expression in the majority of these neurons, mainly in the nucleus, as shown in the inset.
SGK1 has trophic effects on adult SN dopaminergic neurons
To determine whether SGK has the ability to exert trophic effects on SN dopamine neurons, they were transduced with either AAV hSGK1(WT) or a constitutively active form in which the serine residue at position 422 is replaced with a phospho-mimetic aspartate, AAV hSGK1(S422D). The ability of each of these vectors to transduce neurons of the SNpc was confirmed by immunoperoxidase stain of the FLAG epitope in each construct (Fig. 2A), and by immunofluorescence double-labeling for FLAG and TH (Fig. 2A). Transgenic human SGK, like endogenous SGK, was expressed within the nucleus, but it was predominantly expressed in the cytoplasm (Fig. 2A).
Figure 2.

Wild-type and constitutively active forms of hSGK1 have trophic effects on dopaminergic neurons of the SNpc. A, Transduction of neurons of the SNpc (indicated by an arrow) with both hSGK1 constructs is demonstrated in the top by immunoperoxidase staining for the FLAG epitope. No staining is observed on the contralateral, uninjected side (CONTROL). A single neuron shown at high power in the inset demonstrates expression mainly in the cytoplasm, but also in the nucleus (arrow NUC). Double immunofluorescence labeling for TH (red) and FLAG (green) demonstrates expression of the transgenes in dopamine neurons, mainly in the cytoplasm, but also in the nucleus (outlined by a dotted white line). B, Immunoperoxidase stain for TH, with thionin counterstain, at 4 weeks following intranigral injection of AAV hSGK1(S422D) reveals a greater intensity of staining on the injected side as compared with the contralateral, uninjected (CONTROL or C) side. Representative neurons are shown at higher magnification in the bottom, and It can be seen at the cellular level that the increase in staining intensity is due, at least in part, to an increase in the size of the dopaminergic neurons. This increase in size following injection with either AAV hSGK1(WT) or AAV hSGK1(S422D) is shown quantitatively in C. C, Effects on size were highly significant (p < 0.001, one-way ANOVA; Tukey's post hoc analysis as shown; N = 50 neurons on each brain side for each experimental condition) (CON: Control, uninjected side; EXP: Experimental, injected side). Each construct also induced an increase in TH-positive neuron number (p < 0.001, one-way ANOVA; Tukey's post hoc analysis as shown; AAV GFP N = 8, AAV hSGK1(WT) N = 6, AAV hSGK1(S422D) N = 8). This increase in number was not, however, associated with an actual increase in the number of SN neurons stained with the neuronal marker NeuN, for mice treated with AAV hSGK1(S422D) as shown. D, These effects on neuronal cell bodies were associated with a modest increase in striatal immunoperoxidase staining for TH for the hSGK1(S422D) construct, as shown, but not the hSGK1(WT) construct.
At 4 weeks following intranigral injection of the vectors, several trophic effects on dopamine neurons and their axonal projections were observed. Following immunoperoxidase staining for TH on coronal sections of the mesencephalon, there was a macroscopically visible increase in the intensity of chromogen stain on the injected side (Fig. 2B). At a cellular level, two effects on dopamine neurons accounted for this increase. Both hSGK1(WT) and hSGK1(S422D) induced an increase in neuron size. This increase is shown in a representative micrograph in Figure 2B and quantitatively in Figure 2C. hSGK(WT) induced a 1.3-fold increase in the cross-sectional area of TH-positive neurons, and hSGK1(S422D) induced a 1.4-fold increase. There was no significant difference between the two constructs in the magnitude of this effect (p > 0.15, Tukey's post hoc).
In addition to this increase in neuron size, both constructs also induced an increase in the number of TH-positive neurons in the SN, determined by stereologic counts (Fig. 2C). This increase could either be due to an actual increase in number, by an as yet unknown mechanism, or an apparent increase due to augmented expression of TH protein and enhanced detection of neurons by immunoperoxidase stain. To distinguish between these two possibilities, we performed stereologic counts of neurons in the SNpc following peroxidase stain for the general neuronal marker NeuN in mice treated with AAV hSGK1(S422D). This analysis revealed no increase in the actual number of neurons following transduction (Fig. 2C). We therefore conclude that the apparent increase in the number of TH-positive neurons is likely to be due to enhanced detection by immunoperoxidase staining, an effect that we had previously identified for Akt (Ries et al., 2006).
In addition to these effects on dopamine neuron cell bodies, hSGK1(S244D), but not hSGK1(WT), induced a modest (10%), but significant (p = 0.02), increase in immunoperoxidase staining of TH-positive axons and terminals in the striatum, measured as optical density (Fig. 2D).
Constitutively active hSGK1(S422D) protects both cell bodies and axons of SN dopamine neurons from neurotoxin-induced degeneration
To assess the ability of SGK to provide protection from induced neurodegeneration, we tested its ability to forestall both retrograde axon degeneration and cell death from intrastriatal injection of 6-OHDA. This model induces an acute retrograde axonal degeneration that takes place during the first week postlesion (Ries et al., 2008) and a more gradual, progressive loss of neuronal cell bodies over several weeks (Sauer and Oertel, 1994) due to apoptosis (Martí et al., 2002). We assessed the ability of AAV hSGK1(S422D) to prevent neuron loss when administered either before or after 6-OHDA lesion. The latter paradigm is intended to more closely simulate a clinical context in which intervention is possible only after disease onset.
When administered 3 weeks before lesion (Fig. 3A), AAV hSGK1(S422D) afforded substantial protection against acute neuron loss. At 1 week following lesion, whereas AAV GFP-injected control mice showed a 48% loss of TH-positive neurons, AAV hSGK1(S422D)-injected mice demonstrated only an 11% loss (Fig. 3B). To confirm that this difference was indeed due to enhanced survival and not just the prevention of loss of the TH-positive phenotype, we also examined stereologic counts of neurons in the SN stained for NeuN. This analysis demonstrated that while there was a 41% loss of NeuN-positive profiles by stereologic counts in mice treated with AAV GFP, there was only an 8% loss in the mice treated with AAV hSGK1(S422D) (Fig. 3B). To determine whether the protection provided by AAV SGK1(S422D) was robust and lasting, we examined TH-positive neuron counts at 4 weeks postlesion. At this time point as well, while greater neuron loss had occurred in both experimental conditions, protection by AAV hSGK1(S422D) was still observed. In the GFP group there was an 88% loss, and in the hSGK1(S422D) group there was only a 62% loss, a significant difference (Fig. 3C), representing a threefold increase in the number of surviving TH-positive neurons in the AAV hSGK1(S422D)-treated mice.
Figure 3.
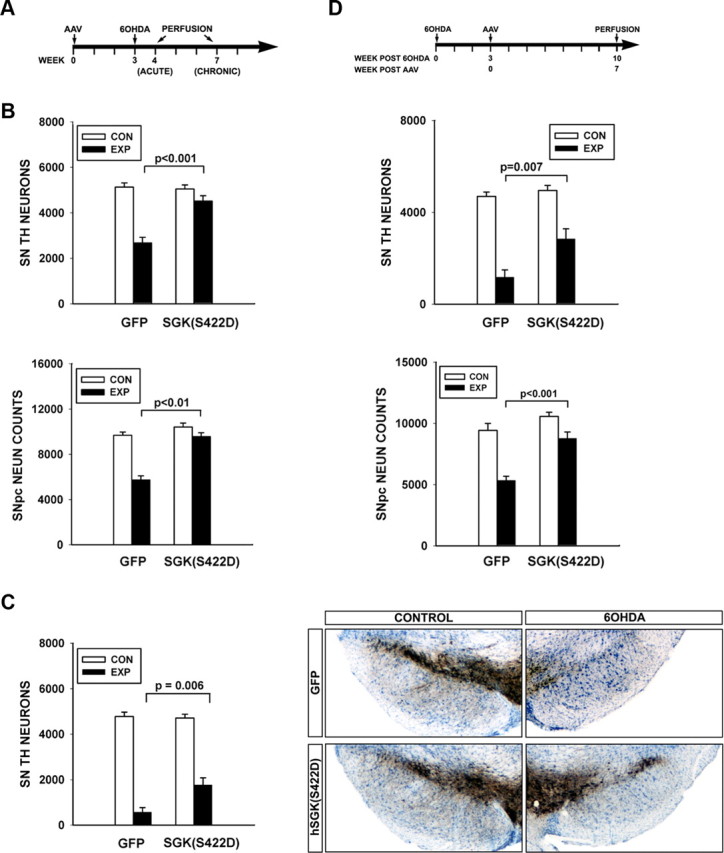
Constitutively active SGK1 protects SN dopaminergic neurons from neurotoxin-induced cell death. A, Mice received a unilateral intranigral injection of AAV hSGK1(S422D) at Time = 0, followed in 3 weeks by an intrastriatal injection, on the same side, of 6-OHDA. B, At 1 week postlesion, there was a greater loss of SN TH-positive neurons in mice receiving an injection control of AAV GFP (N = 7) compared with mice treated with AAV hSGK1(S422D) (N = 6) (p < 0.001, ANOVA; Tukey's post hoc as shown) (CON: Control, uninjected side; EXP: Experimental, injected side). Similarly, there was a greater loss of NeuN-positive profiles in mice treated with AAV GFP (N = 7) than in the mice treated with AAV hSGK1(S422D) (N = 6) (p < 0.001 ANOVA; Tukey's post hoc as shown). C, Survival was assessed at 4 weeks postlesion, and the protective effect of AAV hSGK1(S422D) (N = 5) was still evident compared with the AAV GFP injection control group (N = 6) (p < 0.001, ANOVA; Tukey's post hoc as shown). Right, Representative sections immunostained for TH and thionin counterstained, illustrate this protective effect. D, AAVs were injected at 3 weeks after 6-OHDA, and then neuron survival assessed at 7 weeks after AAV. In this paradigm, while there was a 75% loss of neurons in the GFP condition, there was only a 43% loss in the hSGK1(S422D) condition (p < 0.001, ANOVA; Tukey's post hoc as shown; AAV GFP N = 6, AAV hSGK1(S422D) N = 7). Again, improved survival, rather than just preservation of phenotype, was confirmed by counts of NeuN-positive profiles (p < 0.001, ANOVA; Tukey's post hoc as shown).
In the intrastriatal 6-OHDA model ∼50% of neuron loss has occurred by 3 weeks postlesion, and it continues over the subsequent weeks (Sauer and Oertel, 1994). Therefore, in this model, it is theoretically possible to forestall late neuron degeneration by an intervention at 3 weeks postlesion. We therefore tested the ability of hSGK1(S422D) to provide protection in this context, and found that despite the 3 week delay, there was enhanced survival (Fig. 3D). We again confirmed by counts of NeuN-positive neurons that improved survival, not just preservation of phenotype, had been achieved.
The cellular mechanisms that underlie axon degeneration are distinct from the canonical pathways of programmed cell death that mediate neuron cell soma degeneration (Finn et al., 2000; Raff et al., 2002). Indeed, it has often been observed that experimental interventions that block programmed cell death and are highly protective of dopamine neuron cell bodies are not effective in preventing axon degeneration (Chen et al., 2008; Ries et al., 2008). We have recently reported that another member of the AGC family of kinases, Akt, which shares close functional relationships with SGK (Pearce et al., 2010), is capable of inhibiting retrograde axonal degeneration (Cheng et al., 2011). We therefore sought to determine whether SGK has a similar ability. To perform this assessment, we used TH-GFP mice, because we have previously shown that expression of GFP in the dopaminergic nigrostriatal projection makes it possible to monitor acute axon degeneration following injury (Cheng et al., 2011). Using this approach, we found that hSGK1(S422D) forestalls acute retrograde axon degeneration following intrastriatal 6-OHDA. At 7 d postlesion, control AAV-injected mice show a 36% loss of GFP-positive axons, whereas AAV hSGK1(S422D)-injected mice show only a 15% loss (p = 0.002, t test) (Fig. 4A). To determine whether this protection of axons was lasting, we assessed dopaminergic innervation of the striatum at 4 weeks following 6-OHDA lesion. At that time, AAV hSGK1(S422D)-treated mice showed a >2-fold preservation of striatal TH immunostaining (Fig. 4B).
Figure 4.

Constitutively active SGK1 protects dopaminergic axons from acute neurotoxin-induced retrograde degeneration. A, TH-GFP mice received an intranigral injection of AAV hSGK1(S422D) or AAV dsRed as a control, followed in 3 weeks by an intrastriatal injection of 6-OHDA. At 7 d postlesion, the number of surviving dopaminergic axons in the MFB was determined by confocal optical sectioning. Each individual image represents a maximal projection of 20 0.1 μm optical sections. For each side of the brain, three adjacent maximal projections are shown for the medial, central, and lateral MFB. It can be seen in these representative images that 6-OHDA induces a loss of axons in the mice that received AAV dsRed compared with the CONTROL, contralateral, non-6-OHDA-injected side. However, axons are preserved in mice that received AAV hSGK1(S422D). This effect is shown quantitatively in the graph in which the number of axons remaining on the lesioned (Experimental; E) side of the brain is expressed as a percentage of the contralateral, nonlesioned Control (C) (E%C). A significant protective effect on axons is observed (p = 0.002, t test; AAV dsRed N = 5, AAV hSGK1(S422D) N = 6). B, In a separate experiment mice were killed at 4 weeks postlesion and processed for TH immunoperoxidase staining of the striatum. The AAV hSGK1(S422D)-treated mice demonstrated a significant preservation of striatal dopaminergic TH-positive staining, as shown in the representative coronal sections and by quantitative determination of optical densities as shown in the graph (p = 0.006, t test; AAV GFP N = 6, AAV hSGK1(S422D) N = 5) (CONTROL: contralateral, uninjected side).
hSGK1(S422D) induces restoration of the nigrostriatal dopaminergic projection
At 3 weeks after 6-OHDA lesion, degeneration of neurons remains ongoing, and there exists an opportunity for protection of those that have survived, as shown in Figure 3D. However, the same is not true for axons, because they begin to degenerate by the first postlesion day (Ries et al., 2008), and the process is essentially complete by 1 week (Kim et al., 2011a). Therefore, neuroprotection is no longer possible. However, we have recently shown that in addition to its ability to protect axons from degeneration, the AGC kinase Akt also has the ability to induce dopaminergic axonal regeneration (Kim et al., 2011a). To determine whether SGK can induce a similar phenotype, mice were lesioned with 6-OHDA, and after a 3 week delay they were injected with either AAV hSGK1(S422D) or AAV GFP as an injection control. At 7 weeks following AAV injection dopaminergic re-innervation of the striatum was assessed. At that time, control mice, given an AAV GFP control injection, showed little restoration of striatal TH-positive staining, with optical densities on the lesioned side at 36.8 ± 4.0 (SEM) percent of the contralateral, nonlesioned side (Fig. 5). In mice injected with AAV hSGK1(S422D), however, there was substantial restoration of innervation, with optical densities at 74.3 ± 8.8% (p = 0.004, t test) (Fig. 5).
Figure 5.
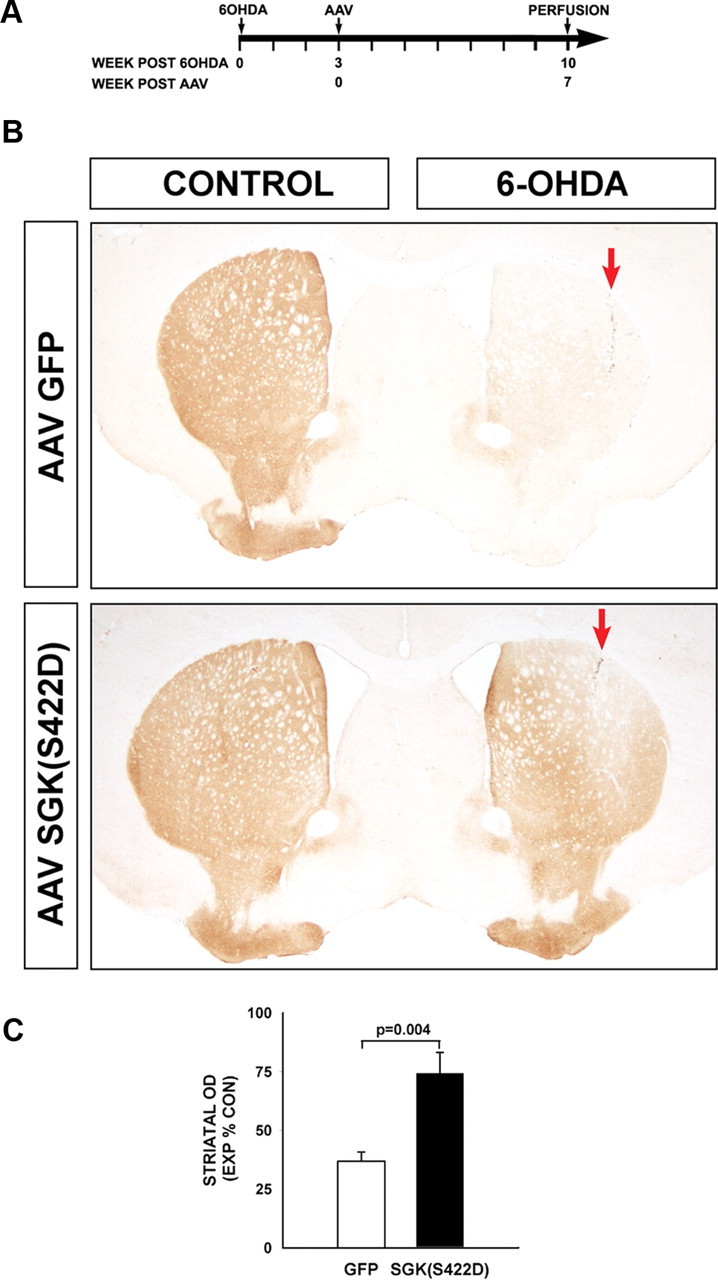
Constitutively active SGK1 induces restoration of the nigrostriatal dopaminergic projection following its destruction by neurotoxin lesion. A, Mice received an intrastriatal 6-OHDA lesion at Time = 0, and then at 3 weeks postlesion received AAV hSGK1(S422D) or AAV GFP injection control. B, At 7 weeks following AAV injection, extensive re-innervation of the striatum is observed, as shown in representative coronal sections stained for TH. In each panel, the red arrow indicates the site of the 6-OHDA injection (CONTROL: Contralateral, uninjected side). C, Restoration of striatal innervation is shown quantitatively as optical density of TH staining, with the Experimental (EXP) lesioned side expressed as a percentage of density in the Control (CON) nonlesioned side (p = 0.004, t test; AAV GFP N = 6, AAV hSGK1(S422D) N = 7).
Discussion
An emerging theme in the neurobiology of mesencephalic dopamine neurons is that not only during development, but also in adulthood, they depend on neurotrophic support and cell survival signaling. Conditional ablation of glial cell line-derived neurotrophic factor (GDNF) in adult mice results in a delayed and progressive loss of dopamine neurons (Pascual et al., 2008), as does selective deletion of the Ret tyrosine kinase, an important mediator of GDNF signaling (Kramer et al., 2007). These observations would suggest that the cellular signaling pathways that mediate neurotrophic support are likewise important for the adult maintenance of these neurons. One such pathway, often identified as a possible mediator of GDNF effects, is that of PI3K/Akt signaling (Soler et al., 1999; Besset et al., 2000; Sawada et al., 2000). While the role of Akt signaling in maintaining viability of adult neurons remains to be fully explored, there is compelling evidence that it is an important regulator of developmental cell death in these neurons (Ries et al., 2009) and critical for axonal maintenance in the adult (Kim et al., 2011b). Given the close relationships between Akt and SGK (Alessi et al., 2009), and previous reports suggesting a possible survival role of SGK in dopamine neurons (Schoenebeck et al., 2005), we hypothesized that SGK-mediated signaling may also be capable of mediating neurotrophic responses in these neurons.
We confirmed the observations of others that SGK mRNA and protein are expressed within the SNpc (Schoenebeck et al., 2005; Stichel et al., 2005) and specifically within dopamine neurons (Stichel et al., 2005). We herein show that almost all SN dopamine neurons express SGK protein. Based on cell fractionation studies Schoenebeck et al. (2005) concluded that SGK protein is principally localized to cell membranes and the nucleus (Schoenebeck et al., 2005), and we have confirmed localization of endogenous SGK in the nucleus of dopamine neurons.
Although SGK shares activation mechanisms and some substrates with Akt (Tessier and Woodgett, 2006), it also phosphorylates many target molecules that are not shared, particularly diverse ion channels and membrane carriers, pumps, and transporters (Lang et al., 2006). We therefore expected that in dopamine neurons SGK would share some cellular phenotypes with Akt, but not all. Contrary to our expectations, we have found that SGK recapitulates every Akt-induced phenotype in dopamine neurons that we examined.
Based on previous studies (Schoenebeck et al., 2005) we expected that SGK, like Akt, would provide neuroprotection from neuron death. In the intrastriatal 6-OHDA model, the principal morphology of death is that of apoptosis (Martí et al., 2002; Ries et al., 2008), and it is inhibited by a constitutively active form of Akt (Ries et al., 2006). Akt has diverse anti-apoptotic effects in multiple canonical pathways of programmed cell death (Brunet et al., 2001; Downward, 2004; Burke, 2008), and SGK shares some of its targets (Tessier and Woodgett, 2006), including the transcription factor FOXO3a (FKHRh1) (Brunet et al., 2001) and the kinase IKKβ in the IKK-NF-KB pathway (Downward, 2004; Zhang et al., 2005).
Like Akt (Ries et al., 2006), SGK induced hypertrophy of dopaminergic neurons. This cellular phenotype has been widely demonstrated for Akt signaling in diverse contexts, and it has been largely attributed to signaling through the mTor kinase (Shioi et al., 2002; Easton et al., 2005; Manning and Cantley, 2007). Unlike the prosurvival phenotype that SGK shares with Akt, there are no obvious targets that SGK shares with Akt for this hypertrophy effect. While SGK is a phosphorylation target of TORC2, it does not, conversely, have known activation effects on mTor signaling, as does Akt. Thus the mediators of the neuronal hypertrophy effects of SGK are unknown. One possible candidate that may merit future investigation is FOXO3a. FOXO3a has been demonstrated to play a role in the regulation of cardiac (Skurk et al., 2005) and skeletal muscle size (Zhao et al., 2007). Akt-mediated regulation of cell size has been proposed to be mediated not only by mTor activation, but also by inhibition of FOXO transcription factors (Skurk et al., 2005; Zhao et al., 2007), so it is therefore possible that SGK may exert similar effects by inhibition of FOXO3a.
A recently reported novel phenotype for Akt in neurons is its ability to forestall retrograde axon degeneration (Cheng et al., 2011). This effect is to be distinguished from the diverse anti-apoptotic effects of Akt, because the mechanisms of axon degeneration are distinct from those of apoptosis (Finn et al., 2000; Raff et al., 2002; Coleman and Freeman, 2010). Indeed, it has often been shown, specifically for dopamine neurons, that experimental approaches that block programmed cell death are highly effective in achieving neuron survival, but not in preserving axons (Chen et al., 2008; Ries et al., 2008). The ability of Akt to forestall retrograde axon degeneration has been attributed to inhibition of macroautophagy, which has been postulated to be due to its ability to activate mTor (Cheng et al., 2011). The mechanisms by which SGK mediates protection from retrograde axonal degeneration are at present unknown. Given that SGK is not known to signal through mTor, an alternative signaling mechanism will need to be identified.
Our studies used an overexpression approach. While we have confirmed the observations of others that SGK mRNA and protein are expressed in SN dopamine neurons, whether endogenous SGK plays a role in regulating the cellular phenotypes that we have examined remains unknown. Our demonstration that increased expression of either wild-type or constitutively active SGK induces these cellular effects does, however, suggest that the necessary endogenous downstream signaling pathways are in place. A central question to be addressed in future studies will be whether endogenous SGK plays an important role in maintaining the viability of dopamine neurons, and, if so, whether this ability is of relevance to the pathogenesis of human disease, such as PD. Intriguingly, in this regard, SGK protein has been identified in Lewy bodies (Schoenebeck et al., 2005), the intraneuronal inclusions that are a hallmark of the disease. Aside from a possible role in PD pathogenesis, we have herein demonstrated that viral vector-mediated expression of SGK in nigral neurons has numerous beneficial neurotrophic effects. These may therefore be explored in the development of neuroprotective and neuroregenerative strategies for the disease.
Footnotes
This work was supported by National Institutes of Health Grants NS26836 and NS38370, the Parkinson's Disease Foundation, and the Parkinson's Alliance (R.E.B.).
The authors declare no competing financial interests.
References
- Alessi DR, Pearce LR, García-Martínez JM. New insights into mTOR signaling: mTORC2 and beyond. Sci Signal. 2009;2:pe27. doi: 10.1126/scisignal.267pe27. [DOI] [PubMed] [Google Scholar]
- Aleyasin H, Rousseaux MW, Marcogliese PC, Hewitt SJ, Irrcher I, Joselin AP, Parsanejad M, Kim RH, Rizzu P, Callaghan SM, Slack RS, Mak TW, Park DS. DJ-1 protects the nigrostriatal axis from the neurotoxin MPTP by modulation of the AKT pathway. Proc Natl Acad Sci U S A. 2010;107:3186–3191. doi: 10.1073/pnas.0914876107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baki L, Neve RL, Shao Z, Shioi J, Georgakopoulos A, Robakis NK. Wild-type but not FAD mutant presenilin-1 prevents neuronal degeneration by promoting phosphatidylinositol 3-kinase neuroprotective signaling. J Neurosci. 2008;28:483–490. doi: 10.1523/JNEUROSCI.4067-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besset V, Scott RP, Ibáñez CF. Signaling complexes and protein-protein interactions involved in the activation of the Ras and phosphatidylinositol 3-kinase pathways by the c-Ret receptor tyrosine kinase. J Biol Chem. 2000;275:39159–39166. doi: 10.1074/jbc.M006908200. [DOI] [PubMed] [Google Scholar]
- Brunet A, Datta SR, Greenberg ME. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol. 2001;11:297–305. doi: 10.1016/s0959-4388(00)00211-7. [DOI] [PubMed] [Google Scholar]
- Burke RE. Programmed cell death and new discoveries in the genetics of parkinsonism. J Neurochem. 2008;104:875–890. doi: 10.1111/j.1471-4159.2007.05106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke RE, Cadet JL, Kent JD, Karanas AL, Jackson-Lewis V. An assessment of the validity of densitometric measures of striatal tyrosine hydroxylase-positive fibers: relationship to apomorphine-induced rotations in 6-hydroxydopamine lesioned rats. J Neurosci Methods. 1990;35:63–73. doi: 10.1016/0165-0270(90)90095-w. [DOI] [PubMed] [Google Scholar]
- Burke RE, Franklin SO, Inturrisi CE. Acute and persistent suppression of preproenkephalin mRNA expression in the striatum following developmental hypoxic- ischemic injury. J Neurochem. 1994;62:1878–1886. doi: 10.1046/j.1471-4159.1994.62051878.x. [DOI] [PubMed] [Google Scholar]
- Chen X, Rzhetskaya M, Kareva T, Bland R, During MJ, Tank AW, Kholodilov N, Burke RE. Antiapoptotic and trophic effects of dominant-negative forms of dual leucine zipper kinase in dopamine neurons of the substantia nigra in vivo. J Neurosci. 2008;28:672–680. doi: 10.1523/JNEUROSCI.2132-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HC, Kim SR, Oo TF, Kareva T, Yarygina O, Rzhetskaya M, Wang C, During M, Talloczy Z, Tanaka K, Komatsu M, Kobayashi K, Okano H, Kholodilov N, Burke RE. Akt suppresses retrograde degeneration of dopaminergic axons by inhibition of macroautophagy. J Neurosci. 2011;31:2125–2135. doi: 10.1523/JNEUROSCI.5519-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman MP, Freeman MR. Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci. 2010;33:245–267. doi: 10.1146/annurev-neuro-060909-153248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 2004;15:177–182. doi: 10.1016/j.semcdb.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VM, Szabolcs M, de Jong R, Oltersdorf T, Ludwig T, Efstratiadis A, Birnbaum MJ. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol. 2005;25:1869–1878. doi: 10.1128/MCB.25.5.1869-1878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Khodor BF, Kholodilov NG, Yarygina O, Burke RE. The expression of mRNAs for the proteasome complex is developmentally regulated in the rat mesencephalon. Brain Res Dev Brain Res. 2001;129:47–56. doi: 10.1016/s0165-3806(01)00181-x. [DOI] [PubMed] [Google Scholar]
- Finn JT, Weil M, Archer F, Siman R, Srinivasan A, Raff MC. Evidence that Wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. J Neurosci. 2000;20:1333–1341. doi: 10.1523/JNEUROSCI.20-04-01333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene LA, Levy O, Malagelada C. Akt as a victim, villain and potential hero in Parkinson's disease pathophysiology and treatment. Cell Mol Neurobiol. 2011;31:969–978. doi: 10.1007/s10571-011-9671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi K, Tsuda M, Wanaka A, Tohyama M, Takagi T. Differential expression of sgk mRNA, a member of the Ser/Thr protein kinase gene family, in rat brain after CNS injury. Brain Res Mol Brain Res. 1994;26:189–196. doi: 10.1016/0169-328x(94)90090-6. [DOI] [PubMed] [Google Scholar]
- Iwata S, Nomoto M, Morioka H, Miyata A. Gene expression profiling in the midbrain of striatal 6-hydroxydopamine-injected mice. Synapse. 2004;51:279–286. doi: 10.1002/syn.10307. [DOI] [PubMed] [Google Scholar]
- Kholodilov N, Yarygina O, Oo TF, Zhang H, Sulzer D, Dauer W, Burke RE. Regulation of the development of mesencephalic dopaminergic systems by the selective expression of glial cell line-derived neurotrophic factor in their targets. J Neurosci. 2004;24:3136–3146. doi: 10.1523/JNEUROSCI.4506-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim RH, Peters M, Jang Y, Shi W, Pintilie M, Fletcher GC, DeLuca C, Liepa J, Zhou L, Snow B, Binari RC, Manoukian AS, Bray MR, Liu FF, Tsao MS, Mak TW. DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell. 2005;7:263–273. doi: 10.1016/j.ccr.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Kim SR, Chen X, Oo TF, Kareva T, Yarygina O, Wang C, During M, Kholodilov N, Burke RE. Dopaminergic pathway reconstruction by Akt/Rheb-induced axon regeneration. Ann Neurol. 2011a;70:110–120. doi: 10.1002/ana.22383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SR, Ries V, Cheng HC, Kareva T, Oo TF, Yu WH, Duff K, Kholodilov N, Burke RE. Age and alpha-synuclein expression interact to reveal a dependence of dopaminergic axons on endogenous Akt/PKB signaling. Neurobiol Dis. 2011b;44:215–222. doi: 10.1016/j.nbd.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Deak M, Morrice N, Cohen P. Characterization of the structure and regulation of two novel isoforms of serum- and glucocorticoid-induced protein kinase. Biochem J. 1999;344:189–197. [PMC free article] [PubMed] [Google Scholar]
- Kramer ER, Aron L, Ramakers GM, Seitz S, Zhuang X, Beyer K, Smidt MP, Klein R. Absence of Ret signaling in mice causes progressive and late degeneration of the nigrostriatal system. PLoS Biol. 2007;5:e39. doi: 10.1371/journal.pbio.0050039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvajo M, McKellar H, Gogos JA. Molecules, signaling, and schizophrenia. Curr Top Behav Neurosci. 2010;4:629–656. doi: 10.1007/7854_2010_41. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang F, Cohen P. Regulation and physiological roles of serum- and glucocorticoid-induced protein kinase isoforms. Sci STKE. 2001;2001:re17. doi: 10.1126/stke.2001.108.re17. [DOI] [PubMed] [Google Scholar]
- Lang F, Böhmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev. 2006;86:1151–1178. doi: 10.1152/physrev.00050.2005. [DOI] [PubMed] [Google Scholar]
- Malagelada C, Jin ZH, Greene LA. RTP801 is induced in Parkinson's disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J Neurosci. 2008;28:14363–14371. doi: 10.1523/JNEUROSCI.3928-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markus A, Zhong J, Snider WD. Raf and akt mediate distinct aspects of sensory axon growth. Neuron. 2002;35:65–76. doi: 10.1016/s0896-6273(02)00752-3. [DOI] [PubMed] [Google Scholar]
- Martí MJ, Saura J, Burke RE, Jackson-Lewis V, Jiménez A, Bonastre M, Tolosa E. Striatal 6-hydroxydopamine induces apoptosis of nigral neurons in the adult rat. Brain Res. 2002;958:185–191. doi: 10.1016/s0006-8993(02)03694-6. [DOI] [PubMed] [Google Scholar]
- Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15:161–170. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- Namikawa K, Honma M, Abe K, Takeda M, Mansur K, Obata T, Miwa A, Okado H, Kiyama H. Akt/protein kinase B prevents injury-induced motoneuron death and accelerates axonal regeneration. J Neurosci. 2000;20:2875–2886. doi: 10.1523/JNEUROSCI.20-08-02875.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual A, Hidalgo-Figueroa M, Piruat JI, Pintado CO, Gómez-Díaz R, López-Barneo J. Absolute requirement of GDNF for adult catecholaminergic neuron survival. Nat Neurosci. 2008;11:755–761. doi: 10.1038/nn.2136. [DOI] [PubMed] [Google Scholar]
- Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Ries V, Henchcliffe C, Kareva T, Rzhetskaya M, Bland R, During MJ, Kholodilov N, Burke RE. Oncoprotein Akt/PKB: Trophic effects in murine models of Parkinson's Disease. Proc Natl Acad Sci U S A. 2006;103:18757–18762. doi: 10.1073/pnas.0606401103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries V, Silva RM, Oo TF, Cheng HC, Rzhetskaya M, Kholodilov N, Flavell RA, Kuan CY, Rakic P, Burke RE. JNK2 and JNK3 combined are essential for apoptosis in dopamine neurons of the substantia nigra, but are not required for axon degeneration. J Neurochem. 2008;107:1578–1588. doi: 10.1111/j.1471-4159.2008.05713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries V, Cheng HC, Baohan A, Kareva T, Oo TF, Rzhetskaya M, Bland RJ, During MJ, Kholodilov N, Burke RE. Regulation of the postnatal development of dopamine neurons of the substantia nigra in vivo by Akt/protein kinase B. J Neurochem. 2009;110:23–33. doi: 10.1111/j.1471-4159.2009.06101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakoda H, Gotoh Y, Katagiri H, Kurokawa M, Ono H, Onishi Y, Anai M, Ogihara T, Fujishiro M, Fukushima Y, Abe M, Shojima N, Kikuchi M, Oka Y, Hirai H, Asano T. Differing roles of Akt and serum- and glucocorticoid-regulated kinase in glucose metabolism, DNA synthesis, and oncogenic activity. J Biol Chem. 2003;278:25802–25807. doi: 10.1074/jbc.M301127200. [DOI] [PubMed] [Google Scholar]
- Sauer H, Oertel WH. Progressive degeneration of nigrostriatal dopamine neurons following intrastriatal terminal lesions with 6 hydroxydopamine a combined retrograde tracing and immunocytochemical study in the rat. Neuroscience. 1994;59:401–415. doi: 10.1016/0306-4522(94)90605-x. [DOI] [PubMed] [Google Scholar]
- Sawada H, Ibi M, Kihara T, Urushitani M, Nakanishi M, Akaike A, Shimohama S. Neuroprotective mechanism of glial cell line-derived neurotrophic factor in mesencephalic neurons. J Neurochem. 2000;74:1175–1184. doi: 10.1046/j.1471-4159.2000.741175.x. [DOI] [PubMed] [Google Scholar]
- Sawamoto K, Nakao N, Kobayashi K, Matsushita N, Takahashi H, Kakishita K, Yamamoto A, Yoshizaki T, Terashima T, Murakami F, Itakura T, Okano H. Visualization, direct isolation, and transplantation of midbrain dopaminergic neurons. Proc Natl Acad Sci U S A. 2001;98:6423–6428. doi: 10.1073/pnas.111152398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenebeck B, Bader V, Zhu XR, Schmitz B, Lübbert H, Stichel CC. Sgk1, a cell survival response in neurodegenerative diseases. Mol Cell Neurosci. 2005;30:249–264. doi: 10.1016/j.mcn.2005.07.017. [DOI] [PubMed] [Google Scholar]
- Shioi T, McMullen JR, Kang PM, Douglas PS, Obata T, Franke TF, Cantley LC, Izumo S. Akt/protein kinase B promotes organ growth in transgenic mice. Mol Cell Biol. 2002;22:2799–2809. doi: 10.1128/MCB.22.8.2799-2809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skurk C, Izumiya Y, Maatz H, Razeghi P, Shiojima I, Sandri M, Sato K, Zeng L, Schiekofer S, Pimentel D, Lecker S, Taegtmeyer H, Goldberg AL, Walsh K. The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J Biol Chem. 2005;280:20814–20823. doi: 10.1074/jbc.M500528200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler RM, Dolcet X, Encinas M, Egea J, Bayascas JR, Comella JX. Receptors of the glial cell line-derived neurotrophic factor family of neurotrophic factors signal cell survival through the phosphatidylinositol 3-kinase pathway in spinal cord motoneurons. J Neurosci. 1999;19:9160–9169. doi: 10.1523/JNEUROSCI.19-21-09160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stichel CC, Schoenebeck B, Foguet M, Siebertz B, Bader V, Zhu XR, Lübbert H. sgk1, a member of an RNA cluster associated with cell death in a model of Parkinson's disease. Eur J Neurosci. 2005;21:301–316. doi: 10.1111/j.1460-9568.2005.03859.x. [DOI] [PubMed] [Google Scholar]
- Tessier M, Woodgett JR. Serum and glucocorticoid-regulated protein kinases: variations on a theme. J Cell Biochem. 2006;98:1391–1407. doi: 10.1002/jcb.20894. [DOI] [PubMed] [Google Scholar]
- Timmons S, Coakley MF, Moloney AM, O' Neill C. Akt signal transduction dysfunction in Parkinson's disease. Neurosci Lett. 2009;467:30–35. doi: 10.1016/j.neulet.2009.09.055. [DOI] [PubMed] [Google Scholar]
- Wang Q, Liu L, Pei L, Ju W, Ahmadian G, Lu J, Wang Y, Liu F, Wang YT. Control of synaptic strength, a novel function of Akt. Neuron. 2003;38:915–928. doi: 10.1016/s0896-6273(03)00356-8. [DOI] [PubMed] [Google Scholar]
- Webster MK, Goya L, Ge Y, Maiyar AC, Firestone GL. Characterization of sgk, a novel member of the serine/threonine protein kinase gene family which is transcriptionally induced by glucocorticoids and serum. Mol Cell Biol. 1993;13:2031–2040. doi: 10.1128/mcb.13.4.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gehrke S, Haque ME, Imai Y, Kosek J, Yang L, Beal MF, Nishimura I, Wakamatsu K, Ito S, Takahashi R, Lu B. Inactivation of Drosophila DJ-1 leads to impairments of oxidative stress response and phosphatidylinositol 3-kinase/Akt signaling. Proc Natl Acad Sci U S A. 2005;102:13670–13675. doi: 10.1073/pnas.0504610102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Cui R, Cheng X, Du J. Antiapoptotic effect of serum and glucocorticoid-inducible protein kinase is mediated by novel mechanism activating I{kappa}B kinase. Cancer Res. 2005;65:457–464. [PubMed] [Google Scholar]
- Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6:472–483. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]