

Summary
Recent studies from our laboratory have indicated that the transcription factor AP-2α plays a critical role in the differentiation of human villous cytotrophoblast cells (CTB) to syncytiotrophoblast cells (STB). However, little is known about the expression of AP-2α in placentas from pathologic pregnancies. This study compares the expression of AP-2α in placentas from high-risk pregnancies to gestational age-matched controls. Paracentral sections from grossly unremarkable areas of 10 placentas from each group of pregnancies complicated by mild preeclampsia (PE), severe PE, diabetes mellitus (DM), chronic hypertension (HTN), fetal growth restriction (FGR) and 10 control cases of placentas from normal pregnancies matched for gestational age (CG) were double immunostained for AP-2α and E-cadherin. The total numbers of CTB and STB and the numbers of AP-2α positive and negative nuclei in both of these cell types were counted by two pathologists blinded to disease status, in ten representative 40x fields (HPF) for each placenta. Abnormal placental maturation in most of pathologic pregnancies was evidenced by a 1.5–1.7-fold lower STB to CTB ratio. AP-2α expression in STB was lower in mild PE, DM, HTN and FGR (p<0.0001 in each instance) and was higher by 2-fold in severe PE, though this increase was not statistically significant (p=0.3). Since AP-2α has been shown to be critical for villous CTB differentiation, our findings suggest that abnormalities in the AP-2α cascade of transcription factors and/or signaling molecules may contribute to the pathogenesis of the abnormal maturation in placentas in certain types of high-risk pregnancies. The different pattern of AP-2α expression in mild and severe PE clearly suggests that these conditions may have two independent pathogenic mechanisms.
Keywords: AP-2α, trophoblast differentiation, mild preeclampsia, severe preeclampsia, immunohistochemistry
1. Introduction
The trophoblast layer of the human placental villus consists of multinucleated syncytiotrophoblast cells (STB) and mononuclear cytotrophoblast cells (CTB) [1]. The STB, which are in direct contact with maternal blood, perform many critical functions throughout pregnancy, including regulation of substrate and gas exchange between mother and fetus and synthesis and secretion of many hormones that modulate maternal and fetal growth and metabolism [1–2]. The underlying mononuclear CTB, which are located between the STB and the basement membrane of the trophoblast layer, fuse with the syncytium, therefore being the source of STB layer [1]. Extravillous CTB, on the other hand, invade the underlying endometrium and maternal vascular bed, migrating up the uterine spiral arteries that supply the implantation site [1].
At present, much is known about the regulation of trophoblast development in the mouse [3], but less about the differentiation of human villous CTB. In particular, the function of transcription factors and signaling molecules that are involved in the terminal differentiation of human villous CTB to a STB phenotype are incompletely understood. Furthermore, while abnormalities in the development of the trophoblast layer have been observed in several pathologic conditions of pregnancy associated with higher maternal and fetal morbidity and mortality, including preeclampsia (PE) and fetal growth restriction (FGR) [4–5], little is known about the transcriptional regulation of trophoblast differentiation in these conditions. Earlier studies from our laboratory demonstrated that the transcription factor AP-2α (activator protein-2 alpha, also called TFAP2A) plays an important role in terminal differentiation of villous CTB to a STB phenotype [6–8]. AP-2α mRNA and protein are induced during the initial stage (first 2 hr.) of in vitro CTB differentiation. Global gene profiling indicated that the majority of the 100 most induced genes during in vitro differentiation of human CTB have one or more putative AP-2α binding sites in the proximal 1 kb of their promoters [8]. AP-2α also transactivates the human placental lactogen (hPL) [9], human chorionic gonadotropin (hCG) α and hCGβ [10] promoters and the promoters of other genes that are induced during spontaneous differentiation of primary cultures of villous CTB. In addition, AP-2α was shown by other investigators to stimulate the expression of the genes for aromatase cytochrome P-450 [11], germ cell alkaline phosphatase [12] and 17β-hydroxysteroid dehydrogenase type 1 [13] in human trophoblast cell lines during CTB differentiation. Silencing of AP-2α activity in primary cultures of villous CTB undergoing spontaneous differentiation by an adenovirus that expresses a dominant/negative AP-2α mutant markedly blocked the expression of the mRNAs of hPL, hCG, corticotropin releasing hormone, pregnancy-specific glycoprotein 1 and other genes normally induced during the differentiation process [8].
Recent findings by Kotani and co-workers [14] indicated that overexpression of AP- 2α or AP-2γ inhibits the migratory and invasive capacities of a transformed cell line of human first trimester trophoblast cells (HTR-8/SVneo cells). The increase in the AP-2α protein in the cells led to changes in protease, metalloproteinase and plasminogen activator inhibitor-1 levels. These in vitro studies suggest that AP-2α also suppresses trophoblast cell migration and invasion. They also reported that the expression of AP- 2α, as well as AP-2, was higher in twelve PE placentas as compared to gestational age-matched control placentas. The increase in the AP-2α proteins was demonstrated by immunohistochemical analysis and immunoblotting of placental tissue. The increase in AP-2α expression was noted in both STB and extravillous trophoblast cells.
In this study, we examined the expression of AP-2α by immunohistochemistry in STB and CTB in placentas from pregnancies complicated by mild PE, severe PE, chronic hypertension (HTN), diabetes mellitus (DM) or FGR and gestational age-matched CG placentas.
2. Methods
2.1. Inclusion criteria
The protocol for this research was approved by the Institutional Review Boards of Cincinnati Children’s Hospital Medical Center and the University of Cincinnati. Archival paraffin blocks of placentas from patients with mild PE, severe PE, chronic HTN, DM, and FGR were selected from the tissue repository of the Department of Pathology of the University of Cincinnati Medical Center. The selected cases for each group were matched for gestational age, but otherwise were randomly selected from the database. A total of 10 placentas from singleton deliveries were chosen from each group according to standard diagnostic criteria. All placental reports and archived hematoxylin and eosin (H&E) slides were re-reviewed and relevant gross, placental weight and histological findings recorded. Clinical data was extracted from electronic medical records and diagnoses confirmed by chart review. The diagnosis of mild PE was based on the presence of blood pressure > 140, but <160 mmHg systolic and > 90 <110 mmHg diastolic, measured in at least two different occasions more than 12 hours apart, in a woman who was normotensive before 20 weeks’ gestation and the presence of proteinuria (> 300 mg and < 5 g/24h) [15]. Severe PE was defined as blood pressure > 160/110 mmHg and urinary protein excretion of > 5g/24h. Chronic HTN was defined as a blood pressure elevation of > 140 mmHg systolic or > 90 mmHg diastolic in the absence of proteinuria that was detected prior to pregnancy or by the twentieth week of gestation [15]. Placentas from diabetic pregnancies included 5 pre-gestational DM and 5 gestational DM, diagnosed according to the White classification [16]. Placentas from infants whose weights were below the tenth percentile for gestational age and whose mothers did not have history of PE, chronic HTN or DM were included in the FGR group [17]. There was no mutual overlap of conditions among the groups studied. Cases of pregnancy-induced hypertension with superimposed PE as well as cases of DM with superimposed PE were not included. The CG consisted of gestational-age matched placentas from non-hypertensive, non-diabetic patients who delivered infants with birth weights appropriate for gestational age.
2.2 Microscopy and immunohistochemistry
Microscopic and immunohistochemical studies were performed using placental paraffin blocks from grossly unremarkable areas of each placenta that had been previously fixed in 10% buffered formaldehyde solution. H&E stained sections were used for routine microscopy. Histological parameters evaluated included villous maturation, histological evidence of acute ascending infection (acute chorioamnionitis, funisitis and/or chorionic plate vasculitis,), evidence of vascular lesions (decidual arteriolopathy, infarction, retroplacental hematoma, thrombosis in fetal vessels, multifocal clusters of avascular villi), and chronic inflammation (decidual plasma cell infiltrates, chronic villitis of unknown etiology) [18]. Recognizing that the placenta responds in various ways to hypoxia and to study the correlation between the different clinical conditions and patterns of diffuse chronic hypoxic injuries, the placentas were classified as pre-uterine, uterine and post-uterine hypoxia, based on previously described criteria [19]. Shortly, the preuterine hypoxic pattern of chronic hypoxic placental injury is secondary to maternal hypoxemia, as in pregnancies at high altitudes, or maternal anemia, and features a homogeneous placental maturation, villous hypervascularity, decreased extracellular matrix of chorionic villi, and increased syncytial knots, villous cytotrophoblasts and Hofbauer cells. The uterine chronic hypoxic pattern is due to uteroplacental malperfusion as in preeclampsia and late onset FGR, and histologically differs form the preuterine pattern by focal villous hypermaturity and hypervascularity. The diffuse chronic postuterine hypoxic pattern, as in stillbirth and early onset FGR, differs histologically from the above patterns by homogeneous villous hypermaturity (terminal villous hypoplasia), hypovascularity, increased villous extracellular matrix, apoptotic syncytial knots [20] and decreased Hofbauer cells. Other placental lesions related to placental hypoxia such as excessive amount of extravillous trophoblasts, microscopic chorionic pseudocysts, clusters of multinucleated giant cells in decidua were classified according to previously described criteria [21].
Double immunostaining with anti-AP-2α and anti-E-cadherin antibodies was performed to evaluate AP-2α expression in the CTB and STB, respectively. Briefly, for dual immunostaining, 0.4 μ sections were mounted on superfrost plus slides and then stained using the automated Ventana immunostainer. The following protocol was used: deparaffinization, cell conditioning with EDTA for 30 min, incubation with dual primary antibodies anti- AP-2α (Genway, San Diego, CA) at dilution 1:25 for 8 min and E-cadherin (Cell Marque, Rocklin, CA) RTU for 52 min, followed by basic Ultraview DAB and RedMap detection systems (Ventana Medical Systems, Tucson, AZ). Slides were then rinsed with reaction buffer and counterstained with hematoxylin for 4 min, followed by post-counterstaining with bluing reagent for 4 min. Appropriate positive and negative controls were used.
2.3. Data analysis
Microscopic examinations were independently performed by two pathologists (RS and JS), who were blinded to the disease status. Ten random 40X high power fields (HPF) from each transmural paracentral placental section without infarctions, intervillous thrombus, or massive perivillous fibrin deposition were examined for the numbers of chorionic villi, syncytial knots, total numbers of CTB and STB and AP-2α positive and negative CTB and STB. Total number of CTB and AP-2α-positive and negative CTB were counted, each nucleus corresponding to one CTB cell. As STB cell borders are indistinct and may not be present in the same plane of section, total number of STB nuclei and AP-2α-positive and negative STB nuclei were counted. Syncytial knots were defined as multinucleated protrusions from the villous surface, with ten or more nuclei [21], and were excluded from the total count. Size or degree of projection of syncytial knots above the villous surface were not measured. E-cadherin immunostaining (red membrane staining) highlights the CTB cell membrane and the inner STB cell membrane, thus distinguishing between the two trophoblastic cell types (STB and CTB) from themselves and from all the other cells of the chorionic villous, such as fibroblasts, endothelial cells and Hofbauer cells, which did not stain. AP-2α-positive CTB (brown nuclear staining) featured AP-2α nuclear and E-cadherin (red) membranous staining in the same cell (double positivity). AP-2α-positive STB featured AP-2α (brown) nuclear and E-cadherin (red) positive staining present only in the inner membrane of the same cell. Cells were interpreted as negative or positive only and intensity of positive staining was not scored. STB: CTB ratio and percentage of AP-2α positive cells per total number of CTB or AP-2α positive STB nuclei per total number of STB were used as endpoints for analysis. Values were expressed as mean or geometric mean, as appropriate, and 95% confidence interval. Data were initially examined for deviation from normality assumption. Total number of villi, syncytial knots; ratios of STB:CTB per HPF; as well as percentage of AP-2α positive STB nuclei or AP-2α positive CTB per total number of STB nuclei or CTB per HPF; were transformed using the natural log for analysis. Untransformed raw data were used for other analyses. The analyses were performed using SAS®, version 9.2 (SAS institute, Cary, NC). Analysis of variance or Fisher’s exact test was used to compare the clinical and placental factors between the groups. Analysis of the quantitative histology involved a mixed model approach, assuming unequal variance, to account for the multiple measures per observer. The multiple post hoc comparisons between groups were accounted for by using the Tukey adjustment. Statistical significance was assumed at p<0.05.
3. Results
3.1. Clinical and pathologic features
The mean gestational age of the pregnancies from the control and pathologic groups was 31.7 ± 5 weeks (range 26–39 weeks). There was no statistical difference on the method of delivery (vaginal vs. caesarean section). In addition, as shown in Table 1, no significant differences were noted in the placental weights between mild PE, severe PE, chronic HTN and CG, but the average weight of the DM placentas was significantly higher than that of the CG (p<0.001). Placentas of the FGR group showed significantly lower weights compared to CG (p<0.01). Histological evidence of chronic hypoxic placental injury of the uterine type was observed in all severe PE and 20% of the mild PE placentas (Table 1, Fig.1). In contrast, patterns of chronic hypoxic placental injury of any type were noted in only 10–20% of the remainder pathologic placentas, and were absent in the CG. The incidence of non-marginal infarction was higher in the mild and severe PE groups compared to CG (p< 0.05); but frequency of infarction did not increase with severity of PE. Mild and severe PE placentas also showed other changes related to uteroplacental vascular pathology or fetal thrombotic vasculopathy, including hypertrophic decidual arteriolopathy and intimal fibrin cushioning of stem or chorionic veins (p<0.001). Plasma cell deciduitis and increased placental site giant cells were also more commonly seen in the PE groups (p<0.05) (Fig.1).
Table 1.
Selected clinical and placental (H&E histology) variables
| Feature | Control No. | Mild PE No. | Severe PE No. | Chronic HTN No. | DM No. | FGR No. | F or χ2 (P)1 |
|---|---|---|---|---|---|---|---|
| Placental weight (g, average ± SD) | 313 ± 41A | 347 ± 34 | 304 ± 43 | 279 ± 56 | 427 ± 65B | 238 ± 35B | 1.9 (0.1) |
| Placenta abruption | 1 | 1 | 2 | 0 | 0 | 0 | 5.3 (0.3) |
| Meconium macrophages in placenta membranes | 1 | 1 | 3 | 3 | 2 | 2 | 2.5 (0.7) |
| Acute chorioamnionitis | 4 | 3 | 2 | 1 | 4 | 0 | 7.4 (0.9) |
| Laminar necrosis of placental membranes | 3 | 1 | 2 | 1 | 0 | 0 | 6.6 (0.2) |
| Non-marginal infarction (> 5% of placental disc) | 1AB | 6A | 3AB | 1AB | 0B | 4AB | 13 (< 0.05) |
| Perivillous fibrin deposition | 1 | 0 | 0 | 1 | 0 | 1 | 3.1 (0.7) |
| Fibrosis of placental tissue | 0 | 1 | 1 | 0 | 0 | 1 | 4.4 (0.5) |
| Intervillous thrombus | 0 | 1 | 1 | 2 | 0 | 2 | 4.4 (0.5) |
| Hypertrophic decidual arteriolopathy | 1B | 8A | 8A | 5AB | 1B | 1B | 18.1 (< 0.001) |
| Acute atherosis of maternal vessels | 1 | 1 | 3 | 2 | 0 | 0 | 6.6 (0.2) |
| Chorangiosis | 1 | 1 | 1 | 0 | 2 | 1 | 2.2 (0.8) |
| Increased placental site giant cells | 0 | 3 | 4 | 0 | 0 | 0 | 8.7 (< 0.05)* |
| Fetal thrombotic vasculopathy | 0 | 1 | 0 | 1 | 0 | 0 | 4.1 (0.5) |
| Intimal cushioning of stem or chorionic veins | 0 | 0 | 4 | 0 | 0 | 0 | 13.1 (<0.001)* |
| Plasma cell deciduitis | 1 | 4 | 4 | 0 | 0 | 0 | 8.6 (< 0.05)* |
| Evidence of global hypoxia | 0A | 4A | 10B | 2A | 2A | 1A | 23.5 (< 0.001) |
| Preuterine | 0 | 2 | 0 | 0 | 1 | 1 | 5.3 (0.4) |
| Uterine | 0A | 2A | 10B | 0A | 1A | 0A | 45 (<0.001) |
| Postuterine | 0 | 0 | 0 | 2 | 0 | 0 | 10 (0.06) |
Descriptive statistics: F (Analysis of variance, single factor) or Fisher’s exact test: those with same superscript letters are not different from each other. A is different from B (p<0.05);
no individual differences; overall distribution difference only.
PE- preeclampsia; HTN- hypertension; DM- diabetes mellitus; FGR- fetal growth restriction
Fig. 1.
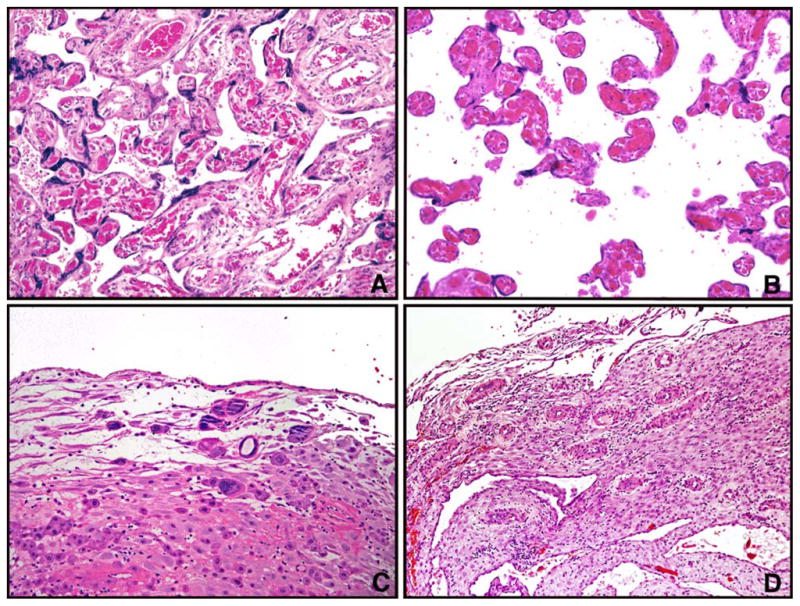
Selected patterns of chronic hypoxic placental injury diagnosed on hematoxylin-eosin conventional histology. A and B, (representative areas from same placenta), Uterine hypoxic pattern, featuring heterogeneous placental maturation with areas of larger intermediate villi with adjacent hypermature terminal villi, severe PE at 35 weeks. C, clusters of multinucleated trophoblastic giant cells in decidua basalis, severe PE at 28 weeks. D, Hypertrophic decidual arteriolopathy, mild preeclampsia at 31 weeks (hematoxylin-eosin, 20X original magnification).
Quantitative histology based on conventional histology and E-cadherin immunostaining in CTB and STB are summarized in Table 2. The number of villi per HPF in all study groups was not significantly different from that of gestational age-matched CG placentas. The syncytial knot count did not reach statistical difference from the CG, except in mild PE (p<0.005). The STB: CTB ratio was 1.6-fold higher in severe PE whereas the other groups of pathologic placentas showed a 1.5–1.7 fold decrease. The lower STB: CTB ratio seen in HTN, DM and FGR resulted from a significant decrease in number of STB, which was most pronounced in the FGR group (P<0.001). The lowest STB: CTB ratio observed in chronic HTN (p<0.005) was a consequence of both lower numbers of STB (p<0.05) and higher numbers of CTB (p<0.05).
Table 2.
Quantitative histology based on H&E and E-cadherin immunohistochemistry
| Group | Villi/HPF1 | Syncytial knots/HPF2 | STB nuclei/HPF3 | CTB/HPF4 | STB: CTB ratio5 |
|---|---|---|---|---|---|
| Control | 24.3 [21.9; 27.0] | 8.2 [6.6; 10.2] | 313 [295; 330] | 28.1 [23.4; 33.8] | 8.9 [7.1; 11.1] |
| Mild PE | 26.9 [23.3; 31.0] | 14.7*** [12.2; 17.6] | 262 [229; 294] | 43.1 [34.9; 53.3] | 5.1 [3.8; 6.8] |
| Severe PE | 28.0 [26.0; 30.1] | 11.9 [9.8; 14.5] | 312 [295; 329] | 17.4 [13.0; 23.3] | 14.2 [10.3; 19.5] |
| Chronic HTN | 22.1 [18.7; 26.2] | 11.8 [9.0; 15.3] | 259* [229; 289] | 43.0* [36.5; 50.5] | 5.1*** [4.4; 6.0] |
| DM | 22.3 [20.1; 24.8] | 10.5 [8.0; 13.6] | 271* [252; 289] | 41.8 [32.1; 54.3] | 5.6 [4.2; 7.5] |
| FGR | 19.8 [17.8; 21.9] | 11.4 [9.6; 13.3] | 233**** [204; 263] | 36.5 [25.9; 51.2] | 5.2 [3.8; 7.3] |
PE- preeclampsia; HTN- hypertension; DM- diabetes mellitus; FGR- fetal growth restriction
NOTE: Data presented as geometric mean [95% confidence interval] except for STB/HPF, which is presented as raw mean [95% confidence interval]. Statistically significant differences from the post-hoc tests of each studied group compared to control group are indicated as:
p<0.05,
p<0.01,
p < 0.005,
p < 0.001 vs. control group.
Average number chorionic villi per HPF.
Average number of syncytial knots per HPF.
Average number of CTB cells per HPF.
Average number of STB nuclei per HPF.
Average number of STB/CTB per HPF.
3.2. AP-2α immunohistochemistry
Fig. 2 and Table 3 demonstrate the results of AP-2α expression in CTB and STB as determined by immunohistochemistry. The percentage of AP-2α positive STB nuclei was significantly lower in mild PE, chronic HTN, DM and FGR placentas compared to CG (p<0.0001). In contrast, a 2-fold increase in AP-2α positive STB nuclei was observed in severe PE placentas, but the difference was not statistically significant (p=0.3). The percentage of AP-2α positive CTB in all groups was not significantly different from the CG. In the CG placentas, the ratio of AP-2α positive STB to AP-2α positive CTB was 1.2. This ratio was higher in severe PE (3.2, p=0.2) and significantly lower in the other groups, with the greatest reduction seen in mild PE (0.06; p< 0.005).
Fig. 2.

Immunohistochemical staining for AP-2α (brown) and E-cadherin (red) in placental villi from A, control group. B, mild PE. C, severe PE. D, chronic HTN. E, DM. F, FGR placentas at 31 weeks gestation (40X lens magnification).
SN, syncytial knot; empty arrowheads, CTB; solid arrowheads, AP-2α positive CTB; empty arrows, STB; solid arrows, AP-2α positive STB.
Table 3.
Quantitative histology based on AP-2α immunohistochemistry
| Group | AP2-2α (+) STB/HPF1 |
AP-2α (+) CTB/HPF1 |
Ratio AP-2α (+) STB: AP-2α(+) CTB2 |
AP-2α (+) STB nuclei/HPF3 |
AP-2α (+) CTB/HPF4 |
|---|---|---|---|---|---|
| Control | 20.8 [15.6; 27.8] | 17.4 [13.4; 22.6] | 1.19 [0.79; 1.77] | 7.2 [5.0; 10.4] | 28.0 [59.8; 73.6] |
| Mild PE | 1.5**** [0.4; 3.5] | 30.4 [22.9; 40.2] | 0.06*** [0; 0.30] | 0.34**** [0.1; 0.9] | 43.1 [67.4; 79.1] |
| Severe PE | 41.2 [23.9; 70.6] | 12.75 [9.0; 17.9] | 3.17 [1.51; 6.54] | 14.9 [8.3; 27.0] | 17.4 [66.0; 81.9] |
| Chronic HTN | 1.1**** [0.4; 2.3] | 30.6* [25.4; 36.9] | 0.13** [0.0.40] | 0.2**** [0.1; 0.5] | 43.0 [68.6; 78.2] |
| DM | 1.8**** [0.6; 4.0] | 26.0 [17.6; 38.4] | 0.10* [0; 0.49] | 0.4**** [0.1; 1.0] | 41.8 [58.5; 76.8] |
| FGR | 1.3**** [0.5; 2.4] | 21.6 [13.0; 35.4] | 0.13** [0; 0.42] | 0.3**** [0.1; 0.6] | 36.5 [55.7; 74.2] |
PE- preeclampsia; HTN- hypertension; DM- diabetes mellitus; FGR- fetal growth restriction
NOTE: Data presented as geometric mean [95% confidence interval]. Statistically significant differences from the post-hoc tests of each studied group compared to control group are indicated as:
p<0.05,
p<0.01,
p < 0.005,
p < 0.001 vs. control group.
Average number of AP-2α-positive STB nuclei or CTB cells per HPF, respectively.
Ratio of AP-2α-positive nuclei in STB per HPF/AP-2α-positive CTB per HPF.
Percentage of AP-2α-positive STB nuclei=number of positive AP-2α-positive nuclei per total number of STB nuclei per HPF.
Percentage of AP-2α-positive CTB= number of positive AP-2α-positive CTB per total number of CTB per HPF.
4. Discussion
Abnormalities in the differentiation of villous trophoblasts have been implicated in the pathogenesis of PE, FGR and other pregnancy complications [4,5,22]. Transcription factor AP-2α has been shown to play a critical role in the induction of trophoblastic differentiation [6–8]. This study demonstrated that AP-2α expression is abnormal in all selected pathologic conditions of pregnancy. In the majority of cases, abnormal AP-2α expression detected by immunohistochemistry preceded the manifestation of placental changes assessed with conventional histology, including evidence of villous hypoxia, which was only significant in severe PE group. Our study indicates that aberrant AP-2α expression may be involved in the disturbances of villous maturation frequently observed in these conditions.
Based on AP-2α expression in villous trophoblasts, the pathologic placentas could be separated into two distinct subgroups. The first group included placentas of pregnancies with mild PE, chronic HTN as well as DM and FGR. The second group was comprised of an outlier, severe PE. The first group shared increased number of CTB and decreased number of STB, leading to a decreased STB: CTB ratio, as well as a lower AP-2α expression in STB. All of these conditions may share significant early gestational insult leading to programming of delayed trophoblast differentiation [31]. Severe PE differs from the first group by an increase in AP-2α positive STB and decreased number of CTB and AP-2α positive CTB. These findings indicate that while all of the discussed pathologic conditions may be affected by hypoxic placental injury, the differences in AP-2α expression in severe PE cannot be attributed to hypoxia alone.
Immunoreactive AP-2α in STB of severe PE was higher by 2-fold but the increase was not statistically significant. Kotani et al (14) also reported higher AP-2α expression in severe PE, which was statistically significant. The lack of statistical difference in our study may be due in part to the fact that in our study, the placenta sections (and controls) were obtained from preterm placentas while Kotani et al studied placentas from term pregnancies. In addition, these investigators measured AP-2α protein in both villous and extravillous trophoblast cells, which were not accounted for in our study. The increase in AP-2α protein levels measured by Western blot analysis of whole PE placenta samples may be due, at least in part, to the elevated AP-2α expression in extravillous trophoblast cells. Also, pregnancies complicated by PE, in particular severe PE, are frequently associated with fetal growth restriction (FGR). Such cases were not included in this study to prevent possible overlap of the two conditions when interpreting the results.
Higher numbers of CTB with lower STB: CTB ratio was observed in mild PE, chronic HTN, DM and FGR but the differences were only significant in chronic HTN placentas. The finding that severe PE placentas have normal numbers of CTB differs from previous studies that showed higher rates of CTB proliferation in PE as well as other pathologic conditions associated with hypoxia [23–27]. The observed pattern of higher CTB density is thought to represent a response to low oxygen levels seen in hypoxic conditions of pregnancy [27], and is most likely due to abnormalities in CTB cell proliferation, fusion and/or STB cell loss [25]. The difference in findings between the present study and earlier investigations may result from differences in patient selection. The earlier studies did not report the severity of the PE in their study groups, which may have also included some mild cases.
The observation of striking differences in AP-2α expression and trophoblastic differentiation between mild and severe PE suggests that these two conditions are not only clinically diverse but may also have different pathophysiologic mechanisms. Previous studies have also noted significant differences in other parameters between the two conditions [28–30]. PE has been proposed to be a “two-stage” disorder resulting from interaction of both reduced placental perfusion and predisposing maternal factors [28]. There are numerous reasons to support this theory. Preterm PE is strongly associated with low birth weight; whereas unaffected infants, low or even large-for-gestational age infants can be seen in term PE (> 37 weeks), indicating variable degrees of placental dysfunction [29]. Recent speculations have proposed that late onset PE is less likely to be associated with low birth weight and placental infarcts than early onset PE [29,30]. However, it is also important to consider that the differences between mild and severe PE found in this study might be due to the fact that at the early gestational ages studied, differences in villous trophoblast profile such as accelerated ischemia, turnover rate and repair may not be significantly changed until later in pregnancy. In addition, later cases could show a different histology or expression pattern due to overlap with features of physiological maturation. We also recognize that matching placentas for GA does not exclude the possibility that the pattern observed for mild PE could progress to that for severe PE, if these pregnancies were allowed to continue until term.
The question whether the differences in AP-2α expression indicate an early intrinsic placental defect or an acquired abnormality secondary to hypoxia, systemic mediators, and/or accelerated maternal hypertension remains to be answered. A recent study on global gene expression profile of four first trimester placentas in patients who eventually developed PE approximately 6 months later in pregnancy revealed significant dysregulation of gene expression [32]. Serum biomarkers of PE have been shown to be altered at as early as 7 weeks gestation, indicating that an intrinsic abnormality of the placental in PE is present much earlier than the onset of decreased flow of maternal blood, when failure of transformation of spiral arteries are thought to occur in PE [28]. However, remarkable differences in levels of placental markers, such as insulin-like growth factor-1 [29] in placentas of mild vs. severe PE indicate that placental dysfunction may not be present in all cases.
In summary, this study showed abnormal AP-2α expression in several pathologic conditions of pregnancy associated with placental hypoxia. Since AP-2α has been shown to be a critical component of the transcriptional pathway that regulates villous CTB differentiation, the abnormal AP-2α in pathologic placentas may contribute to the pathogenesis of the accelerated or delayed maturation that have been previously ascribed to these conditions. The abnormal expression of AP-2α in the pathologic placentas could result from changes in epigenetic regulation and/or upstream factors that regulate AP-2α gene expression or protein degradation or processing. Unknown maternal and/or environmental factors may play a major role in the pathogenesis of severe PE. Further studies are required in order to delineate the precise mechanisms involved in the abnormal AP-2α expression in pathologic placentas, in particular severe PE complicated by FGR and chronic HTN or DM with superimposed PE.
Acknowledgments
This work was supported by NIH grant HD653369 (SH).
Footnotes
Presented at the 2010 Annual Society for Pediatric Pathology Meeting in Washington, DC
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Benirschke K, Kaufmann P. Early development of the human placenta. In: Benirschke K, Kaufmann P, editors. Pathology of the Human Placenta. New York, NY: Springer; 2006. pp. 42–49. [Google Scholar]
- 2.Castellucci M, Scheper M, Scheffen II, Celona A, Kaufmann P. The development of the human placental villous tree. Anat Embryol. 1990;181:117–28. doi: 10.1007/BF00198951. [DOI] [PubMed] [Google Scholar]
- 3.Cross JC. How to make a placenta: mechanisms of trophoblast cell differentiation in mice- a review. Placenta. 2005;26A:S3–9. doi: 10.1016/j.placenta.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 4.Crocker IP, Tansinda DM, Baker PN. Altered cell kinetics in cultured placental villous explants in pregnancies complicated by pre-eclampsia and intrauterine growth restriction. J Pathol. 2004;204:11–8. doi: 10.1002/path.1610. [DOI] [PubMed] [Google Scholar]
- 5.Lunghi L, Ferretti ME, Medici S, Biondi C, Vesce F. Control of human trophoblastic function. Reprod Biol Endocrinol. 2007;5:6. doi: 10.1186/1477-7827-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richardson BD, Cheng YH, Langland RA, Handwerger S. Differential expression of AP-2gamma and AP-2alpha during human trophoblast differentiation. Life Sci. 2001;69:2157–65. doi: 10.1016/s0024-3205(01)01299-1. [DOI] [PubMed] [Google Scholar]
- 7.Aronow BJ, Richardson BD, Handwerger S. Microarray analysis of trophoblast differentiation: gene expression reprogramming in key gene function categories. Physiol Genomics. 2001;6:105–16. doi: 10.1152/physiolgenomics.2001.6.2.105. [DOI] [PubMed] [Google Scholar]
- 8.Cheng YH, Aronow BJ, Hossain S, Trapnell B, Kong S, Handwerger S. Critical role for transcription factor AP-2alpha in human trophoblast differentiation. Physiol Genomics. 2004;18:99–107. doi: 10.1152/physiolgenomics.00181.2003. [DOI] [PubMed] [Google Scholar]
- 9.Richardson BD, Langland RA, Bachurski CJ, Richards RG, Kessler CA, Cheng YH, et al. Activator protein-2 regulates human placental lactogen gene expression. Mol Cell Endocrinol. 2000;160:183–92. doi: 10.1016/s0303-7207(99)00209-9. [DOI] [PubMed] [Google Scholar]
- 10.Johnson W, Albanese C, Handwerger S, Williams T, Pestell RG, Jameson JL. Regulation of the human chorionic gonadotropin alpha and beta subunit promoters by AP-2. J Biol Chem. 1997;72:15405–12. doi: 10.1074/jbc.272.24.15405. [DOI] [PubMed] [Google Scholar]
- 11.Yamada K, Harada N, Honda S, Takagi Y. Regulation of placenta-specific expression of the aromatase cytochrome P-450 gene. Involvement of the trophoblast-specific element binding protein. J Biol Chem. 1995;270:25064–9. doi: 10.1074/jbc.270.42.25064. [DOI] [PubMed] [Google Scholar]
- 12.Wada N, Chou JY. Characterization of upstream activation elements essential for the expression of germ cell alkaline phosphatase in human choriocarcinoma cells. J Biol Chem. 1993;268:14003–10. [PubMed] [Google Scholar]
- 13.Piao YS, Peltoketo H, Vihko P, Vihko R. The proximal promoter region of the gene encoding human 17beta- hydroxysteroid dehydrogenase type 1 contains GATA, AP-2, and Sp1 response elements: analysis of promoter function in choriocarcinoma cells. Endocrinology. 1997;138:3417–25. doi: 10.1210/endo.138.8.5329. [DOI] [PubMed] [Google Scholar]
- 14.Kotani T, Iwase A, Ino K, Sumigama S, Yamamoto E, Hayakawa H, et al. AP-2 impairs the invasion of a human extravillous trophoblast cell line. Endocrinology. 2009;150:4376–85. doi: 10.1210/en.2008-1645. [DOI] [PubMed] [Google Scholar]
- 15.Twickler DM, et al., editors. Williams obstetrics. 23. New York, NY: McGraw-Hill Medical; 2010. [Google Scholar]
- 16.White P. Classification of obstetric diabetes. Am J Obstet Gynecol. 1978;130:228–30. doi: 10.1016/0002-9378(78)90373-3. [DOI] [PubMed] [Google Scholar]
- 17.Alexander GR, Himes JH, Kaufman RB, Mor J, Kogan M. A United States national reference for fetal growth. Obstet & Gynecol. 1997;87:163–168. doi: 10.1016/0029-7844(95)00386-X. [DOI] [PubMed] [Google Scholar]
- 18.Redline RW. Placental pathology: A systematic approach with clinical correlations. Placenta. 2008;22:S86–S91. doi: 10.1016/j.placenta.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 19.Stanek J. Diagnosing placental membrane hypoxic lesions increases the sensitivity of placental examination. Arch Lab Med. 2010;134:989–95. doi: 10.5858/2009-0280-OA.1. [DOI] [PubMed] [Google Scholar]
- 20.Kingdom JC, Kaufmann P. Oxygen and placental villous development origins of fetal hypoxia. Placenta. 1997;18:613–21. doi: 10.1016/s0143-4004(97)90000-x. [DOI] [PubMed] [Google Scholar]
- 21.Kraus FT, Redline RW, Gersell DJ, Nelson DM. Placental Pathology. 1. Washington, DC: American Registry of Pathology and AFIP; 2004. [Google Scholar]
- 22.Huppertz B. Placental origins of preeclampsia. Challenging the current hypothesis. Hypertension. 2008;51:970–5. doi: 10.1161/HYPERTENSIONAHA.107.107607. [DOI] [PubMed] [Google Scholar]
- 23.Mayhew TM. A stereological perspective on placental morphology in normal and complicated pregnancies. J Anat. 2009;215:77–90. doi: 10.1111/j.1469-7580.2008.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayhew TM, Manwani R, Ohadike C, Wijesekara J, Baker PN. The placenta in pre-eclampsia and intrauterine growth restriction: studies on exchange surface areas, diffusion distances and villous membrane diffuse conductances. Placenta. 2007;28:233–8. doi: 10.1016/j.placenta.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 25.Smith SC, Price E, Hewitt MJ, Symonds EM, Baker PN. Cellular proliferation in the placenta in normal human pregnancy and pregnancy complicated by intrauterine growth restriction. J Soc Gynecol Investig. 1998;5:317–23. doi: 10.1016/s1071-5576(98)00035-5. [DOI] [PubMed] [Google Scholar]
- 26.Arnoldt H, Meisel F, Faundrey K, Lohrs U. Proliferation of villous trophoblast of the human placenta in normal and abnormal pregnancies. Virchow Arch B Cell Pathol Incl Mol Pathol. 1991;60:365–72. doi: 10.1007/BF02899568. [DOI] [PubMed] [Google Scholar]
- 27.Genbacev O, Zhou Y, Ludlow J, Fisher SJ. Regulation of human placental development by oxygen tension. Science. 1997;277:1669–72. doi: 10.1126/science.277.5332.1669. [DOI] [PubMed] [Google Scholar]
- 28.Roberts JM, Hubel CA. The two stage model of preeclampsia: variations on the theme. Placenta. 2009:S32–S37. doi: 10.1016/j.placenta.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vatten LJ, Skjaerven R. Is pre-eclampsia more than one disease? BJOG. 2004;111:298–302. doi: 10.1111/j.1471-0528.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- 30.Sebire NJ, Goldin RD, Regan L. Term preeclampsia is associated with minimal histopathological placental features regardless of clinical severity. J Obstet Gynecol. 2005;25:118. doi: 10.1080/014436105400041396. [DOI] [PubMed] [Google Scholar]
- 31.Myatt L. Placental adaptive responses and fetal programming. J Physiol. 2006;572:25–30. doi: 10.1113/jphysiol.2006.104968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Founds SA, Conley YP, Lyons-Weiler JF, Jeyabalan A, Hogge WA, Conrad KP. Altered global gene expression in first trimester placentas of women destined to develop preeclampsia. Placenta. 2009;30:15–24. doi: 10.1016/j.placenta.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]