

Abstract
Peptide-based hydrogels are attractive biological materials. Study of their self-assembly pathways from their monomer structures is important not only for undertaking the rational design of peptide-based materials, but also for understanding their biological functions and the mechanism of many human diseases relative to protein aggregation. In this work, we have monitored the conformation, morphological, and mechanical properties of a hydrogel-forming peptide during hydrogelation in different dimethylsulfoxide (DMSO)/H2O solutions. The peptide shows nanofiber morphologies in DMSO/H2O solution with a ratio lower than 4:1. Increased water percentage in the solution enhanced the hydrogelation rate and gel strength. One-dimensional and two-dimensional proton NMR and electron microscopy studies performed on the peptide in DMSO/H2O solution with different ratios indicate that the peptide monomer tends to adopt a more helical structure during the hydrogelation as the DMSO/H2O ratio is reduced. Interestingly, at the same DMSO/H2O ratio, adding Ca2+ not only promotes peptide hydrogelation and gel strength, but also leads to special shear-thinning and recovery properties of the hydrogel. Without changing the peptide conformation, Ca2+ binds to the charged Asp residues and induces the change of interfiber interactions that play an important role in hydrogel properties.
Introduction
Peptide-based hydrogel is an attractive material for biological applications in tissue engineering, drug delivery, and three-dimensional (3D) cell culture (1–3). Understanding of how do the peptide, conformations, assembly properties, and dynamic behaviors respond to external parameters has led to the rational novel design of various peptide biomaterials (4,5). Recently, most artificial peptide hydrogels have been achieved by synthesizing peptide sequences with alternating charged and noncharged amino acids or blocks of hydrophobic and hydrophilic copolymers (3,6,7). These designs allow the peptide monomers to fold into stable α-helical or β-sheet structures that separate the hydrophobic domain from the hydrophilic domain within a monomer. With hydrophobic association, peptide monomers assemble as nanofibers and further cross-link to a network through interfiber interactions. The nanofiber and hydrogel-like formations in native systems such as collagen, extracellular matrices, and the aggregation of amyloid peptides in Alzheimer’s disease have characteristics similar to those of artificial peptides, but the formations rely on more complex molecular structures (8–12). Studying the self-assembly pathways of these native molecules is a key challenge in understanding their biological functions and the mechanism of many human diseases.
In addition, many novel peptide designs have successfully incorporated biofunctionality, biocompatibility, and biodegradability into peptide hydrogel by modifying amino acid building blocks (13–17). These biological properties have stimulated the development of hydrogels as injectable systems for potential biomaterial applications. The most prevalent injectable hydrogels have been designed as free-flowing solutions. After syringe injection into the target position, the sol-gel transition of these peptide solutions could be triggered by physiological pH, temperature, or even photoinitiated hydrogelation, however, leakage of gel precursor solutions into neighboring tissue or the bloodstream causes unexpected problems with free-flowing injected systems (18,19). Since 2002, Pochan, Schneider, and co-workers have been developing a series of β-hairpin peptide hydrogels with special shear-thinning and rapid recovery properties (16,20,21) that provide an alternate strategy for injectable hydrogel therapies. The hydrogel scaffolds are fractured into small domains under proper shear stress, which leads to the liquid-like gel flowing properties. When the shear ceases, these hydrogel domains immediately percolate and cross-link into a network and recover solid-like behavior (22). Similar physical properties were also found in self-assembling multidomain peptides by Hartgerink’s group (23,24). These peptides contain a series of (Gln/Ser)-Leu repeats that are flanked by positively or negatively charged residues on each side. With the development of the protein engineering, some protein-based hydrogels were also designed with injectable properties for cell culture and the drug delivery system (25). Tirrell and co-workers developed a hydrogel material from recombinant telechelic proteins, which behaves as shear thinning by three orders of magnitude at 500% shear strain but resets to full elastic stiffness in seconds after injection (26). In addition, protein-protein interaction between the complementary peptide domains is also applied as a molecular-recognition gelation strategy for protein-based physical hydrogel development (27). By using two protein components containing WW and proline-rich peptide domain, Heilshorn and co-workers developed a serial of injectable materials named mixing-induced, two component hydrogels (MITCHs) (27).The self-healing rate of MITCH hydrogels are dependent on the binding strength of two components. Although the shear thinning system presents advantages and potential in many biomedical fields, rational design of these materials with controllable physical properties is still limited because of the lack of understanding at molecular level of the assembly pathways and interfiber interactions of hydrogel materials.
Interestingly, we found similar mechanical reversible properties in a native protein sequence-based peptide hydrogel from our laboratory in 2010 (28). The peptide sequence FLIVIGSIIGPGGDGPGGD (h9e) was rationally built from two functional native proteins: the FLIVIGSII segment of human muscle L-type calcium channel (29) and the (GPGGX)n segment of β-spiral motif of spider flagelliform silk protein (30,31). Asp was selected in X positions for Ca2+-sensitive nanofiber formation (5). The h9e peptide could be induced to form two distinct hydrogels by adding Ca2+ and adjusting pH, respectively, and only the h9e Ca2+ hydrogel exhibited the special shear-thinning and rapid recovery properties (28). This phenomenon excluded the effect of sequence difference on hydrogel properties and led us to focus on the effect of the peptide monomer self-assembly pathway and interfiber interactions on hydrogel shear-thinning and rapid recovery properties. The native sequence of this peptide was outside the model of the regular artificial amphiphilic peptide; therefore, predicting a proposed self-assembly pathway from its primary and secondary structure became difficult. Thus, we undertook the conformational study of the peptide monomer to understand the assembly of a peptide hydrogel and its interfiber interactions.
Initially, we tried to use proton NMR spectroscopy to monitor the conformational changes of peptide monomers during the gel-forming process in water-based solutions, but the short gelling time and the semisolid state of the hydrogel caused broadening of proton resonances along with a strong water peak and made the NMR assignment impossible. To solve this problem, dimethylsulfoxide (DMSO) was selected to dissolve h9e peptide in this study. By adding H2O, we slowly converted the h9e DMSO solution into hydrogel and maintained the DMSO/H2O ratio lower than 4:1 (v/v). The hydrogelation time and gel stiffness depended on the DMSO/H2O ratio. Proton NMR spectroscopy, transmission electron microscopy (TEM), and rheological studies of h9e peptide in different DMSO/H2O ratios revealed the conformational, morphological, and mechanical changes during hydrogel formation. In addition, we added Ca2+ to the h9e DMSO/H2O solution to enhance hydrogel formation. Similar to a water-based solution, Ca2+ promoted shear-thinning and rapid recovery properties in the DMSO/H2O solution by exhibiting faster recovery and a higher recovery strength percentage in mechanical studies. Further studies of the peptide hydrogel in different Ca2+ concentrations emphasized that, besides the peptide monomer assembly into the individual nanofibers, interfiber interactions were another important factor contributing to physical properties of peptide hydrogel.
Materials and Methods
Materials
N,N-dimethylformamide (DMF), trifluoroacetic acid (TFA), piperidine, N,N-diisopropylethylamine (DIEA), triisopropylsilane (TIS), calcium chloride (CaCl2), and DMSO were purchased from Sigma-Aldrich (Milwaukee, WI). N-methylpyrrolidinone (NMP), anhydrous ether, and dichloromethane (DCM) were purchased from Fisher Scientific (Pittsburgh, PA). Rink amide MBHA resin, 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), and all protected amino acids were purchased from EMD Biosciences (San Diego, CA). N-hydroxybenzotriazole (HOBT) was purchased from CEM (Matthews, NC). Deuterium oxide (D2O), sodium deuteroxide (NaOD), and deuterated dimethyl sulfoxide (DMSO-D6) were purchased from Cambridge Isotopes Laboratories (Andover, MA).
Peptide synthesis and hydrogel preparation
The h9e peptide was synthesized according to a previously published protocol (28). Briefly, based on the base-labile 9-fluorenylmethoxycarbonyl (Fmoc) strategy, peptides were synthesized on an automated CEM Liberty microwave peptide synthesizer (CEM,) with Rink amide resin and Fmoc-protected amino acids. The N-terminal Fmoc group was the final-deprotected. The resin-bound peptides were side-chain-deprotected and cleaved using TFA/TIS/water (95/2.5/2.5 v/v). After cleavage, peptides were precipitated and washed three times with anhydrous ether. The peptide samples were dried in air overnight and dissolved in acetonitrile and deionized water (50/50 v/v), and then freeze-dried. Molecular weight and purity of the synthesized peptides were confirmed by matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy and high-performance liquid chromatography.
Lyophilized peptide was completely dissolved in 100% DMSO with a peptide concentration of 6 mM. Water and DMSO were added to peptide DMSO solution for the final peptide concentration of 3 mM and DMSO/H2O ratio as 9:1, 4:1, and 7:3 (v/v), respectively. Peptide solutions were hydrogelated at room temperature. To prepare h9e hydrogel with Ca2+, CaCl2 was first dissolved in 100% DMSO, and then added to h9e solution for the finial Ca2+/peptide ratio of 1:1, 2:1, and 10:1, respectively. Afterward, the final DMSO/water ratio was adjusted to 9:1, 4:1, and 7:3 (v/v).
TEM
TEM samples were prepared by a negative staining method as follows: 1), prepare staining solution of 2 wt.% uranyl acetate in water and place a drop (∼100 μl) of the aqueous solution onto a parafilm surface; 2), glow discharge TEM Cu grids that are coated with continuous carbon support film; 3), place a small drop of the sample solution (∼5 μl) onto the top surface of the TEM grids for 1 min; 4), flip the TEM grids upside down and let it float on the top surface of the staining solution for 20 s; 5), pick up the TEM grids and dry in air. The TEM work was carried out using a FEI Tecnai G2 F20 with field emission gun, at a working voltage of 200 kV. All images were acquired as zero-loss images by excluding the inelastic scattering electrons using Gatan Image Filter. The elemental mapping was performed using a jump-ratio method.
NMR
The one-dimensional (1D) and two-dimensional (2D) 1H-1H NMR experiments were performed on a Varian 500 NMR System (Varian, now Agilent Technologies, Palo Alto, CA) equipped with a 5 mm cryogenic triple-resonance inverse detection pulse field gradient probe operating at 499.84 MHz for 1H frequency. 2D- 1H-1H total correlation spectroscopy (TOCSY) and nuclear Overhauser effect spectroscopy (NOESY) data were acquired in phase-sensitive (States-TPPI) mode into 2000 t2 and 256 t1 points with a spectral width of 13 ppm in each dimension, and four transients per increment. Spin-lock time of 80 ms at a B1 field strength of 7.0 KHz was used for TOCSY experiments, and mixing times of 200 and 400 ms were used for NOESY experiments. All experimental data were zero-filled to 4 K data points in the t2 dimension, and, when necessary, the spectral resolution was enhanced by Lorenzian-Gaussian apodization. DMSO-D6 peak (2.5 ppm for 25°C) was considered a reference for chemical shift assignments. Data processing was done using VnmrJ2.2c (Varian, now Agilent Technologies) and analyzed using Sparky - NMR Assignment and Integration software (32).
Structure calculations
Structure calculations were performed using standard methods based on the unambiguously identified and classified NOE crosspeaks. NOE crosspeaks were assigned to interproton distance constraint ranges based on their peak intensities; values given to each category of crosspeaks were strong (1.8–2.7 Å), medium (1.8–3.5 Å), weak (1.8–4.0 Å) (33). Upper distance limits for NOEs involving methyl protons and nonstereo specifically assigned methylene protons were corrected appropriately for center averaging.
The assigned distance constraints were used as input in the calculation of peptide structures starting from an extended structure. The initial extended structure was created from the amino acid sequence using the program Crystallography & NMR System (CNS) version 1.1. CNS uses both a simulated annealing protocol and molecular dynamics to produce low-energy structures with the minimum distance constraint violations. From 100 generated structures, a subset of structures with the lowest overall energies and lesser dihedral angle violation were selected as representative structures.
Dynamic light scattering analysis
A ZetaPALS (Brookhaven Instruments, Holtsville, NY) was used to measure the hydrodynamic diameters of h9e amorphous aggregation in 100% and 90% DMSO solution with 0 and 30 mM Ca2+. 3 mM peptide solution was prepared at room temperature over night and then diluted in HPLC-grade water at 1:300 ratio. The samples were measured at 25°C and 658 nm wavelength.
Rheological tests
A C-VOR 150 rheometer system (Malvern Instruments, Malvern, UK) was used to determine the storage and loss moduli (G′ and G″, respectively) of h9e hydrogels with a 20-mm diameter parallel plate geometry and 500 μm gap size at 25°C. The peptide DMSO/H2O solution was placed on the measuring system immediately after mixing for a gel-forming rate test. Single frequency (1 Hz) and steady shear strain (1%) were selected for a 2-h test. The frequency sweep test was conducted under 1% shear strain with a frequency range from 0.01 to 10 Hz. To determine the linear viscoelastic regime of h9e hydrogel, 3 mM peptide with 0, 3, and 30 mM Ca2+ was prepared in 70% DMSO solution and allowed for gelation for 12 h, and evaluated with an amplitude sweep test with shear strain ranging from 0.01% to 500%. To determine the shear-thinning and recovery capability of the hydrogel, the peptide in 70% DMSO solution was incubated at room temperature overnight for hydrogelation, and then transferred to a lower measuring plate for a 1-h, single-frequency test (1 Hz, 1% strain). The hydrogel was disturbed using 1 Hz frequency and 500% shear strain for 1 min. Afterward, resetting the instrument parameters took 1 min, and the hydrogel moduli during the reassembly period were measured under 1 Hz frequency and 1% shear strain for 1 h.
Results and Discussion
The peptide h9e dissolved very well in 100% DMSO. Adding H2O to the h9e DMSO solution allowed the peptides to pack into their corresponding nanofiber structure in an orderly fashion and induced the self-supported hydrogel formation when the DMSO/H2O ratio was lowered to 4:1. This hydrogelation process was enhanced by increasing the water percentage. For example, when DMSO/H2O ratio was 4:1, hydrogel formation took ∼48 h. This process was completed within 2 h at a DMSO/H2O ratio of 7:3 and lowered to 50 min when the DMSO/H2O ratio reached 1:1. The morphological properties of peptide solutions with different DMSO/H2O ratios observed by TEM confirmed this hydrogelation process. Fig. 1 A shows a diffuse amorphous morphology of h9e peptide in 100% DMSO. The peptides dispersed homogeneously in the visual field without obviously ordered aggregation. This amorphous morphology was maintained with more anisotropic aggregations of individual peptides when DMSO/H2O ratio was 9:1 (Fig. 1 B). Dynamic light scattering (DLS) measurement further confirmed this visible increasing of molecular size by showing the hydrodynamic diameter of peptide amorphous structure 79.65 nm in 100% DMSO and 126.60 nm in 90% DMSO (Fig. S1 in the Supporting Material). The 90% DMSO h9e solution was stable in a free-flowing liquid state for more than 1 month after it was prepared. The nanofiber morphology with ∼5 nm diameter appeared at 80% DMSO h9e solution (Fig. 1 C) and became clearer and denser when the DMSO/H2O ratio was decreased to 7:3 (Fig. 1 D).
Figure 1.

TEM images of h9e peptide in 70–100% DMSO solutions without Ca2+. (A) h9e peptide in 100% DMSO solution. (B) h9e peptide in 90% DMSO solution. (C) h9e peptide in 80% DMSO solution. (D) h9e peptide in 70% DMSO solution.
To further understand the molecular conformation changes during peptide hydrogelation, 1D and 2D proton NMR experiments were performed at the Biomolecular NMR facility, using a 500 MHz NMR instrument equipped with a 5 mm triple resonance inverse detection cryogenic probe, which allowed the assignment of the proton resonances of peptide in DMSO/H2O solution at increasing water concentration. Based on the proton resonance assignments in both 2D 1H-1H TOCSY and NOESY spectra of h9e in 100% DMSO (Fig. S2), the calculated monomeric structure of the h9e peptide (Fig. 2 A) showed a helical conformation with the C-terminal residues bent back into the helix, thus allowing the potentially charged side chains of the Asp residues to come into proximity. Analysis of the predicted surface of the monomeric peptide (Fig. 2 A) structures presented segregation of the hydrophobic (gray color) and hydrophilic side chains (red color). In addition, the Chemical Shift Index (CSI) for the alpha protons of each amino acid residue in different DMSO/H2O ratio solutions was calculated (Fig. 2 B) as an indicator of the peptide backbone conformation. In 100% DMSO, a more β-sheet-like structure appeared for the hydrophobic residues (Leu-2, Ill-3, Val-4, and Ill-5) of the N-terminal compared with the helix or turn-like structure present for the nonhydrophobic residues of the C-terminal. With increasing water concentration, the peptide solution became more and more gel-like, and the backbone conformation of the whole monomeric peptide was inclined to adopt a more helix-like structure. Especially in the N-terminal region, the significant decrease of CSI values for residue Leu-2 to Ile-9 indicated the change of this β-strand-like domain segment into a turn- or twist-like structure. Interestingly, unlike most native or artificial hydrogel peptides in which a stable β-sheet-like molecular structure is preferred for hydrogelation (34–36), this helix-like structure-induced hydrogelation is rarely seen in a native protein sequence-based peptide. Although the segregation of the hydrophobic and nonhydrophobic side chains of the monomeric h9e structure was observed in terminal-to-terminal direction in 100% DMSO solution, the top view (Fig. 2, A and C), of the calculated peptide structure showed that both the side chains of the hydrophobic residues (gray color, Fig. 2 C) and the potentially charged Asp residues (red color, Fig. 2 C) were crowded to one side perpendicular to the terminal-to-terminal direction. We suggest that on adding water to the h9e DMSO solution, along with peptide hydrogelation, the backbone conformational changes caused turning of the hydrophobic domain and bent some of their side-chains perpendicular to the terminal-to-terminal direction from one side to the other and resulted in extension of the terminal-to-terminal distance (Fig. 2 C), which further separates the hydrophobic and potentially charged segment in both directions. The stacked hydrophobic interactions benefited from this further segregation and may have contributed the assembly of the peptide monomers to the nanofiber (see Fig. 2 E). The calculated monomeric structure of the h9e peptide in 100% DMSO was ∼1.67 nm in width and 1.92 nm in length (Fig. 2 D), which could extend to more than 2 nm in length during the hydrogelation. Therefore, according to the hydrophobic stacked peptide assembly pathway, a 4–5 nm nanofiber diameter could be expected. The 5 nm nanofiber diameter observed under TEM (Fig. 1 C) matched this expectation quite well. The rheological tests of h9e DMSO/H2O solutions further demonstrated the morphological and conformational properties of peptide hydrogelation (Fig. 2 F). During the 2-h test, 90% DMSO h9e solution remained in liquid form and showed very weak elastic modulus (G′, ∼0 Pa). With increasing water concentration, the G′ of peptide solution with 70% DMSO increased slowly and reached ∼80 Pa at the end of the test. For h9e peptides in 50% DMSO solution, the G′ reached 100 Pa within 50 min and kept increasing within the test time, indicating the much faster formation of the self-support hydrogel with stronger gel stiffness. Moreover, we prepared peptide/DMSO/water mixtures and held at room temperature for 48 h to stabilize the hydrogel. The frequency sweep test of finial hydrogel materials exhibited that h9e in 90% DMSO was still in low viscous liquid form, G′ of h9e hydrogel in 70% DMSO was stable around 100 Pa, and G′ of h9e hydrogel in 50% DMSO reached over 1000 Pa (Fig. S3).
Figure 2.
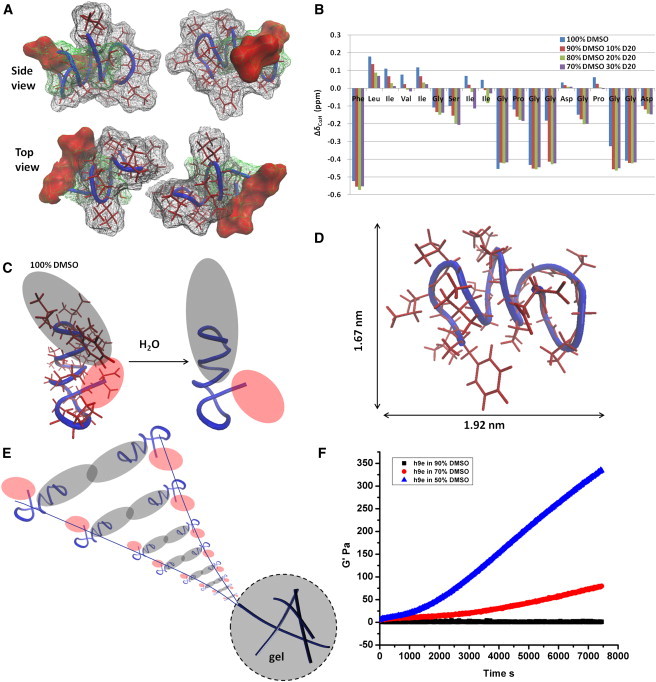
Monomeric study of peptide hydrogelation. (A) Calculated monomeric structure of h9e in 100% DMSO (hydrophobic, nonhydrophobic, and Asp side-chain surfaces colored as gray, green, and red, respectively). (B) Chemical Shift Index for peptide in 70–100% DMSO solution without Ca2+. (C) Increasing water concentration of DMSO/H2O solvent mixture induces the helix turning of peptide backbone, which leads to a further segregation of hydrophobic (gray) and hydrophilic (red) domains. (D) Molecular size of h9e peptide in 100% DMSO. (E) Proper stacked hydrophobic interactions contribute to peptide assembly for nanofiber formation. (F) Mechanical property of peptides in 90%, 70%, and 50% DMSO during the first 2 h of hydrogelation.
Further studies were focused on the shear-thinning and rapid recovery properties of h9e hydrogel with Ca2+ ions. Similar to the water-based h9e solution, peptides in DMSO/H2O solution responded to Ca2+ and exhibited a significant decrease in hydrogelation time with Ca2+. For example, with 50 mM Ca2+, the hydrogelation time of 5 mM h9e peptide in 80% DMSO solution was 30 min compared with 2 days for that without Ca2+. The TEM observation of h9e peptide with Ca2+ (the peptide/Ca2+ ratio is 1:10) in different DMSO/H2O ratio solutions demonstrated that these metal ions triggered nanofiber-forming promotion (Fig. 3). In 100% DMSO solution, there were more anisotropic aggregations of individual peptides with Ca2+ (Fig. 3 A) compared with those without Ca2+ (Fig. 1 A). DLS data supported this TEM observation by showing the hydrodynamic diameter increased from 79.65 to 227.54 nm of peptide amorphous structure in 100% DMSO with and without Ca2+ (Fig. S1). These aggregations became more fiber-like morphologies when DMSO was reduced to 90% (Fig. 3 B). Again, the hydrodynamic diameter of the peptide anisotropic aggregations increased dramatically from 126.60 to 568.17 nm when Ca2+ was added into this peptide DMSO solution (Fig. S1). With the same Ca2+ concentration, the nanofiber morphology turned clearer with the increased water concentration in the DMSO/H2O solvent (Fig. 3, C and D). Fig. 3 E is the calcium elemental map that only shows the Ca2+ distribution. It is evident that the Ca2+ ions did exist and were evenly located in the same area of peptide nanofiber matrix in 70% DMSO solution as Fig. 3 D, which gives a visual evidence of the medal ions involved in hydrogelation.
Figure 3.

Morphological and rheological properties of h9e peptide in DMSO/H2O solution with Ca2+ (A–D) TEM images of 3 mM h9e peptide in 70–100% DMSO solution with 30 mM Ca2+. (E) Calcium elemental map of 3 mM h9e peptide in 70% DMSO with 30 mM Ca2+ (same area as D).
In addition, the Ca2+ induced a faster hydrogelation process and stronger gel stiffness than that without Ca2+ of the identical peptide concentration in the same DMSO/H2O solution. To further explore this hydrogel forming process corresponding to the specific shear-thinning and rapid recovery property of h9e hydrogel, we prepared 3 mM h9e peptide in 70% DMSO solution with Ca2+ concentration 0, 3, and 30 mM, respectively. The hydrogel forming process was monitored under a time sweep test of a rheometer right after the peptide and Ca2+ solutions were mixed. The rheological data (Fig. 4 A) show that, with 3 mM Ca2+ concentration, the peptide had an almost identical hydrogelation rate as that without Ca2+ for the first 2 h (7200 s) of testing time, however, the frequency sweep data of the stabilized gel materials by 12 h presents that the stiffness of 3 mM h9e with 3 mM Ca2+ was around 450 Pa, which was much higher than 100 Pa of that without Ca2+ (Fig. S4). As Ca2+ increased to 30 mM, the h9e peptide showed a dramatically fast hydrogelation rate (Fig. 4 A) and had over 1000 Pa in gel strength, which was confirmed by the frequency sweep test obtained at 12 h gelling time (Fig. S4). In addition, we performed an amplitude sweep test with a shear strain range from 0.01% to 500% to identify the linear viscoelastic regime of the 3 mM h9e peptide hydrogel with 0, 3, and 30 mM Ca2+ (Fig. S5). The rheological data exhibit that the hydrogel was intact when shear strain was smaller than 4% but disturbed as shear strain increased, and was completely destroyed when shear strain was higher than 100% (Fig. S5). According to this result, we further studied the mechanical property of h9e hydrogel with different Ca2+ concentrations with 1% shear strain for 1 h and 500% shear stain for 1 min, and then monitoring the gel recovery process with 1% shear strain for 1 h. There was 1 min waiting time applied between each test phase to reset the shear strains. Fig. 4 B shows that, without Ca2+, only ∼30% of the gel was recovered during a 1 min waiting time (Fig. 4 B). The recovery rate of this hydrogel was very slow, and restored only ∼40% after 1 h. With Ca2+, the gel strength recovered much faster. For example, ∼60% and 75% gel strength was recovered after the 1 min waiting time for the hydrogels with 3 mM and 30 mM Ca2+, respectively. After 1 h, ∼80% gel strength of both hydrogels was recovered. Like hydrogel in an aqueous environment (28), the construction of h9e hydrogel with Ca2+ presents a much faster reassemble capability than that without Ca2+ after mechanical disturbance in DMSO/H2O solution. Interestingly, the effect of Ca2+ on h9e peptide hydrogelation was observed only when the water percentage was high enough to promote peptide assembly into hydrogel. In 100% or 90% DMSO, h9e solution was still in liquid form, even adding enough concentration of Ca2+ with a long incubation period. On the other hand, there was no significant change in the α proton CSI of h9e peptide in 70% DMSO solution with 0, 3, or 6 mM Ca2+ (Fig. 4 C), which suggests that the monomeric structures of peptides are almost identical in these solutions. This phenomenon indicates that the conformational promotion of peptide monomeric structure is the primary requirement for peptide self-assembly into hydrogel. When the required structure is available, h9e peptides would follow a similar pathway for nanofiber assembly, and the surrounding Ca2+ ions may promote the hydrophobic stacked interaction by capturing the charged Asp residues on the opposite site, and further influence the interfiber interactions of peptide hydrogel.
Figure 4.
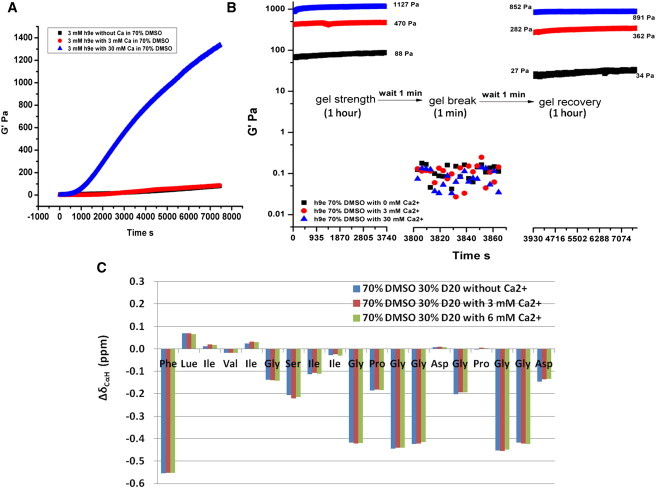
Mechanical and monomeric structural properties of h9e peptide in 70% DMSO solution with/without Ca2+. (A) G′ of h9e hydrogel in 70% DMSO solution with 0, 3, and 30 mM Ca2+. (B) Shear-thinning and recovery properties of h9e hydrogel in 70% DMSO solution with 0, 3, and 30 mM Ca2+. (C) Chemical Shift Index of h9e peptide in 70% DMSO solution with 0, 3, and 6 mM Ca2+.
The effect of Ca2+ charge screening on mechanical properties of other types of peptide hydrogels has been reported (37–39). The research suggests that, besides the strength of individual nanofibers, interfiber interactions, which are relative to the charge screen of Ca2+, were considered important contributors to gel stiffness. Therefore, in addition to the differences in the rheological data presented previously, the morphological properties of interfiber interactions of hydrogel with/without Ca2+ were also observed. Fig. 5 A shows the unbent nanofiber with various lengths of h9e hydrogel in 70% DMSO without Ca2+. Most junction points of nanofibers of this 3D network are static points, where the noncovalent attractive interactions such as hydrogen bonding, hydrophobic, or van der Walls interactions dominate the bunch between nanofibers (see Fig. 5 D). Such junctions cannot reform after they are broken, which may relate to the mechanical irreversibility of the corresponding hydrogel. In contrast, more flexible nanofibers with more uniform long lengths were observed in h9e 70% DMSO hydrogel with 3 mM Ca2+ (Fig. 5 B). Although some static junctions remained, most nanofibers in this hydrogel network were held together by dynamic entanglements (see Fig. 5 E). Compared with the bunch of nanofibers in static points, the entanglement crossings of nanofibers with enough fiber length were healed much more easily after they were broken. Increasing the Ca2+ concentration to 30 mM rendered the dynamic entanglements of nanofibers even more obvious with rare static junctions (Fig. 5, C and F). High-magnification TEM images (Fig. 5, G and H), show that the nanofiber of hydrogel with 3 mM Ca2+ is composed of two individual fibers, which indicates a stack of two fibers (Fig. 5 I). Higher Ca2+ concentration (30 mM) occupied most charged Asp residues on the nanofiber surface, which affected the interfiber interaction and led to individual fibers with a 5 nm diameter (Fig. 5 J). The higher occupation of Ca2+ on the surface may prevent the attracted interactions between nanofibers at static junctions.
Figure 5.
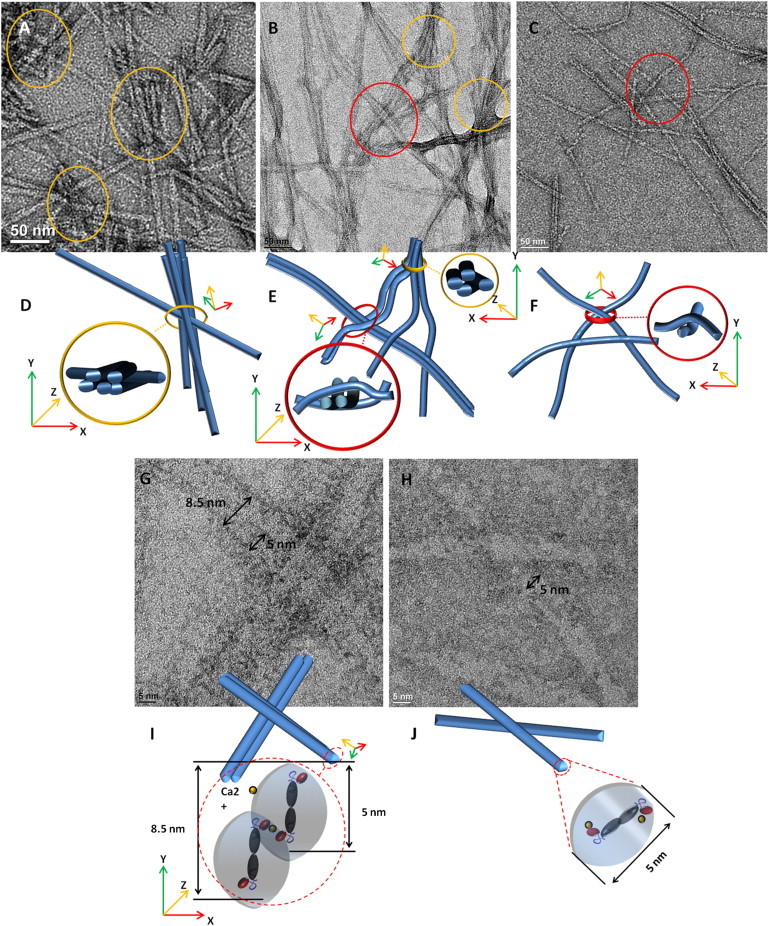
Morphological properties of interfiber interactions of h9e hydrogel in 70% DMSO solution with/without Ca2+. (A and D) Unbent nanofibers with various fiber lengths of hydrogel without Ca2+ showing static junctions (circle, yellow online). (B and E) Both dynamic entanglements (dashed circle, red online) and static junctions (circle, yellow online) were observed in peptide hydrogel with 3 mM Ca2+ with flexible and more uniform long-length nanofibers. (C and F) The peptide network was held by dynamic entanglements (dashed circle, red online) of hydrogel with 30 mM Ca2+. (G and I) Nanofiber of h9e hydrogel with 3 mM Ca2+ is composited by stacking of two individual fibers. (H and J) Ca2+ occupying most charged Asp residue on fiber surface leads to nanofiber with 5 nm diameter of hydrogel with 30 mM Ca2+.
Conclusions
The h9e peptide presents an amphiphilic structural property with C-terminal proximity of potentially charged Asp residues in DMSO solution. With increasing water concentration in a DMSO/H2O solvent mixture, the peptide monomer tends to adopt a more helical structure, which enhances the stacked hydrophobic interactions for nanofiber formation. Peptides self-assemble into clear nanofiber morphology when the DMSO/H2O ratio is lower than 4:1. The hydrogelation rate and gel strength are improved when water percentage is increased. With the same DMSO/H2O ratio, Ca2+ could enhance the hydrogelation rate and stable gel strength and cause the peptide hydrogel to exhibit special shear-thinning and rapid recovery properties. Instead of changing the peptide monomeric structure, Ca2+ occupies the charged Asp residues on the fiber surface and further influences the interfiber interactions of peptide hydrogel. Compared with static junctions of nanofibers of hydrogel in 70% DMSO without Ca2+, entanglement crossings of nanofibers hold the networks of peptide hydrogel with Ca2+, which may play an important role in hydrogel shear-thinning and rapid recovery property. This study helps us understand the assembly pathways of peptide monomers into their corresponding nanofiber structures and may lead to the development of new biological materials with controlled stiffness and shearing-respondent behaviors.
Acknowledgments
We thank the University of Kansas Mass Spectrometry Lab for HPLC and mass spectrometric analysis, and Microscopy and Imaging Center of Texas A&M University for TEM images.
NMR instrumentation at KSU was funded by National Institutes of Health grant (S10-RR 025441). This project was partially supported by Kansas State University Targeted Excellence Program and Kansas State University Research Foundation Scholarship Program. Contribution No. 12-288-J from the Kansas Agricultural Experiment Station.
Contributor Information
Om Prakash, Email: omp@k-state.edu.
Xiuzhi S. Sun, Email: xss@k-state.edu.
Supporting Material
References
- 1.Hauser C.A.E., Zhang S. Designer self-assembling peptide nanofiber biological materials. Chem. Soc. Rev. 2010;39:2780–2790. doi: 10.1039/b921448h. [DOI] [PubMed] [Google Scholar]
- 2.Langer R., Tirrell D.A. Designing materials for biology and medicine. Nature. 2004;428:487–492. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 3.Zhao X., Pan F., Lu J.R. Molecular self-assembly and applications of designer peptide amphiphiles. Chem. Soc. Rev. 2010;39:3480–3498. doi: 10.1039/b915923c. [DOI] [PubMed] [Google Scholar]
- 4.Aluri S., Janib S.M., Mackay J.A. Environmentally responsive peptides as anticancer drug carriers. Adv. Drug Deliv. Rev. 2009;61:940–952. doi: 10.1016/j.addr.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang H., Sun X.S. Rational design of responsive self-assembling peptides from native protein sequences. Biomacromolecules. 2010;11:3390–3394. doi: 10.1021/bm100894j. [DOI] [PubMed] [Google Scholar]
- 6.Luo Z., Wang S., Zhang S. Fabrication of self-assembling D-form peptide nanofiber scaffold d-EAK16 for rapid hemostasis. Biomaterials. 2011;32:2013–2020. doi: 10.1016/j.biomaterials.2010.11.049. [DOI] [PubMed] [Google Scholar]
- 7.Hule R.A., Nagarkar R.P., Pochan D.J. Dependence of self-assembled peptide hydrogel network structure on local fibril nanostructure. Macromolecules. 2009;42:7137–7145. doi: 10.1021/ma9003242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dvir T., Timko B.P., Langer R. Nanotechnological strategies for engineering complex tissues. Nat. Nanotechnol. 2011;6:13–22. doi: 10.1038/nnano.2010.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okuyama K., Hongo C., Bächinger H.P. High-resolution structures of collagen-like peptides [(Pro-Pro-Gly)4-Xaa-Yaa-Gly-(Pro-Pro-Gly)4]: implications for triple-helix hydration and Hyp(X) puckering. Biopolymers. 2009;91:361–372. doi: 10.1002/bip.21138. [DOI] [PubMed] [Google Scholar]
- 10.Eyrich D., Brandl F., Blunk T. Long-term stable fibrin gels for cartilage engineering. Biomaterials. 2007;28:55–65. doi: 10.1016/j.biomaterials.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 11.Härd T. Protein engineering to stabilize soluble amyloid β-protein aggregates for structural and functional studies. FEBS J. 2011;278:3884–3892. doi: 10.1111/j.1742-4658.2011.08295.x. [DOI] [PubMed] [Google Scholar]
- 12.Friedman R. Aggregation of amyloids in a cellular context: modelling and experiment. Biochem. J. 2011;438:415–426. doi: 10.1042/BJ20110369. [DOI] [PubMed] [Google Scholar]
- 13.Ruoslahti E. Targeting tumor vasculature with homing peptides from phage display. Semin. Cancer Biol. 2000;10:435–442. doi: 10.1006/scbi.2000.0334. [DOI] [PubMed] [Google Scholar]
- 14.Yang M., Yamauchi K., Asakura T. Design of silk-like biomaterials inspired by mussel-adhesive protein. Tissue Eng. 2007;13:2941–2947. doi: 10.1089/ten.2006.0448. [DOI] [PubMed] [Google Scholar]
- 15.Bunick C.G., Nelson M.R., Chazin W.J. Designing sequence to control protein function in an EF-hand protein. J. Am. Chem. Soc. 2004;126:5990–5998. doi: 10.1021/ja0397456. [DOI] [PubMed] [Google Scholar]
- 16.Branco M.C., Pochan D.J., Schneider J.P. The effect of protein structure on their controlled release from an injectable peptide hydrogel. Biomaterials. 2010;31:9527–9534. doi: 10.1016/j.biomaterials.2010.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garty S., Kimelman-Bleich N., Gazit D. Peptide-modified “smart” hydrogels and genetically engineered stem cells for skeletal tissue engineering. Biomacromolecules. 2010;11:1516–1526. doi: 10.1021/bm100157s. [DOI] [PubMed] [Google Scholar]
- 18.Haines-Butterick L., Rajagopal K., Schneider J.P. Controlling hydrogelation kinetics by peptide design for three-dimensional encapsulation and injectable delivery of cells. Proc. Natl. Acad. Sci. USA. 2007;104:7791–7796. doi: 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang C.H., Kuo T.F., Liu H.C. Tissue engineering-based cartilage repair with allogenous chondrocytes and gelatin-chondroitin-hyaluronan tri-copolymer scaffold: A porcine model assessed at 18, 24, and 36 weeks. Biomaterials. 2006;27:1876–1888. doi: 10.1016/j.biomaterials.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 20.Schneider J.P., Pochan D.J., Kretsinger J. Responsive hydrogels from the intramolecular folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 2002;124:15030–15037. doi: 10.1021/ja027993g. [DOI] [PubMed] [Google Scholar]
- 21.Nowak A.P., Breedveld V., Deming T.J. Rapidly recovering hydrogel scaffolds from self-assembling diblock copolypeptide amphiphiles. Nature. 2002;417:424–428. doi: 10.1038/417424a. [DOI] [PubMed] [Google Scholar]
- 22.Yan C., Altunbas A., Pochan D.J. Injectable solid hydrogel: mechanism of shear-thinning and immediate recovery of injectable β-hairpin peptide hydrogels. Soft Matter. 2010;6:5143–5156. doi: 10.1039/C0SM00642D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong H., Paramonov S.E., Hartgerink J.D. Self-assembly of multidomain peptides: balancing molecular frustration controls conformation and nanostructure. J. Am. Chem. Soc. 2007;129:12468–12472. doi: 10.1021/ja072536r. [DOI] [PubMed] [Google Scholar]
- 24.Bakota E.L., Wang Y., Hartgerink J.D. Injectable multidomain peptide nanofiber hydrogel as a delivery agent for stem cell secretome. Biomacromolecules. 2011;12:1651–1657. doi: 10.1021/bm200035r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guvendiren M., Lu H.D., Burdick J.A. Shear-thinning hydrogels for biomedical applications. Soft. Matter. 2012;8:260–272. [Google Scholar]
- 26.Olsen B.D., Kornfield J.A., Tirrell D.A. Yielding behavior in injectable hydrogels from telechelic proteins. Macromolecules. 2010;43:9094–9099. doi: 10.1021/ma101434a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong Po Foo C.T., Lee J.S., Heilshorn S.C. Two-component protein-engineered physical hydrogels for cell encapsulation. Proc. Natl. Acad. Sci. USA. 2009;106:22067–22072. doi: 10.1073/pnas.0904851106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang H., Shi J., Sun X.S. Design of a shear-thinning recoverable peptide hydrogel from native sequences and application for influenza H1N1 vaccine adjuvant. Soft Matter. 2011;7:8905–8912. [Google Scholar]
- 29.Shen X., Mo X., Sun X.S. Adhesion and structure properties of protein nanomaterials containing hydrophobic and charged amino acids. J. Nanosci. Nanotechnol. 2006;6:837–844. doi: 10.1166/jnn.2006.126. [DOI] [PubMed] [Google Scholar]
- 30.Hayashi C.Y., Lewis R.V. Spider flagelliform silk: lessons in protein design, gene structure, and molecular evolution. Bioessays. 2001;23:750–756. doi: 10.1002/bies.1105. [DOI] [PubMed] [Google Scholar]
- 31.Lammel A., Schwab M., Scheibel T. Processing conditions for the formation of spider silk microspheres. ChemSusChem. 2008;1:413–416. doi: 10.1002/cssc.200800030. [DOI] [PubMed] [Google Scholar]
- 32.Goddard T.D., Kneller D.G. University of California; San Francisco: 2004. SPARKY 3. [Google Scholar]
- 33.Wüthrich K., Billeter M., Braun W. Pseudo-structures for the 20 common amino acids for use in studies of protein conformations by measurements of intramolecular proton-proton distance constraints with nuclear magnetic resonance. J. Mol. Biol. 1983;169:949–961. doi: 10.1016/s0022-2836(83)80144-2. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Y., Yokoi H., Tan T. Self-assembled pH-responsive hydrogels composed of the RATEA16 peptide. Biomacromolecules. 2008;9:1511–1518. doi: 10.1021/bm701143g. [DOI] [PubMed] [Google Scholar]
- 35.Rapaport H., Grisaru H., Silberstein T. Hydrogel scaffolds of amphiphilic and acidic beta-sheet peptides. Adv. Funct. Mater. 2008;18:2889–2896. [Google Scholar]
- 36.Wu L.C., Yang J., Kopeček J. Hybrid hydrogels self-assembled from graft copolymers containing complementary β-sheets as hydroxyapatite nucleation scaffolds. Biomaterials. 2011;32:5341–5353. doi: 10.1016/j.biomaterials.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kopecek J., Yang J. Peptide-directed self-assembly of hydrogels. Acta Biomater. 2009;5:805–816. doi: 10.1016/j.actbio.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dagdas Y., Tombuloglu A., Guler M. Interfiber interactions alter the stiffness of gels formed by supramolecular self-assembled nanofibers. Soft Matter. 2011;7:3524–3532. [Google Scholar]
- 39.Toksoz S., Mammadov R., Guler M.O. Electrostatic effects on nanofiber formation of self-assembling peptide amphiphiles. J. Colloid Interface Sci. 2011;356:131–137. doi: 10.1016/j.jcis.2010.12.076. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.