

Abstract
The AhR was initially identified as a ligand-activated transcription factor mediating effects of chlorinated dioxins and polycyclic aromatic hydrocarbons on cytochrome P450 1 (CYP1) expression. Recently, evidence supporting involvement of the AhR in cell-cycle regulation and tumorigenesis has been presented. To further define the roles of the AhR in cancer, we investigated the effects of AhR expression on cell proliferation, migration, invasion, and tumorigenesis of MCF-7 human breast cancer cells. In these studies, the properties of MCF-7 cells were compared with those of two MCF-7-derived sublines: AHR100, which express minimal AhR, and AhRexp, which overexpress AhR. Quantitative PCR, Western immunoblots, 17β-estradiol (E2) metabolism assays, and ethoxyresorufin O-deethylase assays showed the lack of AhR expression and AhR-regulated CYP1 expression in AHR100 cells, and enhanced AhR and CYP1 expression in AhRexp cells. In the presence of 1 nM E2, rates of cell proliferation of the three cell lines showed an inverse correlation with the levels of AhR mRNA. In comparison with MCF-7 and AhRexp cells, AHR100 cells produced more colonies in soft agar and showed enhanced migration and invasion in chamber assays with E2 as the chemoattractant. Despite the lack of significant AhR expression, AHR100 cells retained the ability to form tumors in severe combined immunodeficient mice when supplemented with E2, producing mean tumor volumes comparable to those observed with MCF-7 cells. These studies indicate that, while CYP1 expression and inducibility are highly dependent on AhR expression, the proliferation, invasion, migration, anchorage-independent growth, and estrogen-stimulated tumor formation of MCF-7 cells do not require the AhR.
Keywords: Aryl hydrocarbon receptor, breast cancer, MCF-7 xenograft
INTRODUCTION
The aryl hydrocarbon receptor (AhR) is a transcription factor that is activated by a variety of environmental, dietary, and endogenous ligands [1,2]. Xenobiotic AhR ligands include benzo(a)pyrene (BaP) and other polycyclic aromatic hydrocarbons (PAH), and halogenated aromatic hydrocarbons, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). AhR agonist and antagonist activities have been attributed to a number of dietary components, including various flavonoids [1], and a number of endogenous compounds, notably indole derivatives [3-8]. The ligand-bound AhR, in conjunction with its heterodimerization partner, the aryl hydrocarbon nuclear transporter, facilitates transcription of a number of genes including those of cytochromes P450 of the CYP1 family, which catalyze the metabolism of various xenobiotics and endogenous substrates [9]. While the concept of physiologically relevant AhR signaling involving an endogenous ligand has yet to be universally accepted, the rapid, AhR-mediated induction of CYP1A1 observed when cells were placed in suspension [10] established that the AhR can be activated in the absence of an exogenous ligand [10-13]. The recently reported activation of the AhR by the tryptophan-derived ligand, 6-formylindolo[3,2-b]carbazole, in response to ultraviolet irradiation supports the existence of a physiologic mechanism that ultimately leads to AhR-mediated activation of gene transcription [14].
In recent years, functions of the AhR in addition to those relating to the transcriptional regulation of genes encoding phase I and II enzymes have been identified [15-17]. Evidence supporting involvement of the AhR in immune regulation [18-21], control of the cell-cycle and cell proliferation [22-24], cross-talk of hormone and growth-factor signaling pathways, and physical interactions with other nuclear receptors impacting their function and their rates of degradation has been presented [24-26]. Many of the roles of the AhR that have been characterized are dependent on the context of specific tissue and cell type and the presence or absence of specific AhR ligands [27,28]. In MCF-7 human breast cancer cells, nuclear localization of the AhR subsequent to exposure to ligands increases AhR interaction with retinoblastoma protein [29] and induces cell-cycle arrest [30].
The involvement of the AhR in cancer has traditionally been viewed in terms of its role in the regulation of expression of the carcinogen-bioactivating CYP1 enzymes, leading to adductive and oxidative DNA damage and resulting in mutagenesis in the initiation phase of the disease [9,31]. In recent studies, additional roles of the AhR in the promotion and progression stages of breast cancer have been proposed; however, they are not consistent, particularly with regard to the role of the AhR in development of the invasive phenotype [27,32]. In light of the conflicting roles of the AhR that have been proposed in breast cancer, this study was undertaken to determine the involvement of the AhR in the post-initiation phases of estrogen receptor (ER)-positive breast cancer. To further define the roles of the AhR in human breast cancer, we used MCF-7 cells, which express ERα and are classified as luminal A breast cancer, and two MCF-7-derived sublines: AHR100 cells, which express minimal AhR, and AhRexp cells, which overexpress AhR. The effects of differing AhR expression levels on CYP1 induction, metabolism of 17β-estradiol (E2), cell proliferation, migration, invasion, anchorage-independent growth, and tumorigenesis were evaluated. Our results are consistent with the obligatory role of the AhR in CYP1 inducibility, but indicate that the AhR is not required for tumor formation and growth from MCF-7 cells.
MATERIALS AND METHODS
Cell Lines and Culture
MCF-7 cells were those used in our previous studies [33]. AHR100 cells, which are deficient in AhR, were derived from MCF-7 cells after 6 to 9 months exposure to BaP [34]. Stock cultures of AHR100 cells were maintained in 0.8 μM BaP (Sigma, St. Louis, MO), which, unless otherwise indicated, was withdrawn for at least 2 weeks prior to the initiation of the experiments. AhRexp cells were obtained by stable transfection of MCF-7 cells with the AhR-expression construct, pcDNA3.1(+)AhR [35]. AhRexp cells were maintained in 0.3 mg/mL Geneticin (Invitrogen, Carlsbad, CA), although the antibiotic was removed prior to initiation of the experiments. Stock cultures of MCF-7, AHR100, and AhRexp cells were maintained in DF5 medium, which consisted of Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% (v/v) fetal bovine serum (Hyclone, Logan, UT), 100 μM nonessential amino acids, 2 mM L-glutamine, 10 μg/L insulin, 100 U/mL penicillin, and 100 μg/mL streptomycin. Unless otherwise indicated, all experiments were performed using DC5 medium, which has minimal estrogen content [35]. The formulation of DC5 differed from that of DF5 in that it contained 5% (v/v) bovine calf serum (Cosmic calf serum; Hyclone Laboratories, Logan, UT) in place of the fetal bovine serum, and it was prepared with phenol-red-free DMEM. TMX2-28, an ERα-negative subline isolated from a MCF-7 culture that had been subjected to long-term tamoxifen exposure [36], was maintained in DC5 medium. As indicated, cultures were exposed in DC5 medium to the following compounds: IC182,780 (ICI; Tocris, Ellisville, MO), TCDD (Cambridge Isotope Laboratories, Andover, MA), and/or E2 (Sigma) with dimethylsulfoxide (DMSO; Sigma) as the vehicle.
Ethoxyresorufin-O-deethylase (EROD) Assays
EROD assays were performed in 96-well plates as described [37]. Confluent cultures were exposed for 48 or 72 h to 10 nM TCDD, 1 nM E2, and/or 100 nM ICI. After treatments, the medium was replaced with medium (100 μL/well) containing 4 μM ethoxyresorufin and 10 μM dicumarol (Sigma), and the plates were incubated at 37°C for 30 min. Fluorescence measurements were made using a Fusion Universal Microplate Analyzer (Packard Instrument Co., Meriden, CT) with 535-nm excitation and 590-nm emission filters. Data were normalized to total protein, which was determined using the BCA Protein assay reagent (Pierce, Rockford, IL).
Reverse-transcription and Quantitative Real-time PCR (qPCR)
Cultures at confluence in 6-well plates were exposed to 10 nM TCDD with or without 1 nM E2 for 48 h. Isolation of total RNA from cell lysates and tumor homogenates, reverse-transcription of oligo-dT-primed RNA with Superscript III (Invitrogen), and qPCR using the LightCycler System with the FastStart DNA Master SYBR Green I kit (Roche Molecular Biochemicals, Indianapolis, IN) were performed as previously described [35]. Quantification of cDNA was by comparison of cycle numbers for unknowns to those of purified cDNA standards of known concentration. For qualitative analysis of vector-derived AhR cDNA, the AhR-reverse primer was substituted with the BGH-reverse universal primer (5′-TAGAAGGCACAGTCGAGG-3′).
Western Immunoblots
Western immunoblots of total cellular lysates for the analysis of ERα and AhR were performed as described [35]. Samples containing equal amounts of total protein were subjected to electrophoresis in 10% acrylamide NuPAGE Bis-Tris denaturing gels (Invitrogen). Proteins were transferred onto Immobilon-P membranes (Millipore, Bethesda, MD), and blots were probed with rabbit polyclonal anti-human AhR, rabbit polyclonal anti-human ERα, or rabbit polyclonal anti-GAPDH antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). Immunoreactive proteins were detected by using the West Pico Enhanced Chemiluminescence kit (Pierce).
Estrogen Metabolism Assays
Confluent cultures in 6-well plates were exposed to 10 nM TCDD for 48 h. Cultures were then exposed to 1 μM E2 in 2 mL of medium for 6 h. Media were recovered, and 1 mL of each sample was hydrolyzed at 37 °C with sulfatase (SULF) from Helix pomatia (Sigma Type H-1; 24,190 units SULF and >300 units β-glucuronidase) for 6 h. Estrogen metabolites were recovered from the hydrolyzed and non-hydrolyzed samples by solid-phase extraction, converted to pyridyl-3-sulfonyl derivatives and analyzed by liquid chromatography-tandem mass spectrometry [38,39].
Cell Proliferation Assays
The Sulforhodamine B assay [40] was used for the determination of cell proliferation. To initiate the assay, cultures were seeded in 96-well plates at 6000 cells per well. At various times, plates were fixed with 10% trichloroacetic acid and stained for total protein with 0.4% Sulforhodamine B (Sigma, St. Louis, MO) in 1% acetic acid. After rinsing, the dye was dissolved by the addition of 10 mM Tris base, and the absorbance at 490 nm was determined.
Determination of Anchorage-independent Growth
Cells were trypsinized and suspended in DMEM with 0.1% bovine serum albumin and diluted to 3000 cells/mL in 0.3% soft agar (Difco, Becton Dickinson, Sparks, MD). Aliquots of the cell suspension (500 μL) were layered over a base layer of solidified agar (500 μL, 0.5%) in 24-well plates [41]. After solidification of the top layer, 1 mL of medium was added, and colonies were allowed to form for 2 weeks, after which colonies of ≥50 μm in 6 randomly selected fields were counted.
Invasion and Migration Assays
Cell migration and invasion assays were performed using Boyden chambers with inserts having 8.0 μm pores (BD Biosciences, Bedford, MA). Uncoated inserts were used for migration assays, whereas Matrigel-coated inserts were used for invasion assays [42,43]. Cells were suspended in DMEM containing 0.1% bovine serum albumin, and 105 cells per well were seeded in the upper chamber. The lower chamber contained 1 nM E2 as the chemoattractant in DC10, a DMEM-based medium with 10% bovine calf serum. After 40 h, inserts were scrubbed, fixed with methanol for 1 to 2 min, dried, and stained with 0.1% crystal violet for cell counting.
Xenograft Assays for Tumorigenicity
For the xenograft studies, groups of 15 mice received inoculations of cells with or without E2 supplementation [44]. Each inoculation consisted of 1 × 106 cells in 50 μl of culture medium injected into the surgically exposed mammary fat pads of 6-to 8-week-old severe combined immunodeficient (SCID) mice from Taconic Farms (Germantown, NY). E2 supplementation was accomplished by subcutaneous implantation of Silastic tubing capsules (2 mm in length) containing solid E2, inserted on the day of tumor-cell implantation. Under this protocol, these implants produce serum E2 levels of 100 pg/mL [45]. Mice were palpated daily, and emerging tumors were measured with Vernier calipers several times per week for 68 to 71 days. Tumor volume was calculated using the formula V = (π/6)d2D, assuming the tumor shape to be ellipsoid with D as the long axis. The Albany Medical College Animal Care and Use Committee approved all work with animals.
Statistical Evaluations
Statistical evaluations of biochemical determinations were performed in replicates of three or more by analysis of variance and the Bonferroni t-test for multiple comparisons. Statistical evaluations for comparisons of tumor volumes were performed with the Mann-Whitney rank sum test. Proliferation data were fit to the four parameter Chapman model: y = y0 + a(1-e-bx)c using the SigmaPlot program (SPSS). Growth rates were calculated as the first derivative at the inflection point of the sigmoidal curve [46].
RESULTS
Expression of AhR, ERα, and TCDD-inducible EROD activities in AHR100, MCF-7, and AhRexp cells
The AhRexp clone was selected from among several clones as having the highest level of AhR expression. AhR mRNA and protein in MCF-7, AHR100, and AhRexp cells were examinedby PCR and Western immunoblot (Figure 1). When mRNA from AhRexp cells was isolated and analyzed by PCR, but with substitution of the usual reverse primer that was homologous to the coding sequence with a primer that was homologous to the vector-derived 3′ untranslated RNA, the PCR product indicative of vector-derived expression was observed (Figure 1A; lane 2, 614-bp). None of this product was observed when RNA from MCF-7 cells was analyzed (Figure 1A; lane 5). PCR product representing mRNA encoded by the endogenous gene (Figure 1A; lane 4, 377-bp) was obtained from MCF-7 cDNA using the reverse primer homologous to the coding sequence. Total AhR cDNA representing endogenous plus heterologous AhR RNA from AhRexp cells was analyzed using the primers that amplified the AhR coding sequence (Figure 1A; lane 3, 377-bp). The lack of PCR product formed in the negative-control amplification reaction of AhRexp RNA in which reverse transcriptase was omitted (Figure 1A, lane 1) indicates that the heterologous cDNA was not derived from contamination of the RNA with vector DNA. Analysis by Western immunoblot did not show detectable AhR protein in AHR100 cells, whereas AhRexp cells expressed higher levels of AhR protein than MCF-7 cells (Figure 1B). ERα was expressed in the MCF-7-derived sublines, AHR100 and AhRexp. The MCF-7-derived cell line, TMX2-28 [36], was included as an ERα-negative control.
Figure 1.
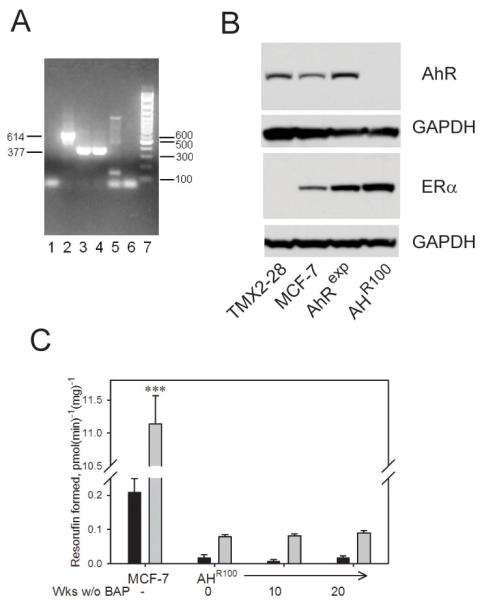
Expression of AhR and ERα in MCF-7 and MCF-7-derived cell lines. (A) Heterologous AhR mRNA expression in AhRexp cells. Total RNA from AhRexp (lanes 1-3) and MCF-7 cells (lanes 4-5) was isolated, and PCR was performed with primers specific for the coding sequence of AhR mRNA representing total AhR mRNA (377-bp product) or specific for the vector-derived mRNA (614-bp product). Lane 1, reverse transcriptase-negative control using vector-specific primers; lanes 2 and 5, PCR with vector-specific primers; lanes 3, 4, and 6, PCR with primers specific for the AhR coding sequence; lane 6, minus-RNA control using primers specific for the AhR coding sequence; lane 7, 100-bp ladder. (B) AhR and ERα protein levels in AHR100, MCF-7, and AhRexp cells. Western blots of whole cell lysates from MCF-7, AhRexp, and AHR100 cells were probed with anti-AhR and -ERα antibodies and detected with enhanced chemiluminescence. Lysates from TMX2-28 cells are included as an ERα-negative control. GAPDH was probed as a loading control for each blot. (C) The loss of TCDD-inducible EROD activity in AHR100 cells is persistent. BaP was withdrawn from AHR100 cultures for 10 or 20 weeks, and compared with cells without BaP withdrawal (designated as 0 weeks without BaP), after which cells were exposed for 72 h to 10 nM TCDD (gray bars) or the vehicle, 0.1% (v/v) DMSO (black bars). CYP1 activity was then measured by EROD assay. EROD activity of MCF-7 cells is shown for comparison; note the difference in scale. Data are presented as mean + SE; n = 5. Significant differences for TCDD-treated cultures in comparison with the TCDD-treated control group (AHR100 cells without BaP withdrawal) are indicated (***P<0.001).
Because AHR100 cells were originally obtained from MCF-7 cultures after continuous exposure to BaP [34], it was necessary to confirm that AhR activity remained at a minimal level after the removal of BaP. The results presented in Figure 1C show that AhR-mediated, TCDD-induced EROD activity in AHR100 cells was less than 1% of that of the TCDD-induced activity of MCF-7 cells. After culture for 10 and 20 weeks in the absence of BaP, AHR100 cells maintained the very low response to TCDD in the EROD assay.
When the three cell lines were directly compared with regard to AhR-mediated CYP1 induction in the EROD assay (Figure 2), AhRexp cells showed approximately twice the level of TCDD-induced CYP1 activity as MCF-7 cells, while AHR100 cells showed approximately 2% of the induced CYP1 activity in the EROD assay compared to that of MCF-7 cells, which is consistent with the results of Trapani et al. [34]. Induction of EROD activity, which is primarily a measure of CYP1A1 activity, was modestly inhibited by E2. This inhibition was reversed by inclusion of the antiestrogen, ICI, indicating involvement of the ER in this effect.
Figure 2.
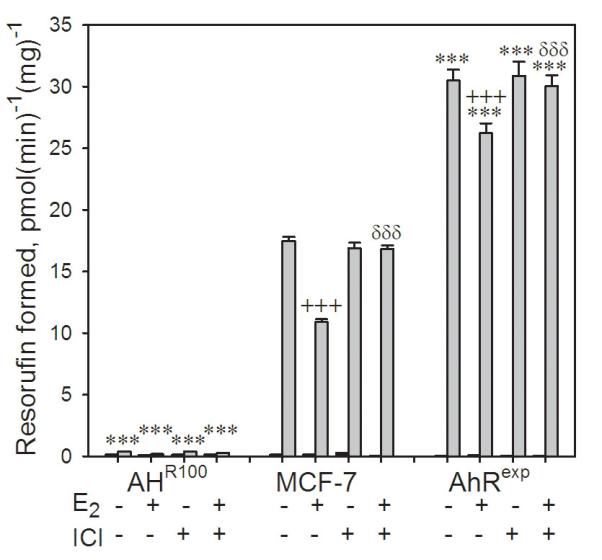
Ah-responsiveness in AHR100, MCF-7, and AhRexp cells as measured by the EROD assay. Confluent cultures of AHR100, MCF-7, and AhRexp cells were exposed for 48 h to 10 nM TCDD (gray bars), or the vehicle, 0.16% (v/v) DMSO (black bars), with or without 1 nM E2 and 100 nM ICI, as indicated. CYP1 activity was then measured by the EROD assay. Data are represented as the mean + SE; n = 8. Significant differences between TCDD-exposed AHR100 or AhRexp cultures and MCF-7 cultures in which all other treatments were identical (***P<0.001); between groups differing only in E2 exposure (+++P<0.001); and between groups differing only in ICI exposure (+++P<0.001) are indicated.
Levels of ERα, CYP1A1, and CYP1B1 mRNAs in MCF-7-derived cell lines
Cultures of MCF-7, AHR100, and AhRexp cells were exposed to 1 nM E2 and/or 10 nM TCDD for 48 h, and RNA was isolated and was analyzed by real-time qPCR for the following transcripts: AhR, ERα, CYP1A1, CYP1B1, and 36B4, which encodes acidic ribosomal phosphoprotein PO and is commonly used as a control mRNA that is not regulated by E2 [47]. These mRNA levels are presented in Figure 3. AhR mRNA levels were 1.8 fold higher in AhRexp cells than in MCF-7 cells, whereas AHR100 cells expressed approximately 2.5% of the AhR mRNA level of MCF-7 cells in the absence of E2 treatment. In the absence of E2 and TCDD, ERα mRNA levels in the three cell lines were consistent with ERα protein levels as determined by Western blot analysis (Figure 1B). In each of the three cell lines, E2 exposure caused a significant down-regulation of ERα mRNA expression. Levels of the CYP1A1 and CYP1B1 mRNAs in TCDD-exposed cells correlated with the levels of expression of the AhR mRNA in the three cell lines. Levels of CYP1B1 mRNA in the absence of E2 and TCDD also correlated with AhR mRNA levels. Interestingly, E2 exposure resulted in a moderate down-regulation of TCDD-induced CYP1B1 mRNA levels in AhRexp cells. Consistent with its effects on TCDD-induced EROD activity (Figure 2), E2 modestly down-regulated TCDD-induced CYP1A1 mRNA levels in MCF-7 and AhRexp cells.
Figure 3.

Effect of AhR expression on ERα mRNA levels and the inducibility of CYP1 mRNAs. Confluent cultures of AHR100, MCF-7, and AhRexp cells were exposed to 10 nM TCDD, 1 nM E2, or the vehicle, 0.16% (v/v) DMSO, for 48 h, and the RNA isolated. qPCR was performed with primers specific for the transcripts as indicated. Data are represented as the average +/- SEM; n=3, and are normalized to total RNA. Significant differences between TCDD-exposed AHR100 or AhRexp cultures in comparison with MCF-7 cultures in which all other treatments were identical (*P<0.05; ***P<0.001); and between E2 exposure and the respective group differing only in the absence of E2 exposure (++P<0.01; +++P<0.001) are indicated.
Metabolism of E2 in MCF-7, AHR100, and AhRexp cells
Having confirmed differential expression of AhR at the protein and mRNA levels in MCF-7, AHR100, and AhRexp cells, we then determined the effect of differential AhR expression on the TCDD-induced metabolism of E2. The rates of formation of 2-, 4-, 6α, and 15α-hydroxyestradiol (OHE2), and 2- and 4-methoxyestradiol (MeOE2), with and without exposure to 10 nM TCDD and with and without hydrolysis of conjugates prior to analysis are shown in Figure 4. In MCF-7 and AhRexp cells, basal E2 metabolism at the C-2 and C-4 positions was detected, and this metabolism was greatly increased by TCDD exposure. Comparison of the levels of metabolites recovered with and without prior SULF treatment indicate that significant amounts of 2- and 4-OHE2, and 2- and 4-MeOE2 were present in conjugated form, whereas 6α- and 15α-OHE2 were not conjugated to an appreciable extent. In contrast to E2 metabolism in MCF-7 and AhRexp cells, AHR100 cells did not show appreciable E2 metabolism, with or without TCDD exposure.
Figure 4.

Basal and TCDD-induced E2-metabolite formation in AHR100, MCF-7, and AhRexp cells. Confluent cultures of AHR100, MCF-7, and AhRexp cells were exposed for 48 h to 10 nM TCDD or the solvent vehicle, 0.1% (v/v) DMSO, as indicated. Metabolite formation was determined with (+SULF) and without (-SULF) prior hydrolysis of conjugates as described in Materials and Methods. Data are represented as the mean + SE and are normalized to total cellular protein; n = 3. Significant differences between TCDD-exposed AHR100 or AhRexp cultures and MCF-7 cultures, where all other treatments were identical (***P<0.001) are indicated; ND denotes that the metabolites were not detected.
Proliferation, anchorage-independent growth, invasion, and migration of AHR100, MCF-7, and AhRexp cells
We next investigated the consequences of differential AhR expression on in vitro indices of tumorigenesis, with and without E2 exposure. Cell proliferation of the two MCF-7-derived lines as compared with that of MCF-7 cells in the presence or absence of 1 nM E2 is shown in Figure 5A. Growth rates derived from the data in Figure 5A are presented in Table 1. Proliferation without E2 supplementation was highest for AHR100 cells and lowest for MCF-7 cells. MCF-7 cells, however, showed the greatest enhancement of proliferation in response to E2 supplementation, and AhRexp cells showed no enhancement of proliferation in response to E2. AhR100 cells showed the highest proliferation among the three cell lines with or without E2 supplementation. In the presence of E2, growth rates of the three cell lines (Table 1) were inversely correlated with AhR mRNA levels (Fig 3); R2= 0.981.
Figure 5.

Proliferation, migration, and invasion of AHR100, MCF-7, and AhRexp cells. (A) Proliferation: AHR100, MCF-7, and AhRexp cells were seeded in 96-well plates at 6000 cells per well, and the media were replenished every 3 or 4 days with DC5 (closed circles) or DC5 containing 1 nM E2 (open circles). Proliferation was measured at the indicated times using the Sulforhodamine B assay. Data are presented as mean + SE; n = 8; however, for all points the error bar falls within the symbol. Curves were obtained by fitting data to the four parameter Chapman model. (B) Anchorage-independent growth: AHR100, MCF-7, and AhRexp cells were suspended in 500 μL 0.3% soft agar in 24-well plates at 1500 cells per well. The cells were cultured for 2 weeks in DC5 or DC5 containing 1 nM E2 as indicted. Anchorage-independent growth was assessed under 10X magnification by counting colonies larger than 50 μm (black bars) or larger than 100 μm (gray bars) in diameter. Significant differences between AHR100 or AhRexp cultures in comparison with MCF-7 cultures when all other treatments were identical (**P<0.01; ***P<0.001) are indicated. Data are represented as mean colonies per field + SE; n = 6. Significant differences for colonies ≥ 50 μm are shown. (C) Migration and invasion: AHR100, MCF-7, and AhRexp cells were suspended in DMEM containing 0.1% bovine serum albumin, and 105 cells per well were seeded in Matrigel-coated chambers for measurement of invasion (black bars) or in control inserts for measurement of migration (gray bars). The lower chamber contained 1 nM E2 in DC10 as a chemoattractant. After 40 h, inserts were fixed and stained with crystal violet. Data are represented as mean cells per insert + SE; n = 3. Significant differences between AHR100 or AhRexp cultures in comparison with MCF-7 cultures (**P<0.01; ***P<0.001) are indicated.
Table 1.
Proliferation rates of AHR100, MCF-7, and AhRexp cells
| Cell line | |||
|---|---|---|---|
| AHR100 | MCF-7 | AhRexp | |
| Medium | |||
| DC5 | 0.382 (0.353-0.429)a | 0.137 (0.129-0.150) | 0.2446 (0.2443-0.2449) |
| DC5 + 1 nM E2 | 0.659 (0.623-0.744) | 0.398 (0.363-0.433) | 0.215 (0.167-0.275) |
Rates are expressed as 105 cells/day, with 95% confidence intervals given in parenthesis.
The three cells lines were then evaluated for colony formation in soft agar in the presence of DC5 and DC5 containing 1 nM E2 (Figure 5B). Among the three cell lines, anchorage-independent growth was greatest for AHR100 cells with each medium tested. We then investigated migration and invasion of the three cell lines using Boyden chambers. The invasion assay, which requires that the cells degrade and transverse a layer of extracellular matrix, analogous to the penetration of the basement membrane by cancer cells in the initial events of metastasis [42], utilized Matrigel-coated inserts, whereas uncoated chamber inserts were used to assay for migration [43]. In both assays, the lower chamber contained medium supplemented with E2 as the chemoattractant. Using these assays we found that AHR100 cells were >30-fold more invasive than MCF-7 or AhRexp cells, and that migration of AHR100 cells was also highly elevated compared with that of AhRexp and MCF-7 cells (Figure 5C).
Tumorigenesis of AHR100, MCF-7, and AhRexp cells in SCID mice
To evaluate in vivo tumorigenicity, AHR100, MCF-7, and AhRexp cells were inoculated into the mammary glands of SCID mice, with and without E2 supplementation as a Silastic implant (Figure 6). In all of these experiments, tumors, although palpable, were not able to grow appreciably without E2 supplementation. Included for comparison are ERα-negative TMX2-28 cells, which, despite the fact that they are highly proliferative and invasive in vitro [48], grew poorly as mammary-gland xenografts with or without E2 supplementation (Figure 6, lower right panel). With E2 supplementation, tumors from implanted AHR100 cells (Figure 6, upper left panel) grew to an extent comparable to MCF-7 cells, indicating that the AhR is not required for tumor formation and growth in SCID mice. AhRexp-cell xenografts showed a trend toward slower tumor growth in comparison with those from MCF-7 cells; however, this difference was not statistically significant. Interestingly, with AhRexp cells, tumors became palpable but did not grow to any significant extent, which was similar to the results found following inoculation of TMX2-28 cells.
Figure 6.
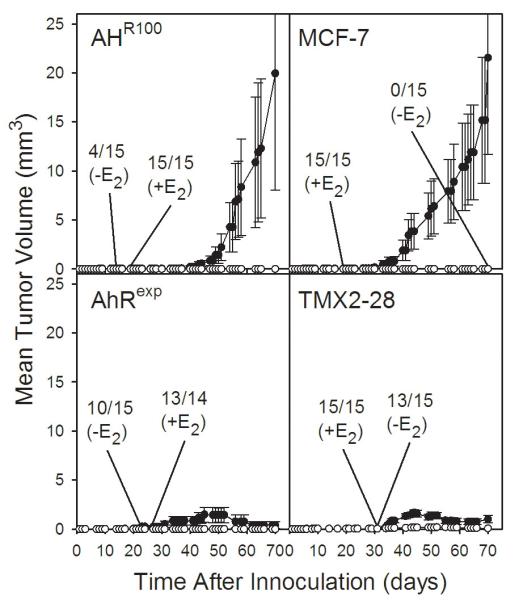
Tumorigenicity of the MCF-7-derived cell lines with differential expression of AhR and ERα. AHR100, MCF-7, AhRexp cells, or TMX2 cells, as indicated, were implanted into the mammary glands of SCID mice, with (closed circles) or without (open circles) E2 supplementation as a Silastic implant. Tumor growth was monitored by palpation at the times indicated. The latency of tumor formation is indicated in the plots by denotation of the day at which the maximal incidence of tumor formation was observed, with the number of mice with palpable tumors/total number of mice. Data are presented as mean tumor volume + SE of the mice with measurable tumors.
To determine whether AHR gene expression in the xenograft tumors was comparable to that in the cells in culture, we examined the level of AhR mRNA in MCF-7 and AHR100 tumors dissected from animals after the final tumor-growth measurements were taken. AhRexp tumors were of insufficient size for dissection. AhR cDNA and GAPDH cDNA for normalization were amplified with human-specific primers, which did not amplify the homologous targets from murine cDNA (results not shown). We found that the levels of AhR mRNA in tumors obtained from AHR100 xenografts remained low, at less than 7% the level in MCF-7 cells.
DISCUSSION
Studies have been conducted in a number of laboratories with the goal of deciphering the roles of the AhR in initiation, promotion, and progression phases of cancer, and from them a variety of roles of the receptor in cancer have been proposed [9,17,27-28,31]. Dependent on the species, tissue, and phase of carcinogenesis, evidence of both oncogenic [9,27,31,49-51] and tumor-suppressing functions [32,52,53] of the AhR have been reported. In this study, we show that, in the MCF-7 model of luminal A breast cancer, CYP1 expression and inducibility by TCDD in cultured cells are highly dependent on AhR expression, whereas cellular proliferation, invasion, migration, anchorage-independent growth, and estrogen-stimulated formation of xenograft tumors do not require the AhR. Our current studies with MCF-7 cells suggest that the AhR may have both pro- and anti-carcinogenic roles within ER-positive luminal breast cells during the various stages of carcinogenesis.
The importance of the AhR in the initiation phase of PAH-induced carcinogenesis is well established. The AhR-mediated induction of the CYP1 enzymes that catalyze the metabolic activation of PAH to DNA adductive forms is integral to the carcinogenic process [9]. AhR-null mice are refractory to PAH-induced carcinogenesis [49], and studies with CYP1 single- and double-knockout animals show the roles of these enzymes in differential organ carcinogenesis subsequent to PAH exposure [31]. These enzymes not only catalyze the metabolic activation of PAHs, but also heterocyclic aromatic amines [54] and endogenous estrogens [55]. The AhR may thus be involved in the initiation events from several exogenous and endogenous carcinogens. In our current study, the inducibility of CYP1A1 and CYP1B1 mRNAs, proteins, and CYP1 activities were consistent with the levels of AhR expression in AHR100, MCF-7, and AhRexp cells in culture. Increased rates of the 2- and 4-hydroxylation pathways of E2 metabolism in response to TCDD exposure, which reflect the activities of CYP1A1 and CYP1B1, respectively [33], and the induction of EROD activity were not observed in the absence of AhR expression. The ultimate metabolites of E2 in MCF-7 cells, the sulfate conjugates of 2- and 4-MeOE2 [56], were not produced at appreciable levels in TCDD-treated AHR100 cells, nor were 6α-OHE2 and 15α- OHE2, which are products of CYP1A1-catalyzed metabolism [57]. These results indicate that the induction of E2 metabolism, including the formation of the potentially carcinogenic 4-OHE2 [55], is reliant on AhR expression in MCF-7 cells.
Distinct from the roles of the AhR in CYP1 induction and the regulation of carcinogen metabolism, the AhR appears to have roles in post-initiation stages of carcinogenesis. Recently, transgenic and gene knockout animals have been used to specifically determine role of the AhR in the post-initiation events in carcinogenesis that do not rely on exogenous AhR ligands or CYP1-catalyzed carcinogen metabolism. Fritz et al. [52] used the transgenic adenocarcinoma of the mouse prostate (TRAMP) model, in which expression of the simian virus 40 large T and small t antigens are under the control of the androgen-dependent probasin gene promoter, to investigate the role of the AhR in tumor formation. They found that when TRAMP mice were crossed with Ahr-/- mice, the Ahr+/- and Ahr-/- TRAMP progeny showed greater prostate tumor incidence than in Ahr+/+ TRAMP mice, indicating an inhibitory effect of AhR expression in this model of prostate carcinogenesis. Evidence of a tumor-suppressor function of the AhR was also reported by Fan et al. [53] in their study of Ahr-/- mice exposed to the direct-acting carcinogen, diethylnitrosamine. A significant increase in liver adenomas, which are thought to be the precursors of hepatocellular carcinomas, was observed in male Ahr-/- but not female mice in comparison with Ahr+/+ mice after exposure to the carcinogen. Our results are consistent with these studies of prostate tumors Ahr-/- TRAMP mice [52] and liver adenomas [53] in Ahr-/- mice, as we found that AhR expression in MCF-7 cells is not necessary for xenograft tumor formation in SCID mice, and may in fact have a tumor-suppressor function in these cells.
A caveat that is associated with the use of AHR100 cells is that these cells were obtained after long-term exposure to BaP. Mutations and epigenetic modifications in AHR100 cells in addition to those directly related to the reduction of AhR expression and their ability to survive and proliferate in the presence of BaP may have occurred. However, in this regard we note that AHR100 cells retain ERα expression and remain dependent on estrogen for tumor growth as xenografts. They also retain epithelial-like morphology in culture. Recently, it was reported that vector-driven re-expression of the AhR in AHR100 cells restored the inducibility of the CYP1A1 and CYP1B1 mRNAs by TCDD [58], which supports the premise that these cells retain many characteristics of the parental cell line. Regardless of the genetic and/or epigenetic changes that may have occurred in the development of AHR100 cells and their loss of AhR expression, our conclusion that expression of the AhR is not required for the proliferation, migration, invasion, or estrogen-dependent tumorigenesis of MCF-7 breast cancer cells is entirely valid.
Rates of cell proliferation and alterations in cell-cycle control are recognized as key factors impacting carcinogenesis, and the AhR is known to affect them, albeit in cell-specific manner [17,24,59]. There are numerous studies indicating that, in the absence of an exogenous ligand, the AhR promotes progression through the cell cycle, whereas ligand activation of the AhR causes cell-cycle arrest in a number of cell types [17,22,29,30]. However, these effects are not universally observed; the role of the AhR in progression through the cell cycle and cell proliferation is dependent on cell type. Expression of the AhR in AhR-defective Hepa1c1 cells enhances the rate of cell proliferation in the absence of an exogenous AhR ligand [11]. Exposure to the AhR ligand, β-naphthoflavone, or overexpression of the AhR in A549 lung carcinoma cells caused enhanced cell proliferation [24]. The reduced rate of cell proliferation observed in AhR-null embryo fibroblasts appears be multi-factorial, but a significant role was attributed to the overproduction of TGF-β [23]. Conversely, our present study showed enhanced cell proliferation of AhR-deficient AHR100 cells, which is consistent with the increased G0/G1 phase progression that was observed when AhR expression was knocked down by small inhibitory RNA in MCF-7 cells [59].
An aspect of AhR activity in breast and other cancers that remains unresolved is the role of the AhR in development of the invasive phenotype that may be indicative of metastatic potential. Several studies suggest that the AhR, through regulation of Slug, initiates the down-regulation of E-cadherin and consequently the epithelial-to-mesenchymal transition and development of the invasive phenotype [27,50,51]. In contrast, Hall et al. [32] reported evidence for inhibitory roles of AhR expression and ligand activation of the AhR in cellular invasion and processes related to metastasis in a panel of breast cancer cell lines representing the major breast-cancer subtypes. Exogenous AhR agonists significantly inhibited cell invasiveness and motility and inhibited colony formation in soft agar regardless of ER, progesterone receptor, or human epidermal growth factor receptor 2 status. Knockdown of AhR expression using a small inhibitory RNA was also shown to be sufficient to increase, rather than decrease, the invasiveness of SKBR3 and MDA-MB-231 cells, and activation of the AhR by TCDD caused decreases in colonization in soft agar of SKBR3, MDA-MB-231, ZR-75-1 and MCF-7 cells [32].
The reasons for the differences in experimental observations regarding the role of the AhR in cellular invasiveness and anchorage-independent growth in breast cancer cells are not known, but they may be influenced by the specific developmental lineage of the tumor. The two main differentiation pathways in the mammary epithelium give rise to the luminal and basal/myoepithelial cell types [60]. We suggest that the AhR may have quite different roles in “basal-like” breast tumors versus “luminal-like” tumors. The epithelial-to-mesenchymal transition appears to occur within the specific genetic context of the basal phenotype [61]. If AhR-mediated regulation of Slug is a property of the basal-like tumor cells, then this could explain the differences in invasive properties of tumor cells that have been reported for the AhR. MCF-7 cells, which are luminal in phenotype, do not express significant levels of Slug [60], and our gene expression studies [44] did not show induction of the Slug transcript by TCDD exposure in MCF-7 cells.
In numerous studies it has been suggested that the AhR could be a therapeutic target in cancer. However, the results of the present study, notably those obtained with AHR100 cells, indicate that breast cancer cells can in some cases obtain a growth advantage when AhR expression is diminished. We found that cell proliferation, migration, invasion, and anchorage-independent growth do not correlate with AhR expression in MCF-7 cells and its sublines, AHR100 and AhRexp. Over-expression of AhR in AhRexp cells did not confer increased anchorage-independent growth, invasiveness, or tumorigenicity. As has been reported in numerous previous studies, tumor formation from MCF-7 xenografts was highly dependent on the presence of E2 in the host animal. Mice without E2 supplementation did not have measurable tumor growth as xenografts from MCF-7, AHR100, or AhRexp cells. Surprisingly, even the highly invasive and proliferative MCF-7-derived cell line, TMX2-28 [36,48], which is unresponsive to E2, was unable to sustain appreciable tumor growth in SCID mice.
In summary, our results show that, in the MCF-7 model of luminal A breast cancer, CYP1 expression and inducibility by TCDD are highly dependent on AhR expression, whereas cellular invasion, migration, anchorage-independent growth, and estrogen-stimulated formation of tumors do not require the AhR. Our experiments with AHR100 cells show that highly proliferative, invasive, and tumorigenic cells can arise from AhR-deficient tumor cells. The studies reported here indicate that the AhR and the AHR gene may represent chemoprevention targets that are focused on reducing the cancer-initiating events due to bioactivation of exogenous and endogenous carcinogens, but they are not likely to represent universal targets in breast cancer therapeutics aimed at inhibiting the growth and invasiveness of the developing tumor.
Acknowledgements
This research was supported by the National Institutes of Health grant CA081243. The authors gratefully acknowledge the use of the Wadsworth Center’s Tissue Culture Facility and the Biochemical and Molecular Genetics Core Facilities. The AHR100 cells line was a generous gift from Dr. Grace Chao Yeh of the National Cancer Institute at Frederick, NIH, Frederick, MD. The TMX2-28 cell line was kindly provided by Dr. John F. Gierthy of the Wadsworth Center. The authors thank Benjamin P. Leard for his assistance in data analysis.
Abbreviations
- AhR
aryl hydrocarbon receptor
- BaP
benzo(a)pyrene
- PAH
polycyclic aromatic hydrocarbons
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- CYP
cytochrome P450
- ER
estrogen receptor
- E2
17β-estradiol
- DMEM
Dulbecco’s modified Eagle’s medium
- ICI
IC1-182,780 or Fulvestrant
- DMSO
dimethylsulfoxide
- EROD
ethoxyresorufin-O-deethylase
- qPCR
quantitative real-time PCR
- SULF
sulfatase
- SCID
severe combined immunodeficient
- MeOE2
methoxyestradiol
- OHE2
hydroxyestradiol
- TRAMP
transgenic adenocarcinoma of the mouse prostate
REFERENCES
- 1.Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol. 2003;43:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol. 2008;21(1):102–116. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wei YD, Bergander L, Rannug U, Rannug A. Regulation of CYP1A1 transcription via the metabolism of the tryptophan-derived 6-formylindolo[3,2-b]carbazole. Arch Biochem Biophys. 2000;383(1):99–107. doi: 10.1006/abbi.2000.2037. [DOI] [PubMed] [Google Scholar]
- 4.Adachi J, Mori Y, Matsui S, et al. Indirubin and indigo are potent aryl hydrocarbon receptor ligands present in human urine. J Biol Chem. 2001;276(34):31475–31478. doi: 10.1074/jbc.C100238200. [DOI] [PubMed] [Google Scholar]
- 5.Song J, Clagett-Dame M, Peterson RE, et al. A ligand for the aryl hydrocarbon receptor isolated from lung. Proc Natl Acad Sci U S A. 2002;99(23):14694–14699. doi: 10.1073/pnas.232562899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nguyen LP, Hsu EL, Chowdhury G, Dostalek M, Guengerich FP, Bradfield CA. D-amino acid oxidase generates agonists of the aryl hydrocarbon receptor from D-tryptophan. Chem Res Toxicol. 2009;22(12):1897–1904. doi: 10.1021/tx900043s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chowdhury G, Dostalek M, Hsu EL, et al. Structural identification of Diindole agonists of the aryl hydrocarbon receptor derived from degradation of indole-3-pyruvic acid. Chem Res Toxicol. 2009;22(12):1905–1912. doi: 10.1021/tx9000418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schroeder JC, Dinatale BC, Murray IA, et al. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry. 49(2):393–400. doi: 10.1021/bi901786x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem. 2004;279(23):23847–23850. doi: 10.1074/jbc.R400004200. [DOI] [PubMed] [Google Scholar]
- 10.Sadek CM, Allen-Hoffmann BL. Cytochrome P450IA1 is rapidly induced in normal human keratinocytes in the absence of xenobiotics. J Biol Chem. 1994;269(23):16067–16074. [PubMed] [Google Scholar]
- 11.Ma Q, Whitlock JP., Jr. The aromatic hydrocarbon receptor modulates the Hepa 1c1c7 cell cycle and differentiated state independently of dioxin. Mol Cell Biol. 1996;16(5):2144–2150. doi: 10.1128/mcb.16.5.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang CY, Puga A. Constitutive activation of the aromatic hydrocarbon receptor. Mol Cell Biol. 1998;18(1):525–535. doi: 10.1128/mcb.18.1.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh SS, Hord NG, Perdew GH. Characterization of the activated form of the aryl hydrocarbon receptor in the nucleus of HeLa cells in the absence of exogenous ligand. Arch Biochem Biophys. 1996;329(1):47–55. doi: 10.1006/abbi.1996.0190. [DOI] [PubMed] [Google Scholar]
- 14.Fritsche E, Schafer C, Calles C, et al. Lightening up the UV response by identification of the arylhydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc Natl Acad Sci U S A. 2007;104(21):8851–8856. doi: 10.1073/pnas.0701764104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujii-Kuriyama Y, Kawajiri K. Molecular mechanisms of the physiological functions of the aryl hydrocarbon (dioxin) receptor, a multifunctional regulator that senses and responds to environmental stimuli. Proc Jpn Acad Ser B Phys Biol Sci. 86(1):40–53. doi: 10.2183/pjab.86.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haarmann-Stemmann T, Bothe H, Abel J. Growth factors, cytokines and their receptors as downstream targets of arylhydrocarbon receptor (AhR) signaling pathways. Biochem Pharmacol. 2009;77(4):508–520. doi: 10.1016/j.bcp.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 17.Puga A, Ma C, Marlowe JL. The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem Pharmacol. 2009;77(4):713–722. doi: 10.1016/j.bcp.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen NT, Kimura A, Nakahama T, et al. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc Natl Acad Sci U S A. 107(46):19961–19966. doi: 10.1073/pnas.1014465107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esser C, Rannug A, Stockinger B. The aryl hydrocarbon receptor in immunity. Trends Immunol. 2009;30(9):447–454. doi: 10.1016/j.it.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Boitano AE, Wang J, Romeo R, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 329(5997):1345–1348. doi: 10.1126/science.1191536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Casado FL, Singh KP, Gasiewicz TA. The aryl hydrocarbon receptor: regulation of hematopoiesis and involvement in the progression of blood diseases. Blood Cells Mol Dis. 44(4):199–206. doi: 10.1016/j.bcmd.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ge NL, Elferink CJ. A direct interaction between the aryl hydrocarbon receptor and retinoblastoma protein. Linking dioxin signaling to the cell cycle. J Biol Chem. 1998;273(35):22708–22713. doi: 10.1074/jbc.273.35.22708. [DOI] [PubMed] [Google Scholar]
- 23.Elizondo G, Fernandez-Salguero P, Sheikh MS, et al. Altered cell cycle control at the G(2)/M phases in aryl hydrocarbon receptor-null embryo fibroblast. Mol Pharmacol. 2000;57(5):1056–1063. [PubMed] [Google Scholar]
- 24.Shimba S, Komiyama K, Moro I, Tezuka M. Overexpression of the aryl hydrocarbon receptor (AhR) accelerates the cell proliferation of A549 cells. J Biochem. 2002;132(5):795–802. doi: 10.1093/oxfordjournals.jbchem.a003289. [DOI] [PubMed] [Google Scholar]
- 25.Wormke M, Stoner M, Saville B, et al. The aryl hydrocarbon receptor mediates degradation of estrogen receptor alpha through activation of proteasomes. Mol Cell Biol. 2003;23(6):1843–1855. doi: 10.1128/MCB.23.6.1843-1855.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohtake F, Baba A, Takada I, et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature. 2007;446(7135):562–566. doi: 10.1038/nature05683. [DOI] [PubMed] [Google Scholar]
- 27.Schlezinger JJ, Liu D, Farago M, et al. A role for the aryl hydrocarbon receptor in mammary gland tumorigenesis. Biol Chem. 2006;387(9):1175–1187. doi: 10.1515/BC.2006.145. [DOI] [PubMed] [Google Scholar]
- 28.Okino ST, Pookot D, Basak S, Dahiya R. Toxic and chemopreventive ligands preferentially activate distinct aryl hydrocarbon receptor pathways: implications for cancer prevention. Cancer Prev Res (Phila Pa) 2009;2(3):251–256. doi: 10.1158/1940-6207.CAPR-08-0146. [DOI] [PubMed] [Google Scholar]
- 29.Puga A, Barnes SJ, Dalton TP, Chang C, Knudsen ES, Maier MA. Aromatic hydrocarbon receptor interaction with the retinoblastoma protein potentiates repression of E2F-dependent transcription and cell cycle arrest. J Biol Chem. 2000;275(4):2943–2950. doi: 10.1074/jbc.275.4.2943. [DOI] [PubMed] [Google Scholar]
- 30.Marlowe JL, Knudsen ES, Schwemberger S, Puga A. The aryl hydrocarbon receptor displaces p300 from E2F-dependent promoters and represses S phase-specific gene expression. J Biol Chem. 2004;279(28):29013–29022. doi: 10.1074/jbc.M404315200. [DOI] [PubMed] [Google Scholar]
- 31.Shi Z, Dragin N, Miller ML, et al. Oral benzo[a]pyrene-induced cancer: two distinct types in different target organs depend on the mouse Cyp1 genotype. Int J Cancer. 127(10):2334–2350. doi: 10.1002/ijc.25222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hall JM, Barhoover MA, Kazmin D, McDonnell DP, Greenlee WF, Thomas RS. Activation of the aryl-hydrocarbon receptor inhibits invasive and metastatic features of human breast cancer cells and promotes breast cancer cell differentiation. Mol Endocrinol. 2010;24(2):359–369. doi: 10.1210/me.2009-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spink DC, Spink BC, Cao JQ, et al. Differential expression of CYP1A1 and CYP1B1 in human breast epithelial cells and breast tumor cells. Carcinogenesis. 1998;19(2):291–298. doi: 10.1093/carcin/19.2.291. [DOI] [PubMed] [Google Scholar]
- 34.Trapani V, Patel V, Leong CO, et al. DNA damage and cell cycle arrest induced by 2-(4-amino-3-methylphenyl)-5-fluorobenzothiazole (5F 203, NSC 703786) is attenuated in aryl hydrocarbon receptor deficient MCF-7 cells. Br J Cancer. 2003;88(4):599–605. doi: 10.1038/sj.bjc.6600722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spink DC, Katz BH, Hussain MM, Pentecost BT, Cao Z, Spink BC. Estrogen regulates Ah responsiveness in MCF-7 breast cancer cells. Carcinogenesis. 2003;24(12):1941–1950. doi: 10.1093/carcin/bgg162. [DOI] [PubMed] [Google Scholar]
- 36.Fasco MJ, Amin A, Pentecost BT, Yang Y, Gierthy JF. Phenotypic changes in MCF-7 cells during prolonged exposure to tamoxifen. Mol Cell Endocrinol. 2003;206(1-2):33–47. doi: 10.1016/s0303-7207(03)00256-9. [DOI] [PubMed] [Google Scholar]
- 37.Spink BC, Hussain MM, Katz BH, Eisele L, Spink DC. Transient induction of cytochromes P450 1A1 and 1B1 in MCF-7 human breast cancer cells by indirubin. Biochem Pharmacol. 2003;66(12):2313–2321. doi: 10.1016/j.bcp.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 38.Xu L, Spink DC. Analysis of steroidal estrogens as pyridine-3-sulfonyl derivatives by liquid chromatography electrospray tandem mass spectrometry. Anal Biochem. 2008;375(1):105–114. doi: 10.1016/j.ab.2007.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cole JR, Zagorevski D, Spink DC. High-resolution analysis of the pyridine-3-sulfonyl derivatives of 17β-Estadiol and its metabolites by Orbitrap-mass spectrometry. J Am Soc Mass Spectrom. 2009;20(5, S1):S12. [Google Scholar]
- 40.Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82(13):1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 41.Rizzino A. Soft agar growth assays for transforming growth factors and mitogenic peptides. Methods Enzymol. 1987;146:341–352. doi: 10.1016/s0076-6879(87)46035-7. [DOI] [PubMed] [Google Scholar]
- 42.Albini A, Iwamoto Y, Kleinman HK, et al. A rapid in vitro assay for quantitating the invasive potential of tumor cells. Cancer Res. 1987;47(12):3239–3245. [PubMed] [Google Scholar]
- 43.Pinkas J, Leder P. MEK1 signaling mediates transformation and metastasis of EpH4 mammary epithelial cells independent of an epithelial to mesenchymal transition. Cancer Res. 2002;62(16):4781–4790. [PubMed] [Google Scholar]
- 44.Spink BC, Bennett JA, Pentecost BT, et al. Long-term estrogen exposure promotes carcinogen bioactivation, induces persistent changes in gene expression, and enhances the tumorigenicity of MCF-7 human breast cancer cells. Toxicol Appl Pharmacol. 2009;240(3):355–366. doi: 10.1016/j.taap.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bennett JA, Zhu S, Pagano-Mirarchi A, Kellom TA, Jacobson HI. Alpha-fetoprotein derived from a human hepatoma prevents growth of estrogen-dependent human breast cancer xenografts. Clin Cancer Res. 1998;4(11):2877–2884. [PubMed] [Google Scholar]
- 46.Tjorve E, Tjorve KM. A unified approach to the Richards-model family for use in growth analyses: why we need only two model forms. J Theor Biol. 2010;267(3):417–425. doi: 10.1016/j.jtbi.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 47.Laborda J. 36B4 cDNA used as an estradiol-independent mRNA control is the cDNA for human acidic ribosomal phosphoprotein PO. Nucleic Acids Res. 1991;19(14):3998. doi: 10.1093/nar/19.14.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gozgit JM, Pentecost BT, Marconi SA, Otis CN, Wu C, Arcaro KF. Use of an aggressive MCF-7 cell line variant, TMX2-28, to study cell invasion in breast cancer. Mol Cancer Res. 2006;4(12):905–913. doi: 10.1158/1541-7786.MCR-06-0147. [DOI] [PubMed] [Google Scholar]
- 49.Shimizu Y, Nakatsuru Y, Ichinose M, et al. Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. 2000;97(2):779–782. doi: 10.1073/pnas.97.2.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Belguise K, Guo S, Yang S, et al. Green tea polyphenols reverse cooperation between c-Rel and CK2 that induces the aryl hydrocarbon receptor, slug, and an invasive phenotype. Cancer Res. 2007;67(24):11742–11750. doi: 10.1158/0008-5472.CAN-07-2730. [DOI] [PubMed] [Google Scholar]
- 51.Dietrich C, Kaina B. The aryl hydrocarbon receptor (AhR) in the regulation of cell-cell contact and tumor growth. Carcinogenesis. 2010;31(8):1319–1328. doi: 10.1093/carcin/bgq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fritz WA, Lin TM, Cardiff RD, Peterson RE. The aryl hydrocarbon receptor inhibits prostate carcinogenesis in TRAMP mice. Carcinogenesis. 2007;28(2):497–505. doi: 10.1093/carcin/bgl179. [DOI] [PubMed] [Google Scholar]
- 53.Fan Y, Boivin GP, Knudsen ES, Nebert DW, Xia Y, Puga A. The aryl hydrocarbon receptor functions as a tumor suppressor of liver carcinogenesis. Cancer Res. 70(1):212–220. doi: 10.1158/0008-5472.CAN-09-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Turesky RJ. Formation and biochemistry of carcinogenic heterocyclic aromatic amines in cooked meats. Toxicol Lett. 2007;168(3):219–227. doi: 10.1016/j.toxlet.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 55.Cavalieri E, Chakravarti D, Guttenplan J, et al. Catechol estrogen quinones as initiators of breast and other human cancers: implications for biomarkers of susceptibility and cancer prevention. Biochim Biophys Acta. 2006;1766(1):63–78. doi: 10.1016/j.bbcan.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 56.Spink BC, Katz BH, Hussain MM, et al. SULT1A1 catalyzes 2-methoxyestradiol sulfonation in MCF-7 breast cancer cells. Carcinogenesis. 2000;21(11):1947–1957. doi: 10.1093/carcin/21.11.1947. [DOI] [PubMed] [Google Scholar]
- 57.Spink DC, Eugster HP, Lincoln DW, 2nd, et al. 17β-estradiol hydroxylation catalyzed by human cytochrome P450 1A1: a comparison of the activities induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin in MCF-7 cells with those from heterologous expression of the cDNA. Arch Biochem Biophys. 1992;293(2):342–348. doi: 10.1016/0003-9861(92)90404-k. [DOI] [PubMed] [Google Scholar]
- 58.Celius T, Matthews J. Functional analysis of six human aryl hydrocarbon receptor variants in human breast cancer and mouse hepatoma cell lines. Toxicology. 2010;277(1-3):59–65.3. doi: 10.1016/j.tox.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 59.Abdelrahim M, Smith R, 3rd, Safe S. Aryl hydrocarbon receptor gene silencing with small inhibitory RNA differentially modulates Ah-responsiveness in MCF-7 and HepG2 cancer cells. Mol Pharmacol. 2003;63(6):1373–1381. doi: 10.1124/mol.63.6.1373. [DOI] [PubMed] [Google Scholar]
- 60.Proia TA, Keller PJ, Gupta PB, et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell. 8(2):149–163. doi: 10.1016/j.stem.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sarrió D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008;68(4):989–997. doi: 10.1158/0008-5472.CAN-07-2017. [DOI] [PubMed] [Google Scholar]