

Abstract
This study focuses on determining whether the combination of NYP-BKM120 (BKM120) and RAD001 exerts enhanced therapeutic effect against lung cancer. The combination of BKM120 and RAD001 exerted synergistic inhibitory effects on the growth of lung cancer cells both in culture and in mouse xenograft model. This combination abrogated RAD001-induced Akt phosphorylation and exerted enhanced suppressive effect on 4EBP1 phosphorylation. Collectively, we suggest that the combination of RAD001 and BKM120 may be an effective regimen for treatment of lung cancer, hence warranting further evaluation of the combination in the clinic.
Keywords: RAD001, BKM120, PI3 kinase, mTOR, lung cancer
1. Introduction
Aberrant activation of the phosphoinositide 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling pathway occurs in many types of cancers including non-small cell lung cancer (NSCLC), largely due to frequent mutations of K-Ras, PTEN, PIK3CA, LKB1 and/or epidermal growth factor receptor (EGFR). Therefore, the PI3K/Akt/mTOR signaling pathway has become an area of intensive research and has attracted extensive attention for drug discovery [1; 2; 3; 4]. Consequently, NVP-BKM120 (BKM120) and RAD001 (Everolimus), two inhibitors of this signaling pathway, among many others, have been developed and are currently used or being evaluated for cancer therapy in the clinic.
BKM120, a 2,6-dimorpholino pyrimidine derivative (Fig. 1A), is a novel, potent and highly selective pan-class I PI3K inhibitor. In preclinical studies, it has been shown to be active in suppressing proliferation and inducing apoptosis of cancer cell lines and in inhibiting the growth of human tumors (xenografts) in mice at tolerated doses [5; 6; 7]. Clinically, BKM120 at the maximum-tolerated dose has been shown to be safe with a favorable pharmacokinetic profile, clear evidence of target inhibition and preliminary antitumor activity in a recently completed phase I trial [8].
Fig. 1. BKM120 (A) effectively inhibits the growth of human NSCLC cells (B) and induces G1 arrest (C).
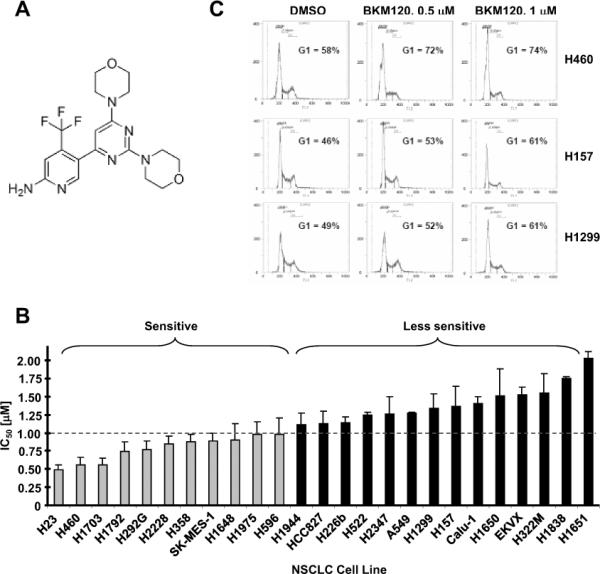
A, Chemical structure of BKM120. B, The panel of human NSCLC cell lines as indicated were seeded in 96-well plates and then treated with different concentrations of BKM120 ranging from 0.125 to 4 μM as indicated on the second day. After 3 days, cell numbers were estimated using SRB assay. IC50s, which were estimated from the growth inhibition curves, are presented as the means of 2–4 independent assays ± SDs. An arbitrary cut-off was used to define BKM120-sensitive (≤ 1 μM) and less sensitive (> 1 μM) cell lines. C, The given cell lines were treated with the indicated concentrations of BKM120 for 48 h and then harvested for cell cycle analysis.
RAD001 (Everolimus or Afinitor) is an analogue of rapamycin with similar function to rapamycin as an allosteric inhibitor of mTOR. In patients with advanced renal cell cancer previously treated with VEGF targeted agents, RAD001 improves progression-free survival [9]. Recently it has also been shown to significantly prolong progression-free survival of patients with progressive advanced pancreatic neuroendocrine tumors with a low rate of severe adverse events [10]. Thus, RAD001 has been approved by the US Food and Drug Administration for these indications. In many other solid organ malignancies, RAD001 and other rapamycin analogues (rapalogs) exert modest anti-cancer effects, which though promising, are not sufficient to warrant monotherapy with these agents [11].
Like rapamycin, RAD001 causes Akt activation in human cancer cells including NSCLC cells and in tumor biopsies while inhibiting mTOR signaling [12; 13]. Akt activation during mTOR inhibition by rapalogs is likely PI3K-dependent and may contribute to attenuation of the therapeutic efficacy of rapalogs [13; 14]. Thus, co-inhibition of both PI3K and mTOR might be an effective therapeutic strategy as we suggested previously [12; 15]. In this study, we focused on determining whether the combination of BKM120 and RAD001 exerts enhanced therapeutic efficacy against NSCLC and found that this combination was more effective than either agent alone in inhibiting the growth of NSCLC cells both in vitro and in vivo.
2. Materials and Methods
2.1, Reagents
RAD001 and BKM120 were supplied by Novartis Pharmaceuticals Corporation (East Hanover, NJ), dissolved in DMSO and stored at −80°C. Rabbit polyclonal anti-actin antibody was purchased from Sigma Chemical Co. (St. Louis, MO). Antibodies against Akt, p-Akt (S473), p-S6 (S235/S236), S6, p-4EBP1 (Thr37/46), 4EBP1, and eIF4E were purchased from Cell Signaling Technology, Inc. (Beverly, MA). Rabbit monoclonal anti p-eIF4E (S209) was purchased from Epitomics (Burlingame, CA). Goat polyclonal anti-p21 antibody was purchased from Santa Cruz Biotechnology, Inc (Santa Cruz, CA).
2.2, Cell lines and cell culture
The human NSCLC cell lines used in this study were either provided by Dr. R. Lotan (M.D. Anderson Cancer Center, Houston, TX) [16] or purchased from the American Type Culture Collection ATCC (Manassas, VA). Except for H157 and A549 cells, which were recently authenticated by Genetica DNA Laboratories, Inc. (Cincinnati, OH) through analyzing short tandem repeat DNA profile, other cell lines have not been authenticated. The genetic alterations in this cell lines were summarized in supplementary table S1. These cell lines were all grown in monolayer culture in RPMI 1640 medium supplemented with 5% fetal bovine serum at 37°C in a humidified atmosphere consisting of 5% CO2 and 95% air.
2.3, Adenoviral infection of cancer cells
Adenovirus harboring an empty vector (Ad-CMV) or a constitutively activated form of Akt (myristoylated Akt; Ad-myr-Akt) and cell infection were described previously [17].
2.4, Measurement of cell numbers
Cells were cultured in 96-well cell culture plates and treated the next day with the agents indicated. Cell number was measured using the sulforhodamine B (SRB) assay, as previously described [16]. Combination index (CI) for drug interaction (e.g., synergy) was calculated using the CompuSyn software (ComboSyn, Inc.; Paramus, NJ).
2.5, Colony formation assay
The effects of the given drugs on colony formation on plates were measured as previously described [18].
2.6, Cell cycle analysis
Cells were harvested after a given treatment and stained with propidium iodide for cell cycle analysis as described previously [19; 20].
2.7, Western blot analysis
Preparation of whole-cell protein lysates and Western blot analysis were described previously [21; 22].
2.8, Lung cancer xenografts and treatments
Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Emory University (DAR-2001130-102714). Five- to six-week old male athymic (nu/nu) mice were ordered from Harlan (Indianapolis, IN) and housed under pathogen-free conditions in microisolator cages with laboratory chow and water ad libitum. A549 cells at 8 × 106 in serum-free medium were injected s.c. into the flank region of nude mice. When tumors reached a size of approximately 100 mm3, the mice were randomized into four groups (n = 6/group) according to tumor volumes and body weights for the following treatments: vehicle control, BKM120 [(15 mg/kg/day, oral gavage (og)], RAD001 (4 mg/kg/day, og), and their combination. Tumor volumes were measured using caliper measurements once every two days and calculated with the formula V = π(length × width2)/6.
2.9, Statistical analysis
The statistical significance of differences between two groups was analyzed with two-sided unpaired Student's t tests (for equal variances) or with Welch's corrected t test (unequal variances) by use of Graphpad InStat 3 software. Results were considered to be statistically significant at P < 0.05.
3. Results
3.1, BKM120 effectively inhibits the growth of human NSCLC cells
The effects of BKM120 on the growth of human NSCLC cells have not been reported. Thus, we first determined whether BKM120 effectively inhibits the growth of NSCLC cells and whether certain genetic alterations (particularly Ras, PIK3CA and/or PTEN) impact cell sensitivity to BKM120. To this end, we treated 25 human NSCLC cell lines with different gene mutations, including K-Ras, p53, LKB1, PIK3CA, PTEN, EGFR and CDKN2A, for 3 days and then measured cell numbers. BKM120 reduced cell numbers in a concentration-dependent manner with IC50s (i.e., a concentration that decreases cell number by 50%) ranging from 0.5 μM to 2 μM (Fig. 1B and supplementary Fig. S1). We failed to identify an obvious association or correlation between the mutation of K-Ras, p53, LKB1, PTEN, EGFR or CDKN2A and cell sensitivity to BKM120. Thus, BKM120 effectively inhibits the growth of NSCLC cells irrespective of these gene mutations. Among these cell lines, there are 3 cell lines (H460, H1975 and H596) carrying a PIK3CA mutation. These cell lines all fell into the BKM120-sensitive cell line group if we used 1 μM IC50 as a cutoff to arbitrarily divide BKM-sensitive and -insensitive cell lines (Fig. 1B).
3.2, BKM120 induces G1 arrest at low concentration ranges
To explore the mechanism by which BKM120 suppresses the growth of NSCLC cells, we then examined the effects of BKM120 on cell cycle and apoptosis. At concentration ranges ≤ 1 μM (e.g., 0.5 and 1 μM), BKM120 did not induce apoptosis (data not shown), but induced G1 arrest (Fig. 1C). At high concentrations (e.g., 2 and 4 μM), we did see that BKM120 induced apoptosis (our unpublished data). Thus, it is clear that BKM120 at low concentration ranges inhibits the growth of human NSCLC cells primarily through inducing G1 arrest.
3.3, BKM120 effectively inhibits the PI3K/Akt/mTOR signaling pathway
Given that BKM120 is a pan PI3K inhibitor, we next determined whether it effectively inhibits the PI3K/Akt/mTOR axis in NSCLC cells by examining the phosphorylation of several key proteins involved in this pathway, including Akt, p70S6K, S6 and 4EBP1. BKM120 at 1 μM reduced the levels of p-Akt, p-p70S6K, p-S6 and p-4EBP1 in all tested cell lines (Fig. 2A). These inhibitory effects could be observed at 3 h post BKM120 treatment and were sustained through the entire treatment period (up to 16 h) (Fig. 2B). Therefore, BKM exerts rapid and potent suppressive effects on PI3K/Akt/mTOR signaling in human NSCLC cells.
Fig. 2. BKM120 effectively inhibits the Akt/mTOR signaling pathway (A and B) and elevated levels of p-Akt decrease cell sensitivity to BKM120 (C and D).

A and B, The given cell lines were treated with 1 μM BKM120 for 24 h (A) or for different times as indicated (B). The cells were then harvested for preparation of whole cell protein lysates and subsequent Western blot analysis. C, Whole cell protein lysates were prepared from the indicated cell lines and used for Western blotting for detect of the indicated proteins. D, H460 cells were infected with the indicated adenoviruses for 48 h. The cells were seeded in 96-well plates and the next day treated with different concentrations of BKM120 for 3 days and subsequent SRB assay. The left cells were also used for preparation of whole-cell protein lysates and subsequent Western blot analysis to detect Akt expression. The results are means of four replicate or triplicate determinations ± SDs. *, P < 0.05 compared with Ad-CMV group.
3.4, High levels of activated Akt may be associated with decreased sensitivity to BKM120
We also compared the basal levels of p-Akt, p-S6 and p-4EBP1 between the cell lines that were most sensitive to BKM120 (H23, H460, H1703, H292, H2228 and H358) and those that were least sensitive (H157, Calu-1, H1650, EKVX, H322, H1838 and H1651). We detected p-Akt in 9 of 14 cell lines, p-S6 in 10 of 14 cell lines and p-4EBP1 in 13 of 14 cell lines (Fig. 2C). We did not identify any apparent differences in the levels of p-S6 and p-4EBP1 between the sensitive and insensitive cell lines. Interestingly, we found that more of the insensitive cell lines (6/7; 86%) than the sensitive cell lines (3/7; 43%) were positive for p-Akt expression. By correlation analysis, we found a trend in which p-Akt levels were inversely correlated with cell sensitivities to BKM120, although this was not statistically significant (P = 0.083).
To further demonstrate the impact of Akt activation on cell sensitivity to BKM120, we enforced activation of Akt by expressing myr-Akt through adenoviral infection in the sensitive cell line H460 and then examined its impact on cell response to BKM120. Compared with cells infected with the control adenovirus Ad-CMV, cells infected with Ad-myr-Akt were clearly less sensitive to BKM120 (Fig. 2D), indicating that high levels of activated Akt indeed reduce cell sensitivity to BKM120. We noted the difference of cell sensitivities between cells infected with Ad-CMV and Ad-myr-Akt was limited albeit being statistically significant. However the difference was reproducible, implying that other unknown factors are also involved in determining cell sensitivity to BKM120.
3.5, The combination of BKM120 and RAD001 synergistically inhibits the growth of NSCLC cells and enhances G1 arrest
We previously demonstrated that the combination of rapamycin or RAD001 with the PI3K inhibitor LY294002 resulted in enhanced growth-inhibitory effects against NSCLC cells both in vitro and in vivo [12; 14]. We then asked whether the combination of BKM120 and RAD001 also exerts augmented anti-cancer activity in NSCLC cells. As shown in Fig. 3, the combination of low concentrations of RAD001 (≤ 1 nM) and BKM120 (≤ 0.5 μM) was more effective than each agent alone in inhibiting the growth of tested NSCLC cell lines. The CIs for most combinations were < 1, indicating synergistic effects on inhibiting the growth of NSCLC cells. In a long-term colony formation assay, which allows us to repeat the treatments for a long time (e.g., 12 days), RAD001 at the tested concentration range (0.25–1 nM) and BKM120 at 0.5 μM alone had partial effects on suppression of colony formation of the NSCLC cells; however the combination either almost eliminated colony formation (e.g., A549) or drastically reduced the colony numbers (Fig. 4A). Thus, it is clear that the combination is much more effective than either single agent in inhibiting the colony formation and growth of NSCLC cells.
Fig. 3. The combination of BKM120 and RAD001 synergistically inhibits the growth of NSCLC cells.

The indicated cell lines were seeded in 96-well plates and then treated next day with different concentrations of BKM120 (BKM), RAD001 (RAD) and their respective combinations as indicated. After 3 days, the cell numbers were estimated using the SRB assay and CIs were calculated with CompuSyn software as labeled inside the graphs. The results are means of four replicate determinations ± SDs.
Fig. 4. The combination of BKM120 and RAD001 exerts enhanced effects on inhibiting colony formation and growth (A) and on inducing G1 arrest (B).

A, The indicated cell lines at a density of approximately 250 cells/well were seeded in 12-well plates. On the second day, the cells were treated with different concentrations of RAD001 (RAD) as indicated, 0.5 μM BKM120 or their combinations. After 12 days, the plates were stained for the formation of cell colonies with crystal violet dye. The colonies were then counted and pictures of the colonies were taken using a digital camera. Columns, means of triplicate determinations; bars ± SDs. *, P < 0.01; **, P < 0.001 compared with both RAD001 alone and BKM120 alone. B, The given cell lines were treated with 1 nM RAD001, 1 μM BKM120 (BKM) and their combination. After 72 h, the cells were harvested for cell cycle analysis.
In agreement, the combination of BKM120 and RAD001 was more potent than each single agent in inducing G1 arrest (Fig. 4B). Under these combinatorial conditions, we did not detect increased apoptosis in any of the tested cell lines (H157, Calu-1 and A549). Thus, we believe that enhanced G1 arrest contributes to augmented growth-inhibitory effects induced by the combination.
3.6, The combination of BKM120 and RAD001 blocks RAD001-induced phosphorylation of Akt and eIF4E and exerts enhanced effects on suppression of 4EBP1 phosphorylation and on p21 induction
To understand the mechanisms by which the combination of BKM120 and RAD001 exert enhanced anticancer activity, we analyzed the effects of the combination on the phosphorylation of Akt and eIF4E, which are known to be induced by rapalogs including RAD001 [12; 14; 23], and on mTOR signaling. As presented in Fig. 5, RAD001 at 1 nM alone increased the levels of both p-Akt and p-eIF4E in the 3 tested cell lines; however, it failed to do so in the presence of 0.5 μM BKM120. RAD001 alone reduced p-S6 levels to undetectable levels in all the tested cell lines; as did BKM120 in Calu-1 and A549 cells. The combination of BKM120 and RAD001 showed similar activity in reducing p-S6 levels as either single agent did. In contrast, both RAD001 and BKM120 at the tested concentrations had no or weak effects on reducing p-4EBP1 levels; however, their combination effectively decreased p-4EBP1 levels. Thus, it is clear that the combination of BKM120 and RAD001 displays enhanced suppressive effects on 4EBP1 phosphorylation.
Fig. 5. Effects of BKM120 and RAD001 combination on Akt phosphorylation, mTOR signaling and p21 expression.

The indicated cell lines were plated in 10 cm-diameter cell culture dishes and treated next day with 1 nM RAD001 in the absence and presence of 1 μM BKM120 for 24 h. The cells were then harvested for preparation of whole-cell protein lysates and subsequent Western blot analysis to detect the indicated proteins.
3.7, The combination of BKM120 and RAD001 exerts augmented activity against the growth of NSCLC xenografts in nude mice
Because of the promising growth-inhibitory effects of the BKM120 and RAD001 combination in NSCLC cells in vitro, we lastly validated the efficacy of the combination against the growth of NSCLC tumors in mice. The combination of BKM120 and RAD001 significantly inhibited the growth of A549 xenografts (P < 0.01 compared with control group), whereas RAD001 or BKM120 alone at the tested doses only minimally or partially inhibited the growth of A549 xenografts as measured by both tumor sizes and weights. Moreover, the combination was significantly more potent than each single agent in inhibiting the growth of the xenografts (P < 0.05 or 0.01 compared with RAD001 or BKM120 group) (Figs. 6A and 6B). These in vivo data further demonstrate that the combination of BKM120 and RAD001 displays augmented anticancer activity. During the treatment, we did not see a significant effect of the combination on body weight loss of the mice (Fig. 6C), suggesting that the combination is well tolerated.
Fig. 6. The combination of BKM120 and RAD001 is significantly more effective than each single agent in suppressing the growth of NSCLC xenografts.

A549 xenografts were treated (once a day) with vehicle control, RAD001 (4 mg/kg, og), BMK120 (15 mg/kg, og) and their combination (BKM + RAD) starting on the same day after grouping. Tumor sizes (A) and body weight (C) were measured once every two days. After 17 days, the mice were sacrificed and the tumors were removed and weighed (B). Each measurement is a mean ± SD (n = 6). Moreover, whole protein cell lysates were also prepared randomly from 3 tumors in each group for Western blotting to detect the indicated proteins (D). *, P < 0.05 and **, P < 0.01 compared with control group; #, P < 0.05 or 0.01 compared with either RAD001 alone or BKM120 alone group; &, P < 0.05 compared with control group.
By analyzing tumor tissues, we detected increased levels of p-Akt in xenograft tumors treated with RAD001, but decreased levels of p-Akt in tumors exposed to either BKM120 or the combination of BKM120 and RAD001 (Fig. 6D), indicating that the presence of BKM120 abrogates the effect of RAD001 on activation of Akt in vivo. Moreover, we found that either BKM120 or RAD001 alone partially decreased p-4EBP1 levels; however, their combination was much more potent than either single agent in reducing p-4EBP1 levels in xenograft tumors (Fig. 6D), demonstrating that the combination of BKM120 and RAD001 also exerts an augmented effect on suppression of 4EBP1 phosphorylation in vivo.
4. Discussion
Using a large panel of human NSCLC cell lines, we have demonstrated that BKM120 effectively inhibits the growth of human NSCLC cells. It has been recently suggested that cancer cell lines with PIK3CA mutation, but not with PTEN or K-Ras mutation, respond better than those without the mutation to BKM120 [5]. In another recent study, it has been suggested that glioma cell lines with wild-type p53 are more sensitive than those with mutant p53 to BKM120 [7]. The 25 NSCLC cell lines used in this panel have known status of genes commonly mutated in cancer including Ras, p53, LKB1, PIK3CA, PTEN, EGFR and CDKN2A. We did not identify clear associations between cell sensitivity to BKM120 and status of K-Ras, p53, EGFR, PTEN, LKB1 or CDKN2A mutation. Among these 25 tested cell lines, only 3 have PIK3CA mutation (H460, H1975 and H596). Interestingly, these cell lines are all in the BKM120-sensitive cell line group. This preliminary data seems in line with the recent finding that cancer cell lines with PIK3CA mutation respond better than those without the mutation to BKM120 [5]. However, we should be aware that the mutation of PIK3CA is rare in human NSCLC [24; 25]. Thus, the impact of PIK3CA on cell response to BKM120 in NSCLC should be limited.
By comparing the basal levels of p-Akt, p-S6 and p-4EBP1 between the NSCLC cell lines that were most and least sensitive to BKM120, we found that more of the sensitive than insensitive cell lines expressed p-Akt, suggesting a trend that higher basal levels of p-Akt are associated with resistance to BKM120, although this correlation was not statistically significant (P = 0.083). Consistently, we found that enforced expression of the constitutively activated Akt, Myr-Akt, attenuated BKM120's growth-inhibitory effects (Fig. 2). These observations may make sense since BKM120 selectively inhibits PI3K/Akt signaling and overactivation of this signaling pathway may counteract the therapeutic efficacy of BKM120. Further study in this regard is warranted.
We noted that BKM120 had comparable potencies across cell lines in inhibiting components of the Akt/mTOR signaling pathway (e.g., suppression of p-Akt, p-S6 and p-4EBP1; Fig. 2), but showed different efficacies in suppressing the growth of NSCLC cells (e.g., H1703 vs. H1651; Fig. 1). Whether this suggests that effective suppression of the Akt/mTOR signaling pathway may not be the exclusive mechanism accounting for BKM120's therapeutic activity also warrants further investigation.
In this study, we have clearly shown that the combination of low concentrations of BKM120 and RAD001 exerts enhanced effects on suppressing the growth of human NSCLC cells in cell culture (Figs. 3 and 4) and on inhibiting the growth of NSCLC xenografts in mice (Fig. 6). To the best of our knowledge, this is the first preclinical study to test the therapeutic efficacy of the BKM120 and RAD001 combination against cancer. These results thus provide a strong rationale and support for our ongoing phase I clinical trial to evaluate the combination of BKM120 and RAD001.
Our previous studies have suggested that increased phosphorylation of both Akt and eIF4E during rapalog treatment is PI3K-dependent. Consequently, this may counteract the therapeutic efficacy of rapalogs and may also be associated with the development of resistance to rapalogs. Accordingly, a reduction in Akt and/or eIF4E phosphorylation enhances the anticancer activity of rapalogs [12; 14; 26]. In this study, BKM120 clearly blocks the RAD001-induced increase in Akt phosphorylation and enhances RAD001's effect on suppressing 4EBP1 phosphorylation both in vitro and in vivo (Figs. 5 and 6D). Moreover, BKM120 inhibits RAD001-induced eIF4E phosphorylation (Fig. 5). Thus, it is reasonable to attribute the augmented anticancer activity of the BKM120 and RAD001 combination to blockage of the RAD001-induced increase in Akt and eIF4E phosphorylation and augmented suppression of 4EBP1 phosphorylation.
In this study, we have clearly demonstrated that the combination of BKM120 and RAD001 augments the arrest of cancer cells in the G1 phase (Fig. 4). This is in agreement with the finding that the BKM120 and RAD001 combination enhances the induction of p21 expression (Fig. 5). Given the critical role of p21 in mediating cell cycle G1 arrest [27], this provides an explanation for the enhanced induction of G1 arrest by the combination. Since we did not detect increased apoptosis in cells exposed to the combination of BKM120 and RAD001, we believe that the combination of BKM120 and RAD001 enhances p21-dependent G1 arrest, leading to augmented inhibitory effects on the growth of NSCLC cells.
In this study, the combination of low concentrations of BKM120 and RAD001 induced G1 arrest, but not apparent apoptosis. Consistently, the combination did slow down the growth of tumor xenografts, but not induce their shrinkage in vivo (Fig. 6). Thus, it is very likely that the combination of BKM120 and RAD001 will be cytostatic for treatment of cancer in the clinic, which should be considered when conducting the clinical trials.
Supplementary Material
Acknowledgments
Acknowledgements We thank Dr. A. Hammond in our department for editing the manuscript. This study is supported by the Georgia Cancer Coalition Distinguished Cancer Scholar award and NIH R01 CA118450 (S-Y Sun), R01 CA160522 (S-Y Sun) and P01 CA116676 (Project 1 to FR Khuri and S-Y Sun). TK Owonikoko, SS Ramalingam, FR Khuri and S-Y Sun are Georgia Cancer Coalition Distinguished Cancer Scholars.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement None
References
- [1].Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- [2].Chen YL, Law PY, Loh HH. Inhibition of PI3K/Akt signaling: an emerging paradigm for targeted cancer therapy. Curr Med Chem Anti-Canc Agents. 2005;5:575–589. doi: 10.2174/156801105774574649. [DOI] [PubMed] [Google Scholar]
- [3].LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat. 2008;11:32–50. doi: 10.1016/j.drup.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chiang GG, Abraham RT. Targeting the mTOR signaling network in cancer. Trends Mol Med. 2007;13:433–442. doi: 10.1016/j.molmed.2007.08.001. [DOI] [PubMed] [Google Scholar]
- [5].Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, Schnell C, Guthy D, Nagel T, Wiesmann M, Brachmann S, Fritsch C, Dorsch M, Chene P, Shoemaker K, De Pover A, Menezes D, Martiny-Baron G, Fabbro D, Wilson CJ, Schlegel R, Hofmann F, Garcia-Echeverria C, Sellers WR, Voliva CF. Identification and Characterization of NVP-BKM120, an Orally Available Pan-Class I PI3-Kinase Inhibitor. Mol Cancer Ther. 2012;11:317–328. doi: 10.1158/1535-7163.MCT-11-0474. [DOI] [PubMed] [Google Scholar]
- [6].Zheng Y, Yang J, Qian J, Zhang L, Lu Y, Li H, Lin H, Lan Y, Liu Z, He J, Hong S, Thomas S, Shah J, Baladandayuthapani V, Kwak LW, Yi Q. Novel phosphatidylinositol 3-kinase inhibitor NVP-BKM120 induces apoptosis in myeloma cells and shows synergistic anti-myeloma activity with dexamethasone. J Mol Med (Berl) 2011 doi: 10.1007/s00109-011-0849-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Koul D, Fu J, Shen R, LaFortune TA, Wang S, Tiao N, Kim YW, Liu JL, Ramnarian D, Yuan Y, Garcia-Echevrria C, Maira SM, Yung WK. Antitumor activity of NVP-BKM120--a selective pan class I PI3 kinase inhibitor showed differential forms of cell death based on p53 status of glioma cells. Clin Cancer Res. 2012;18:184–195. doi: 10.1158/1078-0432.CCR-11-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, Demanse D, De Buck SS, Ru QC, Peters M, Goldbrunner M, Baselga J. Phase I, Dose-Escalation Study of BKM120, an Oral Pan-Class I PI3K Inhibitor, in Patients With Advanced Solid Tumors. J Clin Oncol. 2012;30:282–290. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- [9].Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grunwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, Lebwohl D, Ravaud A. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–456. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- [10].Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, de Vries EG, Tomassetti P, Pavel ME, Hoosen S, Haas T, Lincy J, Lebwohl D, Oberg K. Everolimus for advanced pancreatic neuroendocrine tumors. The New England journal of medicine. 2011;364:514–523. doi: 10.1056/NEJMoa1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Abraham RT, Gibbons JJ. The mammalian target of rapamycin signaling pathway: twists and turns in the road to cancer therapy. Clin Cancer Res. 2007;13:3109–3114. doi: 10.1158/1078-0432.CCR-06-2798. [DOI] [PubMed] [Google Scholar]
- [12].Wang X, Yue P, Kim YA, Fu H, Khuri FR, Sun SY. Enhancing mammalian target of rapamycin (mTOR)-targeted cancer therapy by preventing mTOR/raptor inhibition-initiated, mTOR/rictor-independent Akt activation. Cancer Res. 2008;68:7409–7418. doi: 10.1158/0008-5472.CAN-08-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, Khuri FR. Activation of Akt and eIF4E Survival Pathways by Rapamycin-Mediated Mammalian Target of Rapamycin Inhibition. Cancer Res. 2005;65:7052–7058. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- [15].Wang X, Sun SY. Enhancing mTOR-targeted cancer therapy. Expert Opin Ther Targets. 2009;13:1193–1203. doi: 10.1517/14728220903225008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sun SY, Yue P, Dawson MI, Shroot B, Michel S, Lamph WW, Heyman RA, Teng M, Chandraratna RA, Shudo K, Hong WK, Lotan R. Differential effects of synthetic nuclear retinoid receptor-selective retinoids on the growth of human non-small cell lung carcinoma cells. Cancer Res. 1997;57:4931–4939. [PubMed] [Google Scholar]
- [17].Elrod HA, Lin YD, Yue P, Wang X, Lonial S, Khuri FR, Sun SY. The alkylphospholipid perifosine induces apoptosis of human lung cancer cells requiring inhibition of Akt and activation of the extrinsic apoptotic pathway. Mol Cancer Ther. 2007;6:2029–2038. doi: 10.1158/1535-7163.MCT-07-0004. [DOI] [PubMed] [Google Scholar]
- [18].Wang X, Hawk N, Yue P, Kauh J, Ramalingam SS, Fu H, Khuri FR, Sun SY. Overcoming mTOR inhibition-induced paradoxical activation of survival signaling pathways enhances mTOR inhibitors' anticancer efficacy. Cancer Biol Ther. 2008;7:1952–1958. doi: 10.4161/cbt.7.12.6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sun SY, Yue P, Shroot B, Hong WK, Lotan R. Induction of apoptosis in human non-small cell lung carcinoma cells by the novel synthetic retinoid CD437. J Cell Physiol. 1997;173:279–284. doi: 10.1002/(SICI)1097-4652(199711)173:2<279::AID-JCP36>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- [20].Sun SY, Zhou Z, Wang R, Fu H, Khuri FR. The farnesyltransferase inhibitor Lonafarnib induces growth arrest or apoptosis of human lung cancer cells without downregulation of Akt. Cancer Biol Ther. 2004;3:1092–1098. doi: 10.4161/cbt.3.11.1176. discussion 1099-1101. [DOI] [PubMed] [Google Scholar]
- [21].Liu X, Yue P, Zhou Z, Khuri FR, Sun SY. Death receptor regulation and celecoxib-induced apoptosis in human lung cancer cells. J Natl Cancer Inst. 2004;96:1769–1780. doi: 10.1093/jnci/djh322. [DOI] [PubMed] [Google Scholar]
- [22].Sun SY, Yue P, Wu GS, El-Deiry WS, Shroot B, Hong WK, Lotan R. Mechanisms of apoptosis induced by the synthetic retinoid CD437 in human non-small cell lung carcinoma cells. Oncogene. 1999;18:2357–2365. doi: 10.1038/sj.onc.1202543. [DOI] [PubMed] [Google Scholar]
- [23].Wang X, Yue P, Chan CB, Ye K, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Fu H, Khuri FR, Sun SY. Inhibition of mammalian target of rapamycin induces phosphatidylinositol 3 kinase-dependent and Mnk-mediated eIF4E phosphorylation. Mol Cell Biol. 2007 doi: 10.1128/MCB.00760-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12:175–180. doi: 10.1016/S1470-2045(10)70087-5. [DOI] [PubMed] [Google Scholar]
- [25].Sanders HR, Albitar M. Somatic mutations of signaling genes in non-small-cell lung cancer. Cancer Genet Cytogenet. 2010;203:7–15. doi: 10.1016/j.cancergencyto.2010.07.134. [DOI] [PubMed] [Google Scholar]
- [26].Wang X, Yue P, Chan CB, Ye K, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Fu H, Khuri FR, Sun SY. Inhibition of mammalian target of rapamycin induces phosphatidylinositol 3-kinase-dependent and Mnk-mediated eukaryotic translation initiation factor 4E phosphorylation. Mol Cell Biol. 2007;27:7405–7413. doi: 10.1128/MCB.00760-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.