

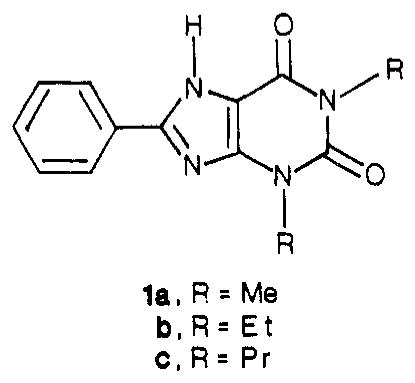
Abstract
Amide derivatives of a carboxylic acid congener of 1,3-dialkylxanthine, having a 4-[(carboxymethyl)oxy]phenyl substituent at the 8-position, have been synthesized in order to identify potent antagonists at A2-adenosine receptors stimulatory to adenylate cyclase in platelets. Distal structural features of amide-linked chains and the size of the 1,3-dialkyl groups have been varied. 1,3-Diethyl groups, more than 1,3-dimethyl or 1,3-dipropyl groups, favor A2 potency, even in the presence of extended chains attached at the 8-(p-substituted-phenyl) position. Polar groups, such as amines, on the chain simultaneously enhance water solubility and A2 potency. Among the most potent A2 ligands are an amine congener, 8-[4-[[[[(2-aminoethyl)amino]carbonyl]methyl]oxy]phenyl]-1,3-diethylxanthine, and its D-lysyl conjugate, which have KB values of 21 and 23 nM, respectively, for the antagonism of N-ethyl-adenosine-5′-uronamide-stimulated adenylate cyclase activity in human platelet membranes. Strategies for the selection and tritiation of new radioligands for use in competitive binding assays at A2-adenosine receptors have been considered.
The synthesis of potent and/or selective analogues of caffeine and theophylline as antagonists at extracellular adenosine receptors remains a challenge. Two adenosine receptor subtypes have been delineated on the basis of inhibition (A1) or stimulation (A2) of adenylate cyclase by adenosine and adenosine analogues. Numerous physiological functions have been shown to be regulated by either or both A1 and A2-adenosine receptors.1 In many organs and cell lines, a single-receptor subtype has been found to occur. For example, in platelets and PC12 pheochromocytoma cells the existing adenosine receptors appear to be all of the A2 subtype.2 Fat cells are considered to contain only the A1-receptor subtype.
Characterization of adenosine receptors with reversibly-binding radioligands has been carried out mainly with brain membranes, due to the high density of A1 receptors in that organ. A number of radioligands for A1 receptors have been reported. Tritiated N6-cycloalkyladenosines3,4 and tritiated (R)-N6-(phenylisopropyl)adenosine5 (PIA) having high specific activity are used regularly in competitive-binding assays to determine affinity of new analogues at A1 receptors. The choice of antagonist radioligands had been limited to [3H]-1,3-diethyl-8-phenylxanthine3 ([3H]-DPX, 1b), which has a Ki value at A1 receptors of about 60 nM. The recently introduced 8-[4-[[[[(2-aminoethyl)-amino]carbonyl]methyl]oxy]phenyl]-1,3-dipropylxanthine ([3H]XAC, 6c), a xanthine amine congener,6,7 has a higher affinity and specific activity than [3H]DPX, and its use overcomes a number of difficulties associated with the use of [3H]DPX as an antagonist radioligand for A1 receptors.
Until recently the screening of analogues at A2 receptors has relied on biological assays of adenylate cyclase, platelet aggregation, or in vivo models such as coronary vasodilation.2,8,14 A competitive binding assay at A2 receptors in striatal tissue using 3H-labeled N-ethyladenosine-5′-uronamide (NECA) as a radiotracer has been introduced.9 Although NECA is one of the most potent A2 agonists reported, it is not selective for that subtype, and it in fact binds with approximately 5–50-fold greater affinity at A1-adenosine receptors.10 Thus, in the striatum, where both adenosine-receptor subtypes occur, it is necessary to block the A1-receptor component of [3H]NECA binding by use of a selective A1 ligand9b or by pretreatment of the membranes with N-ethylmaleimide.9a However, there also is evidence that NECA can bind to sites that have characteristics different from those expected of A2 receptors coupled to adenylate cyclase, particularly in peripheral tissue.13,14 With the goal of finding a more convenient method for the study of A2 receptors, we have applied the functionalized congener approach,6,11 previously used in the design of A1-receptor radioligands, such as [3H]XAC, to the development of potential antagonist A2-receptor radioligands.
Results and Discussion
Although a number of moderately A2-selective antagonists have been reported,15 none are of sufficiently high affinity to serve as radiotracer for competitive-binding studies. For this reason, we approached the development of an A2-antagonist radioligand from the standpoint of maximizing A2 potency regardless of the selectivity ratio. At that point a highly potent candidate should be radiolabeled and its binding properties examined in a membrane preparation such as human platelets in which A2-but not A1-adenosine receptors are present. Subsequently, the platelet assay is to be used to study other potential A2-adenosine ligands by measuring their inhibition of binding of a well-characterized radioligand.
The classical approach to developing new drugs has been to explore the effect of structural modifications at or around a primary pharmacophore. We have developed a “functionalized congener” approach for adenosine-receptor ligands, by which a chemically functionalized chain is incorporated at a point that does not reduce biological activity. The resulting active congener can be joined covalently to a variety of moieties, including amino acids and peptides11 or solid supports for affinity chromatography, through a functional group such as an amine or a carboxylic acid. For xanthines as adenosine antagonists it has been shown6 that distal structural changes on a chain attached through the 8-(p-substituted-phenyl) position can modulate the potency and selectivity of the drugs, perhaps through interactions at sites on the receptor distal to the binding site for the pharmacophore. We now show the validity of this approach for xanthines as antagonists at A2-adenosine receptors.
The Ki values for reversal of 2-chloroadenosine-stimulated cyclic AMP production in guinea pig brain slices for a carboxylic acid congener, 2c, an ethyl ester, 3c, an amine congener, 6c (all R = Pr), and a p-toluidine, 7a (R = Me) were 34, 30, 49, and 20 nM, respectively. Furthermore, within a series of amino acid conjugates of 6c, the D-lysyl conjugate 11c displayed high potency, high water solubility, and stability to enzymatic hydrolysis. Since a dissociation constant in the range of 10−8 M would be acceptable for a radiotracer, these compounds constituted suitable lead compounds. As modifications of these xanthines designed to increases A2 potency we have explored changing the size of the 1,3-dialkyl groups and incorporating distal amino groups in the functionalized 8-phenyl moiety (see Table I for synthesis and characterization).
Table I.
Synthesis and Characterizatione of New Xanthine Derivatives (compound suffixes a, b, and c refer to R = Me, Et, and Pr, respectively)
| no. | % yield | mp, °C | formula | anal. |
|---|---|---|---|---|
| 2b | 67a | >310 | C17H18N4O5·H2O | C, H, N. |
| 3a | 61 | 294–296 | C17H18N4O5·1/4H2O | C, H, N |
| 3b | 86 | 267–269 | C19H22N4O5 | C, H, N |
| 4b | 71 | 275–277 | C20H24N4O5·1/2DMF | C, H, N |
| 5b | 88 | 300–302 | C19H23N5O4·1/2H2O | C, H, N |
| 6a | 99 | 255–258 | C17H20N6O4·1/4H2O | C, H, N |
| 6b | 92 | 233–235 | C19H24N6O4·H2O·DMF | C; H, Nb |
| 7b | 58 | >310 | C24H26N5O4·4/5H2O | C, H, N |
| 8a | 60 | >310 | C24H23N5O6 | H, N; Cc |
| 8b | 92 | 310–313 | C26H27N6O6·1/2H2O | C, H, N |
| 9a | 94 | 280 dec | C25H27N7O5 | H, N; Cd |
| 9b | 77 | 273–278d | C27H31N7O5·3/4H2O | C, H, N |
| 10b | 79 | 215–219 | C38H50N8O9·1/2H2O | C, H, N |
| 10c | 95 | 207–211 | C40H54N8O9 | C, H, N |
| 11b | 100 | 270–275 | C25H36N8O5·2HB·2H2O | C, H, N |
| 11c | 83 | dec begin 190 | C27H40N8O5·3HBr | C, H, N |
Yield calculated from 6-amino-1,3-diethyl-5-nitrosouracil, which was treated with Na2S2O4 and then condensed with [(4-formylphenyl)oxy]acetic acid, and the benzylidene adduct was oxidized with NaIO4 as in ref 12.
H calcd 6.77, found 6.25; N calcd 19.95, found 18.97.
C calcd 60.37, found 52.82.
C calcd 59.40, found 51.49.
Proton NMR resonances (300 MHz) in ppm from Me4Si for selected compounds in (CD3)2SO. 6b: δ 8.15 (t, 1 H, amide NH), 8.05 and 7.05 (each d, J = 8.7 Hz, 2 H, Ar), 4.54 (s, 2 H, CH2O), 4.08 and 3.94 (each q, 2 H, Et methylene), 3.21 (m, 2 H, CH2NHCO), 2.68 (t, J = 6.2 Hz, 2 H, CH2NH2), 1.26 and 1.13 (each t, 2 H, J = 6.8 Hz, CH3). 8a: δ 8.10 (d, 2 H, Ar, meta to O), 7.58 (d, 2 H, Ar, ortho to NH), 7.23 (d, 2 H, Ar, ortho to CH2), 7.13 (d, 2 H, Ar, ortho to O), 4.79 (s, 2 H, CH2O), 3.63 (s, 2 H, CH2Ar), 3.61 (s, 3 H, OCH3), 3.49 and 3.27 (each s, 3 H, NCH3). 9a: δ 8.06 (d, 2 H, Ar, meta to O), 8.02 (1 H, amide NH), 7.56 (d, 2 H, Ar, ortho to NH), 7.21 (d, 2 H, Ar, ortho to CH2), 7.13 (d, 2 H, Ar, ortho to O), 4.75 (s, 2 H, CH2O), 3.48 and 3.24 (each s, 3 H, NCH3), 3.36 (s, 2 H, CH2Ar), 3.07 (m, 2 H, CH2NHCO), 2.60 (t, J = 6.7 Hz, 2 H, CH2NH2).
As reported previously,2 the affinity of XAC (6c) for A2 receptors of human platelets was in close agreement with the value in brain slices, but the potency of 2c showed a discrepancy between these two systems. Since we intended to study binding to A2 receptors in platelets, the effect on platelet adenylate cyclase was adopted as the sole criterion of A2 potency in this study.
In each case the potency of the 1,3-diethyl analogue at platelet A2 receptors was greater than the 1,3-dimethyl analogue and at least as great as that of the 1,3-dipropyl analogue (Table II). The enhancement of A2 affinity by ethyl substituents was more marked for the simple 8-phenyl series, 1, than for the functionalized congeners. In contrast, Bruns et al.9b reported 1,3-dipropyl-8-phenyl-xanthine to be 5-fold more potent than the diethyl analogue in inhibition of [3H]NECA binding to a striatal A2-adenosine receptor.
Table II.
Potencies of Xanthine Derivatives at A2- and A1-Adenosine Receptors
| no. | Ra | A2 receptor:b KB, nM | A1 receptor:c Ki, nM |
|---|---|---|---|
| 1a | Me | 1900 | 70 |
| 1b | Et | 210 | 65 |
| 1c | Pr | 2100 | 13 |
| 2c | Pr | 2400 (1500–3900) | 50 |
| 3b | Et | 170 (75–370) | 140 |
| 3c | Pr | 135 (106–173) | 13 |
| 4b | Et | 73 (54–98) | 90 |
| 5b | Et | 48 (36–65) | 57 |
| 6a | Me | 40 (30–54) | 13 |
| 6b | Et | 21 (18–23) | 12 |
| 6c | Pr | 25 (21–30) | 1.2 |
| 7a | Me | 430 (260–720) | 27 |
| 7b | Et | 100 (60–170) | 9.3 |
| 8b | Et | 210 (190–230) | 58 |
| 9a | Me | 63 (57–68) | 42 |
| 9b | Et | 28 (15–51) | 25 |
| 10b | Et | 230 (100–540) | 180 |
| 11b | Et | 23 (17–30) | 9.4 |
| 11c | Pr | 37 (35–40) | 0.87 |
Substituent at the 1,3-positions (see Figure 1).
Antagonism of NECA-induced stimulation of adenylate cyclase activity in human platelet membranes. Values are means with 95% confidence limits from three separate experiments. Values for 2c, 6c, and 11c from ref 2; value for 1b from ref 14.
Inhibition of [3H]PIA binding to rat cerebral cortex membranes. Values are means from two separate experiments.
While the carboxylic acid congener 2c was not particularly potent, esterification to give compound 3c increased potency markedly at A2 receptors, but not at A1 receptors. A comparison of ethyl and isopropyl esters showed preference of the branched isopropyl group at A2 receptors.
Compounds 7a and 7b, containing the p-toluidine group, were not as potent at A2 receptors as expected on the basis of previous studies. However, when the [(2-aminoethyl)-amino]carbonyl group was attached to the p-methyl group of the toluidine, as in 9, the potency rose. Thus, the potency-enhancing effect of a distal amino group, observed consistently with the A1 receptor particularly with dipropyl analogues (e.g., 6c, 11c),6,11 now appears to apply as well to the A2 receptor. The enhancement of A2 potency by an amino group is also evident in comparing the ethyl amide 5 and the 2-aminoethyl amide 6b.
The potencies of XAC (6c) and the diethyl analogue 6b were nearly identical at A2 receptors. The specific binding of a tritiated 1,3-dipropylxanthine amine congener (XAC) to A2-adenosine receptors in platelets has been reported.13 However, a high degree of nonspecific binding of [3H]XAC will limit the usefulness of this radioligand in screening assays. For an A2 radioligand it would be preferable to use the diethyl analogue, due to the likely lower levels of nonspecific binding with 1,3-diethylxanthines, in comparison to the corresponding 1,3-dipropyl analogues. Although not strictly indicative of the level of potential filter binding, the octanol–water partition coefficients (log p) are as follows: −0.63 (6a), −0.06 (6b), 0.81 (6c), −0.05 (9a), and −1.20 (11b). Thus, there is a large difference in polarity between 1,3-diethyl (e.g., 6b) and 1,3-dipropyl (e.g., 6c) analogues, which is also reflected in maximum aqueous solubility (pH 7.2, 0.01 M phosphate) of 500 and 90 μM, respectively. The lipophilic contribution of an additional ring and amide bond in the chain, as in 9a, is roughly equivalent to substituting 1,3-dimethyl with 1,3-diethyl substituents. Another potential A2 radioligand is 11b, the polar conjugate of D-lysine and diethyl-XAC.
The synthesis of tritiated analogues of 1,3-dipropylxanthines through the catalytic tritiation of 1,3-diallyl precursors has been demonstrated.12 To obtain the corresponding tritiated diethyl analogues in similar fashion would require starting with vinyl substituents. As an alternative we have explored coupling of the diethyl carboxylic congener 2b to ethylenediamine using the water-soluble carbodiimide 1-ethyl-3-[3-(dimethylamino)-propyl]carbodiimide hydrochloride and 1-hydroxybenzotriazole as a catalyst. Coupling reactions using tritiated ethylenediamine to produce the desired radioligand are in progress.
Experimental Section
Xanthine analogues were synthesized as previously reported6,11,12 or by routes shown in Figure 1. HPLC-grade dimethylformamide (Aldrich Chemical Co., Milwaukee, WI) was used without further purification. New compounds were characterized by 300-MHz proton NMR on a Varian 300XL and gave spectra consistent with their structures. Except for compounds 10 and 11, chemicalionization mass spectra (CIMS) were obtained with a Finnigan 1015D spectrometer using ammonia gas with a Model 6000 data collection system. Samples for elemental analysis and biological testing were shown to be homogeneous by thin-layer chromatography (silica, CHCl3/MeOH/HOAc, 85:10:5 and 10:10:1) and, if necessary, recrystallized from solvent mixtures including DMF/ether, DMF/water, or Me2SO/ether.
Figure 1.
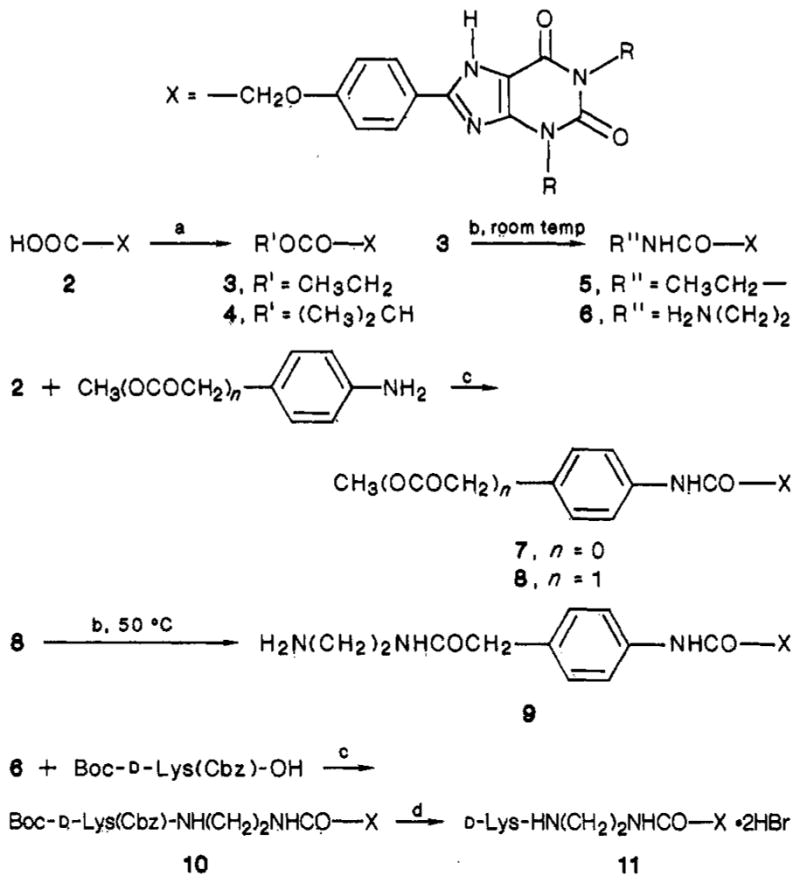
Synthesis of xanthine derivatives. Reagents: (a) EtOH/HCl,7 50 °C (3) or R’OH/DMAP/EDAC (4); (b) 70% ethylamine, aqueous (5) or ethylenediamine, neat6 (6, 9); (c) DCC/HOBt; (d) HBr/HOAc. R = Me, Et, or n-Pr, corresponding to compound suffixes a, b, and c, respectively.
Partition coefficients were determined by addition of a dimethyl sulfoxide solution (5 μL) of the xanthine to an equivolume mixture (2 mL) of 1-octanol and aqueous sodium phosphate, pH 7.2, 0.1 M. After thorough mixing of phases and separation, the ratio of concentrations was determined by comparing absorption at 310 nm of aliquots diluted in methanol (aqueous was filtered). The ±SEM for six determinations corresponded to 0.01 log p unit.
Measurements of the potencies of the xanthines as antagonists of NECA-induced stimulation of adenylate cyclase activity in human platelet membranes and as inhibitors of [3H]PIA binding to rat cerebral cortex membranes were carried out as reported previously.2,3 The slopes of the inhibition curves had Hill coefficients of about 1.0, and all compounds completely inhibited the response to NECA or the specific binding of [3H]PIA.
8-[4-[(Carboxymethyl)oxy]phenyl]-1,3-diethylxanthine 2-Propyl Ester (4b)
Compound 2b (20 mg, 56 μmol) was suspended in a solution of 4-(dimethylamino)pyridine (7 mg) and 2-propanol (0.1 mL) in dimethylformamide (1 mL) and the mixture treated with 1-ethyl-3-[3-(dimethylamino)propyl]-carbodiimide hydrochloride (24 mg, 110 μmol). After several minutes a solution formed, followed by precipitation. After 2 h, water (3 mL) was added, and the white solid was collected, washed with water, and dried, giving 15.8 mg of compound 4; CIMS (NH3), m/e 401 (M + 1)+.
8-[4-[[[[(2-Aminoethyl)amino]carbonyl]methyl]oxy]-phenyl]-1,3-diethylxanthine (6b)
Compound 3b (70 mg, 0.18 mmol) was dissolved in ethylenediamine (2 mL) with stirring. The solvent was evaporated under a stream of nitrogen. The oily residue was triturated with methanol and ether to give compound 6b (67 mg, 92% yield) as a solid; CIMS (NH3), m/e 401 (M +1)+.
Alternately, the carboxylic acid 2b was preactivated with 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide hydrochloride and 1-hydroxybenzotriazole hydrate in dimethylformamide, and this mixture was added slowly to a solution of 1 equiv of ethylenediamine. The product, 6b, was isolated by thin-layer chromatography (CHCl3/MeOH/HOAc, 10:10:1, on silica gel plates) in 44% yield (determined by UV).
8-[4-[[[[4-(Carboxymethyl)anilino]carbonyl]methyl]-oxy]phenyl]-1,3-diethylxanthine Methyl Ester (8b)
Compound 2b (80.8 mg, 0.23 mmol), methyl (p-aminophenyl)acetate hydrochloride (55 mg, 0.27 mmol), HOBt (31 mg, 0.23 mmol), and finally 1-ethyl-3-[3-(dimethylamino)propyl)carbodiimide hydrochloride (98 mg, 0.46 mmol) were combined with stirring in 8 mL of dimethylformamide. Diisopropylethylamine (39 μL, 0.23 mmol) was added, and the mixture was stirred overnight. Water was added, and the product (60 mg) was collected, washed with water, and dried; CIMS (NH3), m/e 506 (M + 1)+.
8-[4-[(Carboxymethyl)oxy]phenyl]-1,3-diethylxanthine 2-(D-Lysylamino)ethylamide Dihydrobromide (11b)
Compound 6b (50 mg, 0.13 mmol) was suspended in 2 mL of dimethylformamide and treated with Nα-Boc-Nε-Cbz-D-lysine (95 mg, 0.25 mmol), HOBt (17 mg, 0.13 mmol), and 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide hydrochloride (54 mg, 0.25 mmol). The mixture was stirred overnight. Aqueous workup, as above, followed by recrystallization from ethyl acetate/hexanes provided 75 mg of compound 10b. The protecting groups were removed with 30% HBr/acetic acid (2 mL), giving compound 11b (72 mg). An analytical sample was prepared by recrystallization from methanol/ether.
Acknowledgments
D. Ukena is on leave from the Pharmakologisches Institut der Universität Heidelberg, with support of the Deutsche Forschungsgemeinschaft (Uk 4/1-1). We thank Dr. D. Ahern of Dupont New England Nuclear for helpful discussions.
Footnotes
Dedicated to Prof. B. Witkop on the occasion of his 70th birthday.
Abbreviations: DPX, 1,3-diethyl-8-phenylxanthine; NECA, N-ethyladenosine-5′-uronamide; XAC, xanthine amine congener (compound 6c); HOBt, 1-hydroxybenzotriazole; EDAC, 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide hydrochloride; PIA, N6-(phenylisopropyl)adenosine.
Registry No. 1a, 961-45-5; 1b, 75922-48-4; 1c, 85872-53-3; 2a, 96865-82-6; 2b, 104576-44-5; 2c, 96865-83-7; 3a, 104576-45-6; 3b, 104576-46-7; 3c, 96865-87-1; 4b, 104576-47-8; 5b, 104576-48-9; 6a, 104576-49-0; 6b, 104598-39-2; 6c, 96865-92-8; 7a, 96865-84-8; 7b, 104576-50-3; 8a, 104576-51-4; 8b, 104576-52-5; 9a, 104576-53-6; 9b, 104576-54-7; 10b, 104576-55-8; 10c, 102255-74-3; 11b, 104576-56-9; 11c, 104576-57-0; 2-PrOH, 67-63-0; H2NCH2CH2NH2, 107-15-3; 4-H2NC6H4CH2CO2Me·HCl, 83528-16-9; Nα-Boc-Nε-Cbz-D-lysine, 55878-47-2; 4-MeC6H4NH2,106-49-0.
References
- 1.(a) Daly JW. J Med Chem. 1982;25:197. doi: 10.1021/jm00345a001. [DOI] [PubMed] [Google Scholar]; (b) Burnstock G, Buckley NJ. In: Methods in Pharmacology. Paton DM, editor. Vol. 6. Plenum; New York: 1985. pp. 193–212. [Google Scholar]
- 2.Ukena D, Daly JW, Kirk KL, Jacobson KA. Life Sci. 1986;38:797. doi: 10.1016/0024-3205(86)90596-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruns RF, Daly JW, Snyder SH. Proc Natl Acad Sci USA. 1980;77:5547. doi: 10.1073/pnas.77.9.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams M, Braunwalder A, Erickson TJ. Naunyn Schmiedeberg’s Arch Pharmacol. 1986;332:179. doi: 10.1007/BF00511410. [DOI] [PubMed] [Google Scholar]
- 5.Schwabe U, Trost T. Naunyn Schmiedeberg’s Arch Pharmacol. 1980;313:179. doi: 10.1007/BF00505731. [DOI] [PubMed] [Google Scholar]
- 6.Jacobson KA, Kirk KL, Padgett WL, Daly JW. J Med Chem. 1985;28:1334. doi: 10.1021/jm00147a038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobson KA, Ukena D, Kirk KL, Daly JW. Proc Natl Acad Sci USA. 1986;83:4089. doi: 10.1073/pnas.83.11.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kusachi S, Thompson RD, Bugni WJ, Yamada N, Olsson RA. J Med Chem. 1985;28:1636. doi: 10.1021/jm00149a016. [DOI] [PubMed] [Google Scholar]
- 9.(a) Yeung SH, Green RD. Naunyn Schmiedeberg’s Arch Pharmacol. 1984;325:218. doi: 10.1007/BF00495947. [DOI] [PubMed] [Google Scholar]; (b) Bruns RF, Lu GH, Pugsley TA. Mol Pharmacol. 1986;29:331. [PubMed] [Google Scholar]
- 10.Ukena D, Padgett W, Olsson R, Daly JW. Can J Physiol Pharmacol. doi: 10.1139/y87-063. in press. [DOI] [PubMed] [Google Scholar]
- 11.Jacobson KA, Kirk KL, Padgett WL, Daly JW. Mol Pharmacol. 1986;29:126. [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobson KA, Ukena D, Daly JW, Kirk KL. J Labelled Compd Radiopharm. 1986;23:519. [Google Scholar]
- 13.Ukena D, Jacobson KA, Kirk KL, Daly JW. FEBS Lett. 1986;199:269. doi: 10.1016/0014-5793(86)80493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ukena D, Böhme; E, Schwabe U. In: Adenosine: Receptors and Modulation of Cell Function. Stefanovich V, Rudolphi K, Schubert P, editors. IRL; Oxford: 1985. pp. 343–349. [Google Scholar]
- 15.Daly JW, Padgett W, Shamin M. J Med Chem. 1986;29:1305. doi: 10.1021/jm00157a035. [DOI] [PubMed] [Google Scholar]