

Summary
Susceptibility to tuberculosis is historically ascribed to an inadequate immune response that fails to control infecting mycobacteria. In zebrafish, we find that susceptibility to Mycobacterium marinum can result from either inadequate or excessive acute inflammation. Modulation of the leukotriene A4 hydrolase (LTA4H) locus, which controls the balance of pro- and anti-inflammatory eicosanoids, reveals two distinct molecular routes to mycobacterial susceptibility converging on dysregulated TNF levels: inadequate inflammation caused by excess lipoxins and hyperinflammation driven by excess leukotriene B4. We identify therapies that specifically target each of these extremes. In humans, we identify a single nucleotide polymorphism in the LTA4H promoter that regulates its transcriptional activity. In tuberculous meningitis, the polymorphism is associated with inflammatory cell recruitment, patient survival and response to adjunctive anti-inflammatory therapy. Together, our findings suggest that host-directed therapies tailored to patient LTA4H genotypes may counter detrimental effects of either extreme of inflammation.
Introduction
Susceptibility to tuberculosis (TB) is typically associated with failed immunity. Genetic deficiencies of immune effectors including Tumor Necrosis Factor (TNF) and interferon-gamma confer increased susceptibility to mycobacterial disease (Fortin et al., 2007). Similarly, a range of non-genetic immunosuppression – Human Immunodeficiency Virus (HIV) infection, TNF-blocking and glucocorticoid treatments - increases susceptibility to TB (Kwan and Ernst, 2011; Lawn and Zumla, 2011). Recently, we found that mutations in the zebrafish gene leukotriene A4 hydrolase (lta4h), which catalyzes the production of the pro-inflammatory eicosanoid LTB4 (Samuelsson et al., 1987)(Figure 1A), were associated with hypersusceptibility to Mycobacterium marinum (Tobin et al., 2010). Functional analyses suggested that reduced LTA4H activity confers hypersusceptibility via an excess production of anti-inflammatory lipoxins (Figure 1A)(Bafica et al., 2005; Chen et al., 2008; Serhan, 2007; Tobin et al., 2010). In humans, two intronic single nucleotide polymorphisms (SNPs) at the LTA4H locus were associated with TB (Tobin et al., 2010). Heterozygosity for the two SNPs was protective, while both homozygous states corresponded to increased disease severity. These findings were surprising for two reasons: heterozygous advantage is unusual, and they implicate both insufficient and excessive inflammation in the pathogenesis of TB (Behr et al., 2010; Tobin et al., 2010). Given the clinical and therapeutic implications of such a dichotomous, genotype-mediated susceptibility, we sought to characterize underlying mechanisms of these two susceptible extremes and to test their relevance in human disease.
Figure 1.
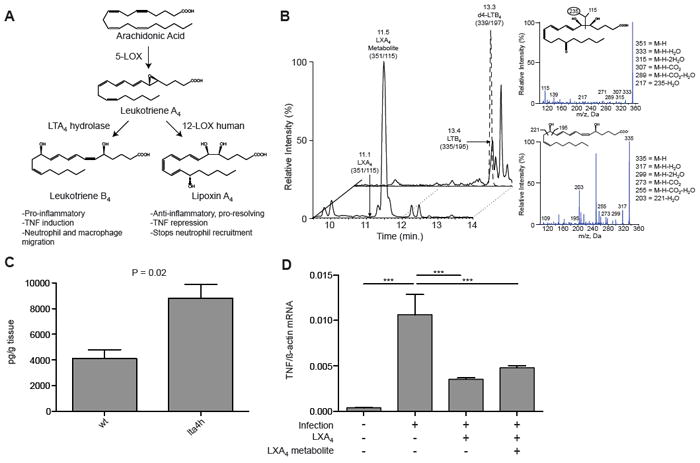
Identification of eicosanoids in zebrafish by LC-MS-MS. (A) Lipoxygenase-derived lipid mediator biosynthesis pathway from arachidonic acid and contribution of LTA4H. (B) LC-MS/MS lipid mediator lipidomics of adult zebrafish. Left: Extracted ion chromatograms of two key lipid mediators, LXA4 metabolite 13,14-dihydro-15-oxo LXA4 (351->115) and LTB4 (335->195). Right: MS/MS fragmentation and structure assignment of lipid mediators. For further MS3 analysis, see Figure S1. (C) LXA4 metabolite levels (mean ± SEM of three animals) in wildtype (wt) and lta4h mutant zebrafish P=0.02 (Student’s unpaired t-test). (D) TNF mRNA levels (mean ± SD) in pooled 3 dpi zebrafish larvae analyzed 7 hours after injection with LXA4 or its metabolite 13, 14-dihydro-15-oxo LXA4 identified using reported criteria (Serhan et al., 1993). ***, P<0.001 (one way ANOVA with Dunnett’s post-test). See also Figure S1.
In the present study, we use zebrafish larvae to uncover the biological basis of LTA4H heterozygous advantage. We show that both excessive pro-inflammatory and anti-inflammatory-pro-resolving eicosanoids namely LTB4 and LXA4 each promote bacterial growth by different pathways that converge on the same endpoint, and identify proof-of-principle genotype-specific therapies intercepting points along the distinct pathways that dominate in the two genotypes. We identify a functional SNP in the human LTA4H promoter, linked to the previously-identified intronic SNPs, that regulates LTA4H transcriptional activity. We confirm its association with disease severity and intra-cerebral inflammatory response in meningeal TB and we show the SNP is associated with response to adjunctive dexamethasone treatment, with only individuals with the high-expression LTA4H genotype deriving benefit from this standard-of-care anti-inflammatory therapy. Together, these results suggest that increased disease severity in humans can also occur for fundamentally opposite reasons: an inadequate host immune response to infection or an excessive one. Thus, the ability to tailor therapies to these divergent inflammatory states and specific LTA4H genotypes could improve patient outcomes.
Results
LTA4H deficiency increases bioactive lipoxins
Lipoxins can impair immunity to Mycobacterium tuberculosis, promoting necrotic death of infected macrophages, and inhibiting initiation of cell-mediated immunity (Chen et al., 2008; Divangahi et al., 2010). Functional studies in zebrafish larvae had suggested a model wherein reduced LTA4H activity increases lipoxin levels, which produce hypersusceptibility by inhibiting transcription of TNF, a key protective cytokine (Figure 1A). Evidence for excess lipoxin function in LTA4H deficiency was discernible even at baseline in uninfected larvae (Tobin et al., 2010). For verification of these functional findings, we used liquid chromatography tandem–mass spectrometry (LC-MS-MS) to identify relevant eicosanoid mediators in uninfected wildtype adult zebrafish and siblings homozygous for an lta4h mutation (a large retroviral insertion in exon 7 that compromises gene function) (Tobin et al., 2010). The expected precursors, products and further metabolites of the lipoxin A4 pathway were identified and quantified, as well as leukotriene B4 using deuterium labeled internal standards with LC-MS-MS (Figures 1B, S1A and S1B). While lipoxin A4 (LXA4) was present in trace amounts, its further metabolite 13,14-dihydro-15-oxo LXA4 (Serhan et al., 1993) was abundant, and its levels were increased 2.1 fold (P=0.02) in lta4h mutants above that in wildtype (Figures 1B, 1C and S1C-E). This LXA4 metabolite displayed functional activity in vivo comparable to LXA4: injection into M. marinum-infected larvae reduced TNF transcription 2.6 fold and 2.3 fold (P<0.05), respectively for LXA4 and its metabolite (Figure 1D). We also identified the pertinent eicosanoids in zebrafish larvae (Figure S1F and S1G), thus validating our functional analyses of infection outcomes at this developmental stage (Tobin and Ramakrishnan, 2008; Tobin et al., 2010). These results identify the conserved eicosanoids in zebrafish and provide definitive evidence that LTA4H deficiency increases functional lipoxin production.
Excess LXA4 and LTB4 activity both promote extracellular bacterial growth
Our work in the zebrafish had identified the cellular mechanism whereby LTA4H deficiency produces susceptibility to M. marinum (Tobin et al., 2010). However, the mechanism behind the association of human susceptibility with the opposite, high-LTA4H expression, homozygous genotype was unclear (Behr et al., 2010; Tobin et al., 2010). So we now used zebrafish larvae to additionally model the high LTA4H activity human genotype. To produce an LTA4H excess state, we injected lta4h RNA into one-cell stage embryos to create what we call LTA4H-high animals. With wildtype animals serving as surrogates for the protected heterozygote state, low-activity LTA4H homozygous genotypes were modeled using a modified antisense oligonucleotide (morpholino) to knock down zebrafish lta4h expression (referred to as LTA4H-low animals) (Tobin and Ramakrishnan, 2008; Tobin et al., 2010). We established the specificity of the effects of both the MO and RNA by showing that lta4h sense but not antisense RNA rescued the increased bacterial burden of the lta4h morphants (Figure S2). The LTA4H-high animals displayed evidence of increased LTB4 activity, both chemoattractant and proinflammatory (Figure 1A) (Goldman et al., 1993; Haeggstrom, 2004; Samuelsson et al., 1987; Tobin et al., 2010). More phagocytes were recruited to M. marinum infection of their hindbrain ventricle (1.7 fold increase over wildtype siblings, P=0.01) and expressed 1.5 fold more TNF mRNA than wildtype (P=0.02) soon after infection ((Figure 2A and 2B).
Figure 2.
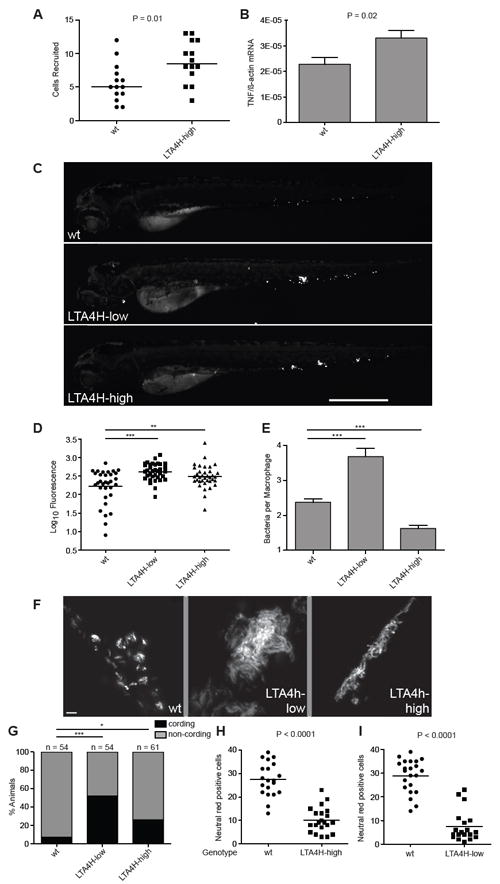
Extremes of LTA4H expression drive hypersusceptibility in zebrafish. (A) Median number of macrophages recruited to the hindbrain ventricle of wildtype and LTA4H-high siblings 4 hrs after injection of 150-200 M. marinum into this space at 30 hpf. P=0.01 (Student’s unpaired t-test). Representative of two independent experiments. (B) TNF mRNA levels (mean ± SEM of three independent experiments) for control and LTA4H-high siblings 1 dpi with 150-200 M. marinum. P=0.02 by Student’s unpaired t-test. (C) Fluorescence images of representative wildtype, LTA4H-low and LTA4H-high larvae 3 dpi with 90-100 M. marinum. (D) Bacterial burden of all larvae from (C) by fluorescence pixel counts (FPC). **P < 0.01; ***P < 0.001. (one-way ANOVA with Tukey’s post-test; all other comparisons not significant). Representative of >7 independent experiments measuring differences in bacterial burden of the three genotypes. (E) Mean (± SEM) number of bacteria per infected macrophage in 11 wildtype larvae, 8 LTA4H-low larvae and 13 LTA4H-high larvae, 40 hpi with 150-200 erp mutant M. marinum. ***P < 0.001 (one way ANOVA with Dunnett’s post-test). Representative of two independent experiments. (F) Fluorescence images showing discrete bacterial clumps indicative of macrophage residence in wildtype versus corded extracellular bacteria in their LTA4H-low and LTA4H-high siblings at 3 dpi with 150-200 M. marinum. (G) Percentage of animals in (F) with cording among wildtype, LTA4H-low and LTA4H-high siblings 4 dpi after infection with 90-100 M. marinum. *, P<0.05; ***, P<0.001 (Fisher’s exact test comparing LTA4H-low and LTA4H-high to wildtype). (H) Quantitation of neutral red positive cells 4 dpi after infection with approximately 100 M. marinum in sibling controls and LTA4H-high animals and (I) sibling controls and LTA4H-low animals. See also Figure S2 and Movies S1-S3.
We next asked if and how their hyperinflammatory state renders the LTA4H-high fish susceptible to infection. While pathological, overexuberant inflammation and immune responses are recognized to cause clinical exacerbations of TB and leprosy, these generally occur in the face of effective mycobacterial control with chemotherapy (Chambers et al., 1984; Hawkey et al., 2005; Kahawita and Lockwood, 2008; Markman and Eagleton, 1981). Therefore these excessive host-detrimental immune responses are thought to be neutral or even favorable to bacterial control. However, soon after infection (by 3 days post infection (dpi)), the LTA4H-high fish had increased bacterial burdens over their wildtype siblings (1.6 fold, P < 0.01), similar to the LTA4H-low fish (Figure 2C and 2D). This result not only supported the human LTA4H heterozygote advantage model but, importantly, linked increased inflammation to increased bacterial growth.
A cellular mechanism by which increased inflammation increases bacterial burdens was revealed by a temporal dissection of infection in LTA4H-high and LTA4H-low siblings. We had found earlier that LTA4H deficiency, like TNF signaling deficiency first produces increased bacterial growth within macrophages followed by necrotic death of the heavily infected macrophages (Clay et al., 2008; Tobin et al., 2010). Macrophage lysis releases the bacteria into their permissive extracellular milieu where they continue to grow even more exuberantly in corded mats. This bacterial cording is a reliable and readily discernible binary phenotype for the extracellular growth that follows upon macrophage lysis (Clay et al., 2008; Tobin et al., 2010). In contrast to LTA4H-low animals, their LTA4H-high siblings displayed an enhanced ability to control intracellular bacterial growth very early in infection. At 40 hours post infection (hpi) when LTA4H-low fish had the expected 1.5 fold (P<0.001) increase in bacterial burdens per macrophage over wildtype, their LTA4H-high siblings restricted intramacrophage bacterial growth even better than wildtype (1.5 fold reduction over wildtype, P<0.001) (Figure 2E). Yet by three to four days, both genotypes had similar increases in bacterial burdens over wildtype and displayed extracellular growth as judged by the cording phenotype (Figure 2F and 2G). To confirm that the bacterial cording phenotype reflected the lysis of infected macrophages, we enumerated the number of macrophages after neutral red staining at 4dpi (Tobin et al., 2010). The LTA4H-high animals had the expected depletion of macrophages associated with cording (2.7-fold fewer than wildtype, P<0.0001) similar to that seen in the LTA4H-low animals (3.9-fold fewer than wildtype, P<0.0001) (Figure 2H and 2I). Thus, despite their increased ability to curtail bacterial growth, the LTA4H-high macrophages underwent lysis, releasing bacteria into the extracellular milieu where their numbers could then reach those of the LTA4H-low fish. For LTA4H deficiency, we had found it to both be associated with and to phenocopy TNF deficiency. Given the increased TNF levels in the LTA4H-high animals, we wondered if TNF excess, like TNF deficiency, also caused necrotic lysis of the infected macrophages, thereby mediating cording-associated hypersusceptibility for both genotypes (Vandenabeele et al., 2010) (Figure 3).
Figure 3.
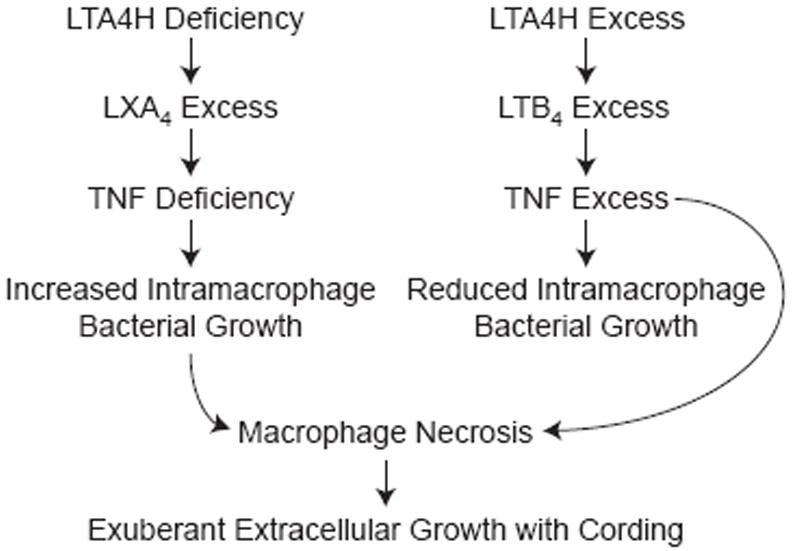
Model of proposed mechanism for susceptibility of LTA4H-low and –high genotypes wherein either TNF deficiency or excess results in macrophage necrosis and exuberant extracellular bacterial growth.
LXA4 and LTB4 excess both produce susceptibility via TNF dysregulation
Our model suggested that opposite deviations in TNF levels from wildtype drive the hypersusceptibility of LTA4H deficiency and excess respectively (Figure 3). If so, then wildtype animals should be rendered hypersusceptible by TNF deviations in either direction i.e. TNF knockdown or addition of exogenous TNF. Moreover, TNF excess should phenocopy the unusual infection phenotype seen for LTA4H-high animals, namely initial improved control of intracellular macrophage growth followed by cell lysis and exuberant extracellular bacterial growth with cording. In order to test this model, we confirmed that recombinant zebrafish TNF (Roca et al., 2008) was functional in our system: we observed a graded reversal of hypersusceptibility of TNF deficiency created by MO knockdown and identified the minimal dose that reversed it completely (Figure S3). We could now compare the infection phenotypes of TNF deficiency (TNF-low) and TNF excess (TNF-high) states by using wildtype animals injected with either the TNF MO or recombinant TNF. TNF-low animals gave the expected phenotype similar to the LTA4H-low animals: diminished control of bacterial growth within macrophages followed by increased bacterial burdens accompanied by macrophage lysis and bacterial cording (Figure 4A-D) (Tobin et al., 2010). In contrast, TNF-high animals displayed the signature phenotype of LTA4H-high animals: initial improved control of bacterial growth within macrophages was then followed by increased bacterial burdens accompanied by macrophage lysis and cording (Figure 4 E-H). These data were consistent with the LTA4H-low and –high phenotypes being due to TNF deficiency and excess, respectively.
Figure 4.
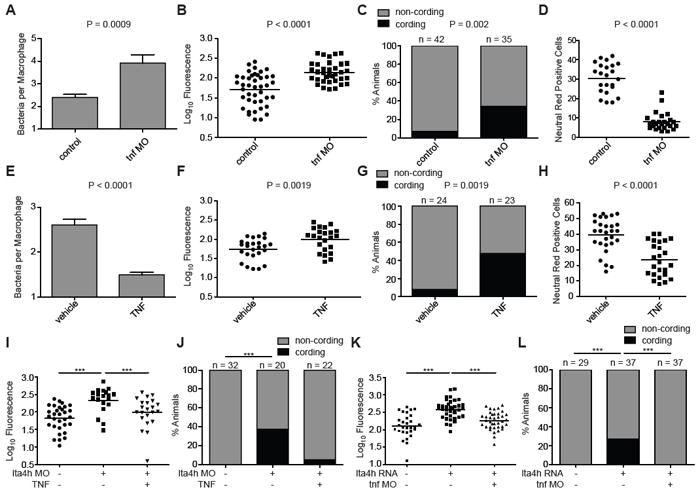
Modulation of TNF levels results in genotype-specific rescue of LTA4H-mediated hypersusceptibility. (A). Mean (± SEM) number of bacteria per infected macrophage 40-44 hpi with 150-200 erp mutant M. marinum of wildtype or TNF morphant (MO) siblings. (B) FPC in control or tnf morphant siblings 3 dpi with 90-100 M. marinum. (C) Quantitation of frequency of bacterial cording of the animals in (B). (D) Quantitation of neutral red positive cells 4 dpi after infection with 100 M. marinum in sibling controls or tnf morphants. (E) Mean (± SEM) number of bacteria per infected macrophage at 40-44 hpi in wildtype animals with or without injection of 0.5 ng rTNF 12 hours after infection with 150-200 erp mutant M. marinum. (F) FPC in control or rTNF injected siblings 3 dpi with 90-100 M. marinum. (G) Quantitation of frequency of bacterial cording of the animals in (F). (H) Quantitation of neutral red positive cells at 4 dpi after infection with 90-100 M. marinum in sibling controls or rTNF injected animals. (I) FPC in wildtype or lta4h morphant siblings 3 dpi with 90-100 M. marinum per animal after injection of 0.5 ng rTNF at 12 hpi. (J) Bacterial cording frequency of the animals in (I). (K) FPC in wildtype animals, LTA4H-high siblings, and LTA4H-high plus TNF morphant animals, at 3 dpi with 90-100 M. marinum. (L) Quantitation of frequency of bacterial cording in the animals in (K). Statistical comparisons in panels (I) and (K) by one-way ANOVA with Tukey’s post-test; in panels (C),(G),(J) and (L) by Fisher’s exact test; (A),(B),(D),(E),(F),(H) by Student’s unpaired t-test. For all panels *, P<0.05; **, P<0.01; ***, P<0.001, all other comparisons not significant. See also Figure S3.
Next, we asked if deviations in TNF do indeed account for the susceptibility of the LTA4H-low and –high states. If TNF deficiency is the principal driver of LTA4H-low susceptibility then it should be corrected by the administration of recombinant TNF. Both increased bacterial burdens and the accompanying bacterial cording were rescued by a single dose of recombinant TNF (Figure 4I and 4J). Conversely, LTA4H-high susceptibility should be corrected by TNF MO blockade of these animals. Again the reduction of bacterial burdens with the MO was accompanied by a reduction in bacterial cording (Figure 4K and 4L). In sum, both hypersusceptible states are mediated substantially, if not entirely, by TNF deviations from an optimal level, in opposite directions for each state. TNF excess, like TNF deficiency, causes macrophage lysis, placing the bacteria in a permissive extracellular niche (Figure 3).
Distinct therapies for hypersusceptibility from LXA4 and LTB4 excess
If poor outcomes in TB can arise from extreme inflammatory states then specific, targeted interventions within these eicosanoid pathways should correct the hypersusceptibility associated with each state while being neutral or even detrimental to the other state (Figure 5A). We tested this first with two agents expected to alter lipoxin levels in opposing directions: reducing lipoxins should benefit the LTA4H-low state while increasing lipoxins should specifically benefit the LTA4H-high state by dampening the excessive pro-inflammatory effects of LTB4 excess (Figure 5A). To reduce lipoxin levels, we used a 15-lipoxygenase inhibitor, shown earlier to reduce lipoxin function in zebrafish larvae (Auerbach et al., 1996; Tobin et al., 2010). This treatment significantly reduced the infection burdens of LTA4H high animals (Figure 5B). In contrast, those of LTA4H-high animals were increased slightly, which was consistent with the removal of lipoxin-mediated TNF repression hence elevating their already high TNF levels further (Figure 5A and 5C). Conversely, we sought to increase endogenous lipoxin function by stimulating the production of aspirin-triggered 15-epi-LXA4 (Claria and Serhan, 1995) which exerts anti-inflammatory effects both on inflammatory cell migration and cytokine production in humans (Morris et al., 2009). We had shown earlier that this aspirin triggered product (15-epi-LXA4) is active in zebrafish larvae where it mimics LXA4 in inhibiting neutrophil migration (Tobin et al., 2010). Here, we identified a concentration of acetylsalicylic acid (ASA), the active ingredient of aspirin, which similarly inhibits neutrophil migration (Figure S4). ASA decreased infection in LTA4H-high animals while increasing it in their LTA4H-low siblings, as would be expected from a further increase in their endogenous lipoxin excess (Figure 5D and 5E).
Figure 5.
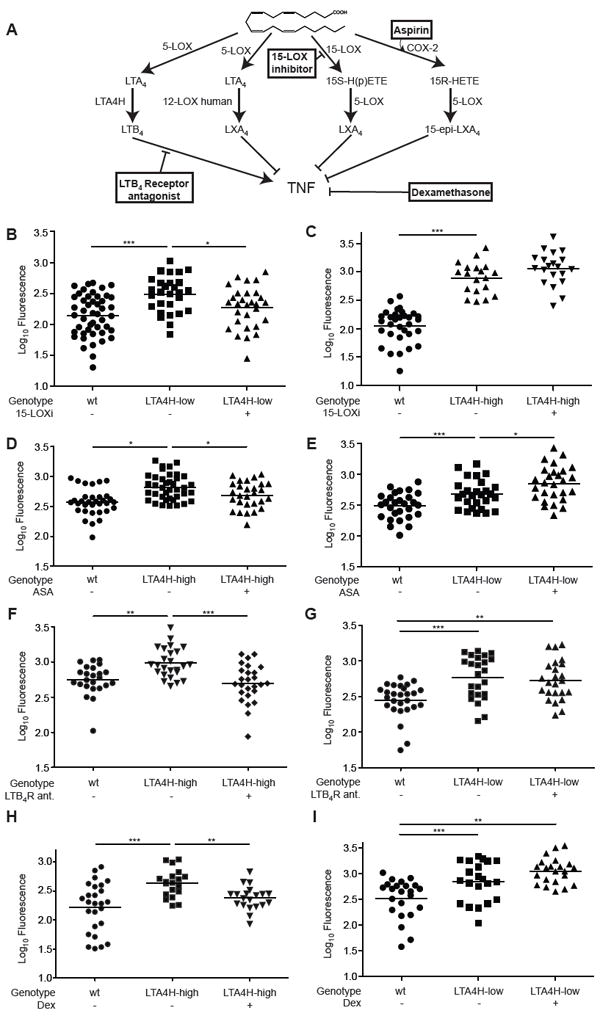
Genotype-dependent therapies rescue hypersusceptibility to M. marinum in the zebrafish. (A) Pathways of LTB4 and LXA4 synthesis highlighting points of pharmacological intervention. (B) FPC of LTA4H-low or wildtype siblings in the presence or absence of 100 nM of PD146176, a 15-lipoxygenase inhibitor (15-LOXi). Representative of 2 independent experiments. (C) FPC as in (B) for LTA4H-high animals or wildtype siblings. Representative of 2 independent experiments. (D) FPC of LTA4H-high or wildtype siblings in the presence or absence of 1 μM ASA. Representative of 2 independent experiments. (E) FPC as in (D) of LTA4H-low or wildtype siblings. (F) FPC of LTA4H-high or wildtype siblings in the presence or absence of 1 μM of U75302, an antagonist of the BLT1 LTB4 receptor (LTB4-R ant). Representative of 3 independent experiments. (G) FPC as in (F) for LTA4H-low or wildtype siblings. (H) FPC of LTA4H-high and wildtype siblings in the presence or absence of 0.75 μM DEX. Representative of 3 independent experiments. (I) FPC as in (H) for LTA4H-low animals or wildtype siblings. Representative of 2 independent experiments. 150-200 M. marinum used for all infections with analyses performed at 3dpi. Statistical comparisons in all panels by one-way ANOVA with Tukey’s post-test. *, P<0.05; **, P<0.01; ***, P<0.001. See also Figures S4 and S5.
We could also specifically overcome the increased bacterial burdens of LTA4H-high animals by directly blocking LTB4 activity with a pharmacological antagonist of the LTB4 receptor BLT1 (Figure 5A and 5F). This antagonist did not alter LTA4H-low bacterial burdens (Figure 5G), a finding consistent with our model that LTB4 deficiency per se does not increase susceptibility (Tobin et al., 2010).
Finally, for each of the two genotypes we assessed the effect of dexamethasone, a glucocorticoid used for its broad anti-inflammatory and immunosuppressive effects in a range of human diseases including certain serious forms of TB (Mayosi et al., 2002; Prasad and Singh, 2008) (Figure 5A). Dexamethasone gave the expected therapeutic result with LTA4H-high animals while this same dose rendered LTA4H-low animals even more susceptible (Figures 5H and 5I).
We tested further the specificity of these therapeutic effects in two ways. That the efficacy of the compounds was specific to the appropriate LTA4H genotype was further validated by assessing their effects on wildtype animals in which none had a salutary effect at these concentrations (Figure S5A-S5D). Second, our model would suggest that therapies specific to the LTA4H-low and LTA4H-high states work by correcting the respective TNF deviations from wildtype. The 5-lipoxygenase inhibitor must work by increasing TNF in the LTA4H deficient state whereas ASA, the LTB4 receptor antagonist and dexamethasone must work by reducing TNF in the LTA4H excess state. If so, then the therapeutic effect of the 5-lipoxygenase inhibitor should be abrogated by the TNF morpholino, and the efficacy of the other compounds abrogated by the addition of TNF. These predictions were borne out for all four compounds (Figure S5E-S5H). Together, these results provide proof-of-principle for genotype-directed interventions targeting pro- and anti-inflammatory lipoxygenase mediator pathways that in turn influence TNF levels, and for their effects in the zebrafish. The genotype-dependent efficacy of dexamethasone therapy in the zebrafish suggests that genotype may also influence the efficacy of this therapy in human TB.
A polymorphism in the human LTA4H promoter regulates transcriptional levels
In order to test these therapeutic principles in the context of human disease, we explored the regulation of LTA4H in humans. Previously we identified intronic SNPs of LTA4H that are associated with incidence, severity, and survival in TB or leprosy (Tobin et al., 2010). These intronic SNPs were unlikely to directly mediate differences in gene expression or activity. In order to identify potential functional polymorphisms, we identified all variants in the LTA4H locus that had become accessible through the 1000 Genomes Project (Durbin et al., 2010). One such SNP, rs17525495 C/T, lies 12 nucleotides upstream of the LTA4H transcription start site at chr12:96,429,377 (hg19). The frequency of allele T of rs17525495 has an unusual worldwide distribution: highest in Asian populations (0.29), intermediate in African populations (0.12) and least common in European populations (0.04) (www.genome.ucsc.edu). Four lines of evidence suggest that rs17525495 genotype influences LTA4H transcription. First, expression profiles of lymphoblastoid cell lines (LCLs) from the International HapMap project (Stranger et al., 2007) indicate that LTA4H mRNA levels differ by rs17525495 genotype, with higher activity associated with the rarer T allele (Figures 6A and S6A). Second, we showed using LCLs from healthy controls that these mRNA differences were reflected in differences in LTA4H protein levels. rs17525495 TT is associated with a 2.3-fold higher LTA4H protein expression than the CC, with intermediate expression in the CT cell lines (P=0.0002) (Figures 6B and S6B). Third, to control for differences in genetic background outside of this locus and examine transcription directly, we isolated mRNA from LCL GM19190, which is heterozygous both at rs17525495 and at a rare coding sequence SNP (rs79510571), which served as a tag to discriminate between transcripts. Transcripts of rs79510571 T were twice as abundant as transcripts of allele C (P=0.03) (Figure 6C). Fourth, as an independent test of whether these associations were due directly to the effect of rs17525495 alleles, we cloned one kb of each of the two promoter genotypes into a luciferase expression vector and transfected each into the murine macrophage cell line RAW264.7. The promoter with rs17525495 T produced 1.3 fold higher luciferase activity than the promoter with genotype C (P=0.006), an increase that was maintained after M. marinum infection of the macrophages (1.2-fold, P=0.02)(Figure 6D). The promoter with rs17525495 T produced a similarly higher luciferase activity than the promoter with genotype C in the human macrophage THP-1 cell line (Figure S6C). Together these data indicate that the LTA4H promoter polymorphism influences the level of transcription in macrophages, a cell type central to the fate of mycobacterial infection, and particularly so in our proposed model of hypersusceptibility (Figure 3).
Figure 6.
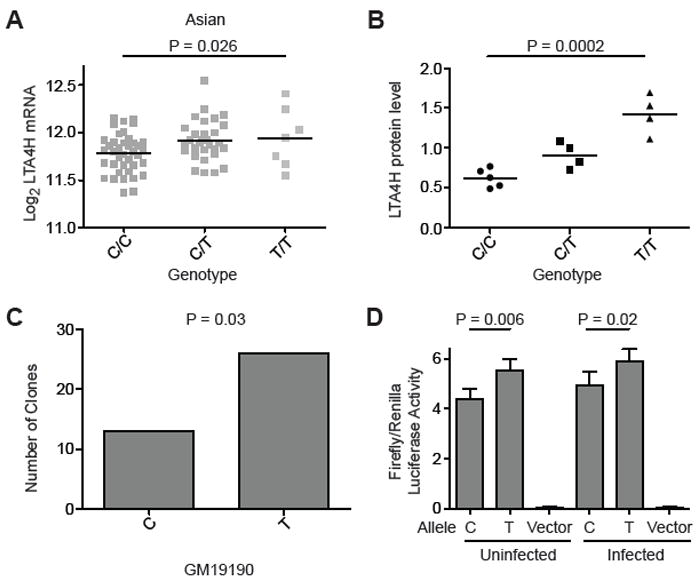
The human LTA4H promoter variant rs17525495 C/T is associated with differences in LTA4H RNA and protein expression. (A) LTA4H mRNA expression in lymphoblastoid cell lines (LCLs) from Asian individuals (CHB+JPT) (from (Stranger et al., 2007)) P=0.026 (one-way ANOVA). Data for all HapMap samples shown in Figure S6A. (B) LTA4H protein expression levels from LCLs of unrelated Han Chinese CC or TT homozygotes or CT heterozygotes, detected by Western blot and normalized to b-tubulin (also see Figure S6B). P=0.0002 (Student’s t-test). Representative of 2 independent experiments for the CC and TT homozygotes; CT heterozygotes were analyzed once in this experiment (C) Number of cDNA clones corresponding to the C or T allele at rs17525495 isolated from the heterozygous LCL GM19190. (D) Luciferase expression (mean ± SEM of 11 independent experiments) transcribed from a one kb promoter fragment immediately upstream of the LTA4H translation start site containing the rs17525495 C and T alleles. P=0.0064 for uninfected cells and P=0.02 for M. marinum-infected cells (paired t-test). See also Figure S6.
The LTA4H promoter polymorphism is associated with treatment response in TB meningitis
In the zebrafish, the two inflammatory poles determined by alternate lta4h genotypes can be distinguished by their responses to targeted pharmacotherapies. We tested whether the human LTA4H polymorphism regulating transcriptional levels also influences response to therapy among patients with TB meningitis. The most severe form of TB, meningitis kills or disables more than half of sufferers despite effective antimicrobial chemotherapy (Prasad and Singh, 2008; Schoeman et al., 1997; Thwaites et al., 2004). Adjunctive anti-inflammatory therapy with glucocorticoids became the standard of care after several clinical trials worldwide suggested that glucocorticoids induce a modest reduction in morbidity and mortality (Prasad and Singh, 2008; Schoeman et al., 1997; Thwaites et al., 2004). Results of the studies of glucocorticoids for treatment of TB meningitis present two conundrums (Donald and Schoeman, 2004; Schoeman et al., 1997). First, it is uncertain why the beneficial effects of glucocorticoids are relatively modest. Second, the mechanisms by which glucocorticoids exert their benefit are unclear; improved survival is not associated with a significant reduction in intracranial pressure, or cerebral vasculitis, which would have been expected to be at the root of the general anti-inflammatory effects of glucocorticoids (Donald and Schoeman, 2004; Schoeman et al., 1997).
Our model addresses these conundrums. Based on our model, we predict that (i) the individuals most severely affected with TB meningitis have opposite inflammatory states governed by their LTA4H genotype and (ii) only patients from the hyperinflammatory pole benefit from glucocorticoids. We tested these predictions in 182 Vietnamese patients with TB meningitis who participated in prior clinical studies in Ho Chi Minh City (Table S1). With respect to our first prediction, individuals with the highest mortality from TB meningitis were homozygous at rs17525495 (Figure 7A). This result was expected, given our previous analysis with the linked intronic SNPs of LTA4H (Tobin et al., 2010) (Figure S7). As an assessment of these patients’ inflammatory state, we evaluated leukocyte counts in cerebrospinal fluid (CSF) samples that were obtained for the diagnosis of TB meningitis prior to initiating treatment. CSF leukocyte counts in heterozygotes were intermediate between those of the homozygous genotypes (Figure 7B). High leukocyte counts in human TT patients mirrored the increased numbers of leukocytes in the hindbrain ventricle of LTA4H-high zebrafish during infection (Figure 2A). In contrast to these differences in cell migration, which is known to be affected by leukotrienes and lipoxins (Figure 1A), nonspecific inflammatory parameters such as CSF opening pressure indicative of intracranial pressure were not associated with rs17525495 genotype (Table S2). Our results suggested that the LTA4H promoter variant influences meningitis outcome and a specific inflammatory marker among the Vietnamese patients, but not in the same way for the two parameters. How can the survival advantage of CT heterozygotes be reconciled with their intermediate white cell counts?
Figure 7.
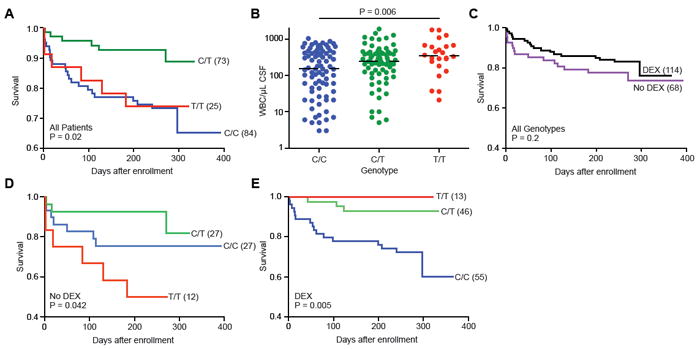
rs17525495 genotype influences TB meningitis survival, inflammation, and treatment response. (A) Mortality from TB meningitis for all patients (treated and untreated with dexamethasone (DEX)), stratified by rs17525495 genotype (P=0.02, log rank test). (B) Median pre-treatment leukocyte counts in cerebrospinal fluid stratified by rs17525495 genotype (P=0.006, one-way ANOVA). (C) Influence of adjunctive dexamethasone treatment on patient survival for all genotypes. Treatment effect is not significant (P=0.2). (D) Survival among patients not treated with dexamethasone, stratified by rs17525495 genotype (P=0.042, log rank test) (E) Survival among patients treated with dexamethasone, stratified by rs17525495 genotype (P=0.005, log rank test). See also Figure S7 and Tables S1 and S2.
To address this question, we tested the second prediction of our model, that the marginal response to dexamethasone among these TB meningitis patients generally (Figure 7C) represented an LTA4H genotype-dependent response, as we had seen in the zebrafish (Figure 5F and 5G). That is, does the effect of dexamethasone depend on a patient’s genotype at rs17525495? Among patients not treated with dexamethasone, those with rs17525495 TT genotype had the highest mortality (Figure 7D). In contrast, among patients treated with dexamethasone, those with genotype rs17525495 TT had the lowest mortality (Figure 7E). These relationships were not due to any interaction between pre-treatment disease severity and genotype (Table S2). These results suggest that LTA4H genotype has a critical influence on response to standard-of-care adjunctive anti-inflammatory therapy in TB meningitis.
Discussion
Our iterative dual approaches in zebrafish and humans lead us to conclude that TB is not only a disease of failed immunity. People succumb for the opposite reasons of an inadequate or excessive immune response. Virulent mycobacteria themselves have evolved to disrupt the fine balance of pro- and anti-inflammatory cues required for host protection (Desvignes and Ernst, 2009; Volkman et al., 2010). Distinct virulence determinants can manipulate macrophage inflammation towards both extremes, inducing anti-inflammatory lipoxin production but also promoting pro-inflammatory TNF (Agarwal et al., 2009; Chen et al., 2008). The host genetic differences in immunoregulation we identify are overlaid on mycobacterial immunomodulation so as to dramatically tilt this balance.
Genetic differences in LTA4H transcription affect inflammatory response, outcome and response to therapy in TB meningitis. The functional promoter variant that causes this differential LTA4H transcription is common in much of the world where TB is endemic, but intriguingly is much less common in Europe. A recent study of TB in a Russian cohort failed to find an association of susceptibility to TB with other polymorphisms at this locus (Curtis et al., 2011). We speculate that the difference between the negative results for the Russian cohort and the positive association that we observed between the functional promoter variant and disease severity for TB in a Vietnamese cohort may be due either to differences in linkage disequilibrium between the functional promoter variant and the other LTA4H SNPs in the Russian cohort compared to Asian cohorts (linkage disequilibrium among the variants is shown for the Vietnamese cohort in Figure S7) and/or to differences in the influence of the functional promoter variant on susceptibility to TB per se versus its influence on severity of disease among infected individuals. In this context, it is noteworthy that LTA4H-mediated differences in disease severity and treatment response that are manifested within hours of infection in the zebrafish larval model are also evident in advanced manifestations of human disease i.e. TB meningitis. The ability of macrophages to contain mycobacterial infection is critical throughout infection, and this ability is dependent on finely tuned LTA4H levels. In addition, because lipoxins and leukotrienes may also impact lymphocyte differentiation (Divangahi et al., 2010; Payan et al., 1984), LTA4H genotype-mediated differences are probably further amplified during later stages of TB when adaptive immunity is in play.
If people succumb to TB for two fundamentally different reasons then it is imperative to design therapeutic strategies that reflect this dichotomy. In evaluating the effects of dexamethasone treatment for TB meningitis without taking into account host genotype, the very substantial benefit of dexamethasone for patients with the high-activity genotype may be diluted by its neutral to possibly detrimental effect for individuals with the low-activity genotype. In this disease with extremely high mortality, a simple genotyping assay for the presence of the high-activity allele could provide a rapid and inexpensive approach to direct adjunctive therapy to the individuals it would benefit. Therefore, clinical studies are urgently needed to determine if genotype-mediated dexamethasone responsiveness is a feature of TB meningitis in other parts of the world. In particular, we need to assess whether or not adjunctive dexamethasone harms those patients with the CC genotype for this would have important implications for clinical practice and suggest alternative treatment strategies for this group.
Adjunctive therapies tailored to LTA4H genotype may improve outcomes for other forms of TB. This work shows that dexamethasone exerts its benefit by intercepting mechanisms fundamental to mycobacterial pathogenesis itself and not solely as a general suppressor of host immune responses gone awry. Thus, glucocorticoids should be therapeutic for other forms of TB as well. In TB pericarditis, another serious disease manifestation, adjunctive glucocorticoids are widely used, but again produce only a modest reduction in mortality and morbidity (Mayosi et al., 2002). In pulmonary TB, the most common form of the disease, some studies have reported that adjunctive glucocorticoids induce faster clearance of bacteria from the sputum, whereas others have failed to find a beneficial effect (Muthuswamy et al., 1995). A recent study in HIV-positive patients found improved bacterial clearing with adjunctive glucocorticoid treatment that was associated with a reduction in TNF levels (Mayanja-Kizza et al., 2005). Therefore, glucocorticoids may improve outcome in all forms of TB associated with high LTA4H activity. In addition, anti-TNF therapies have been reported to accelerate the bacteriological response in pulmonary TB patients (Wallis, 2005). We speculate that the observed modest beneficial effects of these adjunctive therapies in the general patient pool may in fact represent their substantial benefit in the LTA4H-high genotype, as we have found for TB meningitis.
Glucocorticoids are broadly acting drugs with serious side effects. Our work suggests that direct manipulation of eicosanoid lipoxygenase pathways may provide better targeted therapies. For high-activity genotypes these may include therapies that more directly target LTB4 excess, as exemplified by the beneficial effect of LTB4 receptor blockade in the zebrafish. Other therapies for the high-activity genotype that target eicosanoid balance may also prove effective. Aspirin is one of the most widely used, well-tolerated and inexpensive drugs in history, and one of its important effects is to trigger 15-epi-LXA4 (Claria and Serhan, 1995; Morris et al., 2009; Serhan et al., 2010), which in turn can inhibit TNF (Ariel et al., 2003). In the zebrafish, we have found genotype-dependent rescue by the active ingredient of aspirin, a result that is supported by two preliminary human clincal trials in TB meningitis (Misra et al., 2010; Schoeman et al., 2011). In particular, the clinical trial reporting that aspirin may improve outcome from TB meningitis comparably and additively to that induced by glucocorticoids (Misra et al., 2010), is consistent with the synergistic effect of the two compounds predicted by our model. It would be intriguing to evaluate aspirin-responsiveness by genotype. Finally, our data suggesting that TNF excess caused by the LTA4H-high genotype plays a prinicipal role in pathogenesis, makes it important to understand this pathway deeply. In particular, it would be important to identify the cellular sources of TNF that contribute to pathology and the mechanism by which excess TNF produces necrosis of infected cells. Such an understanding may make it possible to tap into drugs that are being developed for the treatment of programmed necrosis in the context of other diseases (Christofferson and Yuan, 2010). Then, for treatment of the low-activity LTA4H genotypes, the development of agents that limit excessive lipoxin production and boost inflammation should improve outcome, as we have uncovered here in the zebrafish.
The basic innate inflammatory pathways affected by this gene are central to many infections. The genotype-directed TB treatment strategies suggested by this work may find use in other serious infections.
Experimental Procedures
Bacterial Strains, Embryo Infection and Husbandry
These were used and performed as described in (Tobin et al., 2010).
Eicosanoid Extraction and LC-MS-MS lipid mediator lipidomics
Adult zebrafish from an incross of lta4hzm5961/+ heterozygotes were identified by PCR as in and samples were extracted and analyzed as described in Extended Experimental Procedures in the Supplemental Information.
Quantitative RT-PCR
This was performed as described in (Tobin et al., 2010) in Extended Experimental Procedures in the Supplemental Information.
Morpholino and RNA injections
Morpholinos were obtained from Genetools (Eugene, OR) and injections were performed as described in (Tobin et al., 2010) in Extended Experimental Procedures in the Supplemental Information.
Recombinant TNF microinjection
Vehicle, 0.5 ng or 1.0 ng of recombinant zebrafish TNF (Roca et al., 2008) was microinjected into the caudal vein of each animal at 16 hpi.
Pharmacological interventions in zebrafish
After infection, small molecules were applied via soaking. Solutions were changed daily. All conditions and controls were standardized to a final concentration of 0.5% DMSO. PD 146176 (BIOMOL) was applied at a final concentration of 100 nM 16 hpi. U75302 (BIOMOL) was applied at a concentration of 1 μM directly after infection. Dexamethasone (Sigma) was applied at a concentration of 0.75 μM directly after infection. Acetylsalicylic acid (Sigma) was applied at a concentration of 1 μM.
A single dose of approximately 5 nL of Lipoxin A4 or its metabolite 13,14-dihydro-15-oxo-LXA4 or vehicle control was microinjected into 3 dpi animals (30 for each condition) at a concentration of 0.2 ng/μL in 10% ethanol. Seven hours later, animals were harvested to measure TNF mRNA levels.
Microscopy
Microscopy was performed on a Nikon E600 equipped with DIC optics, a Nikon D-FL-E fluorescence unit with 100W Mercury lamp and MFC-1000 z-step controller (Applied Scientific Instrumentation) or, for whole animal images, a motorized Nikon inverted Ti-E microscope with further details provided in the Extended Experimental Procedures.
Quantitation of LTA4H protein levels
Western blots from Lymphoblastoid cell lines (LCLs) from Han Chinese in the HapMap project were were probed first with anti-LTA4H (Santa Cruz Biotechnology, 1:50 dilution) and then with anti-b-tubulin (Developmental Studies Hybridoma Bank 1:500) and analyzed as described in Extended Experimental Procedures.
LTA4H promoter luciferase assays
Transfections were performed on RAW 264.7 macrophages using the Amaxa nucleofection system and the ratio of luc2 luciferase activity to Renilla luciferase determined at 18 hours post-transfection. Details of constructs used in Extended Experimental Procedures
Clinical methods
Clinical methods are detailed in Extended Experimental Procedures. All protocols were performed in accordance with human subjects review committees at each site, the Oxford Tropical Research Ethics Committee, the University of Washington (Seattle, WA), and the Western Institutional Review Board (Olympia, WA).
Genomic DNA was prepared via the QIAamp DNA blood kit (Qiagen) from peripheral blood samples. Genotype at rs17525495 was determined after PCR amplification of a 280 bp fragment containing the SNP of interest followed by Qiagen spin column purification and sequencing.
Statistical Analysis
Statistical analysis was performed with Prism (Graphpad Software) and STATA (Statacorp LP, Texas, USA) and is detailed in each figure legend.
Supplementary Material
Acknowledgments
We thank Rachel Brem for advice and providing the HapMap genotypes, Amy Weinmann for advice and help with macrophage transfections, Victor Mulero for recombinant zebrafish TNF, Marcel Wolbers for help with statistical analysis of the TB meningitis cohort, Glenna Peterson and Tiffany Pecor for technical support, James Cameron for fish facility management, Paul Edelstein and John Szumowski for discussion, Paul Edelstein for advice on statistics, and Paul Edelstein, Brendan Cormack and Kevin Urdahl for manuscript review. Funded by grants from the NIH (L.R., C.N.S.), an American Cancer Society postdoctoral fellowship and a Mallinckrodt Scholar Award (D.M.T.), a postdoctoral fellowship from the educational ministry of Spain (F.J.R.), the NWRCE for Biodefense and Emerging Infectious Diseases Research (L.R. and D.C.K.), the Burroughs Wellcome Fund (L.R. and T.R.H.), the Wellcome Trust, UK (G.T., J.F., S.D., T.T.H.C. and N.D.B.), the American Skin Association, the Dermatology Foundation, and an NIH postdoctoral fellowship grant (J.C.V.), and the Chinese Academy of Sciences (Y.Z.). M.C.K. is an American Cancer Society Professor. D.M.T. is a recipient of the NIH Director’s New Innovator Award and L.R. is a recipient of the NIH Director’s Pioneer Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agarwal N, Lamichhane G, Gupta R, Nolan S, Bishai WR. Cyclic AMP intoxication of macrophages by a Mycobacterium tuberculosis adenylate cyclase. Nature. 2009;460:98–102. doi: 10.1038/nature08123. [DOI] [PubMed] [Google Scholar]
- Ariel A, Chiang N, Arita M, Petasis NA, Serhan CN. Aspirin-triggered lipoxin A4 and B4 analogs block extracellular signal-regulated kinase-dependent TNF-alpha secretion from human T cells. J Immunol. 2003;170:6266–6272. doi: 10.4049/jimmunol.170.12.6266. [DOI] [PubMed] [Google Scholar]
- Auerbach BJ, Bisgaier CL, Wolle J, Saxena U. Oxidation of low density lipoproteins greatly enhances their association with lipoprotein lipase anchored to endothelial cell matrix. J Biol Chem. 1996;271:1329–1335. doi: 10.1074/jbc.271.3.1329. [DOI] [PubMed] [Google Scholar]
- Bafica A, Scanga CA, Serhan C, Machado F, White S, Sher A, Aliberti J. Host control of Mycobacterium tuberculosis is regulated by 5-lipoxygenase-dependent lipoxin production. J Clin Invest. 2005;115:1601–1606. doi: 10.1172/JCI23949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr M, Schurr E, Gros P. TB: screening for responses to a vile visitor. Cell. 2010;140:615–618. doi: 10.1016/j.cell.2010.02.030. [DOI] [PubMed] [Google Scholar]
- Chambers ST, Hendrickse WA, Record C, Rudge P, Smith H. Paradoxical expansion of intracranial tuberculomas during chemotherapy. Lancet. 1984;2:181–184. doi: 10.1016/s0140-6736(84)90478-1. [DOI] [PubMed] [Google Scholar]
- Chen M, Divangahi M, Gan H, Shin DS, Hong S, Lee DM, Serhan CN, Behar SM, Remold HG. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J Exp Med. 2008;205:2791–2801. doi: 10.1084/jem.20080767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Current opinion in cell biology. 2010;22:263–268. doi: 10.1016/j.ceb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claria J, Serhan CN. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci U S A. 1995;92:9475–9479. doi: 10.1073/pnas.92.21.9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay H, Volkman HE, Ramakrishnan L. Tumor necrosis factor signaling mediates resistance to mycobacteria by inhibiting bacterial growth and macrophage death. Immunity. 2008;29:283–294. doi: 10.1016/j.immuni.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis J, Kopanitsa L, Stebbings E, Speirs A, Ignatyeva O, Balabanova Y, Nikolayevskyy V, Hoffner S, Horstmann R, Drobniewski F, et al. Association analysis of the LTA4H gene polymorphisms and pulmonary tuberculosis in 9115 subjects. Tuberculosis. 2011;91:22–25. doi: 10.1016/j.tube.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvignes L, Ernst JD. Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity. 2009;31:974–985. doi: 10.1016/j.immuni.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divangahi M, Desjardins D, Nunes-Alves C, Remold HG, Behar SM. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat Immunol. 2010;11:751–758. doi: 10.1038/ni.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donald PR, Schoeman JF. Tuberculous meningitis. N Engl J Med. 2004;351:1719–1720. doi: 10.1056/NEJMp048227. [DOI] [PubMed] [Google Scholar]
- Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin A, Abel L, Casanova JL, Gros P. Host genetics of mycobacterial diseases in mice and men: forward genetic studies of BCG-osis and tuberculosis. Annu Rev Genomics Hum Genet. 2007;8:163–192. doi: 10.1146/annurev.genom.8.080706.092315. [DOI] [PubMed] [Google Scholar]
- Goldman G, Welbourn R, Kobzik L, Valeri CR, Shepro D, Hechtman HB. Lavage with leukotriene B4 induces lung generation of tumor necrosis factor-alpha that in turn mediates neutrophil diapedesis. Surgery. 1993;113:297–303. [PubMed] [Google Scholar]
- Haeggstrom JZ. Leukotriene A4 hydrolase/aminopeptidase, the gatekeeper of chemotactic leukotriene B4 biosynthesis. J Biol Chem. 2004;279:50639–50642. doi: 10.1074/jbc.R400027200. [DOI] [PubMed] [Google Scholar]
- Hawkey CR, Yap T, Pereira J, Moore DA, Davidson RN, Pasvol G, Kon OM, Wall RA, Wilkinson RJ. Characterization and management of paradoxical upgrading reactions in HIV-uninfected patients with lymph node tuberculosis. Clin Infect Dis. 2005;40:1368–1371. doi: 10.1086/429317. [DOI] [PubMed] [Google Scholar]
- Kahawita IP, Lockwood DN. Towards understanding the pathology of erythema nodosum leprosum. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2008;102:329–337. doi: 10.1016/j.trstmh.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Kwan CK, Ernst JD. HIV and Tuberculosis: a Deadly Human Syndemic. Clin Microbiol Rev. 2011;24:351–376. doi: 10.1128/CMR.00042-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawn SD, Zumla AI. Tuberculosis. Lancet. 2011 doi: 10.1016/S0140-6736(10)62173-3. [DOI] [PubMed] [Google Scholar]
- Markman M, Eagleton LE. Paradoxical clinical improvement and radiographic deterioration in anergic patients treated for far advanced tuberculosis. The New England journal of medicine. 1981;305:167. doi: 10.1056/nejm198107163050312. [DOI] [PubMed] [Google Scholar]
- Mayanja-Kizza H, Jones-Lopez E, Okwera A, Wallis RS, Ellner JJ, Mugerwa RD, Whalen CC. Immunoadjuvant prednisolone therapy for HIV-associated tuberculosis: a phase 2 clinical trial in Uganda. J Infect Dis. 2005;191:856–865. doi: 10.1086/427995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayosi BM, Ntsekhe M, Volmink JA, Commerford PJ. Interventions for treating tuberculous pericarditis. Cochrane Database Syst Rev. 2002:CD000526. doi: 10.1002/14651858.CD000526. [DOI] [PubMed] [Google Scholar]
- Misra UK, Kalita J, Nair PP. Role of aspirin in tuberculous meningitis: a randomized open label placebo controlled trial. J Neurol Sci. 2010;293:12–17. doi: 10.1016/j.jns.2010.03.025. [DOI] [PubMed] [Google Scholar]
- Morris T, Stables M, Hobbs A, de Souza P, Colville-Nash P, Warner T, Newson J, Bellingan G, Gilroy DW. Effects of low-dose aspirin on acute inflammatory responses in humans. Journal of immunology. 2009;183:2089–2096. doi: 10.4049/jimmunol.0900477. [DOI] [PubMed] [Google Scholar]
- Muthuswamy P, Hu TC, Carasso B, Antonio M, Dandamudi N. Prednisone as adjunctive therapy in the management of pulmonary tuberculosis. Report of 12 cases and review of the literature. Chest. 1995;107:1621–1630. doi: 10.1378/chest.107.6.1621. [DOI] [PubMed] [Google Scholar]
- Payan DG, Missirian-Bastian A, Goetzl EJ. Human T-lymphocyte subset specificity of the regulatory effects of leukotriene B4. Proc Natl Acad Sci U S A. 1984;81:3501–3505. doi: 10.1073/pnas.81.11.3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad K, Singh MB. Corticosteroids for managing tuberculous meningitis. Cochrane Database Syst Rev. 2008:CD002244. doi: 10.1002/14651858.CD002244.pub3. [DOI] [PubMed] [Google Scholar]
- Roca FJ, Mulero I, Lopez-Munoz A, Sepulcre MP, Renshaw SA, Meseguer J, Mulero V. Evolution of the inflammatory response in vertebrates: fish TNF-alpha is a powerful activator of endothelial cells but hardly activates phagocytes. J Immunol. 2008;181:5071–5081. doi: 10.4049/jimmunol.181.7.5071. [DOI] [PubMed] [Google Scholar]
- Samuelsson B, Dahlen SE, Lindgren JA, Rouzer CA, Serhan CN. Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science. 1987;237:1171–1176. doi: 10.1126/science.2820055. [DOI] [PubMed] [Google Scholar]
- Schoeman JF, Janse van Rensburg A, Laubscher JA, Springer P. The Role of Aspirin in Childhood Tuberculous Meningitis. Journal of child neurology. 2011 doi: 10.1177/0883073811398132. [DOI] [PubMed] [Google Scholar]
- Schoeman JF, Van Zyl LE, Laubscher JA, Donald PR. Effect of corticosteroids on intracranial pressure, computed tomographic findings, and clinical outcome in young children with tuberculous meningitis. Pediatrics. 1997;99:226–231. doi: 10.1542/peds.99.2.226. [DOI] [PubMed] [Google Scholar]
- Serhan C, Ward P, Gilroy D. Fundamentals of Inflammation. Cambridge University Press; 2010. [Google Scholar]
- Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. 2007;25:101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Fiore S, Brezinski DA, Lynch S. Lipoxin A4 metabolism by differentiated HL-60 cells and human monocytes: conversion to novel 15-oxo and dihydro products. Biochemistry. 1993;32:6313–6319. doi: 10.1021/bi00076a002. [DOI] [PubMed] [Google Scholar]
- Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, Ingle CE, Dunning M, Flicek P, Koller D, et al. Population genomics of human gene expression. Nat Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thwaites GE, Nguyen DB, Nguyen HD, Hoang TQ, Do TT, Nguyen TC, Nguyen QH, Nguyen TT, Nguyen NH, Nguyen TN, et al. Dexamethasone for the treatment of tuberculous meningitis in adolescents and adults. N Engl J Med. 2004;351:1741–1751. doi: 10.1056/NEJMoa040573. [DOI] [PubMed] [Google Scholar]
- Tobin DM, Ramakrishnan L. Comparative pathogenesis of Mycobacterium marinum and Mycobacterium tuberculosis. Cell Microbiol. 2008;10:1027–1039. doi: 10.1111/j.1462-5822.2008.01133.x. [DOI] [PubMed] [Google Scholar]
- Tobin DM, Vary JC, Jr, Ray JP, Walsh GS, Dunstan SJ, Bang ND, Hagge DA, Khadge S, King MC, Hawn TR, et al. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell. 2010;140:717–730. doi: 10.1016/j.cell.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Science signaling. 2010;3:re4. doi: 10.1126/scisignal.3115re4. [DOI] [PubMed] [Google Scholar]
- Volkman HE, Pozos TC, Zheng J, Davis JM, Rawls JF, Ramakrishnan L. Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science. 2010;327:466–469. doi: 10.1126/science.1179663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis RS. Reconsidering adjuvant immunotherapy for tuberculosis. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2005;41:201–208. doi: 10.1086/430914. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.