

Abstract
Alzheimer's disease is associated with a disruption of amyloid β (Aβ) homeostasis, resulting in the accumulation and subsequent deposition of Aβ peptides within the brain. The peroxisome proliferator-activated receptor-γ (PPARγ) is a ligand-activated nuclear receptor that acts in a coupled metabolic cycle with Liver X Receptors (LXRs) to increase brain apolipoprotein E (apoE) levels. apoE functions to promote the proteolytic clearance of soluble forms of Aβ, and we found that the synthetic PPARγ agonist, pioglitazone, stimulated Aβ degradation by both microglia and astrocytes in an LXR and apoE-dependent manner. Remarkably, a brief 9 d oral treatment of APPswe/PS1Δe9 mice with pioglitazone resulted in dramatic reductions in brain levels of soluble and insoluble Aβ levels which correlated with the loss of both diffuse and dense-core plaques within the cortex. The removal of preexisting amyloid deposits was associated with the appearance of abundant Aβ-laden microglia and astrocytes. Pioglitazone treatment resulted in the phenotypic polarization of microglial cells from a proinflammatory M1 state, into an anti-inflammatory M2 state that was associated with enhanced phagocytosis of deposited forms of amyloid. The reduction in amyloid levels was associated with a reversal of contextual memory deficits in the drug-treated mice. These data provide a mechanistic explanation for how PPARγ activation facilitates amyloid clearance and supports the therapeutic utility of PPARγ agonists for the treatment of Alzheimer's disease.
Introduction
Alzheimer's disease (AD) is characterized by the accumulation and deposition of amyloid β (Aβ) within the brain because of inefficient clearance of these peptides (Mawuenyega et al., 2010; Querfurth and LaFerla, 2010). Aβ accumulation is associated with perturbations in synaptic function, neuronal loss, and memory and cognitive impairments (Lue et al., 1999; Akiyama et al., 2000b; Palop and Mucke, 2010).
Apolipoprotein E (apoE) is the principal genetic risk factor for sporadic, late-onset AD (Corder et al., 1993; Roses, 1996). apoE is synthesized mainly by astrocytes and plays an isoform-dependent role in modulating Aβ fibrillization and clearance (Holtzman, 2001; Zlokovic et al., 2005; Kim et al., 2009). apoE scaffolds the formation of high-density lipoproteins (HDLs), which traffic cholesterol and phospholipids (Kim et al., 2009). Lipids are transferred to apoE through the transporter, ABCA1 (Wahrle et al., 2004). Importantly, apoE-containing HDL particles facilitate the proteolytic degradation of soluble Aβ (sAβ) (Jiang et al., 2008).
The nuclear receptors, peroxisome proliferator-activated receptor-γ (PPARγ) and Liver X receptors (LXRs) are ligand-activated transcription factors that act as fatty acid and cholesterol sensors (Forman et al., 1997; Kersten et al., 2000). LXR activation induces HDL particle formation through apoe and abca1 expression (Lehmann et al., 1997; Tall, 2008). Sustained PPARγ or LXR activation results in amelioration of AD-related pathophysiology in AD mouse models (Yan et al., 2003; Pedersen and Flynn, 2004; Heneka et al., 2005; Koldamova et al., 2005a; Riddell et al., 2007; Zelcer et al., 2007; Jiang et al., 2008; Donkin et al., 2010; Fitz et al., 2010; Toledo and Inestrosa, 2010). Peripherally, LXRs and PPARγ participate in a coupled metabolic pathway, whereby activation of PPARγ induces expression of LXRα and its target genes abca1 and apoE (Chawla et al., 2001; Seo et al., 2004; Yue and Mazzone, 2009). However, these relationships have yet to be explored in the brain.
Aβ deposition elicits a robust “M1” microglia-mediated inflammatory response contributing to disease pathogenesis (Gordon, 2003; Mosser and Edwards, 2008; Mandrekar-Colucci and Landreth, 2010). It has recently been appreciated that PPARs act as master regulators governing the polarization of macrophages and microglia into “M2” or “alternative” activation states associated with the suppression of inflammation and promotion of phagocytosis and tissue repair (Chawla, 2010; Chinetti-Gbaguidi et al., 2011). While M2 gene expression has been detected in the AD brain, little is known about the mechanisms modulating this phenotypic conversion (Colton et al., 2006; Jimenez et al., 2008; Maier et al., 2008).
We demonstrate that PPARγ activation induces lxrα, apoe, and abca1 expression, promoting Aβ clearance by both microglia and astrocytes. Strikingly, a brief 9 d pioglitazone treatment of APP/PS1 mice reversed pathological and behavioral phenotypes in treated mice. We argue that sAβ clearance results from stimulation of apoE-dependent proteolysis of Aβ, whereas deposited Aβ is removed phagocytically by microglia as a result of a PPARγ-dependent alternative M2 polarization of microglia. These data provide a mechanistic link between PPARγ activation and amyloid clearance and support the therapeutic use of its agonists in AD.
Materials and Methods
Reagents.
The Aβ1–42 peptide was purchased from American Peptide Company, dissolved in DMSO to a final concentration of 1 μg/ml.
Cell culture.
Primary microglia and astrocytes were cultured from postnatal day 0–3 C57BL/6J mice and have been previously described (McDonald et al., 1997). Briefly, pups were decapitated and the meninges and blood vessels were removed from the cortex. The cortices were then minced and trypsinized with 0.05% trypsin-EDTA (Invitrogen) for 20 min at 37°C. DMEM/F12 (Invitrogen) containing 10% heat-inactivated fetal bovine serum (HI-FBS, Atlanta Biologicals) and 1% penicillin-streptomycin (Invitrogen) were then added to stop trypsin digestion. Cells were then triturated and plated in 150 mm dishes (Invitrogen) at a density of half a cortex/plate. Medium was changed the following day to fresh DMEM/F12 with 10% HI-FBS and 1% penicillin/streptomycin and incubated for 14–21 d at 37°C, 5% CO2. Microglia were isolated as described by Saura et al. (2003). Briefly, the microglia were removed by shaking the tissue culture plates to remove loosely adherent microglia for 30 min. Astrocytes were removed next by 0.25% trypsin/HBSS diluted 1:3 in serum-free DMEM/F12 at 37°C for 30 min. The astrocytes were then collected by centrifugation at 2000 rpm for 5 min. The cells were resuspended in DMEM/F12 containing 10% HI-FBS and plated in 24 or 6 well plates (Invitrogen). Astrocyte medium was changed 20 min after plating to remove any microglial contamination of the cultures. Astrocytes were allowed to grow for 3–5 d before experiments. Firmly attached microglia were harvested using 0.25% trypsin/PBS. Loosely and firmly adherent microglia were then combined, and cells were counted and plated for experiments at the appropriate densities in DMEM/F12 medium containing HI-FBS. Medium was changed to serum-free DMEM/F12 overnight before beginning an experiment.
Aβ ELISAs.
Aβ levels were quantified using an Aβ42-specific antibody, 6E10, as the capture antibody and detected using monoclonal 4G8 HRP-conjugated antibodies (Covance). Synthetic Aβ42 was used to generate a standard curve for each experiment. The plates were developed using the TMB substrate kit (Pierce), and the reaction was stopped by the addition of 1 m HCl. The results were read using a SpectraMax colorimetric plate reader (Molecular Devices). Primary mouse microglia or astrocytes were incubated with DMSO or relevant concentrations of pioglitazone (dissolved in DMSO), T0070907 (T0; 10 nm), 22S hydroxycholesterol (22HC; 10 μm) for 24 h at 37°C. Receptor antagonists require a 1–2 h preincubation. Cells were then treated with 2 μg/ml soluble Aβ42 in serum-free medium for 24 h with the specified treatments. Purified human plasma ApoE containing a mixture of all human ApoE isoforms (rPeptide) or human ApoAI was applied at the same time as soluble Aβ42. Cells were washed with PBS and lysed in 1% SDS. Aβ42 levels were measured using ELISA and normalized to total protein.
Western blot analysis.
Protein concentrations of cell lysates or brain extracts were measured using the BCA method (Pierce) and resolved on Bis-Tris 4–12% gels (Invitrogen). The following primary antibodies were used: anti-actin (Santa Cruz Biotechnology); anti-ApoE (Santa Cruz Biotechnology); anti-β-actin (Santa Cruz Biotechnology); anti-ABCA1 (Novus Biologicals); anti-GAPDH (Santa Cruz Biotechnology).
Animals.
APPswe/PS1Δe9 (APP/PS1) transgenic mice [B6C3-Tg(APPswe, PSEN1dE9)85Bdo/J] were obtained from Jackson Laboratories (Jankowsky et al., 2004). APP/PS1 mice or wild-type littermates were gavaged daily for 9 d with 80 mg · kg−1 · d−1 pioglitazone or vehicle (water). Floxed PPARγ (PPARγfl/fl) mice [B6.129-Pparγtm2Rev] were obtained from Jackson Laboratory and were crossed with GFAP-cre mice [B6.Cg-Tg(Gfap-cre)73.12Mvs/J], also obtained from Jackson Laboratories, to generate mice that lacked PPARγ expression in astrocytes and cortical neurons. The animals were then killed and one hemisphere was fixed and processed for immunohistochemistry. The hippocampus and cortex were removed from the other hemisphere and snap-frozen and stored at −80°C until they were subject to RNA and protein extraction. Approximately equal numbers of male and female transgenic and nontransgenic mice were used for all experiments. While the number of mice analyzed for each dataset varies, the male-to-female ratio remained consistent. All experiments involving animals followed approved protocols by the Case Western Reserve University School of Medicine.
Tissue collection and immunohistochemistry.
Postfixed hemispheres were sectioned (10 μm) using a cryostat. Sections were mounted, air-dried, and then stored at −20°C until use. Thioflavin S (thioS) (Sigma) staining was performed by rehydrating sections and staining with 1% thioflavin S. Slides were coverslipped with Prolong Gold (Invitrogen). The numbers of thioS+ plaques were counted by a blinded observer. Alternate sections were stained with 6E10 following pretreatment with 70% formic acid for 3 min. Sections were then blocked with 5% normal goat serum and incubated overnight in primary antibody (6E10, 1:1000, Signet Laboratories; Iba1, 1:300, Wako; CD45, 1:500, Serotec; GFAP, 1:1000, Dako). Slides were incubated with the appropriate Alexa Fluor-conjugated secondary antibodies, followed by DAPI labeling of nuclei. Two sections per slide and 3 slides per animals, spaced evenly from 200 μm anterior to the appearance of the CA3 to the end of the hippocampus, were analyzed. Images were analyzed for the percentage area occupied by 6E10-positive amyloid plaques using Image Pro-Plus software (Media Cybernetics).
Brain homogenates and Aβ ELISA.
Cortices were removed from hemibrains and were homogenized in 800 μl of tissue homogenizing buffer (250 mm sucrose, 20 mm Tris, 1 mm EDTA, 1 mm EGTA in diethylpyrocarbonate water) containing Protease Inhibitor Cocktail (1:100, Sigma), using a glass-on-glass homogenizer at 4°C. The homogenate was centrifuged at 5000 × g for 10 min at 4°C, and supernatants were collected and stored at −80°C for Western blot analysis. For “soluble” Aβ extraction, 400 μl of homogenates were mixed with 400 μl of 0.4% diethylamine, 100 mm NaCl, and the samples were homogenized using a glass-on-glass homogenizer. Samples were centrifuged at 135,000 × g for 1 h at 4°C. Supernatants were collected, 0.5 m Tris-HCl, pH 6.8 was added, and the samples were stored at −80°C for analysis of soluble Aβ. The resulting pellet was sonicated in 70% cold formic acid and centrifuged at 109,000 × g for 1 h at 4°C. The supernatant was collected, the formic acid was neutralized, and the samples were stored at −80°C for the analysis of “insoluble” Aβ. Aβ1–40 and Aβ1–42 ELISAs were performed using 6E10 as the capture antibody and HRP-conjugated antibodies specific to Aβ1–40 and Aβ1–42 (Covance) for detection, and processed as described above. Samples were normalized to protein concentration using the BCA method.
RNA extraction, reverse transcription, and quantitative PCR.
Total RNA was isolated from cortices using RNA-Bee (Tel-Test). Equal amounts of the homogenate and RNA-Bee were combined, chloroform was added, and samples were mixed by vigorous shaking. Samples were incubated on ice for 15 min and then centrifuged at 13,000 × g for 15 min at 4°C. The aqueous phase was removed, combined with an equal amount of 70% ethanol, and applied to an RNeasy Mini Spin Column (Qiagen). RNA was eluted according to the manufacturer's instructions. RNA samples were analyzed for concentration and purity on a Nanodrop 2000 spectrophotometer (Thermo Fischer Scientific). cDNA was synthesized from RNA samples using a QuantiTect Reverse Transcription kit (Qiagen) per the manufacturer's instructions with 0.5 μg total RNA. Fourteen cycles of cDNA preamplification were performed according to the manufacturer's protocol using TaqMan PreAmp Master Mix for select primer sets (Applied Biosystems). Pre-Amplified cDNA was used for qPCR with the StepOne Plus Real Time PCR system (Applied Biosystems) in a 10 μl reaction for 40 cycles. Primers used were labeled with FAM probes including Abca1 (Mm01350760_m1), ApoE (Mm00437573_m1), LXRα (Mm00443451_m1), PPARγ (Mm01184322_m1), GFAP (Mm01253033_m1), Iba1 (Mm00479862_g1), CD45 (Mm01293575_m1), App (Hs00245154_m1), Bace1 (Mm00478664_m1), Psen1 (Mm00501184_m1), Tnfα (Mm99999068_m1), Ccl2 (Mm00441242_m1), Il-1β (Mm01336189_m1), Nos2 (Mm01309902_m1), Cox2 (Mm01307329_m1), Ym1 (Mm00657889_m1), Fizz1 (Mm00445109_m1), Arg1 (Mm00475988_m1), Socs1 (Mm00782550_s1), Tgf-β (Mm01178820_m1) and GAPDH (4352339E-0904021) with a VIC probe from Applied Biosystems. Analysis of gene expression was performed using the comparative Ct method (ΔΔCT), where the threshold cycle for the target genes was normalized to that of GAPDH (ΔCT), and the mRNA expression fold change was calculated using the equation 2−ΔΔCT, where ΔΔCT = ΔCT test sample − ΔCT calibrator sample.
Behavioral analysis; contextual fear conditioning.
In the training phase, mice were individually placed in the shock chamber to freely explore the environment for 2 min. Mice were exposed to the conditioned stimulus (CS: an 85 dB sound at 2800 Hz) for 30 s. After 2 s, the unconditioned stimulus (US: 0.56 mA) was delivered. After the CS/US pairing, the mice were kept in the chamber for another 30 s to measure the immediate freezing response. This process was repeated 4 times. Retention tests were performed 24 h later. Each mouse was returned to the same shock chamber for 5 min for contextual freezing measurements in the absence of tone and the number of freezes was measured. Freezing behavior was monitored by an automated tracking system (Coulbourn Instruments).
Results
PPARγ facilitates the degradation of soluble Aβ through an LXR/ApoE-dependent pathway
Our previous work and that of others has demonstrated a role for LXR activation in stimulating the production of apoE and facilitating the proteolytic degradation of Aβ (Koldamova et al., 2005a; Riddell et al., 2007; Jiang et al., 2008; Donkin et al., 2010; Fitz et al., 2010). PPARγ acts within a common metabolic pathway with LXRs in peripheral macrophages, thus we sought to determine whether activation of PPARγ would activate LXR pathways and induce clearance of Aβ though an apoE-dependent mechanism in the CNS (Chawla et al., 2001; Hong and Tontonoz, 2008). Treatment of primary microglia or astrocytes with the synthetic agonist pioglitazone stimulated the intracellular degradation of soluble Aβ42 in a dose-dependent manner (Fig. 1A,D). Importantly, pioglitazone treatment of primary microglia and astrocytes resulted in the induction of the LXR target genes, abca1 and apoe in both cell types (Fig. 1B–F). Based on the degradation efficiency and gene induction data we chose a pioglitazone dose of 100 nm for microglia and 50 nm for astrocytes for the subsequent in vitro studies. To determine whether degradation of sAβ was dependent on the actions of PPARγ and LXRs, microglia and astrocytes were pretreated with antagonists of these receptors (T0 and 22HC, respectively; Lee et al., 2002; Kase et al., 2007). Both PPARγ and LXR antagonist treatment of microglia and astrocytes inhibited PPARγ-facilitated degradation of sAβ, suggesting that the activation of both PPAR and LXR pathways is necessary for the intracellular degradation of sAβ in these cell types (Fig. 2A,B).
Figure 1.

Activation of PPARγ facilitates the expression of LXR target genes and promotes the degradation of Aβ. Primary microglia (A) or astrocytes (D) were isolated from wild-type mice and pretreated with either DMSO (Ctrl) or increasing concentrations of pioglitazone (Pio) for 24 h. The cells were then incubated with 2 μg/ml Aβ in the presence of DMSO or the indicated dose of pioglitazone for 24 h. Intracellular Aβ levels were quantified using ELISA for Aβ42 and normalized to total protein (mean ±SEM, ***p < 0.001). Primary microglia (B, C) or astrocytes (E, F) were treated with DMSO or increasing doses of Pio for 24 h. Cellular lysates were subjected to SDS-PAGE, and Western analysis for abca1, apoe, or actin was performed (mean ± SEM, Student's t test, *p < 0.05, **p < 0.01). Quantification of three separate experiments has been graphed and representative Western blots have been included with each dataset. These data are represented as a percentage of DMSO-treated control samples.
Figure 2.

PPARγ-mediated degradation of Aβ is dependent on induction of ApoE through the stimulation of LXR and PPAR pathways. Primary wild-type microglia (A) or astrocytes (B) were treated for 24 h with pioglitazone or DMSO followed by the addition of soluble Aβ42 (2 μg/ml) for 24 h. The cells were pretreated with antagonists for PPARγ (T0070907, 10 nm) or LXR (22S hydroxycholesterol, 10 μm) for 2 h. Intracellular Aβ was measured by ELISA. apoe knock-out microglia (C) or astrocytes (D), or wild-type microglia (E) or astrocytes (F) were pretreated for 24 h with DMSO or drug, followed by the addition of soluble Aβ42 and exogenously supplied apoE (1 μg/ml) or apoA1 (2 μg/ml). Remaining intracellular Aβ was measured using ELISA. These data are represented as a percentage of DMSO-treated control samples (mean ± SEM, Student's t test, *p < 0.05, **p < 0.01, ***p < 0.001, n ≥ 3). Cortical homogenates obtained from PPARγ conditional knock-out animals were analyzed by Western blot analysis for ABCA1, apoE, and actin (G) and quantified (H) (mean ± SEM, Student's t test, *p < 0.05, ***p < 0.001, n ≤ 6 animals per genotype).
ApoE has been shown to play a vital role in amyloid clearance and its expression is directly regulated by LXR activation (Beaven and Tontonoz, 2006; Jiang et al., 2008). Given the linkage of PPARγ to the LXR pathway, we sought to determine whether apoE was critically involved in PPARγ-mediated intracellular clearance of sAβ (Chawla et al., 2001). Microglia and astrocytes were obtained from apoe−/− pups and then treated with pioglitazone. Loss of apoe resulted in a significant impairment of intracellular sAβ degradation by both cell types in response to pioglitazone treatment (Fig. 2C,D). This was restored by the addition of exogenously supplied ApoE (a mixture of all human ApoE isoforms) or human apolipoprotein A1 (ApoA1) (Fig. 2C,D). ApoA1 is an HDL-associated apolipoprotein which is lipidated by ABCA1 and acts in a similar fashion to ApoE in the periphery (Koch et al., 2001; Denis et al., 2004; Smith et al., 2004), and has been shown to bind to Aβ (Koldamova et al., 2005b) and promote Aβ degradation (Jiang et al., 2008). Exogenous ApoE or ApoA1 stimulated the clearance of Aβ peptides from the media (Fig. 2E,F). Addition of ApoE or ApoA1 to pioglitazone-treated microglia or astrocytes (wild-type or ApoE−/−) did not have additive or synergistic effects on intracelluar sAβ degradation (Fig. 2E,F). These studies demonstrate that the effect of PPARγ agonists in vitro on sAβ degradation is dependent on the expression of apoE and simply elevating the levels of apolipoproteins are sufficient to facilitate sAβ clearance.
To determine the role of PPARγ in modulating the expression of the LXR target genes, apoE and abca1, mice lacking PPARγ gene expression in astrocytes, as well as most cortical neurons, were obtained by crossing a PPARγ fl/fl mouse with a mouse expressing hGFAP-cre. The PPARγ conditional knock-out (PPARγ CKO) mice display significantly lower levels of pparγ mRNA which was reduced in the cortices of these animals by >70% (data not shown). Six-month-old PPARγ CKO animals displayed more than a 50% reduction in cortical protein levels of abca1 and apoe, demonstrating the requirement for PPARγ in LXR actions in the brain and the normal expression of its target genes apoe and abca1 (Fig. 2G,H). These data document a robust PPARγ-LXR linkage in the brain which has not previously been recognized.
PPARγ activation rapidly stimulates the clearance of Aβ in a mouse model of AD
To determine the effects of PPARγ activation during both early and more advanced stages of amyloid pathogenesis, 6- and 12-month-old APPSwePSEN1ΔE9 (APP/PS1) mice were orally administered pioglitazone (80 mg · kg−1 · d−1) for 9 d. The APP/PS1 transgenic mice begin depositing fibrillar thioS+ plaques at ∼5–6 months of age. Pioglitazone treatment of 6 and 12 month animals resulted in a significant induction in cortical protein levels of both ABCA1 and ApoE (Fig. 3A–D). Additionally, the assessment of LXR target genes by quantitative real-time PCR showed a statistically significant increase in levels of mRNA for abca1, apoE, and lxrα (Fig. 3E,F). Pioglitazone-treated 12 month animals additionally showed an increase in levels of cortical pparγ mRNA (Fig. 3F). These experiments demonstrate that in vivo pioglitazone activates PPARγ and induces the expression of LXRα and its target genes in the AD brain, solidifying the linkage of a PPARγ-LXRα signaling pathway in the CNS. It should be noted that pioglitazone is poorly blood–brain barrier permeable, necessitating administration of the drug at 80 mg · kg−1 · d−1, a dose higher than previously administered (Maeshiba et al., 1997).
Figure 3.

PPARγ activation drives the expression of LXR target genes in vivo. Six (A, C)- and 12 (B, D)-month-old APP/PS1 mice were gavaged orally with 80 mg · kg−1 · d−1 pioglitazone (Pio) or vehicle (H2O) for 9 d. Cortical homogenates were then analyzed by Western blot for levels of abca1, apoe, and actin (A, B) and quantified (C, D). (mean ± SEM, *p < 0.05, **p < 0.01, n ≥ 6 animals/treatment). Quantitative PCR results for LXR target genes from cortices of 6 (E)- or 12 (F)-month-old APP/PS1 animals treated with pioglitazone (Pio) or vehicle (Veh) or wild-type animals treated with vehicle (WT) for 9 d (mean ± SEM, Student's t test, *p < 0.05, **p < 0.01, n ≤ 13 animals/group). Fold change is reported as a percentage of vehicle-treated WT animals.
The brief 9 d treatment of APP/PS1 mice resulted in a striking reduction in cortical amyloid peptide levels and plaque burden. Immunohistochemical analysis of 6E10-immunoreactive plaques in cortical slices from animals of both ages revealed ∼40% and 30% (6 and 12 months, respectively) reduction in total levels of deposited Aβ compared with vehicle-treated APP/PS1 littermates (Fig. 4A–E). There was a parallel loss in the number of thioflavin S+ dense-core amyloid plaques, with an ∼40% and 30% decrease in the number of thioS+ plaques in 6 and 12 month animals, respectively (Fig. 4F). Remarkably, at 6 months of age, only 9 d of pioglitazone treatment reduced soluble and insoluble levels of Aβ by ∼50% (Fig. 4G,H). At 12 months of age, no significant change was seen in levels of soluble Aβ42; however, soluble levels of Aβ40 were reduced by 40% and insoluble levels of both Aβ40 and Aβ42 were reduced by 50% and 40%, respectively (Fig. 4K,L). This striking result suggests that short-term pioglitazone treatment not only reduces levels of sAβ but also results in the clearance of predeposited insoluble dense-core Aβ plaques in 12-month-old transgenic animals.
Figure 4.

Pioglitazone induces the clearance of amyloid in APP/PS1 mice. APP/PS1 mice were gavaged for 9 d with pioglitazone (80 mg · kg−1 · d−1) or vehicle (water). Plaque pathology was evaluated in the cortex of vehicle- or pioglitazone-treated 6 month (A–C, F–H) or 12 month (C–F, K, L) animals by staining with 6E10 (anti-Aβ antibody) (A, B, D, E) or thioflavin S. Quantification of 6E10+ plaque area (C) and number of thioS-positive plaques (F). Soluble and insoluble levels of Aβ40 and Aβ42 were measured by ELISA in 6 (G, H) and 12 (K, L) month animals. Real-time quantification of mRNA levels of app and the APP proteases bace and psen1 were measured in 6 (I) and 12 (M) month animals to determine whether pioglitazone treatment altered the expression of enzymes involved in APP processing. Cortical homogenates were evaluated for protein levels of APP and APP C-terminal fragments to determine whether PPARγ activation affected APP levels or processing in 6 month (J) and 12 month (N) animals (mean ± SEM, Student's t test, *p < 0.05, **p < 0.01, ***p < 0.001, n ≥ 6 animals/group).
Quantitative reverse transcriptase PCR was performed to verify that the changes seen in amyloid pathology in response to PPARγ activation were due to increased proteolysis of the peptide and not to a change in levels of the amyloid precursor protein (APP) or components of the β and γ secretase. mRNA levels of app, bace1, or psen1 or protein levels of APP and APP C-terminal fragments were not altered in response to drug treatment compared with vehicle-treated littermates, suggesting that PPARγ activation does not alter the expression or processing of APP (Fig. 4I,J,M,N). Previous work has also suggested that PPAR activation may regulate the expression of key Aβ proteases, insulin-degrading enzyme (IDE) and neprilysin (Du et al., 2009; Kalinin et al., 2009). We monitored mRNA levels of IDE and neprilysin to determine whether pioglitazone treatment enhanced expression of these proteases and observed no change (data not shown). These data suggest that the changes seen in amyloid deposition were indeed a result of Aβ catabolism and not due to alterations in APP processing or changes in levels of Aβ-degrading enzymes.
Pioglitazone suppresses glial activation and enhances amyloid clearance
The appearance of plaques in the AD brain is associated with a robust induction of astrogliosis and microgliosis (Bornemann et al., 2001; Wegiel et al., 2001). Microglia and astrocytes are found in intimate association with amyloid plaques in both the AD brain and brains of AD mouse models (Itagaki et al., 1989; Wisniewski et al., 1992). The number of glial cells surrounding a plaque directly increases in proportion to the dimensions of the deposits (Stalder et al., 1999; Sasaki et al., 2002). Similarly, the induction of inflammatory mediators parallels amyloid deposition and the phenotypic activation of glial cells in the AD brain (Akiyama et al., 2000a; Wyss-Coray, 2006). Since PPARγ activation has been shown to result in potent anti-inflammatory actions and represses microglia-mediated inflammation, we examined the effects of pioglitazone treatment on glial activation status and their association with amyloid deposits (Combs et al., 2001).
We first assessed association of astrocytes with amyloid plaques in the brains of both vehicle- and pioglitazone-treated 6- and 12-month-old APP/PS1 animals. Pioglitazone treatment significantly reduced the levels of GFAP-immunoreactive astrocytes surrounding amyloid plaques in transgenic animals at both 6 and 12 months of age and seen by immunohistochemistry (Fig. 5A–L). It was not possible to determine astrocyte cell numbers surrounding amyloid deposits with this technique; thus, we measured the mRNA levels of gfap as a surrogate measure of astrocytosis. We found that GFAP was elevated in APP/PS1 animals, but was significantly reduced in both age groups after pioglitazone treatment (Fig. 5N,O). In vehicle-treated animals, we observed astrocytes in close association with 6E10-positive amyloid plaques (Fig. 5C,I). However, after pioglitazone treatment, astrocytes were seen diffusely surrounding Aβ deposits and very little contact was observed between GFAP-positive cells and amyloid plaques (Fig. 5F,L).
Figure 5.
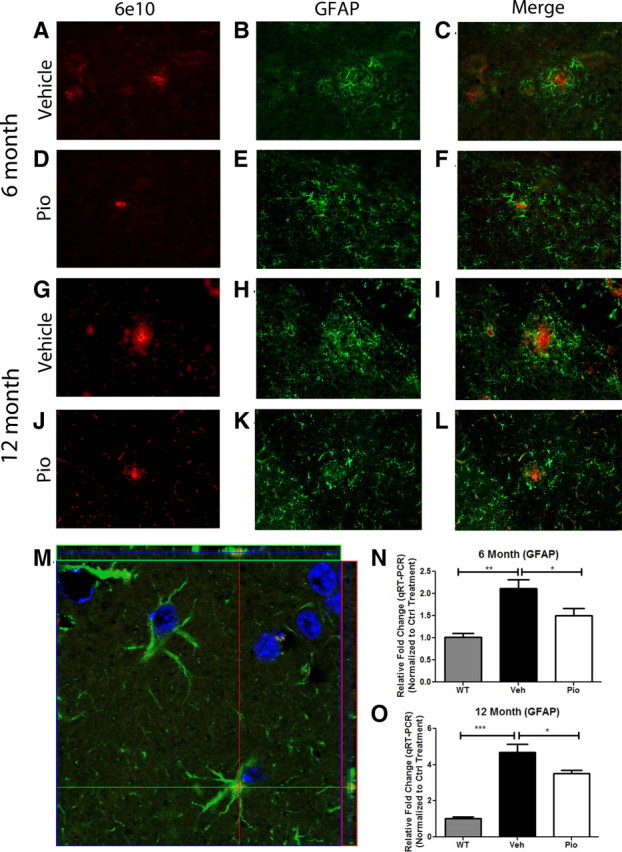
PPARγ suppresses astrocytosis in APP/PS1 animals. GFAP and 6E10 immunochemistry on coronal sections from 6 (A–F)- or 12 (G–L)- month-old APP/PS1 animals treated with pioglitazone (D–F, J–L) or vehicle (A–C, G–I) for 9 d. Magnification 20×. Representative image of astrocytes from the cortex of 12 month pioglitazone-treated animal (M) (red, 6E10; green, GFAP; blue, DAPI; 100× magnification). Quantitative real-time PCR in vehicle-treated WT or APP/PS1 animals or pioglitazone-treated APP/PS1 animals for GFAP in 6 (N)- or 12 (O)-month-old animals (mean ± SEM, Student's t test, *p < 0.05, **p < 0.01, ***p < 0.001, n = 8–13 animals/group).
Surprisingly, internalized Aβ peptides were found only in astrocytes of pioglitazone-treated animals, as seen in z-stack images obtained by confocal microscopy (Fig. 5M). We never detected Aβ-positive astrocytes in vehicle-treated animals (data not shown). It is our impression that the numbers of Aβ-positive astrocytes in drug-treated animals represented only a minor subset of total astrocytes. We were unable to quantitatively evaluate this given the limitations of our astroglial marker, GFAP. These data support the view that PPARγ activation promotes the clearance of insoluble Aβ by astrocytes.
We next evaluated microglial reactivity surrounding amyloid deposits in vehicle- and pioglitazone-treated animals. The association of Iba1+ microglial cells with amyloid plaques was reduced at both 6 and 12 months of age after pioglitazone treatment (Fig. 6D–F,J–L). Based on qualitative immunohistological evaluation, we found abundant microglia closely associated with plaque cores in vehicle-treated animals (Fig. 6A–C,G–I). The mRNA levels for iba1 were significantly lower in pioglitazone-treated animals compared with vehicle-treated animals at 6 months of age and a trend toward a reduction was seen at 12 months of age (Fig. 6S,T). Remarkably, at 12 months of age, we found that most microglia in pioglitazone-treated APP/PS1 animals exhibited robust internalization of Aβ (Fig. 7C,D). Microglia containing small amounts of Aβ were visualized in ∼60% of microglial cells in vehicle-treated animals; however, the amount of Aβ internalized by microglia in vehicle-treated animals paled compared with microglia in the cortex of pioglitazone-treated animals (Fig. 7A–D) It is our qualitative assessment that >90% of the microglia in the cortex of drug-treated animals internalized vast quantities of amyloid. Bolmont et al. (2008) have shown that microglia in the AD brain are capable of taking up Aβs and delivering them to lysosomal compartments. However, robust amyloid phagocytosis in response to drug treatment, as seen here, has to our knowledge never been observed before. It was difficult to assess Aβ uptake by microglia in 6-month-old animals since these animals are at the earlier stages of amyloid deposition, and there are very few Iba1+ cells surrounding Aβ deposits in vehicle-treated animals and fewer still in pioglitazone-treated animals. These data suggest that in addition to the clearance of soluble forms of Aβ, pioglitazone treatment facilitated the removal of both diffuse and dense-core Aβ deposits by microglia and astrocytes, presumably through engaging the phagocytic machinery of these cells.
Figure 6.
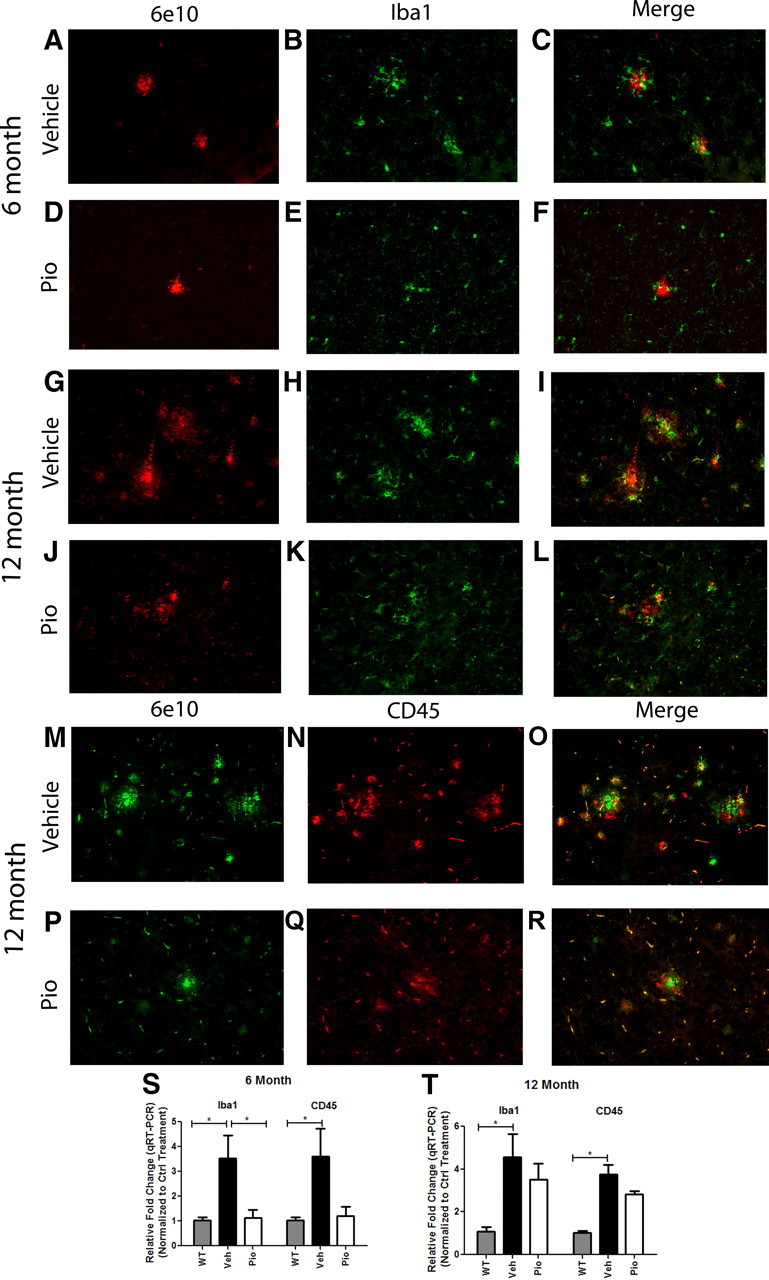
Pioglitazone suppresses microglial activation surrounding amyloid plaques in APP/PS1 animals. Cortical microglia were detected by Iba1 staining and Aβ by 6E10 immunochemistry on coronal sections from 6 (A–F)- or 12 (G–L)-month-old vehicle (A–C, G–I)- or pioglitazone (D–F, J–L)-treated APP/PS1 animals. Cortical CD45 and 6E10 staining in 12-month-old vehicle (M–O)- or pioglitazone (P–R)-treated animals. Real-time PCR quantification of transcript levels of Iba1 or CD45 in 6 (S)- and 12 (T)-month-old pioglitazone or vehicle-treated APP/PS1 animals (mean ± SEM, Student's t test, *p < 0.05, n ≤ 13 animals/group).
Figure 7.
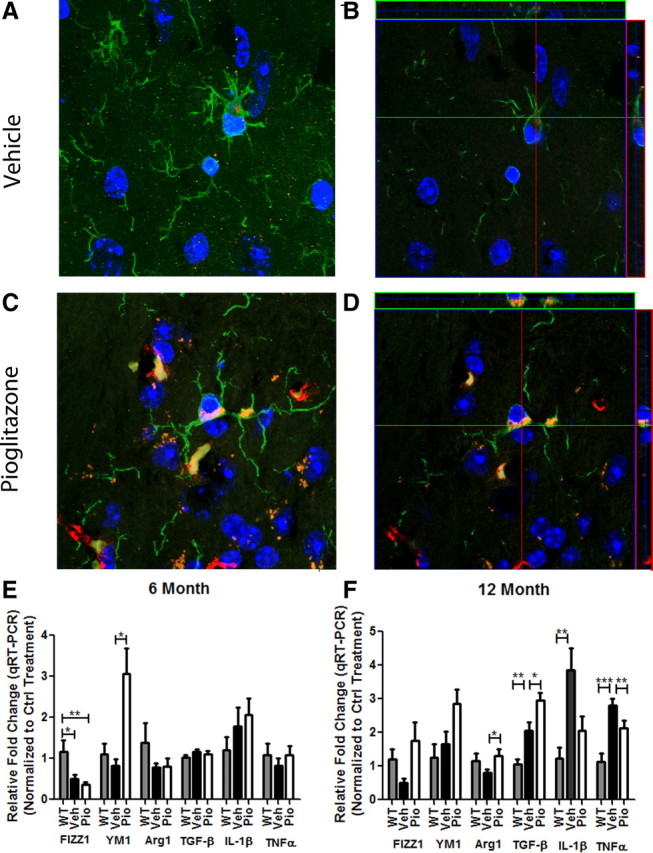
Pioglitazone treatment enhances microglial phagocytosis of Aβ. Representative images from vehicle (A, B)- or pioglitazone (C, D)-treated 12-month-old APP/PS1 animals. A and C represent projection images of z stacks (B, D). Green, Iba1; red, 6E10; blue, DAPI. Images were acquired using a Zeiss LSM 510 confocal microscope, 100× magnification. Six (E)- and 12 (F)-month-old APP/PS1 mice were gavaged orally with 80 mg · kg−1 · d−1 pioglitazone (Pio) or vehicle (Veh) for 9 d and WT animals were gavaged with H2O (WT). Quantitative real-time PCR was performed to assess transcript levels of M1 (classical; IL-1β, TNFα) or M2 (alternative; FIZZ1, YM1, Arg1, TGF-β) activation markers.
We also evaluated the expression by microglia of CD45, a tyrosine phosphatase that plays an integral role in immune signaling (Penninger et al., 2001). CD45 expression is elevated in microglia in the AD brain and is associated with classical M1 microglial activation (Masliah et al., 1991; Wilcock et al., 2001). In 6-month-old vehicle-treated animals, CD45 immunoreactivity was seen surrounding all 6E10-positive amyloid deposits (data not shown). However, only ∼50% of 6E10-positive deposits were associated with CD45+ microglia in pioglitazone-treated animals (data not shown). In 12 month animals, CD45+ microglia were associated with all amyloid deposits regardless of treatment (Fig. 6M–R). However, a trend for a reduction in cd45 mRNA levels was observed in pioglitazone-treated animals of both age groups (Fig. 6S,T). The reduction in levels of GFAP, CD45, and Iba1 in pioglitazone-treated animals may be attributed to the role of PPARγ in suppression of the inflammatory response.
Previous studies have demonstrated a role for PPARγ in polarizing macrophages to an alternative activation phenotype that is anti-inflammatory in nature and induces the phagocytic activity of cells (Mukundan et al., 2009; Chawla, 2010). Thus, we examined markers of M2 or alternative activation, which have been known to exert an anti-inflammatory phenotype and promote tissue repair as well as phagocytosis (Gordon, 2003; Mantovani et al., 2004; Mosser and Edwards, 2008). At 6 months of age, Fizz1 levels were significantly reduced in vehicle- and pioglitazone-treated APP/PS1 animals. Six-month-old APP/PS1 animals showed no differences in levels of Ym1 or Arg1 compared with WT animals. Pioglitazone, however, dramatically increased levels of the alternative activation marker, Ym1 mRNA in 6-month-old APP/PS1 animals (Fig. 7E). At 12 months of age, pioglitazone treatment elevated the expression of all M2 markers assayed, Fizz1, Ym1, and Arg1. Fizz1 levels remained reduced in 12-month-old APP/PS1 animals (Fig. 7F). Brain levels of the inflammatory cytokines, Il-1β and tnfα, were also reduced in 12- but not 6-month-old treated animals, verifying the anti-inflammatory effects of PPARγ activation after amyloid deposition (Fig. 7E,F). Additionally, we monitored an increase in levels of Tgf-β in 12-month-old, but not 6-month-old, APP/PS1 mice. TGF-β is associated with an “acquired deactivation” or “M2c” state that is associated with a suppression of the innate immune system and increased phagocytic capacity that is normally associated with tissue repair (Van Ginderachter et al., 2006; Mandrekar-Colucci and Landreth, 2010). To determine whether increased apoE levels in the brain parenchyma of these animals contributed to M2 polarization of microglia, we treated microglial cells in vitro with apoE or apoA1 and monitored mRNA levels of M2 markers, Ym1, Fizz1, Arg1, and TGF-β. Arg1 and TGF-β mRNA expression remained constant under all treatment paradigms while Ym1 or Fizz1 were not detected in the cultured microglia under any treatment condition (data not shown), suggesting that apoE does not play a role in the M2 polarization of microglial cells that is seen in vivo. These data suggest a conversion of microglial phenotype in APP/PS1 animals, from the classical M1 to an alternative M2 state following PPARγ activation, promoting the phagocytic removal of amyloid deposits by microglial cells.
PPARγ activation ameliorates Aβ-related behavioral deficits
Finally, we examined the effects of PPARγ activation on associative memory in APP/PS1 animals using a contextual fear conditioning assay. Twelve-month-old transgenic animals were treated orally for 9 d and trained and tested for contextual memory following a contextual fear conditioning protocol. Animals were trained for four consecutive training sessions and all groups exhibited learning (Fig. 8B). However, vehicle-treated APP/PS1 animals showed significant impairments in memory retention as evidenced by the low number of freezes in the cued environment (Fig. 8A). Treatment of APP/PS1 mice with pioglitazone resulted in significant behavioral improvement that was not different from wild-type animals. Additionally, pioglitazone treatment reversed noncognitive behavioral deficits seen in APP/PS1 animals and restored distance and speed traveled to levels comparable to wild-type animals in an open field (Lalonde et al., 2005). These results demonstrate that PPARγ activation results in the rapid improvement of memory in an AD mouse model.
Figure 8.

PPARγ activation reverses cognitive deficits in 12-month-old APP/PS1 animals. Contextual fear conditioning was examined in 12-month-old vehicle- or pioglitazone (80 mg · kg−1 · d−1)-treated APP/PS1 animals (A) (mean ± SEM, Student's t test, *p < 0.05, **p < 0.01, n = 7–10 animals/group). Number of freezes is shown as a function of training periods for contextual fear conditioning (two-way ANOVA, Bonferroni post hoc test, **p < 0.01) B–D, Measurement of distance traveled (C) and mean speed (D) in open-field analysis of vehicle-treated WT and APP/PS1- and pioglitazone-treated APP/PS1 animals.
Discussion
We have documented the extraordinary efficacy of PPARγ activation in ameliorating the pathologic and behavioral deficits in an AD mouse model. Importantly, these dramatic changes were elicited after a short 9 d period of drug treatment. A primary finding of this study is the demonstration that PPARγ may act through distinct mechanisms to clear soluble and deposited, fibrillar forms of Aβ. Moreover, we have identified a previously unappreciated role of astrocytes in the removal of amyloid from the brain.
We report that the clearance of soluble forms of Aβ occurs through the ability of PPARγ to induce the expression of apoe through which it promotes sAβ degradation. We have previously documented that apoE acts to facilitate the proteolysis of soluble Aβ peptides both in the interstitial fluid and intracellularly by microglia through the actions of IDE and neprilysin, respectively (Jiang et al., 2008). In microglia, apoE facilitates soluble Aβ degradation through reduction of intracellular cholesterol levels and not through direct contact with the Aβ peptide (Lee et al., 2012). Recently, we have shown that activation of the Retinoid X Receptor, the obligate heterodimeric partner of PPARγ and LXRs, rapidly reverses AD pathology in mouse models of AD, and many of the beneficial effects seen in that study may occur through activation of PPAR signaling pathways (Cramer et al., 2012). The principal conceptual advance in this study was the recognition that in astrocytes and microglia the expression of apoe is robustly regulated through a coupled metabolic pathway regulated by PPARγ and LXRα, and genetic inactivation of PPARγ in astrocytes results in a dramatic reduction in the expression of these LXR target genes (Fig. 2). While this pathway has been well documented in the periphery, it was not recognized to be operative in the brain until now (Chawla et al., 2001).
A significant outcome of this study is a new appreciation for the roles of astrocytes in amyloid clearance. We report that astrocytes have the capacity to take up sAβ and degrade it in an apoE-dependent manner, similar to microglia (Figs. 1, 2; Jiang et al., 2008). Moreover, pioglitazone treatment resulted in intracellular Aβ accumulation in astrocytes, coincident with the reduction in plaque burden (Fig. 5). Astrocytes greatly outnumber microglia in the CNS, and their ability to remove even modest amounts of Aβ in the brain may have a significant impact on amyloid clearance in the brain. Amyloid-laden astrocytes were only found in the cortex of pioglitazone-treated animals and not in vehicle-treated APP/PS1 mice (Fig. 5). Several studies have shown that astrocytes are capable of taking up amyloid peptides (Funato et al., 1998; Matsunaga et al., 2003; Nagele et al., 2003; Lasagna-Reeves and Kayed, 2011), and in vitro this ability was restricted to adult astrocytes (al-Ali and al-Hussain, 1996; Wyss-Coray et al., 2003; Koistinaho et al., 2004; Pihlaja et al., 2008). This study strongly supports the view that PPARγ activation initiates amyloid clearance pathways in astrocytes and suggests a reevaluation for the role of astrocytes in amyloid clearance in AD.
Microglia are the principal immune effector cells in the CNS and are the only professional phagocyte in the CNS. These cells undergo a classical M1 inflammatory activation in response of amyloid deposition, leading to a proinflammatory milieu in the brain (Wyss-Coray, 2006; Heneka et al., 2010). An enigmatic feature of AD is that although microglia are competent phagocytes, they fail to effectively clear deposited Aβ from the brain. The basis of the inactivation of phagocytic function is unknown but is postulated to be the result of autocrine actions of cytokines (Koenigsknecht-Talboo and Landreth, 2005). PPARγ activation has been shown to enhance the phagocytic capabilities of peripheral M2 polarized macrophages (Chinetti-Gbaguidi et al., 2011). One striking finding in this study was the extraordinary ability of pioglitazone to stimulate microglia to phagocytose amyloid deposits from the transgenic mice (Fig. 7). Macrophages/microglia are capable of exhibiting a spectrum of phenotypic activation states which dictate their activities (Gordon, 2003; Mosser and Edwards, 2008; Mandrekar-Colucci and Landreth, 2010). Nuclear receptors have only recently been appreciated to play an crucial role in promoting the phenotypic change of macrophages from a “classically” M1 activated to alternatively activated M2 states (Odegaard et al., 2007, 2008; Zelcer et al., 2007; Bouhlel et al., 2008; Kalinin et al., 2009; Chawla, 2010). Activation of these nuclear receptors suppresses NFκB-mediated inflammatory responses and activates their phagocytic machinery (Zelcer et al., 2007; Mukundan et al., 2009; Glass and Saijo, 2010). In the AD brain, microglia fail to effectively phagocytose Aβ and are also impaired in their ability to degrade Aβ fibrils (Chung et al., 1999; Cameron and Landreth, 2010). Thus, modulating microglial activation status by targeting nuclear receptors appears to restore the competence of these cells to phagocytose and clear amyloid in the AD brain (Mukundan et al., 2009; Chinetti-Gbaguidi et al., 2011). We demonstrate that PPARγ activation in the APP/PS1 animals provoked the conversion of at least a subset of microglia from a classical to alternative activation states, suppressing proinflammatory gene expression (IL-1β, TNFα) and inducing the expression of anti-inflammatory genes (YM1, Fizz1, Arg1, TGF-β) associated with tissue repair (Fig. 7). This latter function is accompanied by expression of genes necessary for phagocytosis and induction of phagocytic activity and this is consistent with our observation of enhanced phagocytosis of fAβ by microglia. Terwel et al. (2011) have recently reported similar findings upon activation of LXRs in a mouse model of AD.
The actions of PPARγ in the brain of murine models of AD have been controversial, largely due to the poor penetrance of its agonists into the brain and active P-glycoprotein-mediated efflux mechanisms (Maeshiba et al., 1997; Hemauer et al., 2010). In the present study we have used a high dose of pioglitazone (80 mg · kg−1 · d−1) that is ∼8 times the clinically approved dose, to obviate this problem. Significantly, we have used a very short treatment interval (9 d), because of the expeditious induction of receptor-mediated gene expression. A number of studies of the thiazolidinedione agonists of PPARγ, pioglitazone (Actos) and rosiglitazone (Avandia), reported salutary effects of these drugs in murine models of AD (Mandrekar-Colucci and Landreth, 2011). Indeed, PPARγ agonists have been shown to effectively lower both soluble levels of Aβ, reduce plaque burden, and improve behavior in some AD mouse models (Yan et al., 2003; Camacho et al., 2004; Heneka et al., 2005; Nicolakakis et al., 2008; Escribano et al., 2010; Toledo and Inestrosa, 2010; Rodriguez-Rivera et al., 2011); however, there has been no consensus on the mechanisms of action (Pedersen and Flynn, 2004; Escribano et al., 2010; Toledo and Inestrosa, 2010). The anti-inflammatory actions of these drugs have been postulated to underlie their beneficial effects. However, other laboratories have explored additional mechanisms through which PPARγ may exert beneficial effects. Pederson and Flynn (2004) have suggested that PPARγ elicits behavioral improvements through its ability reduce peripheral corticosterone levels. Toledo and Inestrosa (2010) suggested that PPARγ activation could promote neuroprotection by activating the wnt signaling pathway. In this paper we have demonstrated yet another mechanism through which PPARγ activation may ameliorate AD pathophysiology.
Because of the efficacy of PPARγ agonist action in mouse models of AD, small clinical trials evaluated the effects of receptor activation and found that pioglitazone treatment improved memory and cognition in patients with mild to moderate AD (Hanyu et al., 2009; Sato et al., 2011). A larger phase II clinical trial showed improvements in memory retention and attention with the treatment of rosiglitazone (6 months) in patients who did not possess an ApoE4 allele (Risner et al., 2006). However, phase III clinical trials, using rosiglitazone, failed to show efficacy for the treatment of AD (Gold et al., 2010). It is important to note that in these trials rosiglitazone was administered at dosages that were much lower than those needed to see beneficial effects on AD pathophysiology in rodent models of the disease.
In this study we have demonstrated the rapid effects of PPARγ activation on amyloid clearance, microglial polarization, and the reversal of cognitive deficits associated with AD. Additionally, we have provided an explanation for how PPARγ modulates AD-related pathology in a mouse model. We have shown a mechanistic linkage between the PPARγ and LXR pathways, which had not previously been documented in the brain, and results in the production of lipidated ApoE particles, facilitating the degradation of soluble Aβ species (Fig. 9). PPARγ activation changes the inflammatory milieu of the brain by phenotypically polarizing microglia to an alternative, M2 phenotype, allowing them to phagocytically remove amyloid deposits (Fig. 9). These data suggest that brain-penetrant PPARγ agonists represent a promising therapeutic approach for the treatment of AD.
Figure 9.

PPARγ activation promotes amyloid clearance from the AD brain. We have shown that PPARγ activation, using the synthetic agonist pioglitazone, promotes the clearance of both soluble and fibrillar amyloid species from the AD brain via distinct mechanisms. PPARγ promotes the degradation of sAβ species through the induction of LXR target genes apoE and abca1. This increases the pool of lipidated apoE particles which then facilitate the proteolytic clearance of sAβ as we have previously described (A; Jiang et al., 2008). Additionally, PPARγ activation induces the phenotypic conversion of the classically activated microglia to an alternative activation state. This conversion suppresses the production of inflammatory cytokines such as IL-1β and TNFα and promotes the expression of genes involved in tissue repair and phagocytosis (FIZZ1, YM1, Arg1, TGF-β), allowing microglial cells to regain their capacity to take up and degrade fibrillar Aβ species (B).
Footnotes
This work was supported by the National Institutes of Health (Grant 5-R01-AG030482). S.M.-C. was supported by a predoctoral Ruth L. Kirschstein National Research Service Award (F31 NS061445). We received support from the Case Western Reserve University (CWRU) Rodent Behavior Core and the Imaging Core Facility of the Department of Neurosciences at CWRU. We thank C.Y. Daniel Lee for his comments on the manuscript and technical assistance.
References
- Akiyama H, Arai T, Kondo H, Tanno E, Haga C, Ikeda K. Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis Assoc Disord. 2000a;14(Suppl 1):S47–S53. doi: 10.1097/00002093-200000001-00008. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000b;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- al-Ali SY, al-Hussain SM. An ultrastructural study of the phagocytic activity of astrocytes in adult rat brain. J Anat. 1996;188:257–262. [PMC free article] [PubMed] [Google Scholar]
- Beaven SW, Tontonoz P. Nuclear receptors in lipid metabolism: targeting the heart of dyslipidemia. Annu Rev Med. 2006;57:313–329. doi: 10.1146/annurev.med.57.121304.131428. [DOI] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornemann KD, Wiederhold KH, Pauli C, Ermini F, Stalder M, Schnell L, Sommer B, Jucker M, Staufenbiel M. Abeta-induced inflammatory processes in microglia cells of APP23 transgenic mice. Am J Pathol. 2001;158:63–73. doi: 10.1016/s0002-9440(10)63945-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhlel MA, Staels B, Chinetti-Gbaguidi G. Peroxisome proliferator-activated receptors—from active regulators of macrophage biology to pharmacological targets in the treatment of cardiovascular disease. J Intern Med. 2008;263:28–42. doi: 10.1111/j.1365-2796.2007.01892.x. [DOI] [PubMed] [Google Scholar]
- Camacho IE, Serneels L, Spittaels K, Merchiers P, Dominguez D, De Strooper B. Peroxisome-proliferator-activated receptor gamma induces a clearance mechanism for the amyloid-beta peptide. J Neurosci. 2004;24:10908–10917. doi: 10.1523/JNEUROSCI.3987-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron B, Landreth GE. Inflammation, microglia, and Alzheimer's disease. Neurobiol Dis. 2010;37:503–509. doi: 10.1016/j.nbd.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A. Control of macrophage activation and function by PPARs. Circ Res. 2010;106:1559–1569. doi: 10.1161/CIRCRESAHA.110.216523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, Liao D, Nagy L, Edwards PA, Curtiss LK, Evans RM, Tontonoz P. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7:161–171. doi: 10.1016/s1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- Chinetti-Gbaguidi G, Baron M, Bouhlel MA, Vanhoutte J, Copin C, Sebti Y, Derudas B, Mayi T, Bories G, Tailleux A, Haulon S, Zawadzki C, Jude B, Staels B. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARgamma and LXRalpha pathways. Circ Res. 2011;108:985–995. doi: 10.1161/CIRCRESAHA.110.233775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, Brazil MI, Soe TT, Maxfield FR. Uptake, degradation, and release of fibrillar and soluble forms of Alzheimer's amyloid beta-peptide by microglial cells. J Biol Chem. 1999;274:32301–32308. doi: 10.1074/jbc.274.45.32301. [DOI] [PubMed] [Google Scholar]
- Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs CK, Bates P, Karlo JC, Landreth GE. Regulation of beta-amyloid stimulated proinflammatory responses by peroxisome proliferator-activated receptor alpha. Neurochem Int. 2001;39:449–457. doi: 10.1016/s0197-0186(01)00052-3. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis M, Haidar B, Marcil M, Bouvier M, Krimbou L, Genest J., Jr Molecular and cellular physiology of apolipoprotein A-I lipidation by the ATP-binding cassette transporter A1 (ABCA1) J Biol Chem. 2004;279:7384–7394. doi: 10.1074/jbc.M306963200. [DOI] [PubMed] [Google Scholar]
- Donkin JJ, Stukas S, Hirsch-Reinshagen V, Namjoshi D, Wilkinson A, May S, Chan J, Fan J, Collins J, Wellington CL. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem. 2010;285:34144–34154. doi: 10.1074/jbc.M110.108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Sun B, Chen K, Fan L, Wang Z. Antagonist of peroxisome proliferator-activated receptor gamma induces cerebellar amyloid-beta levels and motor dysfunction in APP/PS1 transgenic mice. Biochem Biophys Res Commun. 2009;384:357–361. doi: 10.1016/j.bbrc.2009.04.148. [DOI] [PubMed] [Google Scholar]
- Escribano L, Simón AM, Gimeno E, Cuadrado-Tejedor M, López de Maturana R, García-Osta A, Ricobaraza A, Pérez-Mediavilla A, Del Río J, Frechilla D. Rosiglitazone rescues memory impairment in Alzheimer's transgenic mice: mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology. 2010;35:1593–1604. doi: 10.1038/npp.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz NF, Cronican A, Pham T, Fogg A, Fauq AH, Chapman R, Lefterov I, Koldamova R. Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice. J Neurosci. 2010;30:6862–6872. doi: 10.1523/JNEUROSCI.1051-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci U S A. 1997;94:4312–4317. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato H, Yoshimura M, Yamazaki T, Saido TC, Ito Y, Yokofujita J, Okeda R, Ihara Y. Astrocytes containing amyloid beta-protein (Abeta)-positive granules are associated with Abeta40-positive diffuse plaques in the aged human brain. Am J Pathol. 1998;152:983–992. [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol. 2010;10:365–376. doi: 10.1038/nri2748. [DOI] [PubMed] [Google Scholar]
- Gold M, Alderton C, Zvartau-Hind M, Egginton S, Saunders AM, Irizarry M, Craft S, Landreth G, Linnamägi U, Sawchak S. Rosiglitazone monotherapy in mild-to-moderate Alzheimer's disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord. 2010;30:131–146. doi: 10.1159/000318845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Hanyu H, Sato T, Kiuchi A, Sakurai H, Iwamoto T. Pioglitazone improved cognition in a pilot study on patients with Alzheimer's disease and mild cognitive impairment with diabetes mellitus. J Am Geriatr Soc. 2009;57:177–179. doi: 10.1111/j.1532-5415.2009.02067.x. [DOI] [PubMed] [Google Scholar]
- Hemauer SJ, Patrikeeva SL, Nanovskaya TN, Hankins GD, Ahmed MS. Role of human placental apical membrane transporters in the efflux of glyburide, rosiglitazone, and metformin. Am J Obstet Gynecol. 2010;202:383.e1–7. doi: 10.1016/j.ajog.2010.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, O'Banion K, Klockgether T, Van Leuven F, Landreth GE. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1–42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–1453. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- Heneka MT, O'Banion MK, Terwel D, Kummer MP. Neuroinflammatory processes in Alzheimer's disease. J Neural Transm. 2010;117:919–947. doi: 10.1007/s00702-010-0438-z. [DOI] [PubMed] [Google Scholar]
- Holtzman DM. Role of apoe/Abeta interactions in the pathogenesis of Alzheimer's disease and cerebral amyloid angiopathy. J Mol Neurosci. 2001;17:147–155. doi: 10.1385/JMN:17:2:147. [DOI] [PubMed] [Google Scholar]
- Hong C, Tontonoz P. Coordination of inflammation and metabolism by PPAR and LXR nuclear receptors. Curr Opin Genet Dev. 2008;18:461–467. doi: 10.1016/j.gde.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989;24:173–182. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez S, Baglietto-Vargas D, Caballero C, Moreno-Gonzalez I, Torres M, Sanchez-Varo R, Ruano D, Vizuete M, Gutierrez A, Vitorica J. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer's disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci. 2008;28:11650–11661. doi: 10.1523/JNEUROSCI.3024-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinin S, Richardson JC, Feinstein DL. A PPARdelta agonist reduces amyloid burden and brain inflammation in a transgenic mouse model of Alzheimer's disease. Curr Alzheimer Res. 2009;6:431–437. doi: 10.2174/156720509789207949. [DOI] [PubMed] [Google Scholar]
- Kase ET, Thoresen GH, Westerlund S, Højlund K, Rustan AC, Gaster M. Liver X receptor antagonist reduces lipid formation and increases glucose metabolism in myotubes from lean, obese and type 2 diabetic individuals. Diabetologia. 2007;50:2171–2180. doi: 10.1007/s00125-007-0760-7. [DOI] [PubMed] [Google Scholar]
- Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–424. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Donarski N, Goetze K, Kreckel M, Stuerenburg HJ, Buhmann C, Beisiegel U. Characterization of four lipoprotein classes in human cerebrospinal fluid. J Lipid Res. 2001;42:1143–1151. [PubMed] [Google Scholar]
- Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005;25:8240–8249. doi: 10.1523/JNEUROSCI.1808-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med. 2004;10:719–726. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- Koldamova RP, Lefterov IM, Staufenbiel M, Wolfe D, Huang S, Glorioso JC, Walter M, Roth MG, Lazo JS. The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer's disease. J Biol Chem. 2005a;280:4079–4088. doi: 10.1074/jbc.M411420200. [DOI] [PubMed] [Google Scholar]
- Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J Biol Chem. 2005b;280:43224–43235. doi: 10.1074/jbc.M504513200. [DOI] [PubMed] [Google Scholar]
- Lalonde R, Kim HD, Maxwell JA, Fukuchi K. Exploratory activity and spatial learning in 12-month-old APP(695)SWE/co+PS1/DeltaE9 mice with amyloid plaques. Neurosci Lett. 2005;390:87–92. doi: 10.1016/j.neulet.2005.08.028. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Kayed R. Astrocytes contain amyloid-beta annular protofibrils in Alzheimer's disease brains. FEBS Lett. 2011;585:3052–3057. doi: 10.1016/j.febslet.2011.08.027. [DOI] [PubMed] [Google Scholar]
- Lee CY, Tse W, Smith JD, Landreth GE. Apolipoprotein E promotes beta-amyloid trafficking and degradation by modulating microglial cholesterol levels. J Biol Chem. 2012;287:2032–2044. doi: 10.1074/jbc.M111.295451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Elwood F, McNally J, Weiszmann J, Lindstrom M, Amaral K, Nakamura M, Miao S, Cao P, Learned RM, Chen JL, Li Y. T0070907, a selective ligand for peroxisome proliferator-activated receptor gamma, functions as an antagonist of biochemical and cellular activities. J Biol Chem. 2002;277:19649–19657. doi: 10.1074/jbc.M200743200. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, Kliewer SA, Moore LB, Smith-Oliver TA, Oliver BB, Su JL, Sundseth SS, Winegar DA, Blanchard DE, Spencer TA, Willson TM. Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J Biol Chem. 1997;272:3137–3140. doi: 10.1074/jbc.272.6.3137. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshiba Y, Kiyota Y, Yamashita K, Yoshimura Y, Motohashi M, Tanayama S. Disposition of the new antidiabetic agent pioglitazone in rats, dogs, and monkeys. Arzneimittelforschung. 1997;47:29–35. [PubMed] [Google Scholar]
- Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA. Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci. 2008;28:6333–6341. doi: 10.1523/JNEUROSCI.0829-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Landreth GE. Microglia and inflammation in Alzheimer's disease. CNS Neurol Disord Drug Targets. 2010;9:156–167. doi: 10.2174/187152710791012071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Landreth GE. Nuclear receptors as therapeutic targets for Alzheimer's disease. Expert Opin Ther Targets. 2011;15:1085–1097. doi: 10.1517/14728222.2011.594043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, Alford M, Albright T, Terry R, Shapiro P, Sundsmo M, Saitoh T. Immunoreactivity of CD45, a protein phosphotyrosine phosphatase, in Alzheimer's disease. Acta Neuropathol. 1991;83:12–20. doi: 10.1007/BF00294425. [DOI] [PubMed] [Google Scholar]
- Matsunaga W, Shirokawa T, Isobe K. Specific uptake of Abeta1–40 in rat brain occurs in astrocyte, but not in microglia. Neurosci Lett. 2003;342:129–131. doi: 10.1016/s0304-3940(03)00240-4. [DOI] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald DR, Brunden KR, Landreth GE. Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J Neurosci. 1997;17:2284–2294. doi: 10.1523/JNEUROSCI.17-07-02284.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, Goh YP, Eagle AR, Dunn SE, Awakuni JU, Nguyen KD, Steinman L, Michie SA, Chawla A. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med. 2009;15:1266–1272. doi: 10.1038/nm.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagele RG, D'Andrea MR, Lee H, Venkataraman V, Wang HY. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003;971:197–209. doi: 10.1016/s0006-8993(03)02361-8. [DOI] [PubMed] [Google Scholar]
- Nicolakakis N, Aboulkassim T, Ongali B, Lecrux C, Fernandes P, Rosa-Neto P, Tong XK, Hamel E. Complete rescue of cerebrovascular function in aged Alzheimer's disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J Neurosci. 2008;28:9287–9296. doi: 10.1523/JNEUROSCI.3348-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, Chawla A. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, Subramanian V, Mukundan L, Ferrante AW, Chawla A. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008;7:496–507. doi: 10.1016/j.cmet.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen WA, Flynn ER. Insulin resistance contributes to aberrant stress responses in the Tg2576 mouse model of Alzheimer's disease. Neurobiol Dis. 2004;17:500–506. doi: 10.1016/j.nbd.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Penninger JM, Irie-Sasaki J, Sasaki T, Oliveira-dos-Santos AJ. CD45: new jobs for an old acquaintance. Nat Immunol. 2001;2:389–396. doi: 10.1038/87687. [DOI] [PubMed] [Google Scholar]
- Pihlaja R, Koistinaho J, Malm T, Sikkilä H, Vainio S, Koistinaho M. Transplanted astrocytes internalize deposited beta-amyloid peptides in a transgenic mouse model of Alzheimer's disease. Glia. 2008;56:154–163. doi: 10.1002/glia.20599. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Riddell DR, Zhou H, Comery TA, Kouranova E, Lo CF, Warwick HK, Ring RH, Kirksey Y, Aschmies S, Xu J, Kubek K, Hirst WD, Gonzales C, Chen Y, Murphy E, Leonard S, Vasylyev D, Oganesian A, Martone RL, Pangalos MN, et al. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer's disease. Mol Cell Neurosci. 2007;34:621–628. doi: 10.1016/j.mcn.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Risner ME, Saunders AM, Altman JF, Ormandy GC, Craft S, Foley IM, Zvartau-Hind ME, Hosford DA, Roses AD. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer's disease. Pharmacogenomics J. 2006;6:246–254. doi: 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Rivera J, Denner L, Dineley KT. Rosiglitazone reversal of Tg2576 cognitive deficits is independent of peripheral gluco-regulatory status. Behav Brain Res. 2011;216:255–261. doi: 10.1016/j.bbr.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roses AD. Apolipoprotein E alleles as risk factors in Alzheimer's disease. Annu Rev Med. 1996;47:387–400. doi: 10.1146/annurev.med.47.1.387. [DOI] [PubMed] [Google Scholar]
- Sasaki A, Shoji M, Harigaya Y, Kawarabayashi T, Ikeda M, Naito M, Matsubara E, Abe K, Nakazato Y. Amyloid cored plaques in Tg2576 transgenic mice are characterized by giant plaques, slightly activated microglia, and the lack of paired helical filament-typed, dystrophic neurites. Virchows Arch. 2002;441:358–367. doi: 10.1007/s00428-002-0643-8. [DOI] [PubMed] [Google Scholar]
- Sato T, Hanyu H, Hirao K, Kanetaka H, Sakurai H, Iwamoto T. Efficacy of PPAR-γ agonist pioglitazone in mild Alzheimer disease. Neurobiol Aging. 2011;32:1626–1633. doi: 10.1016/j.neurobiolaging.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Saura J, Tusell JM, Serratosa J. High-yield isolation of murine microglia by mild trypsinization. Glia. 2003;44:183–189. doi: 10.1002/glia.10274. [DOI] [PubMed] [Google Scholar]
- Seo JB, Moon HM, Kim WS, Lee YS, Jeong HW, Yoo EJ, Ham J, Kang H, Park MG, Steffensen KR, Stulnig TM, Gustafsson JA, Park SD, Kim JB. Activated liver X receptors stimulate adipocyte differentiation through induction of peroxisome proliferator-activated receptor gamma expression. Mol Cell Biol. 2004;24:3430–3444. doi: 10.1128/MCB.24.8.3430-3444.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JD, Le Goff W, Settle M, Brubaker G, Waelde C, Horwitz A, Oda MN. ABCA1 mediates concurrent cholesterol and phospholipid efflux to apolipoprotein A-I. J Lipid Res. 2004;45:635–644. doi: 10.1194/jlr.M300336-JLR200. [DOI] [PubMed] [Google Scholar]
- Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol. 1999;154:1673–1684. doi: 10.1016/S0002-9440(10)65423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall AR. Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. J Intern Med. 2008;263:256–273. doi: 10.1111/j.1365-2796.2007.01898.x. [DOI] [PubMed] [Google Scholar]
- Terwel D, Steffensen KR, Verghese PB, Kummer MP, Gustafsson JÅ, Holtzman DM, Heneka MT. Critical role of astroglial apolipoprotein E and liver X receptor-α expression for microglial Aβ phagocytosis. J Neurosci. 2011;31:7049–7059. doi: 10.1523/JNEUROSCI.6546-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo EM, Inestrosa NC. Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1DeltaE9 mouse model of Alzheimer's disease. Mol Psychiatry. 2010;15:272–285. 228. doi: 10.1038/mp.2009.72. [DOI] [PubMed] [Google Scholar]
- Van Ginderachter JA, Movahedi K, Hassanzadeh Ghassabeh G, Meerschaut S, Beschin A, Raes G, De Baetselier P. Classical and alternative activation of mononuclear phagocytes: picking the best of both worlds for tumor promotion. Immunobiology. 2006;211:487–501. doi: 10.1016/j.imbio.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Legleiter J, Han X, Fryer JD, Kowalewski T, Holtzman DM. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem. 2004;279:40987–40993. doi: 10.1074/jbc.M407963200. [DOI] [PubMed] [Google Scholar]
- Wegiel J, Wang KC, Imaki H, Rubenstein R, Wronska A, Osuchowski M, Lipinski WJ, Walker LC, LeVine H. The role of microglial cells and astrocytes in fibrillar plaque evolution in transgenic APP(SW) mice. Neurobiol Aging. 2001;22:49–61. doi: 10.1016/s0197-4580(00)00181-0. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Gordon MN, Ugen KE, Gottschall PE, DiCarlo G, Dickey C, Boyett KW, Jantzen PT, Connor KE, Melachrino J, Hardy J, Morgan D. Number of Abeta inoculations in APP+PS1 transgenic mice influences antibody titers, microglial activation, and congophilic plaque levels. DNA Cell Biol. 2001;20:731–736. doi: 10.1089/10445490152717596. [DOI] [PubMed] [Google Scholar]
- Wisniewski HM, Wegiel J, Wang KC, Lach B. Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer's disease. Acta Neuropathol. 1992;84:117–127. doi: 10.1007/BF00311383. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9:453–457. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci. 2003;23:7504–7509. doi: 10.1523/JNEUROSCI.23-20-07504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue L, Mazzone T. Peroxisome proliferator-activated receptor {gamma} stimulation of adipocyte ApoE gene transcription mediated by the liver receptor X pathway. J Biol Chem. 2009;284:10453–10461. doi: 10.1074/jbc.M808482200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, Landreth GE, Vinters HV, Tontonoz P. Attenuation of neuroinflammation and Alzheimer's disease pathology by liver X receptors. Proc Natl Acad Sci U S A. 2007;104:10601–10606. doi: 10.1073/pnas.0701096104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV, Deane R, Sallstrom J, Chow N, Miano JM. Neurovascular pathways and Alzheimer amyloid beta-peptide. Brain Pathol. 2005;15:78–83. doi: 10.1111/j.1750-3639.2005.tb00103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]