

Abstract
Functionalized congeners of the M1-selective muscarinic antagonist telenzepine (4,9-dihydro-3-methyl-4-[(4-methyl-1-piperazinyl)acetyl]-10H-thieno[3,4–b][1,5]benzodiazepin-10-one) were developed and found to bind to the receptor with affinities (Ki values) in approximately the nanomolar range. The derivatives contain a 10-aminodecyl group, which provides a nucleophilic functionality for further derivatization. The attachment of a spacer chain to the distal piperazinyl nitrogen was based on previous findings of enhanced affinity at muscarinic receptors in an analogous series of alkylamino derivatives of pirenzepine [J. Med. Chem. (1991) 34, 2133–2145]. The telenzepine derivatives contain prosthetic groups for radioiodination, protein cross-linking, photoaffinity labeling, and fluorescent labeling and biotin for avidin complexation. The affinity for muscarinic receptors in rat forebrain (mainly m1 subtype) was determined in competitive binding assays vs [3H]-N-methylscopolamine. A (p-aminophenyl)-acetyl derivative for photoaffinity labeling had a Ki value of 0.29 nM at forebrain muscarinic receptors (16-fold higher affinity than telenzepine). A biotin conjugate displayed a Ki value of 0.60 nM at m2-receptors and a 5-fold selectivity versus forebrain. The high affinity of these derivatives makes them suitable for the characterization of muscarinic receptors in pharmacological and spectroscopic studies, for peptide mapping, and for histochemical studies.
INTRODUCTION
Acetylcholine acts as a neurotransmitter through both muscarinic (1) and nicotinic receptors (2), in the central nervous system and at peripheral sites, such as heart and ileum. Five subtypes of muscarinic acetylcholine receptors (m1–m5) have been cloned (3). The receptors belong to the functional and structural class of G-protein-coupled receptors, which contain seven transmembrane helices (4). The binding sites for agonists and antagonists are thought to be nonequivalent, although there is little direct evidence (5) for which amino acid residues are involved in the binding of ligands. The details of the conformational changes in the receptor protein produced by ligand binding are unknown.
In general, structural knowledge of the G-protein-linked receptors has been obtained through site-directed mutagenesis (6) and by using specialized ligands as molecular probes (5, 7–12). Such molecular probes include irreversible affinity labels containing chemically- and photochemically-reactive groups (5, 7–9), fluorescent labels and other spectroscopic reporter groups (10–11), biotinylated probes (10,12). We have developed a functionalized congener approach to the design of muscarinic ligands (13,14), which in this study is extended to sites for the attachment of reporter groups to high-affinity antagonists. We have derivatized the M1-selective antagonist telenzepine (20) in a manner analogous to our previous work with the structurally similar pirenzepine (14). These functionalized ligands are also potentially suited for affinity chromatography and histochemical characterization of the receptors.
EXPERIMENTAL PROCEDURES
1H NMR spectra were recorded using a Varian XL-300 FT-NMR spectrometer and all values are reported in parts per million (ppm, δ) downfield from tetramethylsilane (TMS). Chemical ionization MS using ionized NH3 gas were recorded using a Finnigan 1015D mass spectrometer modified with EXTREL electronics. Fast atom bombardment MS was carried out on a JEOL JMS-SX102 mass spectrometer, and parent ion peaks measured at high resolution are listed in Table II. Thin-layer chromatography (TLC)1 analyses were carried out using EM Kieselgel 60 F254, DC-Alufolien 200-μm plates and were visualized in an iodine chamber and/or with 1% ninhydrin in ethanol. Rf values in two solvent systems are given in Table II. Silica gel columns used MN-Kieselgel 60 (230–400 mesh) silica gel. Elemental analyses were performed by Atlantic Microlabs, Inc. (Atlanta, GA). The term in vacuo refers to a water aspirator (15–30 mmHg) rotary evaporator. Percent yields are rounded to the nearest whole number.
Table II.
Characterization of Compounds Synthesized
| compde | formula |
Rf
|
low resolutiona | high resolutiona
|
||
|---|---|---|---|---|---|---|
| Ad | Bd | calcd | found | |||
| 3 | C28H39BrN4O2S | 0.66 | 577,b 575,b 497, 317, 185 | 575.2055 | 575.2027 | |
| 577.2038 | 577.2029 | |||||
| 4 | C28H41N5O2S | 0.15 | 0 | 512,b M+ (511),c 368,c 327,c 314,c 254c | 512.3059 | 512.3073 |
| 5 | C37H49N5O4S | 0.51 | 0.53 | 660,b 402, 277,185 | 660.3584 | 660.3607 |
| 6 | C36H48N6O3S | 0.41 | 0.32 | 645,b 387, 318, 229 | 645.3584 | 645.3608 |
| 8 | C35H44N8O4S | 0.69 | 0.75 | 674,b 647, 553, 461, 369, 277 | 673.3284 | 673.3344 |
| 9 | C38H55N7O4S2 | 0.39 | 0.26 | 738,b 468, 342, 227 | 738.3835 | 738.3848 |
| 10 | C36H45N7O2S3 | 0.83 | 0.93 | 704,b 688, 307, 242 | 704.2875 | 704.2894 |
| 11 | C37H46N8O3S3 | 0.58 | 747,b 461, 369, 277 | 747.2933 | 747.2964 | |
| 12 | C37H44N8O2S4 | 0.85 | 0.9 | 761,b 729, 553, 461, 369 | 761.2548 | 761.2570 |
| 13 | C36H45N7O2S3 | 0.89 | 0.94 | 704, 553, 461, 369, 277 | 704.2875 | 704.2891 |
| 14 | C49H51N5O8S | 0 | 0.31 | 870,b 613, 461, 309 | 870.3534 | 870.3535 |
| 15 | C49H48Br4N6O7S2 | 0.30 | 1217b | 1212.9838 | 1212.984 | |
Unless noted, by fast atom bombardment mass spectroscopy, using glycerol matrix, in the positive ion mode.
M + 1.
EI mass spectroscopy.
Solvent A = chloroform/methanol/ammonia, 90:10:1; solvent B = chloroform/methanol/acetic acid, 85:10:5.
Refer to Table I for structures. Yields for conjugates of TAC (compounds 5 and 7–13) were generally in the range of 50–80%.
4,9-Dihydro-3-methyl-4-(1-piperazinylacetyl)-10H-thieno[3,4-b][1,5]benzodiazepin-10-one (2)
Telenzepine dihydrochloride (0.527 g, 1.20 mmol; Research Biochemicals Inc., Natick MA) was dried in vacuo over P2O5. Chloroform (7 mL) and N,N-diisopropylethylamine (1.66 mL) were added, and the mixture was cooled in an ice bath and stirred until a solution formed. α-Chloroethyl chloroformate (1.4 g, 9.8 mmol) was added drop-wise, and the solution was warmed for 0.5 h at 40 °C. The CHCl3 was removed in vacuo, and the solid [consisting mainly of the N-(α-chloroethoxycarbonyl) intermediate which was not isolated] was dissolved in methanol and sufficient 1 M HCl in ether to lower the pH to 0–1. The solution was refluxed for another 0.5 h and after cooling immediately basified with aqueous sodium carbonate and washed with ether. The aqueous layer was saturated with sodium chloride and extracted into CHCl3. The organic extracts were evaporated in vacuo, and the residue was purified by silica gel column chromatography (chloroform/ methanol/ammonia, 90:10:1, by vol) to provide 159 mg (37 % yield) of 2: The 1H NMR (CDCl3) spectrum showed a characteristic singlet for the thiophene CH3 (3 H) at δ 2.43 ppm; MS (CI/NH3) m/e 357 (MH+, base).
4,9-Dihydro-3-methyl-4-[[4-(10-aminodecyl)-1-piperazinyl]acetyl]-10H-tbieno[3,4-b][1,5]benzodiazepin-10-one (4)
A solution of 2 (158 mg, 0.43 mmol), 1,10-dibromodecane (1.1 g, 3.6 mmol) and N,N-diisopropyl-ethylamine (0.2 g) was stirred in chloroform (3 mL) at 55 °C for 6 h. The reaction was judged complete by TLC, and all volatile materials were removed in vacuo. The resulting clear mixture was evaported under N2, and the residue was washed several times with petroleum ether to give 4,9-dihydro-3-methyl-4-[[4-(10-bromodecyl)-1-piper-azinyl] acetyl]-10H -thieno [3,4-b] [1,5] benzodiazepin-10-one (3), which was used without further purification.
The powder, 3, was dissolved in 2 mL of methanol, treated with 1 mL of concentrated NH4OH, and heated for 6 h at 50 °C. The reaction mixture was cooled and extracted with ether (6 × 10 mL). The ether extracts were evaporated in vacuo, leaving 0.27 g of the crude product, 4, which was purified on a silica gel column (chloroform/ methanol/ammonia, 90:10:1, by vol). An analytical sample of the trihydrochloride salt of 4 was prepared. Anal. Calcd (C28H44N5Cl3O2S·2H2O): 51.18, C; 7.36, H; 10.66, N. Found: 51.10, C; 7.22, H; 10.48, N.
The purity of 4 was demonstrated using HPLC. With a gradient of 20 to 80 % acetonitrile in water (both solvents containing 0.1 % TFA, LKB Ultrapak C-18 Column, 25 × 0.4 cm, flow rate 1 mL/min, detection at 254 nm, pressure <2K psi), a single peak emerged at 9.9 min. Telenzepine under the same conditions had a retention time of 8.3 min.
4,9-Dihydro-3-methyl-4-[[4-[10-[[3-(4-hydroxyphenyl)propionyl]amino]decyl]-1-piperazinyl]acetyl]-10H-thieno[3,4-b][1,5]benzodiazepin-10-one (5)
Compound 4 (5.0 mg, 7.6 μmol) was dissolved in chloroform (0.7 mL). N-Succinimidyl 3-(4-hydroxyphenyl) propionate (5.5 mg, 21 μmol) was added. The reaction was judged complete by TLC after 6 h at 25 °C. Methanolic HCl was added (pH 1), and the solution was kept in a refrigerator overnight. The precipitate was washed successively with chloroform and dried to give 5. The product, visualized using UV and iodine vapors, was pure by TLC. The product had a retention time of 14 min using HPLC (same conditions as given above for 4).
4,9-Dihydro-3-methyl-4-[[4-[10-[[(4-aminophenyl)-acetyl]amino]decyl]-1-piperazinyl]acetyl]-10H-thieno-[3,4-b][1,5]benzodiazepin-10-one(6)and 4,9-Dihydro-3-methyl-4-[[4-[10-[[[4-[(tert-butoxycarbonyl)-amino]phenyl]acetyl]amino]decyl]-1-piperazinyl]-acetyl]-10H-thieno[3,4-b][1,5]benzodiazepin-10-one (7)
Compound 4 (5.0 mg, 7.6 μmol) was dissolved in benzene/chloroform (0.25 mL, 3:2, by vol). N-Succinimidyl p-[(tert-butoxycarbonyl)amino]phenylacetate was prepared as described in ref 7, dissolved (5.0 mg, 14 μmol) in chloroform (0.5 mL), and added to the solution of 4 with stirring. The reaction was complete after 1 h. Methanolic HCl was added carefully in the cold to acidify the mixture to pH 1–2. Following the addition of 0.2 mL of ether the product precipitated slowly, overnight at 0 °C. A total of two crops of the product was collected, combined, and washed with chloroform. The product, visualized using UV or iodine vapors, was pure by thin-layer chromatography and was used as such for the preparation of 6.
TFA was added slowly to the solid Boc-protected amine derivative 7, and the reaction was stirred for 10 min. The excess TFA was removed under a stream of N2, and the resulting oily residue was washed with ether and dried for 24 h at 50 °C under high vacuum (0.1 mmHg) to yield the free arylamine 6.
4,9-Dihydro-3-methyl-4-[[4-[10-[(4-azido-2-hydroxybenzoyl)amino]decyl]-1-piperazinyl]acetyl]-10H-thieno[3,4-b][1,5]benzodiazepin-10-one (8)
Compound 4 (5.0 mg, 7.6 μmol) was dissolved in benzene/chloroform (0.25 mL, 3:2, by vol). The N-succinimidyl ester of 4-azidosalicylic acid (NHS-ASA, Pierce Chemical Co., Rockford, IL; 6.0 mg, 21.7 μmol) was added. The reaction was kept in the dark and was complete after 0.5 h. Following the addition of 0.2 mL of ether the product precipitated slowly, during 2 days at 0 °C. The product was collected and washed with chloroform and was shown to be pure by reversed-phase HPLC. With a gradient of 20 to 80% acetonitrile in water, both containing 0.1% trifluoroacetic acid (same conditions as given for compound 4), the retention time of 8 was 18 min. An isocratic run at 50% acetonitrile showed a single peak at 5.8 min. A DMSO solution stored at −20 °C in the dark for 2 months remained pure: 1H NMR (CDC13) δ 1.2–1.5 (m, 16 H), 2.4 (s, 3 H), 3.2–3.8 (m, 14 H), 6.5 (m, br d, 1 H), 6.6 (s, 1 H), 7.2–7.5 (m, 4 H), 7.8 (br s, 1 H) ppm.
4,9-Dihydro-3-methyl-4-[[4-[10-[[5-(hexahydro-2-oxo-1H-thieno[3,4-d]imidazol-4-yl)-1-oxopentyl]amino]decyl]-1-piperazinyl]acetyl]-10H-thieno[3,4–b][1,5]-benzodiazepin-10-one (9)
Compound 4 (5.0 mg, 7.6 μmol) was dissolved in benzene/chloroform (0.25 mL, 3:2, by vol). (+)-Biotin N-hydroxysuccinimide ester (Fluka, Ronkonoma, NY; 6.5 mg, 19 μmol, dissolved in 0.4 mL of dimethylformamide) was added. The reaction was stirred for 1 h. Methanolic HCl was added causing the product to precipitate slowly, overnight. The precipitate was washed successively with chloroform and ether to yield 9 (1 mg, 20% yield). The product, which was visualized using UV or iodine vapors, was pure by thin-layer chromatography (alumina 5550, Merck, chloroform/methanol/ ammonia, 90:10:1): 1H NMR (CDC13) δ1.2–1.5 (m, 24 H), 2.4 (s, 3 H), 7.1–7.5 (m, 4 H), 7.8 (s, 1 H) ppm.
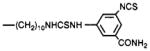
4,9-Dihydro-3-methyl-4-[[4-[10-[[[(3-isothiocyanatophenyl)amino]thiocarbonyl]amino]decyl]-1-piperazinyl]acetyl]-10H-thieno[3,4-b][ 1,5]benzodiazepin-10-one (10)
Compound 4 (2.8 mg, 5.7 μmol, prepared as described in ref 14) was dissolved in DMF (0.2 mL) and treated with 1,3-phenylene diisothiocyanate (6 mg, 31 μmol). After 0.5 h, ethyl acetate (1 mL) and petroleum ether (3 mL) were added, and the cloudy mixture was stored at 4 °C overnight. The supernatant was decanted from the oily residue. The residue solidified upon trituration with dry ether to give 2.0 mg (51 % yield) of pure product: 1H NMR (CDC13) δ 1.2–1.5 (m, 16 H), 2.4 (s, 3 H), 7.1–7.4 (m, 4 H), 7.8 (s, 1 H) ppm.
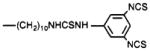
4,9-Dihydro-3-methyl-4-[[4-[10-[[[(3,5-diisothiocyanatophenyl)amino]thiocarbonyl]amino]decyl]-1-piperazinyl]acetyl]-10H-thieno[3,4-b][1,5]benzodiazepin-10-one (12)
Compound 12 was prepared by a method similar to compound 10, except that 1,3,5-benzenetriyl triisothiocyanate (8) was used.
4,9-Dihydro-3-methyl-4-[[4-[10-[[[(4-isothiocyanatophenyl)amino]thiocarbonyl]amino]decyl]-1-piperazinyl]acetyl]-10H-thieno[3,4-b][1,5]benzodiazepin-10-one (13)
Compound 4 (5.0 mg, 7.6 μmol) was dissolved in a mixture of benzene and chloroform (0.25 mL, 3:2, by vol). 1,4-Phenylene diisothiocyanate (13 mg, 67 μmol, dissolved in 0.5 mL of chloroform) was added dropwise and with agitation. The mixture was left for 1 h at 25 °C. The volume was reduced under a stream of nitrogen, and the residue was washed with petroleum ether. The product was pure by thin-layer chromatography: 1H NMR (CDCl3) δ 1.2–1.5 (m, 16 H), 2.4 (s, 3 H), 7.1–7.4 (m, 4 H), 7.8 (s, 1 H) ppm.
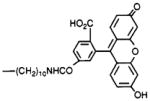
4,9-Dihydro-3-methyl-4-[[4-[10-[[[4-carboxy-3-(6-hydroxy-3-oxo-3H-xanth-9-yl)phenyl]carbonyl]-amino]decyl]-1-piperazinyl]acetyl]-10H-thieno[3,4-b]-[1,5]benzodiazepin-10-one Oxalate (14)
Compound 4 (2.0 mg, 3.0 μmol) was dissolved in benzene/chloroform (0.2 mL, 3:2, by vol). N-Succinimidyl 6-carboxyfluorescein (Research Organics, Inc., Cleveland, OH; 3.0 mg, 6.3 μmol, dissolved in 0.3 mL of dimethylformamide) was added. The reaction was followed by TLC (2 h), and the product, precipitated as an oxalate salt, was pure (silica, chloroform/methanol/acetic acid, 75:25:5, by vol): 1H NMR (CDCl3) δ 1.1–1.3 (m, 16 H), 2.3 (s, 3 H), 6.4 (m, 2 H), 6.5 (s, 1 H), 7.0–7.3 (m, 6 H), 7.6 (s, 1 H), 7.9 (bd, 1 H), 8.1 (bs, 1 H) ppm.
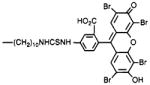
4,9-Dihydro-3-methyl-4-[[4-[10-[[[[4-carboxy-3-(2,4,5,7-tetrabromo-6-hydroxy-3-oxo-3H-xanth-9-yl)-phenyl]amino]thiocarbonyl]amino]decyl]-1-piperazinyl]acetyl]-10H-thieno[3,4-b][1,5]benzodiazepin-10-one (15)
Compound 4 (5.0 mg, 7.6 μmol) was dissolved in benzene/chloroform (0.25 mL, 3:2, by vol). Eosin 5-isothiocyanate (Molecular Probes, Eugene OR; 3.0 mg, 6.3 μmol) dissolved in dimethylformamide (0.25 mL) was added, and the mixture was left at 25 °C for 48 h. The product was precipitated as a hydrochloric acid salt, and purified by chromatography on a thin-layer plate (250 μ) using chloroform/methanol/acetic acid, 85:10:5 as an eluant.
Binding Assays
Crude membrane fractions from rat forebrain (mostly m1-receptors) and from rat heart (m2-receptors) were obtained by homogenizing (Polytron, 3 × 15 s, 75% max) tissue in phosphate-buffered saline at pH 7.2 and centrifuging at 28000g for 20 min. The resulting pellet was washed several more times by resuspension in fresh buffer followed by centrifugation. The final pellet was resuspended at 3 mg/mL and stored at −70 °C until needed.
Inhibition of [3H]NMS binding to membranes from either tissue was determined by incubating (37 °C for 60 min) varying concentrations of the analog with 0.5 nM [3H]NMS in phosphate-buffered saline, pH 7.2, and a quantity of membranes containing 100–300 μg of protein. The total volume in each tube was 1 mL. The mixture was then rapidly filtered over GF/B filters using a Brandel cell harvester, and the filters were equilibrated in scintillation cocktail and counted in a liquid scintillation counter. Nonspecific binding was determined by coin-cubation with 1 μM atropine, and amounted to less than 15 % of the total counts. It was routinely subtracted from the total counts. Competition binding experiments using membranes from transfected A9L cells (m1- and m3-receptors) and from NG108–15 cells (m4-receptors) were carried out as previously described (14).
All competition binding data were analyzed by nonlinear regression using the GraphPAD computer program (GraphPAD, San Diego, CA), and IC50 values were converted to Ki values using the Cheng–Prusoff equation (15). The Kd values, used in these calculations, for [3H]-NMS binding to forebrain muscarinic receptors and to cardiac muscarinic receptors were 0.350 and 0.425 nM, respectively.
To assay for irreversible inhibition, saturation by [3H]-NMS before and after treatment of the membranes with a potential affinity label was measured. Aliquots of brain membranes were incubated with the indicated concentration (typically 100 nM) of an isothiocyanate derivative, freshly diluted from a DMSO stock solution (stored at −20 °C) into phosphate-buffered saline (pH 8.0, 10 mM phosphate, 0.15 M sodium chloride) for 60 min at room temperature, and then centrifuged at 15 000 rpm for 10 min. The resulting pellet was resuspended in 25 mL of fresh phosphate-buffered saline (pH 7.2) and centrifuged as above. The pellet was again resuspended in fresh buffer and centrifuged, and aliquots were taken for [3H]NMS binding experiments as above.
RESULTS
Telenzepine (20) was derivatized with functionalized chains by a scheme analogous to that developed for pirenzepine (14). The N-methyl group was removed from the piperazine ring (Scheme I) and replaced with a terminally functionalized alkyl chain, with the expectation that this modification would not interfere with receptor binding. The demethylation was accomplished using α-chloroethyl chloroformate under basic conditions (14). The secondary amine 2 was purified chromatographically and alkylated with 1,10-dibromodecane, present in excess, to provide the ω-bromoakyl intermediate 3. The chain length of n = 10 was selected on the basis of previously explored structure-activity relationships (SAR) for piren-zepine (14). An aminodecyl derivative of pirenzepine and acylated derivatives were more potent in muscarinic receptor binding assays than shorter homologs. Compound 3 was treated with ammonia in situ, to give a telenzepine amine congener (TAC), 4. This amine derivative was purified and could be readily acylated using active esters or isothiocyanate derivatives, to give amide and thiourea derivatives, respectively, that were subsequently tested for affinity at muscarinic receptors (Table I). These derivatives were characterized using fast atom bombardment mass spectrometry in the high resolution mode for accurate mass determination (Table II).
Scheme Ia. Synthesis of Functionalized Congeners of Telenzepine.
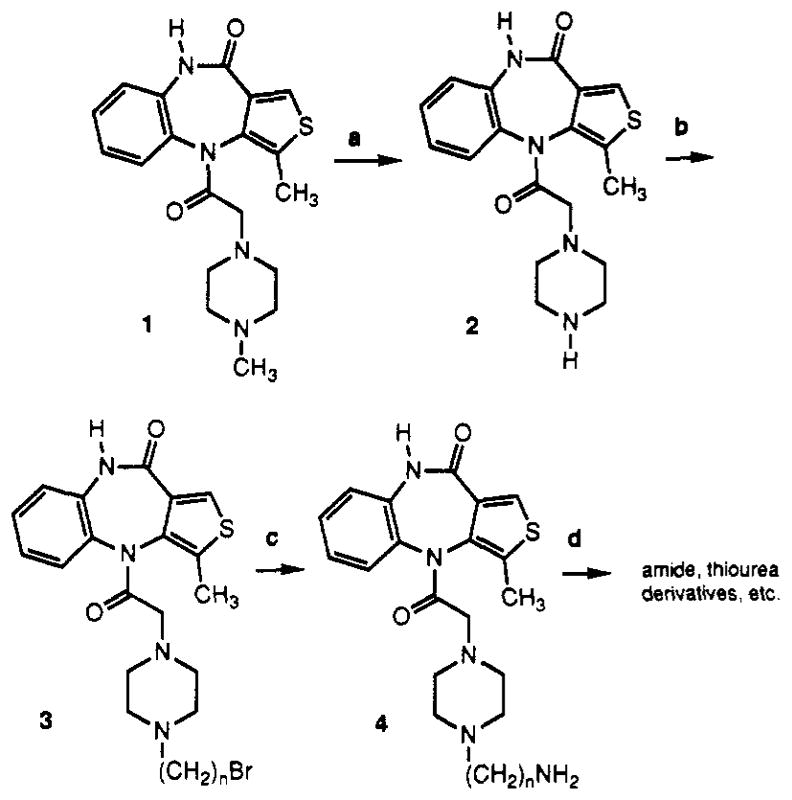
aReagents: (a) N,N -diisopropylethylamine, α-chloroethyl chloroformate; (b) 1,10-dibromodecane, N,N-diisopropylethylamine; (c) NH3; (d) isothiocyanates or activated carboxylic esters.
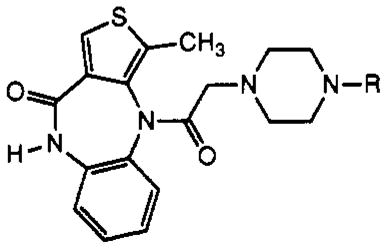
Table I.
Structures of Telenzepine Derivatives Synthesized and Their Affinity at Muscarinic Receptors
| ||||
|---|---|---|---|---|
| no. | R | receptor subtype and Ki valuesa (nM)
|
||
| forebrain | heart | other | ||
| 1 | —CH3 (telenzepine) | 4.67 ± 0.53 | 66.9 ± 32 | m1 (A9L), 1.8; m3, 6.9; m4,17.4 |
| 2 | —H | b | b | |
| 4 | —(CH2)10NH2 | b | 3.7 | m1 (A9L), 2.39; m3, 7.5; m4, 1.3 |
| 5 |
|
2.22 ± 0.48 | 1.75 ± 0.52 | |
| 6 |
|
0.29 ± 0.03 | 0.31 ± 0.07 | |
| 7 |
|
b | b | |
| 8 |
|
1.25 ± 0.12 | 3.7 ± 0.5 | |
| 9 |
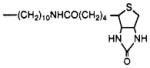
|
3.20 ± 1.33 | 0.60 ± 0.22 | |
| 10 |
|
42.5 ± 10.0 | 43.7 ± 12.2 | |
| 11 |
|
10.5 ± 0.7 | b | |
| 12 |
|
116 ± 17.5 | 82.1 ± 11 | |
| 13 |
|
1.21 ± 0.12 | 1.38 ± 0.15 | |
| 14 |
|
20.1 ± 6.2 | b | |
| 15 |
|
11.8 ± 3.4 | b | |
Ki values for inhibition of binding of [3H]NMS are given. Values are expressed as average for a single experiment run in triplicate or as mean ± SEM for three experiments. Membranes from rat brain (mostly m1-receptors) or rat heart (m2-receptors) were isolated as described in Experimental Procedures. Varying concentrations of compound were incubated with 0.5 nM [3H]NMS and a quantity of membranes containing 100–300 μg of protein for 60 min at 37 °C and then rapidly filtered over GF/B filters. Competition binding experiments using membranes from transfected A9L cells (m1- and m3-receptors) and from NG108–15 cells (m4-receptors) were carried out as previously described (14). The resulting data were analyzed by nonlinear regression, and IC50 values were converted to Ki values using the Cheng–Prusoff equation (15) with the following Kd values for [3H]NMS binding: 0.350 nM with brain membranes and 0.425 nM with cardiac membranes.
Not determined.
N-Hydroxysuccinimide esters were used in the synthesis of compounds 5 [TAC conjugate with Bolton–Hunter reagent (16) for radioiodination], 8 [ASA-TAC (17) for radioiodination and/or photoaffinity labeling of the receptor], 9 [biotinyl-TAC (12)], and 14 [a fluorescein isothiocyanate conjugate of TAC (11)]. Compound 6 [PAPA-TAC, the (p-aminophenyl)acetyl conjugate (18)], for radioiodination and conversion to the azide for photoaffinity labeling, was prepared via the corresponding (tert-butoxycarbonyl)amino derivative (17), 7, which was deprotected in neat trifluoroacetic acid. Compound 7 was synthesized from the amine congener 4 and the appropriate N-hydroxysuccinimide ester (7).
In addition to amide conjugates, thiourea conjugates of TAC were prepared. Compounds 10–13, which contain the isothiocyanate group for chemical affinity labeling, were prepared from the corresponding phenylenediisothio-cyanate (DITC) derivatives (7, 8), present in large stoichiometric excess to avoid formation of bis-adducts. Compound 15, which is a fluorescent eosin derivative intended for the technique of photosensitized labeling of proteins in proximity to the receptor (19), was also prepared from the amine congener 4 and the appropriate isothiocyanate.
The affinity of the analogs for muscarinic receptors was determined in competitive binding assays vs [3H]-N-methylscopolamine (summarized in Table I, selected curves in Figure 1). The selectivities of chosen derivatives were determined by comparison of Ki values in rat forebrain membranes (mainly m1 receptors) and in rat heart membranes (mainly m2 receptors). The affinities generally appeared in the 10−10–10−8 M range, and many of the derivatives were more potent than telenzepine. The compound of highest affinity was an aniline derivative PAPA-TAC, 6, which had a Ki value of 0.29 nM at rat forebrain muscarinic receptors (16-fold higher affinity than telenzepine for these receptors). The 4-azido-2-hydroxybenzoyl derivative (ASA-TAC) 8, also for photoaffinity labeling, displayed nanomolar affinity at forebrain muscarinic receptors. The high affinity of these derivatives make them suitable for the characterization of muscarinic receptors in pharmacological and spectroscopic studies, for peptide mapping, and for histochemical studies. Compounds 4–6 were nonselective for forebrain versus heart receptors. Curiously, compound 9, which displayed a Ki value of 0.60 nM at cardiac muscarinic receptors, was 5-fold selective for m2-muscarinic receptors.
Figure 1.

Inhibition curves for the displacement of [3H]-N-methylscopolamine by PAPA-TAC (6), ASA-TAC (8), biotinyl-TAC (9), and the eosine isothiocyanate conjugate of TAC (15). Specific counts are shown versus concentration of the inhibitor for rat forebrain membranes (●) and rat heart membranes (○). Inhibition of [3H]NMS binding was determined by incubating the analog and membranes with 0.5 nM [3H] NMS in phosphate-buffered saline at pH 7.2 at 37 °C for 60 min. Nonspecific binding was determined by coincubation with 1 μM atropine.
Compounds 10–13 contain an aryl isothiocyanate for labeling of the receptors through the formation of a covalent bond with a nucleophilic residue of the receptor protein (most reactive would be amino or thiol groups). These compounds were first examined under “reversible binding” conditions, and the apparent Ki values are reported in Table I. The para-substituted analog 13, with Ki values of approximately 1 nM at both forebrain and cardiac muscarinic receptors, was clearly more potent than the mono- and di-meta-substituted derivatives.
The ability of compounds 10–13 to irreversibly inhibit muscarinic receptors was examined (Table III). Membranes were incubated with a fixed concentration of each isothiocyanate derivative, then washed exhaustively to remove excess, noncovalently bound ligand, and finally subjected to either radioligand saturation experiments or binding with single concentrations of radioligand. It was found that a 1 h preincubation of rat brain receptors with 10 at a concentration of 1.0 μM resulted in a nearly total loss of the specific receptor binding sites. The detailed pharmacological characterization of the isothiocyanate derivatives will be reported elsewhere.
Table III.
Irreversible Inhibition of Rat Muscarinic Receptors Resulting from Incubation of Membranes with an Isothiocyanate Derivative at a Concentration of 100 nM
| compd |
% decrease in bindinga
|
|
|---|---|---|
| brain | heart | |
| 10 | 43 | 11 |
| 12 | 12 | 15 |
| 13 | 62 | 32 |
Decrease in specific binding of a single concentration (0.50 nM) of [3H]N-methylscopolamine from rat forebrain or rat heart membranes. Membranes were incubated with the indicated isothiocyanate derivative, freshly diluted from a DMSO stock solution (stored at −20 °C) into phosphate-buffered saline (pH 8.0,10 mM phosphate, 0.15 M sodium chloride) for 60 min at room temperature followed by radioligand binding.
Variations in activity dependent on structure were observed for the isothiocyanate derivatives (Table III). The most effective in irreversible inhibition appeared to be the p-DITC conjugate 13, which at a concentration of 100 nM blocked with more than half of the [3H]NMS (0.5 nM) binding sites. Compounds 10 and 12 blocked less than half of these sites.
DISCUSSION
We have shown that telenzepine may be derivatized as a long chain alkylamino derivative (TAC, compound 4), which retains high affinity for muscarinic receptors, albeit with diminished selectivity. Compound 4 has been subsequently coupled to a variety of reporter groups resulting in high-affinity probes for detection and characterization of muscarinic receptors. The new conjugates, in spite of having much higher molecular weights than telenzepine, were potent inhibitors of [3H]NMS binding in rat brain membranes. These probes include analogs for radiolabeling (5,6, and 8), a biotin conjugate (9), ligands for chemical affinity labeling (bearing electrophilic groups, 10–13) and photoaffinity cross-linking (an aryl amine, 6), and fluorescent derivatives (14 and 15) for nonradioactive receptor assays. Compound 5, the conjugate with Bolton-Hunter reagent, is intended for use as an iodinatable radioligand, due to the phenolic prosthetic group. Compound 6, the (p-aminophenyl)acetyl (PAPA) conjugate, is also intended for use as an iodinatable radioligand, with the added feature of potentially being cross-linked to the receptor protein by conversion of the aryl amine to an azide or by use of a photoaffinity cross-linking reagent such as SANPAH. A similar scheme has been developed using ligands bearing the PAPA prosthetic group to affinity label adenosine receptors (18). Compound 8, the ASA conjugate of TAC, is intended for use as an iodinatable photoaffinity ligand radioligand, which is already photoactivated due to the azido group.
The iodination of TAC conjugates bearing phenols or aryl amines could be carried out by first iodinating a precursor of prosthetic group, such as N-succinimidyl 3-(p-hydroxyphenyl)propionate, used to prepare compound 5. Alternately, the final compound may be iodinated through a standard iodination procedure, such as the chloramine T/sodium iodide method (18). Telenzepine itself is unaffected upon brief exposure to a mixture of chloramine T/sodium iodide (data not shown), thus it is expected that the iodination of compounds such as 5 or 6 would occur on phenolic or aniline ring, respectively.
The isothiocyanate derivatives 10–13 have been shown to irreversibly inhibit the binding site of muscarinic receptors. The apparent Ki values under “reversible” conditions (Table I) in most cases indicate the intrinsic affinity of the compound for the antagonist binding site. However, for compound 10 at forebrain muscarinic receptors, this value does not reflect solely “equilibrium” conditions, since the Ki value is similar to the IC50 value in irreversible binding (see Table III). The degree of irreversible binding (Table III) likely reflects a combination of factors, including the intrinsic receptor binding affinity of the compound and the proximity of a nucleophilic group to the NCS of the ligand in the bound state (relating to the intrinsic labeling efficacy).
The para-substituted isothiocyanate 13 was consistently the most potent antimuscarinic compound among the isothiocyanates under apparent “equilibrium” conditions. Under irreversible conditions, at a concentration 83 times its Ki value, 13 resulted in a 62% loss of forebrain [3H]NMS binding. Compound 10 inhibited nearly half of the forebrain [3H]NMS binding at only 2.4 times its Ki value. Compound 12, a diisothiocyanate, was relatively weak in both assays. At roughly half receptor occupancy, it inhibited only a small fraction of [3H] NMS binding. Thus, there is no SAR advantage of the additional NCS group present in 12. Compounds 10 and 13 showed some selectivity for the forebrain versus cardiac receptors in irreversible binding. There is considerable potential for using these affinity labels and related derivatives as pharmacological probes (7,8,22), particularly if selectivity is demonstrated in further testing and structural modification.
This series of telenzepine derivatives includes chemically reactive chains, such as amines (4 and 6), phenols (5 and 8), and isothiocyanates (10–13), potentially of use in anchoring these high-affinity ligands to a solid matrices for isolation of muscarinic receptors by affinity chromatography. In previous studies only the nonselective ligand aminobenztropine (23) has served this purpose.
Fluorescent analogs 14 and 15 retain sufficient affinity for muscarinic receptors to be explored for use in nonradioactive receptor assays (11). These compounds and other fluorescent conjugates of TAC may also be useful for fluorescent histochemical studies to microscopically locate muscarinic receptors in tissue slices.
We have shown that the selectivity in this series of muscarinic antagonists may be modulated to some extent by distal structural changes. The analogues have been shown to be either nonselective or moderately m2-selective, depending on the group with which TAC is acylated. Although this series is based on the m1-selective antagonist telenzepine (here 14-fold selective for forebrain versus cardiac muscarinic receptors), compound 9 displayed a reversal of this selectivity. This may be due to an interaction of the biotin group with a distal site on the receptors that is more energetically favorable in the case of m2-receptors. The isothiocyanate-bearing derivatives were highly variable in measured affinity at rat forebrain receptors, depending on the positions of substitution and substituents of the distal phenyl ring. It appears that a p-isothiocyanate group (compound 13) is favored in that series in competitive binding (Table I), and the m-isothiocyanate group (compound 10) is highly efficient in irreversible inhibition relative to the degree of receptor occupancy (Tables I and III).
Telenzepine is known to consist of two relatively stable enantiomers, which have very divergent affinities at muscarinic receptors, differing by as much as several orders of magnitude (21). The principle potent and selective muscarinic antagonist is the (+)-form, and the (−)-form is of much lower affinity. Nevertheless, in future studies with these compounds it would be desirable to resolve enantiomers in order to maximize potency and to avoid concerns about heterogeneity of biologically active species.
Acknowledgments
We thank Dr. Lewis Pannell and Dr. H. Lee of NIDDK for carrying out mass spectral determinations. We thank Dr. Yossef Raviv of NIDDK for helpful discussions.
Footnotes
Abbreviations used: ASA, 4-azidosalicylic acid; Boc, tert-butyloxycarbonyl; DITC, 1,3-phenylene diisocyanate; DMF, dimethylformamide; NMS, N-methylscopolamine; TFA, trifluoro-acetic acid; TLC, thin-layer chromatography.
LITERATURE CITED
- 1.Brown JH, editor. The Muscarinic Receptors. Humana Press; Clifton, NJ: 1989. [Google Scholar]
- 2.Connolly JG. Structure-function relationships in nicotinic acetylcholine receptors. Comp Biochem Biophys. 1989;93A:221–231. doi: 10.1016/0300-9629(89)90210-7. [DOI] [PubMed] [Google Scholar]
- 3.(a) Bonner TI, Buckley NJ, Young AC, Brann MR. Identification of a family of muscarinic acetylcholine receptor genes. Science. 1987;237:527–532. doi: 10.1126/science.3037705. [DOI] [PubMed] [Google Scholar]; (b) Bonner TI, Young AC, Brann MR, Buckley NJ. Cloning and expression of human and rat m5 muscarinic acetylcholine receptor genes. Neuron. 1988;1:403–410. doi: 10.1016/0896-6273(88)90190-0. [DOI] [PubMed] [Google Scholar]; (c) Peralta EG, Ashkenazi A, Winslow JW, Ramachandran J, Capon DJ. Differential regulation of PI hydrolysis and adenylyl cyclase by muscarinic receptor subtypes. Nature. 1988;334:434–437. doi: 10.1038/334434a0. [DOI] [PubMed] [Google Scholar]
- 4.Dohlman HG, Thorner J, Caron MG, Lefkowitz RJ. Model systems for the study of seven-transmembrane-segment receptors. Ann Rev Biochem. 1991;60:653–688. doi: 10.1146/annurev.bi.60.070191.003253. [DOI] [PubMed] [Google Scholar]
- 5.Curtis CA, Wheatley M, Bansal S, Birdsall NJ, Eveleigh P, Pedder EK, Poyner D, Hulme EC. Propylbenzylcholine mustard labels an acidic residue in transmembrane helix 3 of the muscarinic receptor. J Biol Chem. 1989;264:489–495. [PubMed] [Google Scholar]
- 6.Fraser CM, Wang CD, Robinson DA, Gocayne JD, Venter JC. Site-directed mutagenesis of ml muscarinic receptors: Conserved aspartic acids play important roles in receptor function. Mol Pharmacol. 1989;36:840–847. [PubMed] [Google Scholar]
- 7.Jacobson KA, Barone S, Kammula U, Stiles GL. Electrophilic derivatives of purines as irreversible inhibitors of A1-adenosine receptors. J Med Chem. 1989;32:1043–1051. doi: 10.1021/jm00125a019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boring DL, Ji XD, Zimmet J, Taylor KE, Stiles GL, Jacobson KA. Trifunctional agents as a design strategy for tailoring ligand properties: Irreversible inhibitors of A1 adenosine receptors. Bioconjugate Chem. 1991;2:77–88. doi: 10.1021/bc00008a002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Avissar S, Amitai G, Sokolovsky M. Oligomeric structure of muscarinic receptors is shown by photo-affinity labeling: Subunit assembly may explain high- and low-affinity agonist states. Proc Natl Acad Sci USA. 1983;80:156–159. doi: 10.1073/pnas.80.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobson KA, Ukena D, Padgett W, Kirk KL, Daly JW. Molecular probes for extracellular adenosine receptors. Biochem Pharmacol. 1987;36:1697–1707. doi: 10.1016/0006-2952(87)90056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCabe RT, de Costa BR, Miller RL, Havunjian RH, Rice KC, Skolnick P. Characterization of benzodiazepine receptors with fluorescent ligands. FASEB J. 1990;4:2934–2940. doi: 10.1096/fasebj.4.11.2165950. [DOI] [PubMed] [Google Scholar]
- 12.Wilchek M, Bayer E, editors. Methods in Enzymology. Vol. 184. Academic Press; New York: 1990. Avidin-Biotin Technology. [DOI] [PubMed] [Google Scholar]
- 13.Jacobson KA, Bradbury BJ, Baumgold J. A functionalized congener approach to muscarinic ligands. In: Meyer E, Simpkins J, Yamamoto J, editors. Novel Treatments and Models for Alzheimer’s Disease. Plenum Press; New York: 1990. pp. 1–10. [Google Scholar]
- 14.Karton Y, Bradbury BJ, Baumgold J, Paek R, Jacobson KA. Functionalized congener approach to muscarinic antagonists: Analogues of pirenzepine. J Med Chem. 1991;34:2133–2145. doi: 10.1021/jm00111a032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzyme reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 16.Bolton AE, Hunter WM. The labelling of proteins to high specific radioactivities by conjugation to a 126I-containing acylating agent. Biochem J. 1973;133:529–539. doi: 10.1042/bj1330529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shanahan MF, Wadzinski BE, Lowndes JM, Ruoho AE. Photoaffinity labeling of the human erythrocyte monosaccharide transporter with an aryl azide derivative of D-glucose. J Biol Chem. 1985;260:10897–10900. [PubMed] [Google Scholar]
- 18.Barrington WW, Jacobson KA, Stiles GL. Demonstration of distinct agonist and antagonist conformations of the A1 adenosine receptor. J Biol Chem. 1989;264:13157–13164. [PMC free article] [PubMed] [Google Scholar]
- 19.Raviv Y, Pollard HB, Bruggemann EP, Pastan I, Gottesman MM. Photosensitized labeling of a functional multidrug transporter in living drug-resistant tumor cells. J Biol Chem. 1990;265:3975–3980. [PubMed] [Google Scholar]
- 20.Eltze M, Gönne S, Riedel R, Schlotke B, Schudt C, Simon WA. Pharmacological evidence for selective inhibition of gastric acid secretion by telenzepine, a new antimuscarinic drug. Eur J Pharmacol. 1985;112:211–224. doi: 10.1016/0014-2999(85)90498-4. [DOI] [PubMed] [Google Scholar]
- 21.Schudt C, Boer R, Eltze M, Riedel R, Grundler G, Birdsall NJM. The affinity, selectivity, and biological activity of telenzepine enantiomers. Eur J Pharmacol. 1989;165:87–96. doi: 10.1016/0014-2999(89)90773-5. [DOI] [PubMed] [Google Scholar]
- 22.Newman AH. Irreversible ligands for drug characterization. Annual Reports in Medicinal Chemistry. 1990;25:271–280. [Google Scholar]
- 23.Haga K, Haga T. Affinity chromatography of the muscarinic acetylcholine receptor. J Biol Chem. 1983;258:13575–13579. [PubMed] [Google Scholar]