

Abstract
There is increased interest in the Bn-receptor family because they are frequently over/ectopically-expressed by tumors and thus useful as targets for imaging or receptor-targeted-cytotoxicity. The synthetic Bn-analog,[D-Tyr6,β-Ala11,Phe13,Nle14]Bn(6-14)[Univ.Lig] has the unique property of having high affinity for all three human BNRs(GRPR,NMBR,BRS-3), and thus could be especially useful for this approach. However, the molecular basis of this property is unclear and is the subject of this study. To accomplish this, site-directed mutagenesis was used after identifying potentially important amino acids using sequence homology analysis of all BnRs with high affinity for Univ.Lig compared to the Cholecystokinin-receptor(CCKAR), which has low affinity. Using various criteria 74 amino acids were identified and 101 mutations made in GRPR by changing each to those of CCKAR or to alanine. 22 GRPR mutations showed a significant decrease in affinity for Univ.Lig(>2-fold) with 2 in EC2[ D97N,G112V], 1 in UTM6[Y284A], 2 in EC4[R287N,H300S] showing >10-fold decrease in Univ.Lig affinity. Additional mutations were made to explore the molecular basis for these changes. Our results show that high affinity for Univ.Lig by human Bn-receptors requires positively charged amino acids in extracellular (EC)-domain 4 and to a lesser extent EC2 and EC3 suggesting charge-charge interactions may be particularly important for determining the general high affinity of this ligand. Furthermore, transmembrane amino acids particularly in UTM6 are important contributing both charge-charge interactions as well as interaction with a tyrosine residue in close proximity suggesting possible receptor-peptide cation-pi or H–bonding interactions are also important for determining its high affinity.
Keywords: gastrin-releasing peptide, neuromedin B, bombesin, BRS-3, receptor-mutagenesis
1. Introduction
The mammalian bombesin receptor family comprises three G protein-coupled heptahelical receptors: the neuromedin B (NMB) receptor (BB1), the gastrin-releasing peptide (GRP) receptor (BB2), and the orphan receptor bombesin receptor subtype 3 (BRS-3) (BB3)[1]. The GRP and NMB receptors are widely distributed, especially in the gastrointestinal (GI) tract and central nervous system (CNS), and the receptors have a wide range of effects in both normal physiology and pathophysiological conditions [2,3]. These include potent effects in the CNS (regulation of circadian rhythm, thermoregulation, satiety); in peripheral tissues (regulation of immune function, gastrointestinal hormone release, motility, development) and in pathological processes such as tumor differentiation and growth [1,3,4]. All of the mammalian Bn receptors have a high degree (approximately 50%) of structural homology [1]. BRS-3 is an orphan receptor present in both the central nervous system and peripheral tissues [1,3,5–10]. Its roles in normal physiology or pathological conditions are largely unknown, although studies suggest it may play important roles in tumor growth, energy homeostasis, motility, insulin secretion and appetite control [1–3,5,6,9–12]. A recent study reports its disruption leads to obesity, diabetes, and hypertension [6,10]. Because of its 51 and 47% amino acid homology with the human bombesin (Bn) receptors, NMBR and GRPR, it is classified in this receptor family [1,5].
The mammalian bombesin receptor family including BRS-3, is receiving increased attention, because not only is it important in a number of GI and CNS processes, but also because it is one of the G-protein coupling receptor families most frequently ectopically or overexpressed by a different tumors, including prostate cancer, small cell lung cancer, breast cancer, CNS tumors, and carcinoids (intestinal, thymic, and bronchial) [1,3,13,14]. This tumoral ectopic/overexpression of Bn receptor subtypes is not only important in mediating growth/differentiation effects of the tumor, frequently in an autocrine fashion, but also for possible diagnosis and possible novel therapeutic approaches [1–3,15]. Bn receptor overexpression can be used as a molecular target for specific delivery of bombesin analogues either for diagnosis with radiolabeled analogues or specific delivery of cytotoxic bombesin analogues (i.e. coupled to radiolabeled analogues or cytotoxic agents) [15,16]. The utility of this approach is supported by the results using a similar strategy with radiolabeled somatostatin which is now widely used for diagnosis, tumor localization and treatment of various endocrine tumors which frequently overexpress somatostatin receptors [15,17]. Unfortunately, most of the common tumors do not over-express somatostatin receptors, but do over-express bombesin receptors, so there is interest in applying this approach to these tumors with radiolabeled/cytotoxic Bn analogues.
Because of the widespread involvement of Bn receptors in so many processes and their potential usefulness in various diseases, especially in relation to tumor growth or tumor localization, a number of different classes of synthetic and naturally occurring Bn receptor agonists, as well as synthetic receptor antagonists have been described [1,1,18,19]. Many of these have high affinity or selectivity for the GRP or NMB receptor [1,20,21]. Recently, a synthetic Bn analogue, [D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6–14) [Univ.Lig] has been described that has the unique property of functioning as a high affinity ligand for all 3 human BnR’s subtypes [neuromedin B receptor (NMB-R), gastrin-releasing peptide receptor (GRPR) and orphan receptor (BRS-3)][16,22–24]. There is particular interest in this Bn analogue because of its unique high affinity for all 3 Bn subtypes, and thus it could be particularly valuable as a targeting agent for a number of different human tumors, which ectopically/overexpress different combinations of hGRPR, NMBR and hBRS-3 [13–15]. However, the molecular basis of this unique property is unknown.
The aim of this study is to determine the molecular basis for the high affinity of the Univ.Lig for all human BnR’s. The approach used was to perform site-directed mutagenesis on receptor amino acids potentially involved in determining high affinity for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6–14) [Univ.Lig] which were identified using detailed comparative sequence homology analysis of different bombesin receptors which had high affinity for Univ.Lig compared to other G-protein coupled receptors that did not have high affinity for Univ.Lig. This approach was used because it has been successfully used in studies of the molecular pharmacology of a number of receptors and their ligands [25–29].
2. Material and Methods
2.1 Materials
The mammalian expression vector, pcDNA3, custom primers, 0.25% Trypsin-EDTA were from Invitrogen (Carlsbad, CA). GENETICIN selective antibiotic (G418 Sulfate) was from Cellgro (Manassas, VA). QuikChange Site-Directed Mutagenesis Kit was from Agilent Technologies (Santa Clara, CA). Lipofectamine 2000, Dulbecco’s minimum essential medium (DMEM), phosphate-buffered saline (PBS), fetal bovine serum (FBS), penicillin-streptomycin were from Life Technologies (Grand Island, NY). CHOP cells (Polyoma large T antigen-expressing Chinese hamster ovary cells) were a gift from James W. Dennis (Samuel Lunenfeld Research Institute, Toronto, Canada). Gastrin-Releasing Peptide (GRP) was from Bachem (Torrance, CA). Na125I (2,200 Ci/mmol) was from Amersham Biosciences (Piscataway, NJ). 1,3,4,6-tetrachloro-3α, 6α-diphenylglucoluril (IODO-GEN) and dithiothreitol (DTT) were from Pierce Biotechnology Inc. (Rockford, IL). Bovine serum albumin fraction V (BSA) was from ICN Pharmaceutical Inc. (Costa Mesa, CA. All other chemicals were of the highest purity commercially available.
2.2 Methods
2.2.1 Strategy used to identify amino acids important for Univ.Lig affinity
To identify possible amino acids that might be important for determining high affinity of all human Bn receptors, as well as GRPRs or NMBR’s from all species, and the amphibian receptor BB4, for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14) [Univ.Lig] [1,18,22,24], a modification of the method of sequence homology described previously was used [26,27]. Briefly, the extracellular and transmembrane domains of all known members of the Bn family of receptors with high affinity for Univ.Lig were identified using hydropathy plots [26,27]. Specifically, hGRPR (GenBank accession no. P30550) [30,31], mGRPR (GenBank accession no. P21729) [32], rGRPR (GenBank accession no. P52500), hNMBR (GenBank accession no. P28336) [30,31], mNMBR (GenBank accession no. O54799), rNMBR (GenBank accession no. P24053) [33], hBRS-3 (GenBank accession no. P32247) [5,22], rhBRS-3 (GenBank accession no. AY350447), and fBB4 (GenBank accession no. P47751) [34] were included. Each of these receptors has been shown to have high affinity for Univ.Lig [1,18,22,24,34,35] in various studies. In addition, human CCKAR, which has a low affinity for Univ.Lig (Fig. 1) was also included. The receptor amino acids were aligned using CLUSTAL-W(URL:http://molbio.info.nih.gov/molbiolgcglite/clusta117.html). The default protein gap parameters used were: residue-specific penalties = on; hydrophilic penalties = on; hydrophobic residues = GPSNDQEKR; gap separation distance = 8; and end gap separation penalty = off. The hGRPR was selected for comparative studies and mutagenesis because it has been well studied and its pharmacology differs markedly from that of hBRS-3 for >50 synthetic or natural Bn-related ligands [1,21,36,37]. From the hydropathy plots, 74 amino acids were identified that might be potentially important for high affinity of Univ.Lig interaction using a number of selection criteria and 101 hGRPR mutants were made using these criteria. The following criteria were used to select the amino acids to be studied. First, the amino acid was predicted to lie in either the outer one-third of the transmembrane (TM) helix or in the extracellular domain of the receptor, because the initial interaction of peptide ligands generally occurs in these receptor regions. Second, because previous studies showed the N-terminus of the GRP or NMB receptors are not essential for high affinity GRP or NMB binding, respectively [38–40], only the receptor regions corresponding to the receptor region from TM2 to the carboxyl terminus of the Bn receptors were investigated. Third, amino acids, which were in the same position from TM2 to the carboxyl terminus of the aligned receptors, which had high affinity for Univ.Lig. and either were similar or different from those in comparable positions in hCCKAR were identified. Initially, 41 mutations were made in 38 amino acid positions in hGRPR which differ from those in a comparable position in hCCKAR by mutating the amino acid in hGRPR to either the corresponding hCCKAR amino acid, alanine or an unrelated amino acid (Group I). Subsequently, 13 mutations were made in 12 amino acids in hGRPR, which were present in similar positions in all Bn receptors with high affinity for Univ.Lig (Group II) and in hCCKAR, by mutating the amino acids in hGRPR by replacing it with alanine or a related amino acid. Fourth, 25 single mutants in amino acids in 24 separate positions in the extracellular region of hGRPR were made in which either the positions differed between the BnR’s themselves and were also different from those in hCCKAR in a similar position (n=13), or that the amino acids in all BnR’s had difference properties from that in CCKAR (n=11)(Group III). Finally, 22 mutations were made in 15 locations in hGRPR with amino acids with different characteristics substituted to examine the molecular basis for the importance of the effects of some of the amino acid replacements on receptor affinity found in Groups I-III (Group IV).
Fig. 1.
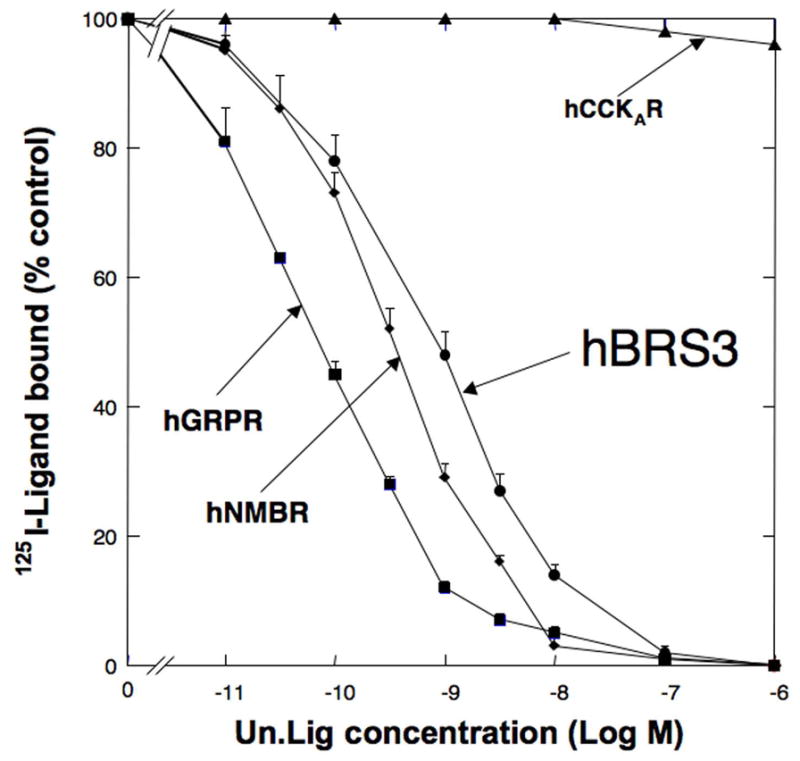
Comparison of the affinity of [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig] for all human BnR’s and hCCKAR. CHOP cells were transiently transfected with hGRPR, hNMBR, hBRS-3 or hCCKAR as described in Methods. Subsequently, the transfected cells were incubated with 50 pM 125I- [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig] for 1 h at 21°C with or without various concentrations of [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig]. Saturable binding was determined as described in Methods, and the results are expressed as the percentage of saturable binding without [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig] present. Results are mean ± SEM of at least 3 separate experiments and each point was determined in duplicate.
2.2.2 Construction of mutant GRP receptors
The cDNA of the human GRP receptor (hGRP receptor) was used, which was identical to that described previously [30,38]. hGRP receptor point mutants were constructed by using the Quick-Change Site-Directed Mutagenesis Kit, following the manufacturer’s instructions with minor modifications as described previously [26,40]. Each amino acid which was possibly important for Univ.Lig high affinity binding identified as described using the above selection criteria was mutated one at a time to the comparable amino acids in either hCCKAR, alanine or another amino acid depending on the criterion used. The cDNA of the hGRP receptor in pCD2 was a gift from Eduardo Sainz and James F. Battey (Laboratory of Molecular Biology, NIDCD, National Institutes of Health). The hGRPR cDNA was inserted into pcDNA3.1 (+) at the EcoR I site. Nucleotide sequence analysis of the entire coding region was performed using an automated DNA sequencer on the wild type receptor and all mutant receptors (Applied Biosystems Inc., Foster City, CA).
2.2.3. Growth and maintenance of cells
CHOP cells were grown in DMEM containing 10% (v/v) FBS, 100 units/ml of penicillin, 100 mg/ml of streptomycin and 200 μg/ml of G418. All cells were maintained at 37°C in a 5% CO2 atmosphere. Cells were split every 3–4 days at confluence after detaching the cells with trypsin/versene solution.
2.2.4. Cell transfections
CHOP cells, which contain no native GRP receptors, were seeded in a 10-cm tissue culture dish at a density of 2.5 × 106 cells/dish and grown overnight at 37°C in growth medium [26,41]. On the following morning, 2.5 μg of plasmid DNA was transfected to CHOP cells by cationic lipid-mediated method using 30 μl of Lipofectamine 2000 in OPTI-MEM I Reduced-Serum Medium for 3 h at 37°C. At the end of the incubation period, the medium was replaced with new growth medium. Cells were maintained at 37°C in a 5% CO2 atmosphere and were used 48 h later for binding assays.
2.2.5. Preparation of 125I-[D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14)
125I-[D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14) at a specific activity of 2200 Ci/mmol was prepared by a modification of methods described previously [22]. Radiolabeled peptides were separated using a Sep-Pak (Waters Associates, Milford, MA) and high-pressure liquid chromatography as described previously [22,42]. Radioligands were stored with 0.5% BSA at 20°C.
2.2.6. Whole cell radioligand binding assays
Binding studies to expressed plasmid DNAs were performed as described previously [32,37,39,42]. For the wild type GRP and mutant GRP receptors, 125I-[D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14) (125I-[Univ.Lig]) was used as the ligand to assess receptor affinity. Briefly, 48 h after transient transfection with Lipofectamine 2000, disaggregated transfected cells were incubated for 1 h at 21°C in 250 μl of binding buffer containing 24.5 mM HEPES (pH 7.4), 98 mM NaCl, 6 mM KCl, 2.5 mM KH2PO4, 5 mM sodium pyruvate, 5 mM sodium fumarate, 5 mM sodium glutamate, 2 mM glutamine, 11.5 mM glucose, 0.5 mM CaCl2, 1.0 mM MgCl2, 0.01% (w/v) soybean trypsin inhibitor, 0.2% (v/v) amino acid mixture, 0.2% (w/v) BSA, and 0.05% (w/v) bacitracin with 50 pM of 125I-[D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14) (2200 Ci/mmol) in the presence of the indicated concentration of unlabeled peptides. In a previous study [43] we demonstrated that hGRP receptor expression varying as much as 160-fold did not alter the receptor affinity for Bn related peptides under the binding conditions used in this study. As an additional precaution to correct for any possible differences that might occur during the binding assays due to possible differential ligand degradation with the different receptors or to slight differences in receptor expression with different mutant receptors resulting in any differences in the ligand bound seen with different mutant hGRP receptors, binding results with each mutant hGRP receptor were compared only to results with wild type hGRP receptor-containing cells binding similar amounts of ligand as described previously [26]. This was accomplished by was adjusting the cell concentration between 0.05 and 4 × 106 cells/ml for each mutant receptor, so that less than 15% of the total added radioactive ligand was bound during the incubation and the results compared to cells transfected with wild type hGRP receptor adjusted in concentration to bind a similar amount of ligand. The amount of radioactivity bound to the cells was measured in a Cobra II Gamma counter (Packard Instruments). Binding was expressed as the percentage of total radioactivity that was associated with the cell pellet. All binding values represented saturable binding (i.e., total binding minus nonsaturable binding). Nonsaturable binding was defined as the amount of binding that occurred with 1 μM 125I-[D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14) in the incubation solution. Nonsaturable binding was <15% of the total binding in all experiments. Each point was measured in duplicate, and each experiment was replicated at least four times. Calculation of affinity was performed by determining the IC50 (the GRP concentration causing half-maximum inhibition of binding), using the curve-fitting program KaleidaGraph (Synergy Software, Luton, London). Statistical analysis was performed with Statview version 4.02 (BrainPower, Inc. Calabasas, CA). with an analysis of variance used to determine the statistical significance of differences in affinity of each hGRP receptor mutant compared with its own wild type hGRP receptor control, binding similar amounts of radioligand and therefore in all statistical analysis only two variables (i.e. GRPR mutant and its wild type control binding similar amounts of ligand) were analyzed.
2.2.7. Modeling of the GRP receptor
The molecular modeling was performed as described previously [26]. Briefly, the primary sequence of the hGRPR-receptor was input into the Swiss-PDB Viewer [44], and a suggested homology modeling template 2Y00b [45], a crystal structure of beta1-adrenergic receptor at 2.5 Å resolution established from a blast search. The model of hGRPR was imported into SYBYL–X 1.3 (Tripos L.P., St Louis, MO). The folded loop region EC3 blocking access to the receptor cavity was removed and the ends of the helical transmembrane regions capped. The putative binding site of the model was visualized by the SiteID module in SYBYL-X (Tripos L.P., St Louis, MO). The model was solvated with water and solvent molecules capable of defining a continuous cluster within 5 Å of the model were visualizes as a surface, which is shown in grey in Fig. 7. Solvent-accessible amino acid residues within interaction distance (5 Å) of this interior space are shown in Fig. 7, with the residues identified in this work as important for binding as ball and stick models.
Fig. 7.

Three-dimensional model of the putative binding site of the human GRP receptor. The three-dimensional model of the human GRP receptor is shown in which the helical domains are indicated as backbone helices that are colored red for helix 1, orange for helix 2, yellow for helix 3, green for helix 4, blue for helix 5, indigo for helix 6, and violet for helix 7. The fourth extracellular domain is colored gray (helix 6 – helix 7). The model and binding pocket were visualized by the SiteID module in SYBYL, as described in Methods. The putative binding pocket is depicted as a dark-gray space-filled structure. 38 amino acids examined in this study were within 5Å of the putative binding pocket. The location and identity of the seven amino acids within 5Å of the binding pocket that were most important for determining high affinity for Univ.Lig are shown in yellow and included D97, L121, I283, Y284, R287, H300, V302. The location and identity of three amino acids (T92, G112, E293), which also contributed to a lesser degree in determining high affinity for Univ.Lig (1.5 to5-fold) but which were >5Å from the binding pocket are shown in green.
3. Results
[D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig] bound with high affinity to each human Bn receptor subtype, [i.e.. hGRPR, hNMBR, and hBRS-3] [IC50 =0.07±0.01 nM, 0.85±0.05 nM, 1.5±0.3nM, respectively] (Fig. 1) as described in other studies [14,22,24,37]. In contrast, Univ.Lig didn’t interact with hCCKAR, even at very high concentrations [IC50 >10,000 nM](Fig. 1). We used this difference in affinity for these two receptor groups, to identify 74 amino acids using the criteria outlined in METHODS, which might be important for determining the selective high affinity of [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig] for all human Bn receptors, GRPRs and NMBRs from all species, and the amphibian Bn receptor (BB4). The hGRPR was selected for comparative studies and mutagenesis because it has been well studied and its pharmacology differs markedly from that of hBRS-3 for >50 synthetic or natural Bn-related ligands [1,21,36,37].We made 41 mutants (Group I) in 38 amino acids in hGRPR which were similar in all the Bn receptors with high affinity for Univ.Lig, but differed in hCCKAR and met the other selection criteria (mutants #1-41, Table 1) (Fig. 2). Of these 22 amino acids were in the EC regions and 19 in the upper TM regions (UTM) (Fig. 2). Eleven of the 22 EC amino acids mutated were in the EC2 of the hGRP receptor (from D97N to G112V), six in the EC3 (from F193A to M212F), and five in the EC4 (from R287N to S304A) (Table 1, Fig. 2). For the 19 amino acids in the upper TM regions (UTM) identified using the above mentioned criteria, mutations were made in three in UTM2 (from L90A toV96A), four in UTM3 (from L115A to L121A), four in UTM4 (from A172T to A176G), two in UTM5 (F215A and F218A), four in UTM6 (from N280A to Y284A), and two in UTM7 (I305A and R308A) (Fig. 2, Table 1). Twelve of the 41 point mutations in Group I resulted in > 2-fold decrease of affinity (T92A, D97N, D104K, G112V, L121A, H281A, I283A, Y284A, R287N, M298P, H300S and R308A)(Table 1, Fig. 3 and 4). Four of these [D97N, G112V, Y284A and R287N] demonstrated > 10-fold decease of affinity for Univ.Lig [IC50 =1.24±0.01nM, 2.98±0.15nM, 5.78±0.33nM, 7.07±0.47nM] (Table 1, Fig. 2 and 4), and two resulted in a small, but significant decrease in affinity (1.5–1.8 fold) [I116A, E175Q][Table 1]. Three additional mutants were made in two extracellular amino acid positions (Y101A, R110A, R110D) and each of these didn’t alter Univ.Lig affinity (Table 1).
Table 1.
Affinities of hGRPR point mutants in Group I for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig]
| No. | LOCATION | POINT MUTATION | Univ.Lig Affinity (nM ± SEM) |
|---|---|---|---|
| Wild type hGRPR 0.1×106 | - | 0.100 ± 0.006 | |
| 1 | UTM2 | L90A | 0.053 ± 0.003 |
| 2 | T92A | 0.53 ± 0.03* | |
| 3 | V96A | 0.13 ± 0.42 | |
| 4 | EC2 | D97N | 1.24 ± 0.01* |
| 5 | A98L | 0.031 ± 0.007 | |
| 6 | S99I | 0.010 ± 0.013 | |
| 7 | R100A | 0.094 ± 0.004 | |
| 8 | Y101N | 0.038 ± 0.001 | |
| 9 | Y101A | 0.120 ± 0.01 | |
| 10 | D104K | 0.20 ± 0.01* | |
| 11 | R110S | 0.074 ± 0.009 | |
| 12 | R110A | 0.087 ± 0.003 | |
| 13 | R110D | 0.026 ± 0.001 | |
| 14 | G112V | 2.98 ± 0.15* | |
| 15 | UTM3 | L115A | 0.050 ± 0.031 |
| 16 | I116A | 0.18 ± 0.01* | |
| 17 | I119A | 0.11 ± 0.01 | |
| 18 | L121A | 0.48 ± 0.02* | |
| 19 | UTM4 | A172T | 0.078 ± 0.009 |
| 20 | I173T | 0.066 ± 0.008 | |
| 21 | E175Q | 0.15 ± 0.01* | |
| 22 | A176G | 0.12 ± 0.01 | |
| 23 | EC3 | F193A | 0.078 ± 0.011 |
| 24 | Y199L | 0.069 ± 0.003 | |
| 25 | P200L | 0.064 ± 0.004 | |
| 26 | S202N | 0.056 ± 0.003 | |
| 27 | L205A | 0.096 ± 0.004 | |
| 28 | M212F | 0.10 ± 0.01 | |
| 29 | UTM5 | F215A | 0.063 ± 0.005 |
| 30 | F218A | 0.074 ± 0.001 | |
| 31 | UTM6 | N280A | 0.037 ± 0.003 |
| 32 | H281A | 0.22 ± 0.01* | |
| 33 | I283A | 0.47 ± 0.02* | |
| 34 | Y284A | 5.78 ± 0.33* | |
| 35 | EC4 | R287N | 7.07 ± 0.47* |
| 36 | Y289A | 0.063 ± 0.008 | |
| 37 | M298P | 0.22 ± 0.01* | |
| 38 | H300S | 0.99 ± 0.03* | |
| 39 | S304A | 0.10 ± 0.01 | |
| 40 | UTM7 | I305A | 0.14 ± 0.01 |
| 41 | R308A | 0.28 ± 0.02* |
Group I (n=41) comprise point mutants made in a hGRPR amino acids which are present in all BnR’s with high affinity for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig] but which different from that in a similar position in hCCKAR identified as described in Methods. The affinities (IC50) were measured in whole cell radioligand binding assays using 50 pM 125I- [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig] as described in Methods and Fig. 3 legend. Each point mutation’s affinity for Univ.Lig was compared to that of cells expressing wild type GRPR which bound comparable amounts of radiolabeled ligand. Data are partially from the dose-inhibition curves of representative mutants shown in Fig. 3. IC50’s, the concentration causing half-maximal inhibition, were calculated from the dose-inhibition curves of [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig] as described in Methods. Values are means ± SEM from three experiments for those mutants showing no affinity change and 4 to 6 experiments for any showing differences from control and in each experiment each point was measured in duplicate.
(in bold)=p<0.05 compared to wild type hGRPR expressed at similar expression levels.
Fig. 2.

Transmembrane hydropathy plot of the hGRPR. Transmembrane hydropathy plot of the hGRPR showing amino acid residues examined by site-directed mutagenesis in the present study. The transmembrane areas of the hGRPR were identified by hydropathy plots as described in Methods. This diagram shows the extracellular side above, and the cytoplasmic side below, with the three glycosylation sites shown by the Y-like symbols. The 74 amino acids identified as described in Methods from comparative alignment of bombesin receptors as possibly having an important role in determining high affinity for Univ.lig. and that were mutated in the present study are shown in the black circles.
Fig. 3.
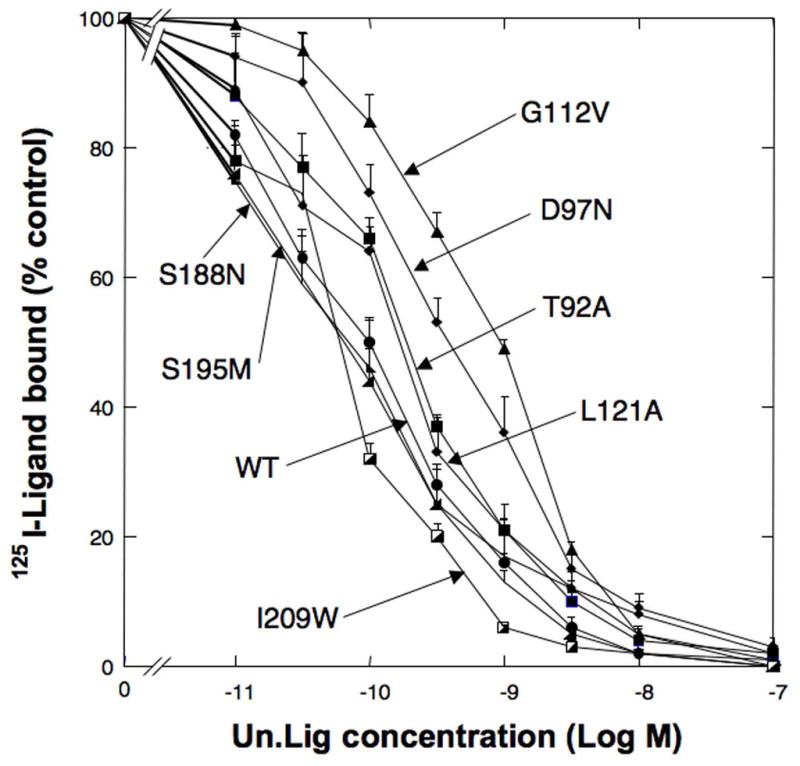
Comparison of the affinity of representative point mutants for Univ.Lig from hGRPR receptor region UTM2 to EC3. Shown are representative results from 7 point mutations made from hGRP receptor regions between UTM2 to EC2 [L90 to M212]. T92A refer to the replacement of tyrosine 92 in the hGRPR (see Fig. 2) by alanine. Experiments were performed as described in Fig. 1 legend. Results are expressed as the percentage of saturable binding without [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig] present. Results are mean ± SEM of at least 3 separate experiments and each point was determined in duplicate. This figure includes representative results of both Group I (T92A, D97N, G112V, L121A) and Group 3 (S188N, S195M, I209W) mutants.
Fig. 4.

Comparison of the affinity of point mutants for Univ.Lig from hGRPR receptor region in UTM5 to UTM7. Shown are representative results from 8 point mutations in hGRPR receptor region UTM5 to UTM7 [F215 to R308]. Experiments were performed as described in Fig. 1 legend. Results are expressed as the percentage of saturable binding without [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig] present. Results are mean ± SEM of at least 3 separate experiments and each point was determined in duplicate. This figure includes representative results of both Group I (I283A, Y284A, R287N, H300S), Group 2 (F301A) and Group 3 (E293R, V302I, T303L) mutants.
We next identified twelve amino acids which were similar in all BnR’s with high affinity for Univ.Lig and in a similar position in hCCKAR (Group II) and met the other selection criteria described in Methods. Thirteen mutants were made in these positions (mutations #42–54, Table 2). Each of these amino acids was in the EC receptor regions (Table 2, Fig. 2). Five of these point mutations [L107A, K114A, V294L, S297A, F301A] demonstrated a small decrease in affinity (1.5–2-fold), with the remainder demonstrating no change in affinity [L102A, W106A, F108A, G109A, F178A, S211A, S288T](Fig. 2 and 4, Table 2). In position 297 of hGRPR, which contains a serine (Fig. 2) an additional point mutant S297T was made and it resulted in no change in affinity (Table 2). We next examined a final group (Group III) of 24 amino acids, each in the EC receptor domains, which showed some differences between all the BnR’s with high affinity for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig], but all the amino acids in the BnR’s differed from those in hCCKAR in a similar position (Fig. 2). Twenty-five point mutants were made in these 24 positions in hGRPR (mutants #55–79, Table 3). Seven of these point mutations showed greater than 2-fold decrease in affinity; [E186T (x2.1), S188N (x2.2), S195M (x2), I209W (x2.7), E293R (x4.2), V302I (x3.5), T303L (x2.3)] (Table 3, Fig. 3 and 4).
Table 2.
Affinities of hGRPR point mutants in Group II for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig]
| LOCATION | POINT MUTATION | Univ.Lig Affinity (nM ± SEM) | |
|---|---|---|---|
| Wild type hGRPR 0.1×106 | - | 0.10 ± 0.01 | |
| 42 | EC2 | L102A | 0.069 ± 0.003 |
| 43 | W106A | 0.083 ± 0.006 | |
| 44 | L107A | 0.17 ± 0.02* | |
| 45 | F108A | 0.14 ± 0.01 | |
| 46 | G109A | 0.050 ± 0.002 | |
| 47 | K114A | 0.15 ± 0.01* | |
| 48 | EC3 | F178A | 0.060 ± 0.032 |
| 49 | S211A | 0.11 ± 0.01 | |
| 50 | EC4 | S288T | 0.12 ± 0.02 |
| 51 | V294L | 0.17 ± 0.01* | |
| 52 | S297T | 0.091 ± 0.001 | |
| 53 | S297A | 0.18 ± 0.02* | |
| 54 | F301A | 0.17 ± 0.01* |
Group II (n=13) comprise point mutants made in a hGRPR amino acids which are present in all BnR’s with high affinity for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig] and in also in a similar position in hCCKAR, which has low affinity for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig] identified as described in Methods. Experiments were performed as described in Table 1 and Fig. 1 legend. The affinities (IC50) were calculated as described in Methods. Values are means ± SEM from three experiments for those mutants showing no affinity change and 4 to 6 experiments for any showing differences from control and in each experiment each point was measured in duplicate.
(in bold)=p<0.05 compared to wild type hGRPR expressed at similar expression levels.
Table 3.
Affinities of hGRPR point mutants in Group III for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig]
| LOCATION | POINT MUTATION | Univ.Lig Affinity (nM ± SEM) | |
|---|---|---|---|
| Wild type hGRPR 0.1×106 | - | 0.100 ± 0.006 | |
| 55 | EC3 | H182A | 0.11 ± 0.01 |
| 56 | H185- | 0.11 ± 0.01 | |
| 57 | E186T | 0.21 ± 0.01* | |
| 58 | E187K | 0.072 ± 0.003 | |
| 59 | S188N | 0.22 ± 0.02* | |
| 60 | T189N | 0.088 ± 0.004 | |
| 61 | I194N | 0.054 ± 0.014 | |
| 62 | S195M | 0.20 ± 0.01* | |
| 63 | A197R | 0.050 ± 0.012 | |
| 64 | P198F | 0.078 ± 0.007 | |
| 65 | H201A | 0.14 ± 0.01 | |
| 66 | E204A | 0.12 ± 0.01 | |
| 67 | H206A | 0.13 ± 0.01 | |
| 68 | K208S | 0.003 ± 0.001 | |
| 69 | K208A | 0.092 ± 0.032 | |
| 70 | I209W | 0.27 ± 0.01* | |
| 71 | EC4 | H290S | 0.078 ± 0.004 |
| 72 | Y291A | 0.13 ± 0.01 | |
| 73 | S292E | 0.12 ± 0.02 | |
| 74 | E293R | 0.42 ± 0.06* | |
| 75 | D295S | 0.18 ± 0.02* | |
| 76 | T296G | 0.10 ± 0.01 | |
| 77 | L299I | 0.081 ± 0.019 | |
| 78 | V302I | 0.35 ± 0.01* | |
| 79 | T303L | 0.23 ± 0.02* |
Group III (n=25) include hGRPR point mutants made in extracellular receptor positions in which the BnR’s and hCCKAR differ identified as described in Methods. Experiments were performed as described in Table 1 and Fig. legends. The affinities (IC50) were calculated as described in Methods. Values are means ± SEM from three experiments for those mutants showing no affinity change and 4 to 6 experiments for any showing differences from control and in each experiment each point was measured in duplicate. [H185-] indicates a hGRPR mutant with histidine deleted from position 185.
(in bold)=p <0.05 compared to wild type hGRPR expressed at similar expression levels.
In Group I–III, we identified 26 amino acids [T92, D97, D104, L107, G112, K114, I116, L121, E175, E186, S188, S195, I209, H281, I283, Y284, R287, E293, V294, S297, M298, H300, F301, V302, T303, R308] in hGRPR, which could contribute to [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig], high affinity (Table 1–4, Fig. 2). To provide possible insights into the molecular basis for these amino acids determining high affinity for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig], we made additional substitutions in 22 amino acid positions, by substituting amino acids with differing properties (mutants #80–101, Table 4, Fig. 5). In position D97 in EC2 (Fig. 2), the substituting of glutamic acid (D97E) for the aspartic acid in hGRPR, had minimum effect on Univ.Lig affinity (1.9× decrease), whereas substitution of an the unrelated amino acid, alanine or asparagine (D97A, D97N) caused a marked decrease (>10-fold) in affinity (Fig. 5A and 6, Table 4, mutants #4, 80, 81). Substitution of a positively charge amino acid, D97K resulted in no binding of Univ.Lig. In position 104, the substitution for aspartic acid in hGRPR of a positively charged amino acid [D104K] or amino acids without charge [D104A, D104N], all resulted in a 2–3-fold decrease in affinity for Univ.Lig [mutants# 10, 82–83, Table 4]. In position 112 of hGRPR, the substitution of alanine for glycine caused a small, but significant decrease in affinity (1.5-fold), whereas the substitution of valine caused a 30-fold decrease (mutants #14, 84, Table 4). In position 116, the replacement of isoleucine in hGRPR by alanine or lysine [I116A, I116K] caused a 2-fold decrease in affinity, however, replacement by leucine or methionine [I116L, I116M] had no effect on Univ.Lig affinity (mutants #16, 85, 87, Table 4). In position 121 (Fig. 2), the replacement of leucine in hGRPR, by alanine or lysine each resulted in 3–5 fold decrease in affinity, whereas replacement by methionine or isoleucine had no effect (mutants #18, 88–91). In position 209, the replacement of isoleucine by either a tryptophan or alanine [I209W, I209A] had a similar effect of reducing affinity for Univ.Lig by 3-fold (mutants #70, 91, Table 4). In position 283, the replacement of the isoleucine by lysine or alanine [I283K, I283A] caused a 5- to 17-fold decrease in affinity for Univ.Lig in hGRPR, but replacement by leucine or methionine, [I283L, I283M], didn’t alter Univ.Lig affinity (Fig. 5B and 6, Table 4). In position 284, replacement of the tyrosine in hGRPR by alanine, leucine or tryptophan (Y284A, Y284L, Y284W) caused a marked decrease in affinity (17–42-fold) for Univ.Lig (Fig. 5C, Table 4). However, replacement with a phenylalanine (Y284F) showed only 1.5-fold decrease affinity (Fig. 5C, Table 4). In position 287, replacement of the arginine in hGRPR, by a lysine (R287K) resulted in 10-fold decrease affinity for Univ.Lig, whereas substitution of asparagine (R287N) caused a 70-fold decreasing affinity. Two additional 287 position mutants, R287A and R287D resulted no binding (Fig. 5D and 6, Table 4). In position 295, the replacement of aspartic acid in hGRPR by a serine (D295S, Table 4) (present in hCCKAR) caused a 2-fold decrease in Univ.Lig affinity, whereas replacement by alanine caused a 5-fold decrease in affinity for Univ.Lig (mutants #75, 97, Table 4). In position 300, replacement of histidine in hGRPR by alanine [H300A] caused a small, but significant decrease in Univ.Lig. affinity (1.5-fold), which is less than the 10-fold decrease by replacing it by serine [H300S], which is in a similar position in hCCKAR (mutants #38, 100, Table 4). Lastly, in position 302, replacement of valine in hGRPR by alanine [V302A] had no effect in affinity for Univ.Lig. In contrast, the replacement by isoleucine [V302I], which is in a similar position in hCCKAR, caused a 4-fold decrease in affinity (mutants #78, 101, Table 4).
Table 4.
Affinities of hGRPR point mutants in Group IV for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig]
| LOCATION | POINT MUTATION | Univ.Lig Affinity (nM ± SEM) | |
|---|---|---|---|
| Wild type hGRPR 0.1×106 | - | 0.10 ± 0.01 | |
| 4 | EC2 | D97N | 1.24 ± 0.01* |
| 80 | D97A | 1.13 ± 0.06* | |
| 81 | D97E | 0.19 ± 0.02* | |
| 10 | D104K | 0.20 ± 0.01* | |
| 82 | D104A | 0.20 ± 0.01* | |
| 83 | D104N | 0.30 ± 0.02* | |
| 14 | G112V | 2.98 ± 0.15* | |
| 84 | G112A | 0.15 ± 0.01* | |
| 16 | UTM3 | I116A | 0.18 ± 0.01* |
| 85 | I116K | 0.22 ± 0.01* | |
| 86 | I116L | 0.055 ± 0.010 | |
| 87 | I116M | 0.051 ± 0.004 | |
| 18 | L121A | 0.48 ± 0.02* | |
| 88 | L121I | 0.067 ± 0.012 | |
| 89 | L121K | 0.29 ± 0.01* | |
| 90 | L121M | 0.13 ± 0.01 | |
| 70 | EC3 | I209W | 0.27 ± 0.01* |
| 91 | I209A | 0.27 ± 0.01* | |
| 33 | I283A | 0.47 ± 0.02* | |
| 92 | UTM6 | I283K | 1.75 ± 0.11* |
| 93 | I283L | 0.22 ± 0.01 | |
| 94 | I283M | 0.36 ± 0.01 | |
| 34 | Y284A | 5.78 ± 0.33* | |
| 95 | Y284F | 0.16 ± 0.01* | |
| 96 | Y284L | 2.53 ± 0.14* | |
| 97 | Y284W | 4.22 ± 0.26* | |
| 35 | R287N | 7.07 ± 0.47* | |
| 98 | R287K | 1.02 ± 0.05* | |
| 75 | EC4 | D295S | 0.18 ± 0.02* |
| 99 | D295A | 0.46 ± 0.01* | |
| 38 | H300S | 0.99 ± 0.03* | |
| 100 | H300A | 0.15 ± 0.01* | |
| 78 | V302I | 0.35 ± 0.01* | |
| 101 | V302A | 0.060 ± 0.001 |
Group IV (22 new mutations) include hGRPR point mutants made to explore the important properties of the amino acid replacement made in that position that was found to alter the affinity for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig] and identified in Tables 1–3. Experiments were performed as described in Table 1, and Fig. 1, 3 and 4 Figure legends. The affinities (IC50) were calculated as described in Methods. Each point mutation’s affinity for Univ.Lig was compared to that of cells expressing wild type GRPR. Values are means ± SEM from three experiments for those mutants showing no affinity change and 4 to 6 experiments for any showing differences from control and in each experiment each point was measured in duplicate.
(in bold)=p<0.05 compared to wild type hGRPR expressed at similar expression levels.
Fig. 5.

Importance of the properties of specific amino acid replacements in hGRPR in position D97, I283, Y284 and R287 for determining affinity for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig]. Experiments were performed as described in Fig. 1 legend. Results are expressed as the percentage of saturable binding without [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig] present. Results are mean ± SEM of at least 3 separate experiments and each point was determined in duplicate.
Fig. 6.

Summary of effect of important amino acids positions in hGRPR for determining affinity for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig]. The four important amino acid positions are shown in the enlarged black circles. Transmembrane hydropathy plot of the hGRPR showing amino acid residues examined by site-directed mutagenesis.
To attempt to gain additional insights into why the different amino acids in the human GRP receptor transmembrane and extracellular domains identified in this study are important for determining high affinity for Univ.Lig, three-dimensional modeling of the GRP receptor was performed (Fig. 7). This was accomplished by starting from a three-dimensional structure of beta1-adrenergic receptor established at 2.5 Å resolution [45]. After hGRPR was imported into SYBYL–X 1.3, the folded loop region EC3 blocking access to the receptor cavity was removed and the ends of the helical transmembrane regions capped. The putative binding site of the model was visualized by the SiteID module in SYBYL-X. The model was solvated with water and solvent molecules capable of defining a continuous cluster within 5 Å of the model were visualizes as a surface, which is shown in grey in Fig. 7. The ends of the transmembrane regions in this 3D model are slightly different from those determined by hydropathy plots. However, all the residues found to be important in the mutagenesis studies are located within the helical transmembrane domains near the interface with the extracellular loop domains in the 3D model. The x-ray crystallographic model of beta1-adrenergic receptor used for the homology modeling of hGRPR contains the small ligand R-dobutamine and the extracellular loop 3 (EC3) is folded down into the body of the receptor. Since this loop blocked the approach to the solvent accessible region of the receptor, it was excised from the 3D model to permit access to the solvent accessible binding site by the much larger bombesin Univ.Lig ligand. The extracellular loop regions are less conserved than the transmembrane domains of the G-protein coupled receptors and also exhibit greater flexibility. Since the mutagenesis studies had shown residues within the transmembrane domains to be important for receptor recognition, the removal of the loop was not expected to adversely influence the potential binding site. 38 amino acids in the exposed transmembrane (TM) residues were within a 5 Å interaction distance of the water occupying the site: TMII; V96 D97, R100, Y101, TMIII; C113, T122 V124, G125, TMIV: L171, A172, I173, E175, A176, V177, TMV; H210, S211, M212, A213, S214, F215, V217, F218, Y219, TMVI; W277 I283, Y284, L285, R287, S288, TMVII; H300, V302, T303, C306, A307, R308, L309, A311 F312. TMI was not located near the binding site. Many of the above amino acids within 5 Å of the binding pocket where not found to be important for Univ.Lig affinity. Seven residues (D97, L121, I283, Y284, R287, H300, V302) identified by the mutagenesis study to be important for high affinity binding of Univ.Lig (>5- to 71-fold) (Table 1 and 3, Fig. 3–6) were within 5 Å of the binding pocket identified on the modeling. These are shown in yellow in the modeling figure (Fig. 7). Three amino acids (T92,G112,E293) which also contributed to a lesser degree in determining high affinity for Univ.Lig (1.5 to 5-fold) (Tables 1 and 3, Fig. 3 and 4), occupied locations which were >5Å from the binding pocket (green, Fig. 7).
4. Discussion
The bombesin receptor family include four G-protein-coupled receptors; the gastrin-releasing peptide receptor (GRPR), the neuromedin B receptor (NMBR), the orphan bombesin receptor, bombesin receptor subtype 3 (BRS-3) and the amphibian receptor, bombesin receptor subtype 4 (BB4) [1,5,34,46,47]. Of >50 natural occurring and synthetic Bn-related ligands (selective, nonselective- agonists/antagonists), [1,21,36,37], only the Bn analog, [D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14) [Univ.Lig] has the unique property of functioning as a high affinity agonist at all 3 human BnR’s [i.e. hNMB-R, hGRPR and hBRS-3], GRPR’s and NMBR’s in all species examined and the amphibian Bn receptor, BB4 [14,16,22,24,35,48]. However, the molecular basis for this unique property is largely unknown. It is also unclear whether it is similar to any of the molecular properties responsible for the high affinity selective binding of GRP to the GRP receptor, NMB to the NMB receptor, or various selective peptide antagonists or peptoid antagonists to either GRPR or NMBR [26–28,38,40,42,49,50]. Recent studies provide a few limited insights [25–28]. One study [25] using a chimera/site-directed mutagenesis approach, demonstrated that the selectivity and high affinity for hBRS-3 of three selective BRS-3 peptide agonists, which were derived from structure-function studies of [D-Tyr6, β-Ala11, Phe13, Nle14]Bn(6-14) [Univ.Lig], was due to interactions with amino acid residues primarily in the extracellular domain 2 (EC2) of hBRS-3 and to the lesser extent in the adjacent upper TMs. This study [25] compared the selectivity of these peptides for hBRS-3 and other Bn receptors by making replacements between them, therefore, because [D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14) [Univ.Lig] has high affinity for each, it provided minimum insight into the molecular basis of high affinity of [D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14) [Univ.Lig] for each Bn receptor. Three other studies identified important amino acids differences between the GRP receptor and BRS-3 [26–28] and between BRS-3 and NMBR, [28,40], which determine selectivity of GRP for the GRP receptor or selectivity of NMB for the NMB receptor over BRS-3. Two of these studies included no data with [D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14) [Univ.Lig] and the third [26], reported that only alterations in two of the extracellular amino acids studied which corresponded to R100 in EC2 and P198 in EC3 of the GRPR, had a small effect on [D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14) [Univ.Lig] affinity. Lastly, one study [51] using a chimeric/site-directed mutagenesis approach, investigated the basis for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14) [Univ.Lig] having low affinity for the mouse and rat BRS-3, in contrast to the human BRS-3. This study [51] demonstrated that this BRS-3 species’ difference in affinity for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14) [Univ.Lig] was due to multiple differences in amino acids in the mid/distal extracellular domain 4 (EC4) regions of the human/rat and mouse BRS-3. While this study, demonstrated amino acid differences in mid/distal EC4 could affect affinity for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14) [Univ.Lig], it provided no insights into whether these amino acids contributed to the high affinity of [D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14) [Univ.Lig] for other Bn receptors.
Therefore, the aim of our study was to attempt to determine the molecular basis for the high affinity of the universal ligand(Univ.Lig) for most Bn receptors. To accomplish this the method of analysis of structural homologies from comparative alignment of related and unrelated G-protein-coupled receptors using hydropathy plots was used [26–28]. Recent studies [26–28] show this approach has been very useful to identify amino acids in related and unrelated G-protein-coupled receptors, important for determining ligand selectivity or high affinity. Specifically, in the present study amino acids in a given position in various Bn-receptors, with high affinity for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14) [Univ.Lig], were compared to each other and also to those in a compatible position in CCKAR, which has low affinity for [D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14) [Univ.Lig]. Using a number of criteria, possible amino acids that could contribute to [D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14) [Univ.Lig] high affinity for most Bn receptors were identified and studied in detail using site-directed mutagenesis.
Using this approach, 79 amino acids were identified in hGRPR that might contribute to its high affinity for Univ.Lig affinity and each was studied by making point mutations in these positions. We found that 27 point mutations (19 in EC’s and 8 in UTM’s regions) resulted in a >1.5-fold decrease in Univ.Lig affinity and, especially, 4 point mutations [D97N (EC2), G112V (EC2), Y284A (UTM6), R287N (EC4)] resulted in a >10-fold decrease. These results show that both extracellular receptor domains as well as upper transmembrane domains are important in determining high affinity for Univ.Lig. Our results have both similarities and differences from studies of the molecular basis of affinity of various gastrointestinal peptide hormone/neurotransmitters for other G-protein-coupled receptors, as well as GRP/NMB for their mammalian Bn receptors. Our results agree with the general conclusion that various peptide gastrointestinal hormone/neurotransmitter interactions with either EC’s, UTM regions or both are most important for determining the ligand’s high affinity, which is in contrast to bioactive amines, such as catecholamines, whose high affinity is determined primarily by amino acids in the TM region [25,26,40,52]. Our results showing both EC and UTM domains are important for high affinity for Univ.Lig differ from findings with a number of G-protein coupled receptor peptide ligands for other GPCRs, where only interaction with EC domains is primarily important including substance P for the NK1 receptor [53], neuropeptide S for neuropeptide S receptor(NPS) [54], gastrin for the CCKB receptor and CCK-8 for CCKAR [55]. Similarly, our results differ from studies reporting primarily interaction with UTM receptor regions are important for peptide selectivity/high affinity as seen with endothelin interaction with the endothelin type A receptor (ETA) [56], ET-3 with the endothelin type B receptor(ETB) [57] and nociceptin with orphanin FQ receptors(N/OFQ) [29]. Our results are similar to those reported for the peptide agonist, substance P with NK1 receptors [52], and CCK-8 with CCKB receptors [55], where interaction with both EC and TM receptor domains are important for these peptide agonists’ selectivity. Our results demonstrating the importance of amino acids in UTM6 being important for Univ.Lig affinity are similar to results with interaction of the peptide agonist des-Arg9 bradykinin with the B1 bradykinin receptor, in which the ligand’s interaction with multiple amino acids in the UTM6 is required for high affinity interaction and selectivity [58]. Similarly, our results demonstrating the importance of EC4 amino acids for Univ.Lig affinity are similar to reported for high affinity interaction of bradykinin with the bradykinin B1 or B2 receptor [59]; CCK-8 with the CCKBR [55]; and CCK-9 related peptides with the CCKAR [60] (R336 in EC4). However, the characteristics of the important amino acids differ in these different receptors. Compared with the other two mammalian bombesin family receptors, NMBR and GRPR, our results are similar in that for selectivity of NMB or GRP, respectively, interaction is required with both EC and UTM receptor regions of the NMBR or GRPR, respectively [26,28,38,40,49]. In terms of the specific EC or UTM receptor domains important for ligand high affinity/selectivity, our results with the Univ.Lig showing amino acids particularly in EC2, EC4 and UTM6 are the most important, is in contrast to NMB selectivity/high affinity for NMBR, where amino acids in EC3 and EC4 [28] are the most important or with GRP selectivity/high affinity for GRPR, where amino acids in the EC3 region [49] and to a lesser degree in EC4, and EC2 of GRPR [26,49] are important. These results demonstrate that even with closely related natural ligands interacting with closely related receptors, the receptor regions determining high affinity/selectivities can vary markedly.
In our study of the 27 point mutations (19 in EC’s and 8 in UTM’s regions) that showed a significant decrease in Univ.Lig affinity, we found 21 point mutations resulted in a >1.6–4.9-fold decrease, two point mutations [T92A (UTM2), H300S (EC4)] resulted in a 5–9.9 fold decrease and four point mutations [D97N (EC2), G112V (EC2), Y284A (UTM6), R287N (EC4)] resulted in a >10-fold decrease in Univ.Lig affinity. Our results show both similarities and differences for the specific amino acids we identified important for high affinity of the Univ.Lig for all Bn receptors, compared to that reported for various peptide ligands of other Bn receptors or other peptide ligands with other gastrointestinal hormone/neurotransmitter receptors. Previous studies [28,38,40] demonstrate that the important amino acids for determining high affinity/selectivity for the natural occurring agonist, NMB for the NMBR, are R127(UTM3); I199, Q203, S205(EC3) and S315(UTM7) [equivalent to Q120(UTM3); A197, H201, Y199(EC3), R308(UTM7) in GRPR] and for the selective NMBR peptoid antagonist, PD168368, are the presence of Y220 in UTM5 [equivalent to F218 (UTM5) in mGRPR] [42]. In contrast, for Univ.Lig. only one of these amino acids, S315 in UTM7, occurring in a comparable position in hGRPR (R308, UTM7), contributed modestly (3-fold increase affinity) to its high affinity for hGRPR. Similarly, investigation of the molecular basis of GRP’s high affinity/selectivity for the GRPR demonstrated that the presence in the mGRPR of Q121 in the UTM3, F185 in EC3 and R288 in EC4 are particularly important (corresponding to Q120, R287, F185 in hGRPR), whereas K101(EC2), A198(EC3), P199(EC3), S293(EC4), T297(EC4) and A308(UTM7) have a less important, but significant effect [26,27,49] [corresponding to R100(EC2), A197, P198(EC3), S292, T296(EC4), A307(UTM7) in hGRPR]. Only one of these equivalent amino acids in hGRPR, R287 in EC4 was important for Univ.Lig high affinity. Similarly, with the highly selective GRPR peptide antagonists, JMV594 and JMV614 [39], only three amino acids in the mGRPR EC4 region (T297, F302, S305) [equivalent to T296, F301, S304 in the hGRPR] were important for GRPR selectivity/affinity, whereas in our study with Univ.Lig, we found only F301 in EC4 had a small effect on affinity (2-fold decrease). These results demonstrate that even though Univ.Lig has a high structural homology to GRP and NMB, sharing 6 of 9 amino acids in the COOH terminus, which is the biologically active portion of the peptide [1,61,62]; the determinants of Univ.Lig’s molecular basis of interacting with high affinity with mammalian Bn receptors differ markedly, from these receptor’s natural ligands or that of various selective peptide analogues for these two receptors.
Our results also demonstrate marked differences in the molecular determinants of high affinity of Univ.Lig for other Bn receptors and for the orphan Bn-related receptor, BRS-3. A recent study [63] analyzing only amino acids proposed to be inward facing into the proposed binding pocket of BRS-3, identified 5 amino acids which were important for determining high potency of Univ.Lig for activating hBRS which included L123 (UTM3); N287, Y291 (UTM6); T312 (EC4) and R316 (UTM7), [equivalent to I116, N280, Y284, S304, R308 in hGRPR][63]. Particularly important, affecting Univ.Lig potency by >400 fold in that study, were L123 (UTM6), Y291 (UTM6), R316 (UTM7), whereas N287 (UTM6) and T312 (EC4) had lesser important (i.e. 16–30-fold affinity). In contrast, in the present study with hGRPR, none of the equivalent amino acids in hGRPR were important for its high affinity for Univ.Lig affinity, except Y284 (UTM6) (which is equivalent to Y291 in hBRS3) which had a moderate effect on Univ.Lig affinity (60-fold decrease). Similarly, two other studies [25,51] identified amino acids important for Univ.Lig affinity for hBRS3 in EC2/adjacent UTMs (V101, H107, G112, R127 of hBRS-3) [equivalent to A94, R100, R105 and Q120 in hGRPR] and in EC4 (Y298, E299, S300, D306, V307, P308 of mBRS-3) [equivalent to Y291, S292, N/A, M298, L299, H300 in hGRPR], but none of these were important determinants in high affinity for Univ.Lig for other Bn receptors. These results demonstrate that even for a given ligand (i.e. Univ.Lig), the molecular determinants of its high affinity for closely related receptors (i.e., BRS-3, GRPR) differ markedly, not only in which particular amino acid is important, but also in its location in the different receptors.
Our results of the specific amino acids involved in determining high affinity of Univ.Lig with Bn receptors has similarities and differences compared to findings with other GI peptide/neurotransmitter receptors. In our study the presence of D97 and G112 in EC2; Y284 in UTM6; R287 and to a lesser extent, H300 in EC4 were the most important determinants of high affinity for Univ.Lig. An arginine in EC4 is important for high affinity for the peptide CCKAR agonist, CCK-9 and the peptide antagonist, JMV179 interaction with the CCKAR [60]. Similarly an arginine is important for high affinity of the mouse TRH receptor for the peptide agonist, TRH and the human ETA-R for the nonpeptide antagonist, bosentan [56]. A tyrosine residue in UTM6 is important for the peptide agonist TRH for mouse TRH receptor [64]; the m2 muscarinic receptor for agonists, acetylcholine, carbachol, oxotremorine M and pilocarpine [65]; and the AT1a angiotensin II-R(rat) for the agonist angiotensin II [66]. Whereas, in our study both glycine and aspartic acid in EC2, were important for Univ.Lig. no other studies of GI hormone/neurotransmitter receptors reported their importance for ligand affinity in this receptor region. However, an aspartic acid in EC4 of the rat B2 bradykinin receptor is needed for high affinity for bradykinin [59] and an aspartic acid in TM2 and TM3 of the and m1 muscarinic acetylcholine receptor for carbachol [67] is important for high affinity. A glycine in TM1 in the human ETA-R is important for high affinity for ET1 [56] and in UTM6 in the human Mel1a melatonin receptor is important for high affinity for melatonin [68]. Lastly, similar to our findings with hGRPR for high affinity Univ.Lig interaction, histidine important in a number of other G-protein coupled receptors for high affinity interaction with various peptide/nonpeptide agonists and/or antagonists. Specifically, histidine in EC3 of CCKBR is necessary for high affinity of CCK [55]; in TM3 of human CRF-R1 for nonpeptide CRF-R1 antagonist, NBI27914 affinity [69]; in TM5 and TM6, human NK1-R for nonpeptide antagonist, L-161, 664 affinity [70]; in TM6 of the D2L receptor for high affinity for 1,4-disubstituted phenylpiperazines [71] and in UTM7 area of NPY Y1-R for affinity for the peptide antagonist, BIBP3226 [72].
To understand the molecular basis for importance of each of these amino acids for high affinity Univ.Lig binding, a series of point mutations were made substituting amino acids with differing characteristics. In various GPCR-ligand studies, the presence of a tyrosine may be needed for high affinity/selectivity of various ligands because of its ability to interact by cation-π binding, π-π binding; and by participating in hydrogen bonding [60,64–66,70]. A tyrosine in the UTM3 domain of the thyrotropin-releasing hormone receptor binds the pyroglutamyl moiety of thyrotropin-releasing hormone through hydrogen bonding [42,64], and a tyrosine in the UTM6 domain of the m2 muscarinic receptor is a critical for the interaction with nonpeptide agonists (acetylcholine, carbachol, oxotremorine M, and pilocarpine) [73], likely by a hydrogen bonding mechanism. In our study, the substitution of Y284 by phenylalanine had little effect on Univ.Lig affinity, whereas the substitution of alanine, leucine or tryptophan caused a markedly decrease in affinity. Beside tyrosine, other aromatic amino acids, including phenylalanine, may interact with ligands through cation- π binding or π-π binding [74–77]. The cation- π binding or π-π binding usually occurs through the side chains of phenylalanine, tyrosine, or tryptophan [75]. Because in our study the substitution of phenylalanine for tyrosine position 284 in hGRPR resulted in almost no change in affinity for Univ.Lig, our result suggest that the interaction of the tyrosine hydroxyl with Univ.Lig is less likely mediated by hydrogen bonding, and is likely mediated through cation-π binding. However, tyrosine is predicted to have a higher cation-π binding potential than phenylalanine secondary to the negative electrostatic potential of the oxygen [76]. Therefore, whether the tyrosine hydroxyl is involved in increasing negative electrostatic potential due to cation-π binding site interaction or to hydrogen bonding cannot be completely resolved by our studies. Furthermore, in our studies because the substitution of tryptophan for tyrosine 284 markedly decreased Univ.Lig affinity, this suggests that steric factors at this position and proper side-chain orientation/placement could be important factors for determining Univ.Lig affinity at this position. With respect to the charged amino acids, D97 and R287 in EC2 of hGRPR and in EC4, our results are similar to that reported with the neuropeptide S receptor [54], where aspartic acid in EC2 is important for high affinity. These results suggest that high affinity binding of Univ.Lig to hGRPR may inquire a negative charge in this location. This conclusion is supported by our finding that substitution of D97E had only a small effect on Univ.Lig affinity, whereas substitution of alanine or asparagine caused a marked decrease in affinity and substitution of a positively charge amino acid, D97K resulted in no binding of Univ.Lig. Similarly our results with substitutions for R287 in EC4 support a positively charged moiety at this receptor location may play an important role in determining Univ.Lig high affinity. This conclusion is supported by the finding the substitution of a lysine for arginine resulted in a 10-fold decrease affinity for Univ.Lig, however substitution of the uncharged residue, asparagine caused a 70-fold decreasing affinity and either alanine or the negatively charged amino acid, aspartic acid resulted in no binding. Our results showing a marked decrease in affinity with the substitution of valine for glycine 112 suggest that steric factors could be particularly important in this position. This proposal is supported by the fact that G112 in EC2 is adjacent to C113 which can form a disulfide bond with C196 in EC3, and therefore insertion of larger residues at this site may introduce less flexibility here.
In conclusion, in the present study, we identified important amino acids for determining Univ.Lig binding affinity for the hGRPR receptor exist in EC and UTM regions; particularly the presence of Asp97 and Gly112 in EC2, Tyr284 in UTM6, Arg287, and His300 in EC4. Detailed substitutions at these locations demonstrate that in the EC area, particularly important is the presence of a positively charged amino acid in EC4 (positive 287) and negatively charged amino acid in EC2 (position 97) suggesting charge-charge interactions at these locations play an important for determining the high affinity of this ligand. On the other hand, in regard to the TM areas, our results show that the unique ability of the Univ.Lig to bind to all human BnR’s with high affinity, is partially dependent on interactions with transmembrane amino acids particularly located in TM6. These results coupled with the fact showing Univ.Lig.-receptor high affinity also requires interaction with a positively charged [Arg287] in EC4 at the top of TM6, demonstrates that Univ.Lig-high affinity interaction requires both charge-charge interaction as well as interaction with a tyrosine residue in close proximity suggesting possible receptor-peptide cation-π and possibly to a lesser extent, H–bonding interactions are also important for determining high affinity.
The results of the mutagenesis analysis to identify key amino acids responsible for the high affinity of Univ.Lig for the Bn family of receptors are supported by the results of our molecular modeling of the human GRP receptor which demonstrated that each of the seven amino acids identified as a major determinants of Univ.Lig affinity for this receptor (D97N, L121A, I283A, Y284A, R287N, H300S, V302I), are within 5Å of the proposed binding pocket and could therefore interact with Univ.Lig. Furthermore, in the molecular model the side-chains of most of these important amino acids are oriented inward toward the proposed binding pocket, which increases the probability they can interact with the ligand. The results from this study allow us to obtain a more detailed picture of the receptor-ligand interaction of a agonist whose ability to widely interact with Bn receptors and behave as a universal agonist was discovered only by chance [22,24,78]. The availability of this model could be of use for helping to identify additional synthetic ligands that could universally or perhaps selectively activate this family of receptors.
Acknowledgments
This work was partially supported by intramural funds of NIDDK, NIH.
Abbreviations
- β-Ala
β-alanine replacement
- BB1
neuromedin B receptor
- BB2
gastrin-releasing peptide receptor
- BB4
bombesin receptor subtype 4 (amphibian)
- Bn
bombesin
- BRS-3
bombesin receptor subtype 3
- BnR
member of bombesin receptor family
- BSA
bovine serum albumin fraction V
- CCKAR
cholecystokinin receptor type A
- CNS
central nervous system
- DMEM
Dulbecco’s minimum essential medium
- DTT
dithiothreitol
- EC
extracellular
- FBS
fetal bovine serum
- GI
gastrointestinal
- GRP
gastrin-releasing peptide
- GPCR
G-protein coupled receptor
- GRPR
gastrin-releasing peptide receptor
- IC50
peptide concentration causing half-maximal inhibition of radiolabeled ligand binding, a measure of receptor affinity
- Nle
norleucine
- NMB
neuromedin B
- NMBR
neuromedin B receptor
- PBS
phosphate-buffered saline
- TM
transmembrane region
- Univ.Lig
universal ligand for Bn receptors ([D-Tyr6, β-Ala11, Phe13, Nle14]Bn (6-14))
- UTM
upper transmembrane regions
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jensen RT, Battey JF, Spindel ER, Benya RV. International Union of Pharmacology. LVIII. Mammalian Bombesin Receptors: Nomenclature, distribution, pharmacology, signaling and functions in normal and disease states. Pharmacol Rev. 2008;60:1–42. doi: 10.1124/pr.107.07108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gonzalez N, Moody TW, Igarashi H, Ito T, Jensen RT. Bombesin-related peptides and their receptors: recent advances in their role in physiology and disease states. Curr Opin Endocrinol Diabetes Obes. 2008;15:58–64. doi: 10.1097/MED.0b013e3282f3709b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weber HC. Regulation and signaling of human bombesin receptors and their biological effects. Curr Opin Endocrinol Diabetes Obes. 2009;16:66–71. doi: 10.1097/med.0b013e32831cf5aa. [DOI] [PubMed] [Google Scholar]
- 4.Jensen JA, Carroll RE, Benya RV. The case for gastrin-releasing peptide acting as a morphogen when it and its receptor are aberrantly expressed in cancer. Peptides. 2001;22:689–99. doi: 10.1016/s0196-9781(01)00380-1. [DOI] [PubMed] [Google Scholar]
- 5.Fathi Z, Corjay MH, Shapira H, Wada E, Benya R, Jensen R, et al. BRS-3: novel bombesin receptor subtype selectively expressed in testis and lung carcinoma cells. J Biol Chem. 1993;268(8):5979–84. [PubMed] [Google Scholar]
- 6.Ohki-Hamazaki H, Watase K, Yamamoto K, Ogura H, Yamano M, Yamada K, et al. Mice lacking bombesin receptor subtype-3 develop metabolic defects and obesity. Nature. 1997;390(6656):165–9. doi: 10.1038/36568. [DOI] [PubMed] [Google Scholar]
- 7.Sano H, Feighner SD, Hreniuk DL, Iwaasa H, Sailer AW, Pan J, et al. Characterization of the bombesin-like peptide receptor family in primates. Genomics. 2004;84:139–46. doi: 10.1016/j.ygeno.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 8.Porcher C, Juhem A, Peinnequin A, Bonaz B. Bombesin receptor subtype-3 is expressed by the enteric nervous system and by interstitial cells of Cajal in the rat gastrointestinal tract. Cell Tissue Res. 2005;320:21–31. doi: 10.1007/s00441-004-1032-1. [DOI] [PubMed] [Google Scholar]
- 9.Guan XM, Chen H, Dobbelaar PH, Dong Y, Fong TM, Gagen K, et al. Regulation of Energy Homeostasis by Bombesin Receptor Subtype-3: Selective Receptor Agonists for the Treatment of Obesity. Cell Metab. 2010;11:101–12. doi: 10.1016/j.cmet.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 10.Majumdar ID, Weber HC. Biology and pharmacology of bombesin receptor subtype-3. Curr Opin Endocrinol Diabetes Obes. 2012;19:3–7. doi: 10.1097/MED.0b013e32834ec77d. [DOI] [PubMed] [Google Scholar]
- 11.Feng Y, Guan XM, Li J, Metzger JM, Zhu Y, Juhl K, et al. Bombesin Receptor Subtype-3 (BRS-3) Regulates Glucose-Stimulated Insulin Secretion in Pancreatic Islets across Multiple Species. Endocrinology. 2011;152:4106–15. doi: 10.1210/en.2011-1440. [DOI] [PubMed] [Google Scholar]
- 12.Guan XM, Metzger JM, Yang L, Raustad KA, Wang SP, Spann SK, et al. Antiobesity effect of MK-5046, a novel bombesin receptor subtype-3 agonist. J Pharmacol Exp Ther. 2011;336:356–64. doi: 10.1124/jpet.110.174763. [DOI] [PubMed] [Google Scholar]
- 13.Jensen RT, Moody TW. Bombesin-related peptides and neurotensin: effects on cancer growth/proliferation and cellular signaling in cancer. In: Kastin AJ, editor. Handbook of Biologically active peptides. Amsterdam: Elsevier; 2006. pp. 429–34. [Google Scholar]
- 14.Reubi JC, Wenger S, Schumuckli-Maurer J, Schaer JC, Gugger M. Bombesin receptor subtypes in human cancers: detection with the universal radoligand (125)I-[D-TYR(6), beta-ALA(11),PHE(13), NLE(14)] bombesin(6-14) Clin Cancer Res. 2002;8:1139–46. [PubMed] [Google Scholar]
- 15.Sancho V, Di Florio A, Moody TW, Jensen RT. Bombesin receptor-mediated imaging and cytotoxicity: review and current status. Curr Drug Deliv. 2011;8:79–134. doi: 10.2174/156720111793663624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moody TW, Mantey SA, Pradhan TK, Schumann M, Nakagawa T, Martinez A, et al. Development of high affinity camptothecin-bombesin conjugates that have targeted cytotoxicity for bombesin receptor-containing tumor cells. J Biol Chem. 2004;279:23580–9. doi: 10.1074/jbc.M401938200. [DOI] [PubMed] [Google Scholar]
- 17.Kwekkeboom DJ, Kam BL, Van Essen M, Teunissen JJ, van Eijck CH, Valkema R, et al. Somatostatin-receptor-based imaging and therapy of gastroenteropancreatic neuroendocrine tumors. Endocr Relat Cancer. 2010;17:R53–R73. doi: 10.1677/ERC-09-0078. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez N, Mantey SA, Pradhan TK, Sancho V, Moody TW, Coy DH, Jensen RT. Characterization of putative GRP- and NMB-receptor antagonist’s interaction with human receptors. Peptides. 2009;30:1473–86. doi: 10.1016/j.peptides.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Schrenck T, Wang LH, Coy DH, Villanueva ML, Mantey S, Jensen RT. Potent bombesin receptor antagonists distinguish receptor subtypes. Am J Physiol. 1990;259:G468–G473. doi: 10.1152/ajpgi.1990.259.3.G468. [DOI] [PubMed] [Google Scholar]
- 20.Uehara H, Gonzalez N, Sancho V, Mantey SA, Nuche-Berenguer B, Pradhan T, et al. Pharmacology and selectivity of various natural and synthetic bombesin related peptide agonists for human and rat bombesin receptors differs. Peptides. 2011;32:1685–99. doi: 10.1016/j.peptides.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang LH, Coy DH, Taylor JE, Jiang NY, Moreau JP, Huang SC, et al. Des-Met carboxyl-terminally modified analogues of bombesin function as potent bombesin receptor antagonists, partial agonists, or agonists. J Biol Chem. 1990;265(26):15695–703. [PubMed] [Google Scholar]
- 22.Mantey SA, Weber HC, Sainz E, Akeson M, Ryan RR, Pradhan TK, et al. Discovery of a high affinity radioligand for the human orphan receptor, bombesin receptor subtype 3, which demonstrates it has a unique pharmacology compared to other mammalian bombesin receptors. J Biol Chem. 1997;272(41):26062–71. doi: 10.1074/jbc.272.41.26062. [DOI] [PubMed] [Google Scholar]
- 23.Mantey SA, Coy DH, Pradhan TK, Igarashi H, Rizo IM, Shen L, et al. Rational design of a peptide agonist that interacts selectively with the orphan receptor, bombesin receptor subtype 3. J Biol Chem. 2001;276:9219–29. doi: 10.1074/jbc.M008737200. [DOI] [PubMed] [Google Scholar]
- 24.Pradhan TK, Katsuno T, Taylor JE, Kim SH, Ryan RR, Mantey SA, et al. Identification of a unique ligand which has high affinity for all four bombesin receptor subtypes. Eur J Pharmacol. 1998;343:275–87. doi: 10.1016/s0014-2999(97)01527-6. [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez N, Hocart SJ, Portal-Nunez S, Mantey SA, Nakagawa T, Zudaire E, et al. Molecular basis for agonist selectivity and activation of the orphan bombesin receptor subtype 3 receptor. J Pharmacol Exp Ther. 2008;324:463–74. doi: 10.1124/jpet.107.132332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakagawa T, Hocart SJ, Schumann M, Tapia JA, Mantey SA, Coy DH, et al. Identification of key amino acids in the gastrin-releasing peptide receptor (GRPR) responsible for high affinity binding of gastrin-releasing peptide (GRP) Biochem Pharmacol. 2005;69:579–93. doi: 10.1016/j.bcp.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 27.Akeson M, Sainz E, Mantey SA, Jensen RT, Battey JF. Identification of four amino acids in the gastrin-releasing peptide C receptor that are required for high affinity agonist binding. J Biol Chem. 1997;272:17405–9. doi: 10.1074/jbc.272.28.17405. [DOI] [PubMed] [Google Scholar]
- 28.Sainz E, Akeson M, Mantey SA, Jensen RT, Battey JF. Four amino acid residues are critical for high affinity binding of neuromedin B to the neuromedin B receptor. J Biol Chem. 1998;273:15927–32. doi: 10.1074/jbc.273.26.15927. [DOI] [PubMed] [Google Scholar]
- 29.Meng F, Taylor LP, Hoversten MT, Ueda Y, Ardati A, Reinscheid RK, et al. Moving from the orphanin FQ receptor to an opioid receptor using four point mutations. J Biol Chem. 1996;271:32016–20. doi: 10.1074/jbc.271.50.32016. [DOI] [PubMed] [Google Scholar]
- 30.Benya RV, Kusui T, Pradhan TK, Battey JF, Jensen RT. Expression and characterization of cloned human bombesin receptors. Mol Pharmacol. 1995;47:10–20. [PubMed] [Google Scholar]
- 31.Fathi Z, Way JW, Corjay MH, Viallet J, Sausville EA, Battey JF. Bombesin receptor structure and expression in human lung carcinoma cell lines. J Cell Biochem Suppl. 1996;24:237–46. doi: 10.1002/jcb.240630519. [DOI] [PubMed] [Google Scholar]
- 32.Benya RV, Fathi Z, Pradhan T, Battey JF, Kusui T, Jensen RT. Gastrin-releasing peptide receptor-induced internalization, down-regulation, desensitization and growth: Possible role of cAMP. Mol Pharmacol. 1994;46(2):235–45. [PubMed] [Google Scholar]
- 33.Benya RV, Wada E, Battey JF, Fathi Z, Wang LH, Mantey SA, et al. Neuromedin B receptors retain functional expression when transfected into BALB 3T3 fibroblasts: analysis of binding, kinetics, stoichiometry, modulation by guanine nucleotide-binding proteins, and signal transduction and comparison with natively expressed receptors. Mol Pharmacol. 1992;42(6):1058–68. [PubMed] [Google Scholar]
- 34.Katsuno T, Pradhan TK, Ryan RR, Mantey SA, Hou W, Donohue PJ, et al. Pharmacology and cell biology of the bombesin receptor subtype 4 (BB4-R) Biochemistry (Mosc) 1999;38:7307–20. doi: 10.1021/bi990204w. [DOI] [PubMed] [Google Scholar]
- 35.Ryan RR, Katsuno T, Mantey SA, Pradhan TP, Weber HC, Battey JF, Jensen RT. Comparative pharmacology of a nonpeptoid neuromedin B antagonist PD 168368. J Pharmacol Exp Ther. 1999;290:1202–11. [PubMed] [Google Scholar]
- 36.Jensen RT, Coy DH. Progress in the development of potent bombesin receptor antagonists. Trends Pharmacol Sci. 1991;12(1):13–9. doi: 10.1016/0165-6147(91)90483-9. [DOI] [PubMed] [Google Scholar]
- 37.Sancho V, Moody TW, Mantey SA, Di Florio A, Uehara H, Coy DH, Jensen RT. Pharmacology of putative selective hBRS-3 receptor agonists for human bombesin receptors (BnR): Affinities, potencies and selectivity in multiple native and BnR transfected cells. Peptides. 2010;31:1569–78. doi: 10.1016/j.peptides.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fathi Z, Benya RV, Shapira H, Jensen RT, Battey JF. The fifth transmembrane segment of the neuromedin B receptor is critical for high affinity neuromedin B binding. J Biol Chem. 1993;268(#20):14622–6. [PubMed] [Google Scholar]
- 39.Tokita K, Katsuno T, Hocart SJ, Coy DH, Llinares M, Martinez J, Jensen RT. Molecular basis for selectivity of high affinity peptide antagonists for the gastrin-releasing peptide receptor. J Biol Chem. 2001;276:36652–63. doi: 10.1074/jbc.M104566200. [DOI] [PubMed] [Google Scholar]
- 40.Gonzalez N, Nakagawa T, Mantey SA, Sancho V, Uehara H, Katsuno T, Jensen RT. Molecular basis for the selectivity of the mammalian bombesin peptide, neuromedin B, for its receptor. J Pharmacol Exp Ther. 2009;331:265–76. doi: 10.1124/jpet.109.154245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benya RV, Fathi Z, Battey JF, Jensen RT. Serines and threonines in the gastrin-releasing peptide receptor carboxyl terminus mediate internalization. J Biol Chem. 1993;268:20285–90. [PubMed] [Google Scholar]
- 42.Tokita K, Hocart SJ, Katsuno T, Mantey SA, Coy DH, Jensen RT. Tyrosine 220 in the fifth transmembrane domain of the neuromedin B receptor is critical for the high selectivity of the peptoid antagonist PD168368. J Biol Chem. 2001;276:495–504. doi: 10.1074/jbc.M006059200. [DOI] [PubMed] [Google Scholar]
- 43.Tsuda T, Kusui T, Hou W, Benya RV, Akeson MA, Kroog GS, et al. Effect of gastrin-releasing peptide receptor number on receptor affinity, coupling, degradation and receptor modulation. Mol Pharmacol. 1997;51(5):721–32. doi: 10.1124/mol.51.5.721. [DOI] [PubMed] [Google Scholar]
- 44.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-Pdb Viewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–23. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 45.Warne T, Moukhametzianov R, Baker JG, Nehme R, Edwards PC, Leslie AG, et al. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature. 2011;469:241–4. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagalla SR, Barry BJ, Creswick KC, Eden P, Taylor JT, Spindel ER. Cloning of a receptor for amphibian [Phe13]bombesin distinct from the receptor for gastrin-releasing peptide: Identification of a fourth bombesin receptor subtype (BB4) Proc Natl Acad Sci U S A. 1995;92:6205–9. doi: 10.1073/pnas.92.13.6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wada E, Way J, Shapira H, Kusano K, Lebacq-Verheyden AM, Coy D, et al. cDNA cloning, characterization, and brain region-specific expression of a neuromedin B-preferring bombesin receptor. Neuron. 1991;6 (#3):421–30. doi: 10.1016/0896-6273(91)90250-4. [DOI] [PubMed] [Google Scholar]
- 48.Ryan RR, Weber HC, Hou W, Sainz E, Mantey SA, Battey JF, et al. Ability of various bombesin receptor agonists and antagonists to alter intracellular signaling of the human orphan receptor BRS-3. J Biol Chem. 1998;273:13613–24. doi: 10.1074/jbc.273.22.13613. [DOI] [PubMed] [Google Scholar]
- 49.Tokita K, Hocart SJ, Coy DH, Jensen RT. Molecular basis of the selectivity of the gastrin-releasing peptide receptor for gastrin-releasing peptide. Mol Pharmacol. 2002;61:1435–43. doi: 10.1124/mol.61.6.1435. [DOI] [PubMed] [Google Scholar]
- 50.Tseng MJ, Coon S, Stuenkel E, Struk V, Logsdon CD. Influence of second and third cytoplasmic loops on binding, internalization, and coupling of chimeric bombesin/m3 muscarinic receptors. J Biol Chem. 1995;270:17884–91. doi: 10.1074/jbc.270.30.17884. [DOI] [PubMed] [Google Scholar]
- 51.Liu J, Lao ZJ, Zhang J, Schaeffer MT, Jiang MM, Guan XM, et al. Molecular basis of the pharmacological difference between rat and human bombesin receptor subtype-3 (BRS-3) Biochemistry (Mosc) 2002;41:8954–60. doi: 10.1021/bi0202777. [DOI] [PubMed] [Google Scholar]
- 52.Li H, Hsu P, Sachais BS, Krause JE, Leeman SE, Boyd ND. Identification of the site in the substance P (NK-1) receptor for modulation of peptide binding by sulfhydryl reagents. J Biol Chem. 1996;271:1950–6. doi: 10.1074/jbc.271.4.1950. [DOI] [PubMed] [Google Scholar]
- 53.Fong TM, Yu H, Strader CD. Molecular basis for the species selectivity of the neurokinin-1 receptor antagonist CP-96, 345 and RP67580. J Biol Chem. 1992;267:25668–71. [PubMed] [Google Scholar]
- 54.Clark SD, Tran HT, Zeng J, Reinscheid RK. Importance of extracellular loop one of the neuropeptide S receptor for biogenesis and function. Peptides. 2010;31:130–8. doi: 10.1016/j.peptides.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Silvente-Poirot S, Escrieut C, Wank SA. Role of the extracellular domains of the cholecystokinin receptor in agonist binding. Mol Pharmacol. 1998;54:364–71. doi: 10.1124/mol.54.2.364. [DOI] [PubMed] [Google Scholar]
- 56.Breu V, Hashido K, Broger C, Miyamoto C, Furuichi Y, Hayes A, et al. Separable binding sites for the natural agonist endothelin-1 and the non-peptide antagonist bosentan on human endothelin-A receptors. Eur J Biochem. 1995;231:266–70. [PubMed] [Google Scholar]
- 57.Krystek SR, Jr, Patel PS, Rose PM, Fisher SM, Kienzle BK, Lach DA, et al. Mutation of peptide binding site in transmembrane region of a G protein-coupled receptor accounts for endothelin receptor subtype selectivity. J Biol Chem. 1994;269:12383–6. [PubMed] [Google Scholar]
- 58.Ha SN, Hey PJ, Ransom RW, Bock MG, Su DS, Murphy KL, et al. Identification of the critical residues of bradykinin receptor B1 for interaction with the kinins guided by site-directed mutagenesis and molecular modeling. Biochemistry (Mosc) 2006;45:14355–61. doi: 10.1021/bi060673f. [DOI] [PubMed] [Google Scholar]
- 59.Novotny EA, Bednar DL, Connolly MA, Connor JR, Stormann TM. Mutation of aspartate residues in the third extracellular loop of the rat B2 bradykinin receptor decreases affinity for bradykinin. Biochem Biophys Res Commun. 1994;201:523–30. doi: 10.1006/bbrc.1994.1733. [DOI] [PubMed] [Google Scholar]
- 60.Gigoux V, Escrieut C, Fehrentz JA, Poirot S, Maigret B, Moroder L, et al. Arginine 336 and asparagine 333 of the human cholecystokinin-A receptor binding site interact with the penultimate aspartic acid and the C-terminal amide of cholecystokinin. J Biol Chem. 1999;274:20457–64. doi: 10.1074/jbc.274.29.20457. [DOI] [PubMed] [Google Scholar]
- 61.Broccardo M, Erspamer GF, Melchiorri P, Negri L, DeCastiglione R. Relative potency of bombesin-like peptides. Br J Pharmacol. 1976;55:221–7. doi: 10.1111/j.1476-5381.1975.tb07631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin JT, Coy DH, Mantey SA, Jensen RT. Comparison of the peptide structural requirements for high affinity interaction with bombesin receptors. Eur J Pharmacol. 1996;294:55–69. doi: 10.1016/0014-2999(95)00510-2. [DOI] [PubMed] [Google Scholar]
- 63.Gbahou F, Holst B, Schwartz TW. Molecular basis for agonism in the BB3 receptor: an epitope located on the interface of transmembrane-III, -VI, and -VII. J Pharmacol Exp Ther. 2010;333:51–9. doi: 10.1124/jpet.109.162131. [DOI] [PubMed] [Google Scholar]
- 64.Perlman JH, Thaw CN, Laakkonen L, Bowers CY, Osman R, Gershengorn MC. Hydrogen bonding interaction of thyrotropin-releasing hormone (TRH) with transmembrane tyrosine 106 of the TRH receptor. J Biol Chem. 1994;269:1610–3. [PubMed] [Google Scholar]
- 65.Funk CD, Furci L, Moran N, Fitzgerald GA. Point mutation in the seventh hydrophobic domain of the human thromboxane A2 receptor allows discrimination between agonist and antagonist binding sites. Mol Pharmacol. 1993;44:934–9. [PubMed] [Google Scholar]
- 66.Hunyady L, Bor M, Balla T, Catt KJ. Critical role of a conserved intramembrane tyrosine residue in angiotensin II receptor activation. J Biol Chem. 1995;270:9702–5. doi: 10.1074/jbc.270.17.9702. [DOI] [PubMed] [Google Scholar]
- 67.Fraser CM, Wang CD, Robinson DA, Gocayne JD, Venter JC. Site-directed mutagenesis of m1 muscarinic acetylcholine receptors: conserved aspartic acids play important roles in receptor function. Mol Pharmacol. 1989;36:840–7. [PubMed] [Google Scholar]
- 68.Gubitz AK, Reppert SM. Chimeric and point-mutated receptors reveal that a single glycine residue in transmembrane domain 6 is critical for high affinity melatonin binding. Endocrinology. 2000;141:1236–44. doi: 10.1210/endo.141.3.7356. [DOI] [PubMed] [Google Scholar]
- 69.Liaw CW, Grigoriadis DE, Lorang MT, De Souza EB, Maki RA. Localization of agonist- and antagonist-binding domains of human corticotropin-releasing factor receptors. Mol Endocrinol. 1997;11:2048–53. doi: 10.1210/mend.11.13.0034. [DOI] [PubMed] [Google Scholar]
- 70.Cascieri MA, Shiao LL, Mills SG, MacCoss M, Swain CJ, Yu H, et al. Characterization of the interaction of diacylpiperazine antagonists with the human neurokinin-1 receptor: identification of a common binding site for structurally dissimilar antagonists. Mol Pharmacol. 1995;47:660–5. [PubMed] [Google Scholar]
- 71.Ehrlich K, Gotz A, Bollinger S, Tschammer N, Bettinetti L, Harterich S, et al. Dopamine D2, D3, and D4 selective phenylpiperazines as molecular probes to explore the origins of subtype specific receptor binding. J Med Chem. 2009;52:4923–35. doi: 10.1021/jm900690y. [DOI] [PubMed] [Google Scholar]
- 72.Du P, Salon JA, Tamm JA, Hou C, Cui W, Walker MW, et al. Modeling the G-protein-coupled neuropeptide Y Y1 receptor agonist and antagonist binding sites. Protein Eng. 1997;10:109–17. doi: 10.1093/protein/10.2.109. [DOI] [PubMed] [Google Scholar]
- 73.Vogel WK, Sheehan DM, Schimerlik MI. Site-directed mutagenesis on the m2 muscarinic acetylcholine receptor: the significance of Tyr403 in the binding of agonists and functional coupling. Mol Pharmacol. 1997;52:1087–94. doi: 10.1124/mol.52.6.1087. [DOI] [PubMed] [Google Scholar]
- 74.Serrano L, Bycroft M, Fersht AR. Aromatic-aromatic interactions and protein stability. Investigation by double-mutant cycles. J Mol Biol. 1991;218:465–75. doi: 10.1016/0022-2836(91)90725-l. [DOI] [PubMed] [Google Scholar]
- 75.Dougherty DA. Cation-pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science. 1996;271:163–8. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- 76.Mecozzi S, West AP, Jr, Dougherty DA. Cation-pi interactions in aromatics of biological and medicinal interest: electrostatic potential surfaces as a useful qualitative guide. Proc Natl Acad Sci U S A. 1996;93:10566–71. doi: 10.1073/pnas.93.20.10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gallivan JP, Dougherty DA. Cation-pi interactions in structural biology. Proc Natl Acad Sci U S A. 1999;96:9459–64. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mantey SA, Gonzalez N, Schumann M, Pradhan TK, Shen L, Coy DH, Jensen RT. Identification of bombesin receptor subtype-specific ligands: effect of N-methyl scanning, truncation, substitution, and evaluation of putative reported selective ligands. J Pharmacol Exp Ther. 2006;319:980–9. doi: 10.1124/jpet.106.107011. [DOI] [PubMed] [Google Scholar]