

Summary
Heat shock response (HSR) that protects cells from proteotoxic stresses is downregulated in aging, as well as upon replicative senescence of cells in culture. Here we demonstrate that HSR is suppressed in fibroblasts from the patients with segmental progerioid Werner Syndrome, which undergo premature senescence. Similar suppression of HSR was seen in normal fibroblasts, which underwent senescence in response to DNA damaging treatments. The major DNA-damage-induced signaling (DDS) pathways p53–p21 and p38-NF-kB-SASP contributed to the HSR suppression. The HSR suppression was associated with inhibition of both activity and transcription of the heat shock transcription factor Hsf1. This inhibition in large part resulted from the downregulation of SIRT1, which in turn was because of decrease in the expression of the translation regulator HuR. Importantly, we uncovered a positive feedback regulation, where suppression of Hsf1 further activates the p38–NF-κB-SASP pathway, which in turn promotes senescence. Overexpression of Hsf1 inhibited the p38–NFκB-SASP pathway and partially relieved senescence. Therefore, downregulation of Hsf1 plays an important role in the development or in the maintenance of DNA damage signaling-induced cell senescence.
Keywords: heat shock response, Hsp70, HuR, inflammation, p38, p53, SIRT1
Introduction
Aging is the most important risk factor in the development of neurodegenerative and cardiovascular diseases, diabetes, osteoporosis, and many types of cancer. Various age-associated disorders are characterized by accumulation of damaged proteins (Jana et al., 2000; Koyama et al., 2006; Rahman et al., 2010; Sakellariou et al., 2011). Accordingly, the reduced capacity to handle misfolded protein has been implicated in the etiology of the disease process (Sherman & Goldberg, 2001). Protein aggregation and accumulation of damaged species in aging reflect the collapse in protein homeostasis (proteostasis; Ben-Zvi et al., 2009; Taylor & Dillin, 2011), which in part results from the reduced ability to induce heat shock proteins that function as molecular chaperones in protein folding and degradation (Wu et al., 1993; Heydari et al., 1994). In turn, malfunction of the heat shock response (HSR) can shorten lifespan of the organism (Hsu et al., 2003). In fact, in Caenorhabditis elegans model, suppression of HSR by downregulation of the heat shock transcription factor Hsf1 shortens the lifespan, while overexpression of Hsf1 extends it (Morley & Morimoto, 2004; Ben-Zvi et al., 2009). As Hsf1 has been implicated both in aging and in disease, understanding its regulation as well as the effects of Hsf1 on aging and disease is of primary importance.
A NAD+-dependent deacetylase SIRT1 has been implicated in control of Hsf1. SIRT1 was found to deacetylate Hsf1 and keep it associated with the promoter region of heat shock proteins, thus enhancing the overall transcription from the Hsf1-responsive promoters (Westerheide et al., 2009). In line with this finding, caloric restriction, which delays aging by increasing SIRT1 activity, increased HSR in aging rats (Heydari et al., 1993). This finding provides a link between the organism aging and downregulation of HSR.
Certain features of the aging paradigm can be recapitulated in cell culture, where cells undergo replicative senescence. For example, downregulation of HSR was demonstrated in fibroblasts that underwent senescence following multiple passages in culture (Lee et al., 1996). Interestingly, SIRT1 levels decrease in replicatively senescent cells (Sasaki et al., 2006), suggesting that this factor may be involved in downregulation of HSR in senescence. Among various factors that regulate SIRT1, HuR binds to SIRT1 mRNA and increases its stability, and ultimately its translation (Fan & Steitz, 1998; Abdelmohsen et al., 2007). Levels of HuR also diminish in senescent cells, suggesting that HuR may mediate the effects of senescence on SIRT1, and consequently on Hsf1.
Cell senescence results from the activation of DNA damage signaling (DDS) (Vaziri et al., 1997; Robles & Adami, 1998; Chang et al., 1999). Among DDS pathways, the p53–p21 pathways trigger the senescence, while the so-called senescence-associated secretion phenotype (SASP) is responsible for senescence maintenance (Rodier et al., 2009; Freund et al., 2011). SASP represents secretion of a variety of signaling molecules, for example, IL-6, IL-1 or IL-8, controlled by the activation of p38MAPK and NF-κB (Acosta et al., 2008; Orjalo et al., 2009). In addition, p38MAPK regulates a distinct senescence factor p16 (Du et al., 2009).
As a model of aging, we chose fibroblasts from patients with Werner Syndrome (WS). WS is caused by a mutation in the WRN gene that presents with premature aging (Goto, 1997). Fibroblasts from patients with WS demonstrate premature senescence and show accumulation of DNA damage (Wyllie et al., 2000).
Here, we investigated the role of DNA damage in the suppression of HSR. Specifically, we probed the main DDS pathways as potential modulators of HSR. We found that Hsf1 activity decreases in DDS-induced senescence in part because of the changes in SIRT1 and HuR. Furthermore, we found a new role of Hsf1 as a modulator of senescence phenotype (Experimental procedures are given in Supporting information).
Results
DNA-damage-induced senescence is associated with suppression of the HSR
To understand how HSR is modulated in aging, we investigated HSR in skin fibroblasts isolated from prematurely aging patients with WS. We used fibroblasts isolated from three patients with WS aged between 19 and 30 at passages 4 to 11 and fibroblasts from normal individuals as age-matched controls, ages between 22 and 27 at passage 14 to 16. Previously, WS fibroblasts were found to undergo premature senescence (Goto, 1997). We observed the typical senescent morphology, including enlargement, flattening, and vacuolization, in WS fibroblasts, which was of a stark contrast to fibroblasts from an age-matched subject (Fig. S1). To assess the HSR in WS, cells were incubated at 43 °C for 35 min, left to recover for 6 h, and expression of the major inducible heat shock protein Hsp70 was measured by immunoblotting. These conditions for HSR were used throughout the study. Compared to fibroblasts from age-matched healthy subjects, WS fibroblasts demonstrated strong decrease in the induction of Hsp70 (Fig. 1A). Unlike finite lifespan WS fibroblasts, hTERT immortalized WS cells did not show suppressed HSR compared with normal hTERT-immortalized fibroblasts (Fig. S2). Therefore, we suggested that downregulation of the HSR in WS fibroblasts could be associated with premature senescence.
Fig. 1.

Activation of p53 pathway caused suppression of HSR. (A) WS fibroblasts (passage 4) and age-matched C fibroblasts (passage 15) were heat shocked and were collected for immunoblotting. (B) TIG-1 fibroblasts were treated with 100 nM of Dox overnight or 10 Gy IR. After 6 days, cells were heat shocked and incubated for 6 h before collection. (C) Same cells from (B) were immunoblotted for indicated proteins. (D) Cells that were induced senescent by 5 days of 10 μM nutlin-3 treatment were heat shocked, and Hsp70 levels were measured. (E) Hsp70 accumulation after heat shock in cells that overexpress p21. (F) TIG-1 cells were infected with empty or shp53 retroviruses, and nutlin-3 was added for additional 5 days. Cells were collected 6 h after heat shock and immunoblotted for Hsp70. Interestingly, we observed an increase in basal level and after heat shock level of Hsp70 upon p53 depletion. (G) Control or p53-depleted cells were treated with 6 Gy IR and cultured for 5 days. Cells were collected 6 h after heat shock and immunoblotted for Hsp70. Abbreviations: C, age-matched healthy control; CT, control.
The premature aging phenotype of WS, which is caused by the mutation in the DNA helicase WRN gene, is associated with DNA instability (Rossi et al., 2010). Diminished induction of Hsp70 in WS fibroblasts could be connected to the DNA damage. Accordingly, we tested whether DNA damaging treatments in normal cells could have a similar suppressive effect on the HSR. Normal human diploid fibroblasts TIG-1 in early passage (between p12 and p15) were treated with 100 nM doxorubicin (Dox) overnight or exposed to 10 Gy γ–irradiation (IR) and then cultured for 5 days. By day 4, such treatments led to withdrawal from the cell cycle, as indicated by reduced Ki-67 staining, and increased senescence-associated β-galactosidase activity (SA-β-gal). These features corresponded to the appearance of senescence phenotype, well in agreement with previous reports (Chang et al., 1999; Fig. S2A,B). As observed with WS fibroblasts, normal fibroblasts exposed to DNA damaging agents showed significantly suppressed induction of Hsp70 (Fig. 1B). Therefore, similar to replicative senescence, premature senescence by DNA damage causes suppression of the HSR in both normal and disease, and establishes a useful model to investigate the relationship between senescence and the HSR (Fig. 6I).
Fig. 6.

Changes in Hsf1 levels modulate senescence phenotype. Retroviral control or shRNA for Hsf1 and/or p53 were expressed in early passage TIG-1 fibroblasts and selected with puromycin. The 10 μM nutlin-3 was added for 5 days. (A) Cells were fixed and stained with SA-β-gal. The means and ±SD are from two independent experiments with 150 cell counts from four different fields. Cells were counted using images taken from bright phase microscope. (B) p53 and p21 were measured in Hsf1- depleted and nutlin-3-treated cells. (C) Control and Hsf1-depleted cells were treated with SB. The levels of p38MAPK and Hsp27 phosphorylation were measured by immunoblotting. (D) Effects of Hsf1 depletion on NF-κB. Cells were first infected with lentiviral NFκB luciferase reporter and then with retroviral empty vector or shRNA against Hsf1. First lane indicates basal luciferase activity reading without retroviral infection. (E) Control or Hsf1-depleted cells were treated with 10 μM nutlin-3, SB or vehicle for additional 5 days and IL-6 mRNA was measured. The mean and ±SEM were of three independent experiments. (F) Control of Hsf1-overexpressing cells were treated with 10 Gy IR and cultured for 6 days, and immunoblotted for indicated proteins. (G) IL-6 mRNA was measured in the same set of cells as in (F). The mean and ±SEM were from triplicates of two independent experiments. (H) Control of Hsf1-overexpressing cells was treated with 8 Gy IR and cultured for 6 days, then fixed and stained with SA-β-gal. The means and ±SD are from two independent experiments with 150 cell counts from four different fields. Cells were counted using images taken from bright phase microscope. (I) A model of relations between Hsf1 and senescence pathways. Radiation or DNA damage in WS causes activation of p53 and p38MAPK pathways. p53 induces p21 which activates senescence. Activation of p38MAPK stimulates NFκB pathway, which maintains senescence by increased production of SASP cytokines. Established senescence causes decrease in HuR, SIRT1, and Hsf1. Downregulation of Hsf1 in a feedback loop inhibits p38MAPK, which further promotes senescence and reduces levels of HuR and SIRT1.
DNA-damage-induced signaling (DDS) pathways regulate HSR
DNA damage can activate the p53 and the p38MAPK signaling pathways, both of which contribute to the development of senescence (Rodier et al., 2009; Freund et al., 2011). Here, we tested the contribution of each pathway in the suppression of HSR. By day 5 post-treatment, γ-irradiated TIG-1 cells showed strong activation of p53 pathway (Fig. 1C), as well as activation of p38MAPK pathway (Fig. 2A).
Fig. 2.
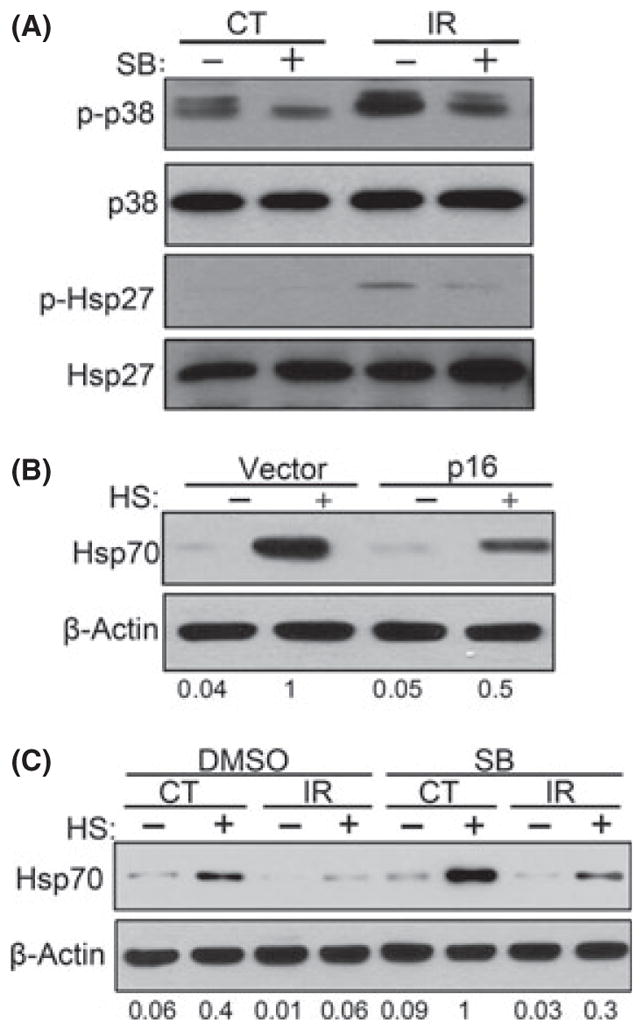
Activation of p38MAPK in DNA-damage-induced senescence contributes to suppression of HSR. (A) Early passage cells were pre-treated with p38MAPK inhibitor SB for 6 h prior to 10 Gy IR. After IR, cells were incubated for 6 days before collection. Levels of p–p38 and p-Hsp27 were measured by immunoblotting. (B) Hsp70 was measured after heat shock in cells with overexpression of p16. (C) Same cells as from (A) were heat shocked and incubated for 6 h before collection. Addition of SB led to partial restoration of Hsp70 induction level after heat shock.
To further characterize the role of the p53 signaling pathway in the suppression of HSR, we up regulated p53 without DNA damage by treating the cells with 10 μM nutlin-3, a compound that stabilizes and activates p53 (Vassilev, 2004). By day 4, early passage cells exhibited increased p53 and p21 levels, and increased SA-β-gal staining, indicating induction of the senescent phenotype (Fig. S4A,B). Induction of Hsp70 by heat shock was strongly reduced in cells treated with nutlin-3 compared to control cells (Fig. 1D), indicating that upregulation of p53 and downstream activation of the DDS even without DNA damage are sufficient for the HSR suppression.
Then we sought to determine the effects of upregulating p21, a downstream target of p53, and an important regulator of cell senescence. The p21 expression by retroviral infection in early passage cells led to the development of the senescence phenotype, which appeared 5–6 days after retroviral infection (Fig. S4A,B). As in the experiment with nutlin-3, expression of recombinant p21 caused significant decrease in induction of Hsp70 (Fig. 1E). All together, these results demonstrated that prolonged activation of the p53–p21 signaling is sufficient to suppress the HSR, (see the scheme on Fig. 6I).
To test the possibility that inhibition of cell cycle suppresses HSR, we tested HSR in cells that have been growth arrested at G1 without activating DDS pathway, and without senescence. Accordingly, early passage TIG-1 was treated with reversible inhibitors of cyclin D/CDK4 and 6 complexes, 1 μM of CDK4 inhibitor (2-Bromo-12,13-dihydro-5H-indolo[ 2,3-a]pyrrolo[3,4-c]carbazole-5,7(6H)-dione) and 10 μM of CDK4/6 inhibitor IV (trans-4-((6-(ethylamino)-2-((1-(phenylmethyl)-1H-indol-5-yl)amino)-4-pyrimidinyl)amino)-cyclohexanol). After 3 days, treatments with either inhibitor led to G1 arrest (as judged by the G1 reporter (Sakaue-Sawano et al., 2008) (Fig. S5). Similar cell cycle arrest was seen in cells overexpressing p21 or treated with nutlin-3 (Fig. S5). However, unlike the latter, cells treated with CDK inhibitors did not demonstrate SA-β-gal staining, indicating that they are not senescent (Fig. S6). Importantly, these growth-arrested but not senescent cells showed normal HSR (Fig. S7)
To examine whether the p53–p21 signaling was necessary for suppression of HSR by DDS, we depleted either of these proteins by retroviral shRNA expression. First, p53-depleted TIG-1 cells were treated with nutlin-3, as described earlier, and HSR was assessed. p53 depletion prevented p21 accumulation and the SA-β-Gal staining (Fig. S4A,C) and prevented suppression of HSR following exposure to nutlin-3 (Fig. 1F). Therefore, the inhibitory effect of nutlin-3 on HSR requires p53. More importantly, we observed reversion of HSR suppression in cells that were exposed to IR (Fig. 1G). These data demonstrated that p53 is an important factor in suppression of HSR in DNA-damage-induced senescent cells. Interestingly, p53-depleted fibroblasts showed both higher basal levels of Hsp70 and enhanced HSR compared to control empty-vector-infected cells. In contrast, depletion of p21 did not reverse the effect of nutlin-3 on HSR (Fig. S8), while, as shown earlier, expression of recombinant p21 had effectively suppressed HSR. This apparent contradiction indicated that p53 could affect HSR both via p21-dependent and p21-independent mechanisms. The p21-independent effect of p53 on HSR could result from the crosstalk between the p53 and p38MAPK pathways. In fact, we observed that nutlin-3 treatment alone caused activation of p38MAPK pathway (Fig. S9).
As such, we further investigated the contribution of the p38MAPK pathway in the suppression of HSR. In addition to the p53 pathway, IR of TIG-1 cells activated the p38MAPK signaling pathway, which plays a major role in maintenance of senescent phenotype (Rodier et al., 2009). By day 5 following exposure to 10 Gy IR, we observed increased phosphorylation of p38MAPK and phosphorylation of its substrate Hsp27 (Fig. 2A). Activation of p38MAPK led to the activation of its downstream target NFκB, as measured by the NFκB luciferase reporter (Fig. S10A), which in turn caused a dramatic rise (8- to 10-fold) in mRNA levels of IL-6 and IL-8 cytokines (Fig. S10B), which are necessary for senescence maintenance. Another downstream target of the p38MAPK pathway, the cyclin-dependent kinase inhibitor p16, was also induced in TIG-1 cells following DNA damage (Fig. S11A). In line with this observation, expression of recombinant p16 led to an increased SA-β-gal staining and suppression of Hsp70 induction (Fig. 2B; Figs S3B and S11B).
To determine the role of p38MAPK in DDS-induced HSR downregulation, we tested whether this downregulation is affected by inhibition of p38MAPK activity. TIG-1 cells were exposed to 10 Gy IR and cultured for 6 days in the presence or absence of specific p38MAPK inhibitor SB203580 (SB) (Davies et al., 2000). SB blocked p38MAPK kinase activity induced by IR as demonstrated by suppression of Hsp27 phosphorylation and IL-6 and IL-8 transcription (Fig. 2A; Fig. S10B). Importantly, inhibition of p38MAPK significantly improved HSR in IR-induced senescent cells (Fig. 2C). Similar to the effects on Hsp70 levels after p53 depletion, p38MAPK inhibition led to significant increase in the basal levels of Hsp70, as well. Together these data indicate that both the p53–p21 and p38MAPK–SASP pathways contribute to the downregulation of the HSR in DDS-induced senescence (see Fig. 6I).
Role of SIRT1 in regulation of Hsf1 in senescence
Next we explored which step of HSR was compromised in DDS-induced senescent cells. Activation of HSF1 and its binding to heat shock elements (HSE) is the major event to induce the transcription of Hsp70 and other heat shock proteins. To assay this activity, lentiviral reporter construct was prepared by inserting six tandem repeats of HSE upstream of firefly luciferase controlled by minimal CMV promoter. To control efficiency of infection, along with the HSE luciferase virus, we co-infected the cells with retrovirus expressing GFP under the constitutive CMV promoter. Accordingly, the ratio between HSE luciferase activity and GFP expression represented the Hsf1 activity. Luciferase activity increased by almost 30-fold in heat shocked cells (Fig. 3A). However, Dox or IR treatment prior to HS dramatically reduced (by 65–80%) the Hsf1-dependent luciferase activity (Fig. 3A). Moreover, upregulation of one of the elements in the DDS pathways, namely p53, p21 or p16, similarly suppressed the Hsf1-driven luciferase activity (Fig. 3A). Hence, suppression of HSR in DDS-induced senescence is caused by the downregulation of Hsf1.
Fig. 3.

DNA-damage-induced senescence suppresses Hsf1 activity by decreasing SIRT1. (A) TIG-1 fibroblasts were infected with the HSE-luciferase and treated with 100 nM Dox overnight and cultured for 6 days; 10 Gy IR and cultured for 6 days; 10 μM nutlin-3 for 5 days or retroviral expression of p21 or p16 for 6 days. After heat shock, cells were incubated for 6 h before collection, and luciferase activity was measured. The means and ±SEM indicate three independent experiments. (B) FLAG-tagged wild-type Hsf1 was expressed with retrovirus in TIG-1 fibroblasts and treated with 10 μM of nutlin-3 for 5 days to induce senescence or depleted of SIRT1 by lentiviral shRNA. Cells were collected promptly after heat shock at 43 °C for 1 h, and immunoprecipitated with anti-FLAG antibody. The precipitates were immunoblotted for SIRT1. (C and D) Early passage TIG-1 fibroblasts were treated as in (A) and immunoblotted for SIRT1. (E) TIG-1 fibroblasts exposed to conditions mentioned (A) were collected for RNA, and qRT-PCR was performed using SIRT1 and GAPDH (housekeeping gene for control) mRNA-specific primers. The means and ±SEM are from three independent experiments. (F) SIRT1 mRNA half-life was measured in control (CT) and nutlin-3-induced senescent cells (Nut) by incubating with 5 μg mL−1 of actinomycin D and collecting RNA after 45 and 90 min. SIRT1 mRNA was normalized for GAPDH mRNA. The means and ±SEM were calculated from triplicates of two independent experiments. (G) Early passage TIG-1 fibroblasts were exposed to conditions mentioned in (A), and HuR protein levels were measured by immunoblotting. (H) HuR mRNA levels were measured by qRT-PCR, as in (F). The means and ±SEM are from three independent experiments. (I) Same set of cells as in Fig. 1(A) were immunoblotted for SIRT1 and HuR.
Here we assessed Hsf1 activity and its levels. The histone decetylase SIRT1 was previously shown to form complex with Hsf1 during heat shock and regulate its activity (Westerheide et al., 2009). We hypothesized that in senescent TIG-1 fibroblasts, Hsf1 transcriptional activity was decreased because of reduced association with SIRT1.
Accordingly, we expressed FLAG-tagged Hsf1 using retroviral expression system. Hsf1 was immunoprecipitated (IP) with anti-FLAG antibody from lysates of heat-shocked control and nutlin-3-treated cells. The amount of SIRT1 in complex with Hsf1 assessed by immunoblotting with anti-SIRT1 antibody was dramatically reduced in nutlin-3-treated fibroblasts compared to control cells (Fig. 3B). This reduction was associated with significant decrease in the SIRT1 levels (by 90%) in the nutlin-3-treated cells (Fig. 3C). Similarly, a remarkable decrease in the SIRT1 levels was observed in senescent cells induced by p21 or p16 expression and in cells subjected to IR (Fig. 3D; Fig. S12A). Thus, upon premature senescence induced by DDS, expression levels of the critical Hsf1 regulator SIRT1 are diminished.
To understand how SIRT1 is downregulated in premature senescence, we compared SIRT1 mRNA levels measured by qRT-PCR. Indeed, SIRT1 mRNA levels were reduced by 60% in the DDS-induced senescent cells compared to control (Fig. 3E; Fig. S12B), indicating that transition to senescence affects either SIRT1 transcription or stability of its mRNA. We further assessed decay of this mRNA following addition of actinomycin D to inhibit transcription. SIRT1 mRNA half-life was >5 h in control cells while in nutlin-3-induced senescent cells, half-life dropped to <45 min (Fig. 3F). This indicated that the reduction in the SIRT1 levels in prematurely senescent cells is associated with decreased mRNA stability.
Role of HuR in SIRT1-mediated regulation of Hsf1 activity in senescence
The RNA binding protein HuR (ELAV1) is the major regulator of SIRT1 mRNA stability (Abdelmohsen et al., 2007). Using the shRNA approach, we confirmed that depletion of HuR in TIG-1 cells led to a dramatic drop in SIRT1 levels (Fig. S13). Because HuR levels decrease in replicative senescence (Marasa et al., 2010), we hypothesized that HuR expression may also be reduced in cells that underwent senescence in response to DNA damage signaling. Indeed, HuR protein levels were significantly reduced in TIG-1 cells following treatments with Dox, IR or nutlin-3 as well as expression of p21 or p16 (Fig. 3G; Fig. S14A). In all these models of premature senescence, HuR downregulation resulted from decrease in mRNA levels (Fig. 3H; Fig. S14B). There was no significant change in half-life of HuR mRNA (Fig. S15), suggesting that effects of senescence on HuR were mostly because of transcription inhibition.
Furthermore, as shown in Fig. 3I, we observed a dramatic drop of both HuR and SIRT1 levels in WS fibroblasts suggesting that pathways of suppression of the HSR are similar in DDS-induced senescence and in the disease.
Hsf1 levels are decreased in DNA-damage signaling-induced senescence
To determine the contribution of SIRT1 in the HSR regulation in TIG-1 fibroblasts, we evaluated effects of the SIRT1 inhibitor nicotinamide on activity on Hsf1 in early passage cells. In line with previous reports, SIRT1 inhibition led to nearly 40% decrease in the Hsf1 activity measured with the HSE luciferase reporter (Fig. 4A), or by immunoblotting for Hsp70 (Fig. S16). Similarly, depletion of SIRT1 strongly (by 60%) inhibited Hsf1 activity (Fig. 4A). Analogous effect was seen in cells with depleted HuR (Fig. 4A), demonstrating that the HuR–SIRT1 pathway regulates Hsf1 in this system. Of note, treatment with nicotinamide or SIRT1 depletion did not cause premature senescence as measured by SA-β-gal or p53 and p21 (Figs S17 and S18A,B).
Fig. 4.

Decrease in Hsf1 level in senescence is related to SIRT1 downregulation. (A) HSE-luciferase lentiviral construct was expressed in early passage cells and treated with 5 mM NAM overnight, nutlin-3 for 5 days, or infected with shRNA for SIRT1 or HuR. After heat shock, cells were incubated for 6 h before collection, and luciferase activity was measured. The means and ±SEM indicate three independent experiments. (B) Control vector or lentiviral shRNA for either SIRT1 or HuR was expressed in early passage TIG-1 fibroblasts. Cells were selected with puromycin and treated with 10 μM nutlin-3 for 5 days. Levels of Hsf1 were measured by immunoblotting. (C) Lysates of cells described in Fig. 1(A,B) were immunoblotted for Hsf1. (D) Early passage TIG-1 fibroblasts were treated with nutlin-3 to induce senescence. Cells were collected and Hsf1 mRNA levels were measured by qRT-PCR. (E) Hsf1 mRNA half-life was analyzed as in Fig. 3E. The means and ±SEM were calculated from triplicates of two independent experiments. (F) Early passage cells were infected with retroviral empty vector or shRNA against p53 and then treated with 10 μM nutlin-3 for 5 additional days. Cells lysates were immunoblotted for HuR, SIRT1, and Hsf1. (G) Early passage cells were incubated with 10 μM SB and 10 μM nutlin-3 for 5 days, and levels of HuR, SIRT1, and Hsf1 were measured. (H) 10 μM of SB was added for 24 days to WS fibroblasts, and levels of HuR, SIRT1 and Hsf1 were measured. (I) 10 μM of SB was added for 24 days to WS fibroblasts and heat shocked for 35 min at 43 °C. After 6 h incubation, cells were collected and immunoblotted for Hsp70.
Beyond regulating Hsf1 activation, depletion of either HuR or SIRT1 consistently caused reduction (about 20%) of Hsf1 protein levels, suggesting that the histone deacetylase activity of SIRT1 contributes to Hsf1 expression (Fig. 4B). In line with these data, we discovered that Hsf1 expression levels were significantly decreased in TIG-1 fibroblasts treated with DNA damaging agents or nutlin-3, and in WS fibroblasts (Fig. 4B,C). In contrast to senescent fibroblasts, lack of HSR downregulation in growth-arrested fibroblasts correlated with the lack of downregulation of HuR, SIRT1, and Hsf1 (Fig. S19). Similar correlation was seen in control and hTERT-immortalized WS fibroblasts (Fig. S20). These data further demonstrate that both Hsf1 activity and the levels are regulated in senescence in HuR–SIRT1-dependent manner. Importantly, nutlin-3 treatment had a stronger inhibitory effect on HSF1 activity (90% inhibition) and levels (40% downregulation) compared to SIRT1 or HuR depletions (Fig. 4A,B). Furthermore, combination of either of these depletions and nutlin-3 treatment did not inhibit the HSR beyond the inhibition by nutlin-3 alone (Fig. 4A,B). These results indicate that nutlin-3 treatment downregulates Hsf1 via both HuR–SIRT1-dependent and HuR–SIRT1-independent mechanisms, and the latter mechanism(s) is likely to affect Hsf1 levels.
The effects of nutlin-3 on HSR were associated with reduction of the Hsf1 mRNA (Fig. 4D), but not the stability of mRNA (Fig. 4E) or protein (not shown). Taken together, these results demonstrate that the stress-induced senescence downregulates Hsf1 via multiple mechanisms, including HuR–SIRT1 pathway, which ultimately leads to suppression of the HSR (see Fig. 6I).
Major DNA-damage signaling pathways are involved in regulation of HuR, SIRT1, and Hsf1
As DDS-induced senescence led to decreased HuR–SIRT1–Hsf1 levels, we tested whether inhibition of either p53 or p38MAPK pathways can restore levels of HuR–SIRT1 and expression of Hsf1. Depletion of p53 significantly increased the levels of HuR, SIRT1 and Hsf1 in naïve cells. Moreover, p53 depletion averted downregulation of the levels of these proteins after nutlin-3 or IR treatments (Fig. 4F; Fig. S21), indicating that p53 significantly contributed to the suppression of the HuR–SIRT1–Hsf1 pathway in senescence (see Fig. 6I). Similarly, we inhibited the p38MAPK pathway with SB and tested HuR, SIRT1 and Hsf1 levels. Cells were treated with nutlin-3 or Dox with or without SB, Dox was removed after 2 days, and all samples were grown for 5 days. Figure 4G and Figure S22(A) show that inhibition of p38MAPK increased the levels of HuR, SIRT1 and Hsf1 in naïve cells and partially restored these levels in DDS-induced senescent cells. In line with these data, we observed restoration of levels of Hsp70 (Fig. S22B,C), which further indicated that HuR–SIRT1–Hsf1 pathway can be regulated by the p38MAPK–SASP signaling (see Fig. 6I).
To investigate the relevance of these findings to the disease, we asked whether HuR–SIRT1–Hsf1 pathway can be restored in WS fibroblasts by the p38MAPK inhibition. This possibility was in line with the previously published study showing that extended inhibition of p38MAPK improved proliferation and reduced indicators of premature senescence in WS fibroblasts (Fleming et al., 2005). We observed that incubation of WS fibroblasts with SB for 24 days led to a restoration of the levels of HuR, SIRT1 and Hsf1 (Fig. 4H). In these cells, we also observed an impressive restoration of HSR measured by induction of Hsp70 (Fig. 4I). These data indicate that the p38MAPK–SASP pathway contributes to suppression of Hur–SIRT1–Hsf1 pathway in WS.
HSF1 regulates HuR–SIRT1 pathway via a positive feedback
Using C. elegans, it was demonstrated that Hsf1 plays a major role in progression of aging: knockdown of Hsf1 accelerated this process, while overexpression delayed it (Hsu et al., 2003; Ben-Zvi et al., 2009; Anckar & Sistonen, 2010). The finding that SIRT1 is involved in the regulation of Hsf1 in senescence suggests that Hsf1 may act downstream and contribute to anti-aging effects of SIRT1. Alternatively, there may be bi-directional relationship between SIRT1 and Hsf1. To address these possibilities, we tested the effects of Hsf1 depletion on the levels of SIRT1. Indeed, depletion of Hsf1 caused a decrease in SIRT1 protein levels by 60% and its mRNA levels by 50%, similar to the effects seen in the senescent cells (Fig. 5A,B). Furthermore, similar to its effects on SIRT1, Hsf1 depletion caused a dramatic decrease in both protein and mRNA levels of HuR (Fig. 5A,B). Thus, Hsf1 regulates the HuR–SIRT1 pathway (see Fig. 6I).
Fig. 5.

Hsf1 depletion downregulates SIRT1 and HuR. (A) Retroviral control or Hsf1 shRNA were expressed in early passage TIG-1 fibroblasts. Puromycin selected cells were treated with nutlin-3 for 5 days to induce senescence. Levels of SIRT1 and HuR were measured by immunoblotting. (B) Levels of SIRT1 and HuR mRNA were measured in cells described in (A). (C and D) Brain and soleus muscles were harvested from 12 months old Hsf1 null (−/−) mice (n = 2) and age-matched control mice (n = 3) and preserved in RNA stable buffer. RNA from whole brain and soleus muscle were isolated, and qRT-PCR was performed with mouse HuR-, SIRT1-, and GAPDH-specific primers. (E) SIRT1 and HuR protein levels were measured by immunoblotting in samples used in C and D.
To investigate whether the discovered Hsf1 signaling occurs in vivo, we compared the levels of HuR and SIRT1 in wild-type and Hsf1 null mice. Twelve months old Hsf1 null mice and age-matched controls were sacrificed, and protein and mRNA levels of SIRT1 and HuR were assessed in various tissues. Indeed, the levels of both SIRT1 and HuR mRNA in brain and muscle were significantly decreased in the Hsf1 null mice compared to the wild-type animals (Fig. 5C,D). Consistent with the mRNA results, Hsf1 knockout strongly decreased the levels of HuR and SIRT1 protein in the muscle and to a lesser degree in the brain (Fig. 5E). These data demonstrate that feedback regulation of SIRT1 and HuR by Hsf1 occurs at the level of organisms.
Altogether these data demonstrate that decrease in HuR–SIRT1 leads to the suppression of Hsf1, while Hsf1 suppression in turn causes downregulation of HuR and SIRT1, thus creating a positive feedback loop.
Hsf1 plays a role in regulation of DNA-damage signaling-induced senescence
How does Hsf1 signal back to regulate HuR and SIRT1 levels? Because the effects of HSF1 depletion on SIRT were similar to those seen in DDS-induced senescence, we explored effects of Hsf1 on p53 and p38MAPK signaling pathways upstream of SIRT1 and on overall senescence development. We found that depletion of Hsf1 increased a fraction of SA-β-gal-positive cells, similar to the one observed after nultin-3 treatment (Fig. 6A). Moreover, Hsf1 depletion caused senescence associated heterochromatin foci formation, further evidencing senescence (Fig. S23). Importantly, depletion of p53 did not reverse SA-β-gal staining caused by Hsf1 depletion (Fig. 6A). Furthermore, even though we observed an upregulation of p53 and p21 in Hsf1-depleted cells, their levels were significantly lower than the levels seen in DDS-induced senescent cells (Fig. 6B). These data suggested that the development of the senescence phenotype in Hsf1 depleted cells was independent of p53 activity and that the alternative p38MAPK–SASP pathway may be an important contributor to the feedback loop. As an indicator of p38MAPK pathway activation, levels of p16 were elevated after Hsf1 depletion (Fig. 6B).
To test whether senescence in Hsf1 depletion was mediated by increased p38MAPK activity and SASP, we explored the role of Hsf1 in regulation of this pathway. Previously, it has been demonstrated that Hsf1 can regulate IL-6 and other chemokines and that LPS-stimulated levels of IL-6 were higher in Hsf1 knockout mice than in wild-type mice (Takii et al., 2010). Here, we observed a strong increase in p38MAPK activity after Hsf1 depletion, measured by both phosphorylation of p38MAPK and phosphorylation of its substrate Hsp27 (Fig. 6C). This activation was comparable to the activation of p38MAPK by nutlin-3 (Fig. S24). Furthermore, the levels of IκB phosphorylation, which indicate activation of NFκB, as well as NFκB firefly luciferase reporter activity were increased significantly upon Hsf1 depletion (Fig. 6D; Fig. S25). As a corollary, we detected significantly increased transcription of NFκB target IL-6 (Fig. 6E). Interestingly, treatment with the p38MAPK inhibitor SB partially reversed SA-β-gal staining following Hsf1 depletion (Fig. S26). These data indicate that downregulation of Hsf1 following DNA damage signals back to the p38MAPK–SASP pathway to facilitate the maintenance of senescence (see Fig. 6I).
To further explore the role of Hsf1 in control of DDS-induced senescence pathways, we tested whether Hsf1 can suppress the activation of the p38MAPK–SASP pathway and the establishment of senescence phenotype after DNA damaging stress. Accordingly, we stably expressed FLAG-tagged Hsf1 in early passage TIG-1 cells using retroviral expression system and exposed them to IR. As a control, we used TIG-1 cells expressing an empty vector. Hsf1 strongly suppressed the IR-induced activation of p38MAPK, measured by reduced phosphorylation levels of both p38MAPK and Hsp27 (Fig. 6F). Similarly, it strongly inhibited production of IL-6 and IL-8 in the cells exposed to radiation (Fig. 6G; Fig. S27). These effects culminated in suppression of the DDS-induced senescence. In fact, we observed about 40% decrease in the fraction of SA-β-gal-positive cells in the irradiated samples (Fig. 6H). Taken together, these data demonstrate that Hsf1 plays an important role in DDS-induced senescence signaling via a positive feedback loop to regulate p38MAPK–SASP pathway (see Fig. 6I).
Discussion
This investigation provided an insight into the mechanisms of HSR suppression in DDS-induced senescence, including the multiple levels of Hsf1 regulation. Our data demonstrate that in prematurely senescent cells from patients with WS, the suppression of HSR is caused by DDS. Moreover, the activation of DDR signaling that induces senescence in early passage cells leads to significant downregulation of Hsf1 levels and activity. Here, in the first endeavor to understand the effects of senescence signaling on HSR, we demonstrated that both p53–p21 and p38MAPK–SASP pathways significantly contribute to regulation of Hsf1. Both pathways affect Hsf1 activity and levels in large part via regulation of SIRT1 levels, which in turn are controlled by the translation regulator HuR (see Fig. 6I). These effects were seen both in normal fibroblasts exposed to DNA damage and in fibroblasts from patients with WS.
A significant advance in this work is the finding that SIRT1 and Hsf1 regulate each other in a positive feedback loop. In fact, depletion of SIRT1 or HuR caused downregulation of Hsf1, while depletion of Hsf1 caused downregulation of HuR and SIRT1. As SIRT1 and Hsf1 were implicated in aging, these feedback relations may explain multiple effects of both proteins on the aging process. Importantly, the discovery of the same regulation in tissues of Hsf1 knockout mice enables us to extrapolate the implications of DNA damage at the cellular level to an organismal level.
This study also established the role of Hsf1 in regulation of the senescence- associated secretory phenotype. SASP represents a chronic inflammatory response, some components of which, for example, IL-6 or IL-1, were reported to control cell senescence. Here, we indeed demonstrate that one of the endpoints of SASP is downregulation of HuR–SIRT1, and subsequent downregulation of Hsf1. In line with these data, chronic inflammation mediated by p38MAPK was reported to play an important role in cessation of growth of WS fibroblasts, and possibly the overall acceleration of aging of patients with WS (Davis et al., 2005). Here we found that inhibition of p38MAPK partially restores Hsf1, as well as HuR and SIRT1. It is possible that such restoration may significantly improve patients’ conditions.
Previously it was reported that the transcription factor C/EBPβ, which responds to SASP, can directly inhibit Hsf1 in human monocytes (Xie et al., 2002). In our system, effects of SASP are certainly distinct, because we could not detect significant effects of C/EBPβ depletion on Hsf1 activity (not shown), even though SASP was clearly associated with downregulation of HuR–SIRT1 axis.
Importantly, the feedback regulation of HuR–SIRT1 upon Hsf1 depletion is mediated by the p38MAPK–SASP pathway. This finding is consistent with previous reports that suggest enhanced inflammatory response in Hsf1 knockout animals (McMillan et al., 1998). In fact, such mice were more sensitive to LPS, and demonstrated stronger upregulation of TNF in response to LPS (Xiao et al., 1999; Takii et al., 2010). In this study, we have found the inextricable link between pro-inflammatory cytokines and Hsf1 activity. In addition to keeping the proteome in balance, Hsf1 prevents establishment of microenvironment that adversely affects the organism. This study introduces a novel perspective on the relations between proteostasis and aging. Previous reports indicated the necessity of functional Hsf1 in extension of lifespan or in decreasing onset of protein misfolding diseases, implicating the importance of proteostasis in these conditions. On the other hand, as pro-inflammatory cytokines, especially IL-6 has been linked to age-related diseases and certain types of cancer, our findings indicate that at least in part effects of Hsf1 could be associated with regulation of signaling pathways involved in chronic inflammation.
A significant finding in this work is that Hsf1 by the feedback signaling can suppress cell senescence. In fact, depletion of Hsf1 facilitated senescence, while overexpression of Hsf1 reduced DNA-damage-induced senescence. In other words, DDS via downregulation of HuR–SIRT1 pathway suppresses Hsf1, which in turn enhances the p38MAPK–SASP pathway, thus supporting chronic inflammation and senescence phenotype (Fig. 6I). These findings uncover unexpected relations between the DNA damage and proteostasis; in normal cells, the proteostasis regulator Hsf1 controls cessation of proliferation of cells with damaged DNA. It is conceivable that failure of this control in cancer cells contribute to the loss of growth cessation of cells with DNA damage/instability.
Supplementary Material
Data S1 Materials and methods.
Fig. S1 Werner Syndrome fibroblasts (passage 4) exhibited prematurely senescent phenotype when compared to age matched controls (passage 15).
Fig. S2 HSR was compared in two lines of hTERT-immortalized control (CT1 and CT2) and WS (WS1 and WS2) fibroblasts. Cells were exposed to heat shock and immunoblotted for Hsp70. The normalized density of the Hsp70 band is shown below.
Fig. S3 (A) Early passage TIG-1 fibroblasts were treated with 10 Gy IR and cultured for 6 days. Cells were fixed, stained with rabbit anti-Ki-67 antibody and anti-rabbit IgG conjugated with Texas-Red secondary antibody. Images were acquired with Axiovert 200 (Carl Zeiss, Oberkochen, Germany) microscope with an ×100 objective using the manufacturer’s software. Radiation decreased % of cells with Ki-67 staining. (B) Early passage cells were treated with 100 nM Dox or 10 Gy IR and cultured for 5 additional days. DNA damage caused increased SA-β-gal staining.
Fig. S4 Cells were infected with empty vector, retroviral shRNA for p53 or recombinant p21. Two days post infection, 10 μM nutlin-3 was added for additional 5 days. (A) nutlin-3 treatment or expression of p21 caused increased staining with SA-β-gal, whereas p53 depletion prevented staining in nutlin-3 treated cells. (B) Nutlin-3 treatment led to accumulation p53 and p21 without directly causing DNA damage, and did not cause p16 upregulation. (C) Depletion of p53 prevented p21 accumulation after nutlin-3 or 6 Gy IR treatment.
Fig. S5 Early passage TIG-1 was infected with empty vector or p21 retrovirus. Next day, vector expressing cells were treated with 10 μM of nultin-3 for 3 days or left untreated. Then all cells were infected with lentiviral cell cycle reporters, green fluorescent tagged mutant Cdt for G1 and red fluorescent tagged mutant Geminin for G2. Next day, untreated vector cells were exposed to 1 μM CDK4 inhibitor or 10 μM CDK4/6 inhibitor IV. After 3 days, cells were fixed. Fluorescent cells under RFP indicate G2 and FITC indicate G1.
Fig. S6 Cells treated with 1 μM CDK4 inhibitor or 10 μM CDK4/6 inhibitor IV for 3 days were fixed and stained with SA-β-gal.
Fig. S7 Same cells from S6 and nutlin-3 treated cells were collected 6 h after heat shock and immunoblotted for Hsp70.
Fig. S8 Early passage TIG-1 was infected with empty vector or shRNA against p21 and 2 days later treated with 10 μM nutlin-3. p21 depletion did not restore the level of Hsp70 accumulation after nutlin-3.
Fig. S9 Cells treated with nutlin-3 was incubated with or without 10 μM SB for 5 days. Cell lysates were immunoblotted for phosphorylated forms of p38MAPK and Hsp27.
Fig. S10 (A) Cells with lentiviral NFκB luciferase reporter was treated with 10 Gy IR and 3 days later infected with either lentiviral empty vector or shRelA and selected. 6 days after IR, cells were collected for luciferase assay. The means and ±SEM indicate three independent experiments. (B) Cells treated as (A) were collected for RNA. qRT-PCR was performed using IL-6, IL-8 and GAPDH mRNA. IL-6 and IL-8 mRNA dramatically increased in IR treated cells and SB inhibited the induction. The mean and ±SEM were from triplicates of two independent experiments.
Fig. S11 (A) Cells treated with 10 Gy IR was lysed and immunoblotted for p16. IR increased p16 levels. (B) Same cells as (A) were fixed and stained for SA-βgal.
Fig. S12 (A) Cells were treated with 100 nM Dox overnight and cultured for 6 days or retroviral expression of p21 or p16 for 6 days, and immunoblotted for SIRT1. SIRT1 decreased after DNA damage induced senescence. (B) Same set of cells as (A) were collected for RNA and qRT-PCR was performed using SIRT1 and GAPDH (housekeeping gene for control) mRNA specific primers. As in protein, SIRT1 mRNA was significantly decreased in senescent cells. The means and ±SEM are from three independent experiments. Abbreviations: CT, control; Vec, vector.
Fig. S13 Early passage TIG-1 was infected with lentiviral empty vector or shRNAs against HuR and SIRT1. Two days post infection, 10 μM nutlin-3 was added for additional 5 days. Nutlin-3 treatment alone decreased SIRT1 and HuR. HuR depletion caused SIRT1 decrease, whereas SIRT1 depletion did not cause HuR decrease (not shown).
Fig. S14 (A) Cells were treated with 100 nM Dox overnight and cultured for 6 days or retroviral expression of p21 or p16 for 6 days, and immunoblotted for HuR. HuR decreased after DNA damage induced senescence. (B) Same set of cells as (A) were collected for RNA and qRT-PCR was performed using HuR and GAPDH (housekeeping gene for control) mRNA specific primers. As in protein, HuR mRNA was significantly decreased in senescent cells. The means and ±SEM are from three independent experiments. Abbreviations: CT, control; Vec, vector.
Fig. S15 HuR mRNA half-life was measured in control and nutlin-3 induced senescent cells by incubating with 5 μg mL−1 of actinomycin D and collecting RNA after 45 and 90 min. HuR and GAPDH mRNA were measured qRT-PCR and normalized by GAPDH mRNA. Data is represented by percentage of HuR mRNA measured at time 0 min (prior to adding actinomycin D), in semi-logorithmic scale. Half-life is calculated as time need for 50% reduction of mRNA. The means and ±SEM was calculated from triplicates of two independent experiments.
Fig. S16 Early passage TIG-1 was treated with 10 μM nutlin-3 for 5 days and 5 mM nicotinamide was added overnight before heat shock. Cell lysates were immunoblotted for Hsp70. Nicotinamide suppressed Hsp70 induction in early TIG-1 fibroblasts as nutlin-3 treatment.
Fig. S17 Cells from S16 were immunoblotted for HuR, SIRT1, p53 and p21. Overnight nicotinamide treatment did not cause decrease HuR and SIRT1, and did not induce senescence by p53–p21.
Fig. S18 (A) Early passage TIG-1 was infected with lentiviral shRNA against SIRT1 and 2 days later incubated with 10 μM nutlin-3 for 5 additional days. SIRT1 depletion alone did not cause increase in p53 and p21. (B) Same set of cells as (A) were fixed and stained with SA-β-gal. SIRT1 depletion alone did not cause increased staining of SA-β-gal.
Fig. S19 Same set of cells as S7 were probed for SIRT1, HuR and Hsf1.
Fig. S20 Same set of cells as S2 were probed for SIRT1, HuR and Hsf1.
Fig. S21 TIG-1 cells were infected with retroviral empty vector or shRNA against p53 and treated with 6 Gy IR. After 5 days post IR, cells were lysed and immunoblotted for HuR, SIRT1 and Hsf1. p53 depletion increased basal level of HuR, SIRT1 and Hsf1, and prevented senescence associated decrease of those proteins.
Fig. S22 (A) Early passage cells were pre-incubated with 10 μM SB for 6 h then co-incubated with 100 nM Dox for 2 days. After removing Dox, SB was added for the additional 4 days. Administration of SB to Dox treated cells led to partial restoration of HuR, SIRT1 and Hsf1. (B) Same set of cells as (A) were heat shocked and incubated for 6 h before collection for immunoblotting for Hsp70. Administration of SB to Dox treated cells led to partial restoration of Hsp70 induction. (C) Early passage cells were incubated with 10 μM SB and 10 μM nutlin-3 for 5 days. Cells were heat shocked the incubated for 6 h before collection for immunoblotting for hsp70. Concomitant administration of SB and nutlin-3 led to partial restoration of Hsp70 induction. Abbreviations: CT, control; Nut, Nutlin-3.
Fig. S23 Early passage TIG-1 was infected with retroviral empty vector or shRNA against Hsf1. Two days later vector cells were treated with 10 μM nutlin-3 or 100 nM Dox, and cultured for 5 additional days. Cells were fixed and stained with DAPI. Arrows indicate cells with punctate DNA foci containing cells, which are senescence associated heterochromatin foci (SAHF).
Fig. S24 Early passage TIG-1 was infected with retroviral empty vector or shRNA against Hsf1, and 2 days later 10 μM nutlin-3 was added to the vector for 5 additional days. Hsf1 depletion increased phosphorylation of Hsp27, similar to nutlin-3 treated cells. Abbreviations: CT, control; Nut, Nutlin-3.
Fig. S25 Early passage TIG-1 was infected with retroviral empty vector or shRNA against Hsf1, and 2 days later 10 μM SB was added 3 additional days, and collected for immunoblotting. Hsf1 depletion increased phosphorylation of IκB. SB treatment decreased phosporylation of IκB in both vector and Hsf1 depleted cells.
Fig. S26 Early passage TIG-1 was infected with retroviral empty vector or shRNA against Hsf1, and 2 days later 10 μM SB was added for 3 additional days. Hsf1 depletion increased SA-β-gal staining, which was decreased by SB treatment.
Fig. S27 Cell expressing empty or FLAG-tagged wild-type Hsf1 were treated with 10 Gy of IR and cultured for 6 days. Cells were collected for RNA. qRT-PCR was performed using IL-8 and GAPDH mRNA. IL-8 mRNA dramatically increased in DNA damage induced senescent cells. This increase was significantly suppressed in Hsf1 expressed cells. The mean and ±SEM were from triplicates of two independent experiments.
List of abbreviations
- HSR
heat shock response
- DDS
DNA damage-induced signaling
- SASP
senescence-associated secretion phenotype
- WS
Werner Syndrome
- Dox
doxorubicin
- IR
γ-irradiation
- SA-b-gal
senescence-associated b-galactosidase activity
- HSE
heat shock elements
- SB
SB203580 compound
Footnotes
Supporting information
Additional supporting information may be found in the online version of this article. As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
References
- Abdelmohsen K, Pullmann R, Lai A, Kim HH, Galban S, Yang XL, Blethrow JD, Walker M, Shubert J, Gillespie DA, Furneaux H, Gorospe M. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol Cell. 2007;25:543–557. doi: 10.1016/j.molcel.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, di Fagagna FD, Bernard D, Hernando E, Gil J. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2010;80:1089–1115. doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- Ben-Zvi A, Miller EA, Morimoto RI. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Nat Acad Sci USA. 2009;106:14914–14919. doi: 10.1073/pnas.0902882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang BD, Xuan YZ, Broude EV, Zhu HM, Schott B, Fang J, Roninson IB. Role of p53 and p21(waf1/cip1) in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene. 1999;18:4808–4818. doi: 10.1038/sj.onc.1203078. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanismof action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis T, Baird DM, Haughton MF, Jones CJ, Kipling D. Prevention of accelerated cell aging in Werner syndrome using a p38 mitogen-activated protein kinase inhibitor. J Gerontol A Biol Sci Med Sci. 2005;60:1386–1393. doi: 10.1093/gerona/60.11.1386. [DOI] [PubMed] [Google Scholar]
- Du ZX, Zhang HY, Meng X, Gao YY, Zou RL, Liu BQ, Guan Y, Wang HQ. Proteasome inhibitor MG132 induces BAG3 expression through activation of heat shock factor 1. J Cell Physiol. 2009;218:631–637. doi: 10.1002/jcp.21634. [DOI] [PubMed] [Google Scholar]
- Fan XHC, Steitz JA. Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J. 1998;17:3448–3460. doi: 10.1093/emboj/17.12.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming JB, Shen G-L, Holloway SE, Davis M, Brekken RA. Molecular consequences of silencing mutant K-ras in pancreatic cancer cells: justification for K-ras-directed therapy. Mol Cancer Res. 2005;3:413–423. doi: 10.1158/1541-7786.MCR-04-0206. [DOI] [PubMed] [Google Scholar]
- Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30:1536–1548. doi: 10.1038/emboj.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto M. Hierarchical deterioration of body systems in Werner’s syndrome: Implications for normal ageing. Mech Ageing Dev. 1997;98:239–254. doi: 10.1016/s0047-6374(97)00111-5. [DOI] [PubMed] [Google Scholar]
- Heydari AR, Wu B, Takahashi R, Strong R, Richardson A. Expression of heat shock protein 70 is altered by age and diet at the level of transcription. Mol Cell Biol. 1993;15:2909–2918. doi: 10.1128/mcb.13.5.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heydari AR, Takahashi R, Gutsmann A, You S, Richardson A. Hsp70 and aging. Experientia. 1994;50:1092–1098. doi: 10.1007/BF01923466. [DOI] [PubMed] [Google Scholar]
- Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- Jana NR, Tanaka M, Wang GH, Nukina N. Polyglutamine length–dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum Mol Genet. 2000;9:2009–2018. doi: 10.1093/hmg/9.13.2009. [DOI] [PubMed] [Google Scholar]
- Koyama S, Arawaka S, Chang-Hong R, Wada M, Kawanami T, Kurita K, Kato M, Nagai M, Aoki M, Itoyama Y, Sobue G, Chan PH, Kato T. Alteration of familial ALS-linked mutant SOD1 solubility with disease progression: Its modulation by the proteasome and Hsp70. Biochem Biophys Res Commun. 2006;343:719–730. doi: 10.1016/j.bbrc.2006.02.170. [DOI] [PubMed] [Google Scholar]
- Lee YK, Manalo D, Liu AYC. Heat shock response, heat shock transcription factor and cell aging. Biol Signals. 1996;5:180–191. doi: 10.1159/000109187. [DOI] [PubMed] [Google Scholar]
- Marasa BS, Srikantan S, Martindale JL, Kim MM, Lee EK, Gorospe M, Abdelmohsen K. MicroRNA profiling in human diploid fibroblasts uncovers miR-519 role in replicative senescence. Aging (Albany NY) 2010;2:333–343. doi: 10.18632/aging.100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan DR, Xiao X, Shao L, Graves K, Benjamin IJ. Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J Biol Chem. 1998;273:7523–7528. doi: 10.1074/jbc.273.13.7523. [DOI] [PubMed] [Google Scholar]
- Morley JF, Morimoto RI. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell. 2004;15:657–664. doi: 10.1091/mbc.E03-07-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1 alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Nat Acad Sci USA. 2009;106:17031–17036. doi: 10.1073/pnas.0905299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MM, Stuchlick O, El-Karim EG, Stuart R, Kipreos ET, Wells L. Intracellular protein glycosylation modulates insulin mediated lifespan in C. elegans. Aging (Albany NY) 2010;2:678–690. doi: 10.18632/aging.100208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles SJ, Adami GR. Agents that cause DNA double strand breaks lead to p16(INK4a) enrichment and the premature senescence of normal fibrolasts. Oncogene. 1998;16:1113–1123. doi: 10.1038/sj.onc.1201862. [DOI] [PubMed] [Google Scholar]
- Rodier F, Coppe JP, Patil CK, Hoeijmakers WAM, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:U973–U142. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi ML, Ghosh AK, Bohr VA. Roles of Werner syndrome protein in protection of genome integrity. DNA Repair (Amst) 2010;9:331–344. doi: 10.1016/j.dnarep.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, Imamura T, Ogawa M, Masai H, Miyawaki A. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132:487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- Sakellariou GK, Pye D, Vasilaki A, Zibrik L, Palomero J, Kabayo T, McArdle F, Van Remmen H, Richardson A, Tidball JG, McArdle A, Jackson MJ. Role of superoxide–nitric oxide interactions in the accelerated age-related loss of muscle mass in mice lacking Cu, Zn superoxide dismutase. Aging Cell. 2011;10:749–760. doi: 10.1111/j.1474-9726.2011.00709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Maier B, Bartke A, Scrable H. Progressive loss of SIRT1 with cell cycle withdrawal. Aging Cell. 2006;5:413–422. doi: 10.1111/j.1474-9726.2006.00235.x. [DOI] [PubMed] [Google Scholar]
- Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- Takii R, Inouye S, Fujimoto M, Nakamura T, Shinkawa T, Prakasam R, Tan K, Hayashida N, Ichikawa H, Hai T, Nakai A. Heat shock transcription factor 1 inhibits expression of IL-6 through activating transcription factor 3. J Immunol. 2010;184:1041–1048. doi: 10.4049/jimmunol.0902579. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Dillin A. Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a004440. pii:a004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT. Small-molecule antagonists of p53-MDM2 binding: research tools and potential therapeutics. Cell Cycle. 2004;3:419–421. [PubMed] [Google Scholar]
- Vaziri H, West MD, Allsopp RC, Davison TS, Wu YS, Arrowsmith CH, Poirier GG, Benchimol S. ATM-dependent telomere loss in aging human diploid fibroblasts and DNA damage lead to the post-translational activation of p53 protein involving poly(ADP-ribose) polymerase. EMBO J. 1997;16:6018–6033. doi: 10.1093/emboj/16.19.6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerheide SD, Anckar J, Stevens SM, Jr, Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323:1063–1066. doi: 10.1126/science.1165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Gu MJ, Heydari AR, Richardson A. The effect of age on the synthesis of 2 heat-shock proteins in the Hsp70 family. J Gerontol. 1993;48:B50–B56. doi: 10.1093/geronj/48.2.b50. [DOI] [PubMed] [Google Scholar]
- Wyllie FS, Jones CJ, Skinner JW, Haughton MF, Wallis C, Wynford-Thomas D, Faragher RGA, Kipling D. Telomerase prevents the accelerated cell ageing of Werner syndrome fibroblasts. Nat Genet. 2000;24:16–17. doi: 10.1038/71630. [DOI] [PubMed] [Google Scholar]
- Xiao X, Zuo X, Davis AA, McMillan DR, Curry BB, Richardson JA, Benjamin IJ. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J. 1999;18:5943–5952. doi: 10.1093/emboj/18.21.5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Chen CM, Stevenson MA, Hume DA, Auron PE, Calderwood SK. NF-IL6 and HSF1 have mutually antagonistic effects on transcription in x3monocytic cells. Biochem Biophys Res Commun. 2002;291:1071–1080. doi: 10.1006/bbrc.2002.6562. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Materials and methods.
Fig. S1 Werner Syndrome fibroblasts (passage 4) exhibited prematurely senescent phenotype when compared to age matched controls (passage 15).
Fig. S2 HSR was compared in two lines of hTERT-immortalized control (CT1 and CT2) and WS (WS1 and WS2) fibroblasts. Cells were exposed to heat shock and immunoblotted for Hsp70. The normalized density of the Hsp70 band is shown below.
Fig. S3 (A) Early passage TIG-1 fibroblasts were treated with 10 Gy IR and cultured for 6 days. Cells were fixed, stained with rabbit anti-Ki-67 antibody and anti-rabbit IgG conjugated with Texas-Red secondary antibody. Images were acquired with Axiovert 200 (Carl Zeiss, Oberkochen, Germany) microscope with an ×100 objective using the manufacturer’s software. Radiation decreased % of cells with Ki-67 staining. (B) Early passage cells were treated with 100 nM Dox or 10 Gy IR and cultured for 5 additional days. DNA damage caused increased SA-β-gal staining.
Fig. S4 Cells were infected with empty vector, retroviral shRNA for p53 or recombinant p21. Two days post infection, 10 μM nutlin-3 was added for additional 5 days. (A) nutlin-3 treatment or expression of p21 caused increased staining with SA-β-gal, whereas p53 depletion prevented staining in nutlin-3 treated cells. (B) Nutlin-3 treatment led to accumulation p53 and p21 without directly causing DNA damage, and did not cause p16 upregulation. (C) Depletion of p53 prevented p21 accumulation after nutlin-3 or 6 Gy IR treatment.
Fig. S5 Early passage TIG-1 was infected with empty vector or p21 retrovirus. Next day, vector expressing cells were treated with 10 μM of nultin-3 for 3 days or left untreated. Then all cells were infected with lentiviral cell cycle reporters, green fluorescent tagged mutant Cdt for G1 and red fluorescent tagged mutant Geminin for G2. Next day, untreated vector cells were exposed to 1 μM CDK4 inhibitor or 10 μM CDK4/6 inhibitor IV. After 3 days, cells were fixed. Fluorescent cells under RFP indicate G2 and FITC indicate G1.
Fig. S6 Cells treated with 1 μM CDK4 inhibitor or 10 μM CDK4/6 inhibitor IV for 3 days were fixed and stained with SA-β-gal.
Fig. S7 Same cells from S6 and nutlin-3 treated cells were collected 6 h after heat shock and immunoblotted for Hsp70.
Fig. S8 Early passage TIG-1 was infected with empty vector or shRNA against p21 and 2 days later treated with 10 μM nutlin-3. p21 depletion did not restore the level of Hsp70 accumulation after nutlin-3.
Fig. S9 Cells treated with nutlin-3 was incubated with or without 10 μM SB for 5 days. Cell lysates were immunoblotted for phosphorylated forms of p38MAPK and Hsp27.
Fig. S10 (A) Cells with lentiviral NFκB luciferase reporter was treated with 10 Gy IR and 3 days later infected with either lentiviral empty vector or shRelA and selected. 6 days after IR, cells were collected for luciferase assay. The means and ±SEM indicate three independent experiments. (B) Cells treated as (A) were collected for RNA. qRT-PCR was performed using IL-6, IL-8 and GAPDH mRNA. IL-6 and IL-8 mRNA dramatically increased in IR treated cells and SB inhibited the induction. The mean and ±SEM were from triplicates of two independent experiments.
Fig. S11 (A) Cells treated with 10 Gy IR was lysed and immunoblotted for p16. IR increased p16 levels. (B) Same cells as (A) were fixed and stained for SA-βgal.
Fig. S12 (A) Cells were treated with 100 nM Dox overnight and cultured for 6 days or retroviral expression of p21 or p16 for 6 days, and immunoblotted for SIRT1. SIRT1 decreased after DNA damage induced senescence. (B) Same set of cells as (A) were collected for RNA and qRT-PCR was performed using SIRT1 and GAPDH (housekeeping gene for control) mRNA specific primers. As in protein, SIRT1 mRNA was significantly decreased in senescent cells. The means and ±SEM are from three independent experiments. Abbreviations: CT, control; Vec, vector.
Fig. S13 Early passage TIG-1 was infected with lentiviral empty vector or shRNAs against HuR and SIRT1. Two days post infection, 10 μM nutlin-3 was added for additional 5 days. Nutlin-3 treatment alone decreased SIRT1 and HuR. HuR depletion caused SIRT1 decrease, whereas SIRT1 depletion did not cause HuR decrease (not shown).
Fig. S14 (A) Cells were treated with 100 nM Dox overnight and cultured for 6 days or retroviral expression of p21 or p16 for 6 days, and immunoblotted for HuR. HuR decreased after DNA damage induced senescence. (B) Same set of cells as (A) were collected for RNA and qRT-PCR was performed using HuR and GAPDH (housekeeping gene for control) mRNA specific primers. As in protein, HuR mRNA was significantly decreased in senescent cells. The means and ±SEM are from three independent experiments. Abbreviations: CT, control; Vec, vector.
Fig. S15 HuR mRNA half-life was measured in control and nutlin-3 induced senescent cells by incubating with 5 μg mL−1 of actinomycin D and collecting RNA after 45 and 90 min. HuR and GAPDH mRNA were measured qRT-PCR and normalized by GAPDH mRNA. Data is represented by percentage of HuR mRNA measured at time 0 min (prior to adding actinomycin D), in semi-logorithmic scale. Half-life is calculated as time need for 50% reduction of mRNA. The means and ±SEM was calculated from triplicates of two independent experiments.
Fig. S16 Early passage TIG-1 was treated with 10 μM nutlin-3 for 5 days and 5 mM nicotinamide was added overnight before heat shock. Cell lysates were immunoblotted for Hsp70. Nicotinamide suppressed Hsp70 induction in early TIG-1 fibroblasts as nutlin-3 treatment.
Fig. S17 Cells from S16 were immunoblotted for HuR, SIRT1, p53 and p21. Overnight nicotinamide treatment did not cause decrease HuR and SIRT1, and did not induce senescence by p53–p21.
Fig. S18 (A) Early passage TIG-1 was infected with lentiviral shRNA against SIRT1 and 2 days later incubated with 10 μM nutlin-3 for 5 additional days. SIRT1 depletion alone did not cause increase in p53 and p21. (B) Same set of cells as (A) were fixed and stained with SA-β-gal. SIRT1 depletion alone did not cause increased staining of SA-β-gal.
Fig. S19 Same set of cells as S7 were probed for SIRT1, HuR and Hsf1.
Fig. S20 Same set of cells as S2 were probed for SIRT1, HuR and Hsf1.
Fig. S21 TIG-1 cells were infected with retroviral empty vector or shRNA against p53 and treated with 6 Gy IR. After 5 days post IR, cells were lysed and immunoblotted for HuR, SIRT1 and Hsf1. p53 depletion increased basal level of HuR, SIRT1 and Hsf1, and prevented senescence associated decrease of those proteins.
Fig. S22 (A) Early passage cells were pre-incubated with 10 μM SB for 6 h then co-incubated with 100 nM Dox for 2 days. After removing Dox, SB was added for the additional 4 days. Administration of SB to Dox treated cells led to partial restoration of HuR, SIRT1 and Hsf1. (B) Same set of cells as (A) were heat shocked and incubated for 6 h before collection for immunoblotting for Hsp70. Administration of SB to Dox treated cells led to partial restoration of Hsp70 induction. (C) Early passage cells were incubated with 10 μM SB and 10 μM nutlin-3 for 5 days. Cells were heat shocked the incubated for 6 h before collection for immunoblotting for hsp70. Concomitant administration of SB and nutlin-3 led to partial restoration of Hsp70 induction. Abbreviations: CT, control; Nut, Nutlin-3.
Fig. S23 Early passage TIG-1 was infected with retroviral empty vector or shRNA against Hsf1. Two days later vector cells were treated with 10 μM nutlin-3 or 100 nM Dox, and cultured for 5 additional days. Cells were fixed and stained with DAPI. Arrows indicate cells with punctate DNA foci containing cells, which are senescence associated heterochromatin foci (SAHF).
Fig. S24 Early passage TIG-1 was infected with retroviral empty vector or shRNA against Hsf1, and 2 days later 10 μM nutlin-3 was added to the vector for 5 additional days. Hsf1 depletion increased phosphorylation of Hsp27, similar to nutlin-3 treated cells. Abbreviations: CT, control; Nut, Nutlin-3.
Fig. S25 Early passage TIG-1 was infected with retroviral empty vector or shRNA against Hsf1, and 2 days later 10 μM SB was added 3 additional days, and collected for immunoblotting. Hsf1 depletion increased phosphorylation of IκB. SB treatment decreased phosporylation of IκB in both vector and Hsf1 depleted cells.
Fig. S26 Early passage TIG-1 was infected with retroviral empty vector or shRNA against Hsf1, and 2 days later 10 μM SB was added for 3 additional days. Hsf1 depletion increased SA-β-gal staining, which was decreased by SB treatment.
Fig. S27 Cell expressing empty or FLAG-tagged wild-type Hsf1 were treated with 10 Gy of IR and cultured for 6 days. Cells were collected for RNA. qRT-PCR was performed using IL-8 and GAPDH mRNA. IL-8 mRNA dramatically increased in DNA damage induced senescent cells. This increase was significantly suppressed in Hsf1 expressed cells. The mean and ±SEM were from triplicates of two independent experiments.