

Abstract
Oxytricha trifallax — an established model organism for studying genome rearrangements, chromosome structure, scrambled genes, RNA-mediated epigenetic inheritance, and other phenomena — has been the subject of a nomenclature controversy for several years. Originally isolated as a sibling species of O. fallax, O. trifallax was reclassified in 1999 as Sterkiella histriomuscorum, a previously identified species, based on morphological similarity. The proper identification of O. trifallax is crucial to resolve in order to prevent confusion in both the comparative genomics and the general scientific communities. We analyzed nine conserved nuclear gene sequences between the two given species and several related ciliates. Phylogenetic analyses suggest that O. trifallax and a bona fide S. histriomuscorum have accumulated significant evolutionary divergence from each other relative to other ciliates such that they should be unequivocally classified as separate species. We also describe the original isolation of O. trifallax, including its comparison to O. fallax, and we provide criteria to identify future isolates of O. trifallax.
Keywords: Oxytricha fallax, Oxytricha trifallax, Sterkiella histriomuscorum, ciliate, spirotrich, hypotrich, evolution, phylogeny, concatenated tree
Introduction
The unicellular eukaryote Oxytricha trifallax is the most extensively studied ciliate of the spirotrichs (Doak et al. 2003). Like most ciliates, O. trifallax contains two types of nuclei: a somatic macronucleus (MAC) and a germline micronucleus (MIC). The MIC participates in sexual conjugation, but is otherwise transcriptionally silent (Narasimharao and Prescott 1967). During conjugation, O. trifallax destroys approximately 95% of its MIC genome and reorganizes the remaining pieces to generate over 20,000 highly processed MAC chromosomes, or nanochromosomes, typically bearing only a single gene (Prescott 2000; Swanton et al. 1980).
One of the first described organisms with scrambled genes (Greslin et al. 1989), Oxytricha sp. are often used as model systems for studying extensive gene rearrangements (Doak et al. 2003; Prescott 2000). Its sibling species, O. fallax, has also been used to study telomeres (e.g. Dawson and Herrick 1984; Pluta et al. 1982), transposon evolution (e.g. Witherspoon et al. 1997) and regulation of cell morphology (e.g., Grimes and Aufderheide 1991). Because it is fertile (see below) and undergoes post-zygotic nuclear development, O. trifallax has recently permitted the discovery of several novel features, including the involvement of non-coding, maternal RNAs in epigenetic programming of DNA rearrangements (Nowacki et al. 2008) and chromosome copy number (Nowacki et al. 2010) during development, as well as the involvement of germline-encoded transposases in this process (Nowacki et al. 2009). These and other studies underscore the importance of Oxytricha biology for understanding eukaryotic genome plasticity.
Ciliates such as Oxytricha are also important for evolutionary studies, providing a foil for comparative genomics of Tetrahymena (Coyne et al. 2008) and Paramecium (Arnaiz et al. 2007), as well as related apicomplexan parasites, such as Plasmodium (Doak et al. 2003), and other microbial eukaryotes. All Oxytricha studies require a clear understanding of its evolutionary history and accurate relationships among the Spirotrichea. Current evolutionary relationships among spirotrichs are complex and confusing, and species of the Oxytricha genus rarely form a single clade in published trees (Bernhard et al. 2001; Paiva et al. 2009; Schmidt et al. 2007), emphasizing that morphology alone cannot distinguish ciliate genera, as already argued by Hoffman et al. (1997a).
O. trifallax was originally isolated and characterized by a set of protocols that guaranteed the recovery of closely related sibling species of a formally identified and characterized strain of O. fallax. This species, isolated in the Sonneborn laboratory, was identified as Oxytricha fallax Stein, 1859 by the stichotrich specialist Arthur C. Borror, based both on protargol-stained slides and living specimens, using the current classification (Borror 1972). This identification was based both on the pattern and stomatogenesis of the ciliature and the lateral flexibility of the organism, and Grimes published a detailed description of this organism (Grimes 1972). Lateral flexibility was an essential feature in the separation of the genus Oxytricha Ehrenberg, 1831 (flexible) from the genus Histriculus Corliss,1960 (inflexible; Borror 1972). In our experience, O. trifallax and other members of the Oxytricha genus show flexibility under all growth conditions that we are aware of (e.g. Nowacki et al. 2008), and this is easily contrasted with genera such as Stylonychia that are quite inflexible. Berger in his book “Monograph of the Oxytrichidae” recognized flexibility as an important trait and, furthermore, suggested that the O. fallax described by Grimes was likely to be O. hymenostoma (a species of Oxytricha that Borror considered synonymous with O. fallax) (Berger 1999, p. 140). Prior to Corliss, the genus Histriculus was referred to as Histrio and the name traces its origin to Sterki in 1878. Within the genus Histriculus, a number of species that were inflexible were subsequently transferred to the new genus Sterkiella that Foissner created in 1991 (Foissner et al. 1991), based on the presence of caudal cirri.
The original O. fallax strain was adopted by Spear (Rae and Spear 1978) and Herrick (Cartinhour and Herrick 1984; Dawson and Herrick 1984; Herrick et al. 1985) and used in most of the original studies of Oxytricha molecular biology. Cysts of the original strain of O. fallax are cryogenically stored and viable, but by the early 1980’s the strain had become senescent and infertile. To develop a functional and viable genetic system in Oxytricha, two of us (R.L.H. and G.H.) initiated in 1985 an extensive isolation and screening program for new Oxytricha strains, using DNA probes to ensure the isolation of only very closely related sibling species of O. fallax. One group of these sibling species formed a mating group complex called Oxytricha trifallax, following Sonneborn’s naming system for the Paramecium aurelia complex (Sonneborn 1975). Adl and Berger (1997, 2000) characterized the cell cycle and conjugation stages of O. trifallax.
Foissner and Berger (1999) then reclassified O. trifallax stocks as the preexisting species S. histriomuscorum (Foissner et al. 1991), based on morphological similarity, and declared O. trifallax a nomen nudum, according to articles 13 and 15 of the International Code of Zoological Nomenclature (ICZN 1999), suggesting that no original studies described morphological characteristics of the species. Therefore, upon analysis of its morphological similarity to S. histriomuscorum BA and O. nova, now S. nova (Foissner and Berger 1999), Foissner and Berger identified all three species as members of the S. histriomuscorum complex, indicating that “the populations… cannot be reliably separated by morphological and morphometric criteria because the extreme values highly overlap in most cases.” However, Foissner and Berger (1999) also noted that S. histriomuscorum (= O. trifallax) and S. nova have many published genetic differences (e.g., Hoffmann and Prescott 1997a, 1997b). Nonetheless, to our knowledge, no molecular phylogenetic analysis has explicitly included both strains that are currently classified as S. histriomuscorum. While Schmidt et al. (Schmidt et al. 2007) supported the reclassification of O. trifallax using nuclear ribosomal RNA gene sequence analysis, their study did not include S. histriomuscorum strain BA, used in the present study. Many other publications include sequences from O. fallax, O. trifallax, or S. histriomuscorum, and the community should be aware that the species labels on GenBank sequences can be misleading.
Because conclusions drawn from a single gene are often poorly resolved and subject to lateral gene transfer, hidden paralogy, or tree reconstruction artifacts (Philippe et al. 2004), here we perform phylogenetic analyses on a suite of highly conserved nuclear genes, in order to infer the relationship between O. trifallax and a previously identified S. histriomuscorum isolate. Our dataset, which is based upon multiple, concatenated, protein-coding gene sequences, overcomes problems arising from a single gene tree and increases support for the overall species tree (Rokas et al. 2003). We examined five closely related ciliates (see below) and this analysis clearly suggests that O. trifallax is a species separate from S. histriomuscorum. We also provide a description of the original isolation and characteristics of O. trifallax, as part of its species description, including a comparison of the original O. fallax to O. trifallax, and we provide a reliable method for the identification of new isolates as O. trifallax.
Results
The analysis in this study includes 42 new complete gene sequences from the following nine protein-coding genes: actin I, cct-γ, ef-1, hsp70, rL10, sucA, tbp, turgor, and Ubc, surveyed in five stichotrichous species, including O. trifallax (strain JRB310), O. nova (now S. nova), S. histriomuscorum (strain BA), Stylonychia lemnae (strain 8×2), and the anaerobic heterotrich Nyctotherus ovalis as an outgroup (Ricard et al. 2008). The complete dataset contains 50 gene (complete nanochromosome) sequences, and we performed phylogenetic analysis on the concatenated gene tree, as well as each of the nine individual gene trees. In addition, each analysis was performed for both full nanochromosome sequences (FNS) corresponding to all the sequence between two telomeres, including the noncoding and highly variable subtelomeric regions, and coding sequence (CDS) regions, corresponding to the protein-coding sequences. Both Bayesian and maximum likelihood trees were generated, with bootstrap resampling for each unique dataset.
Sequence comparisons
The concatenated CDS pairwise sequence alignments of O. trifallax strain JRB310 and S. histriomuscorum strain BA are 14.7% divergent at the nucleotide level, suggesting extensive evolutionary divergence. This is significantly greater than the typical allelic variation observed either within O. trifallax isolates, which is ~3% in genome-wide surveys (Swart et al., unpubl. observ.), or between O. fallax and O. trifallax alleles (Seegmiller et al. 1996). Even at the amino acid level for individual gene comparisons, the predicted protein translations differ by at least 2.4% (ActinI), with an average of 8.7% amino acid divergence between O. trifallax strain JRB310 and S. histriomuscorum strain BA over all nine comparisons, again more consistent with between-species divergence than with allelic variation.
The concatenated CDS multiple sequence alignment (MSA) was 44.7% identical across all five species. The average GC content across the MSA in the coding regions ranged from 39.2% (tbp) to 47.8% (ef-1, hsp70) and the concatenated CDS MSA was 44.2% GC, consistent with previously published results (Cavalcanti et al. 2004).
Concatenated protein-coding sequence phylogeny
The concatenated CDS phylogeny (Fig. 1) is congruent with the concatenated FNS phylogeny (not shown), and in each case S. histriomuscorum is monophyletic with S. nova. O. trifallax appears to diverge earlier than this clade, and S. lemnae is more divergent than O. trifallax. N. ovalis, an earlier diverging, anaerobic heterotrich (class Armophorea; Lynn and Small 2002) that also bears nanochromosomes, was used as an outgroup (Ricard et al. 2008). The S. histriomuscorum and S. nova clade is supported by bootstrap values of 100% in both Bayesian and maximum likelihood analysis, and the O. trifallax earlier-diverging branch is also very strongly supported (bootstrap values of 100% in Bayesian analysis and 98% with maximum likelihood; Fig. 1).
Figure 1.
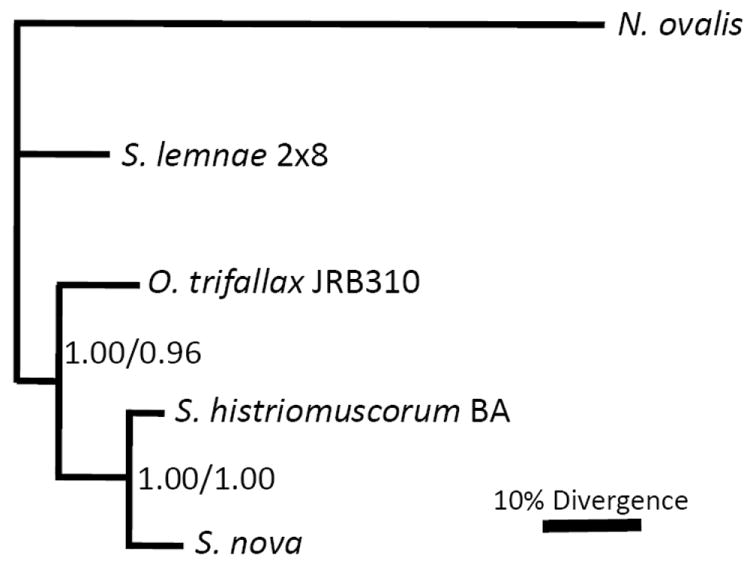
Bayesian and maximum likelihood phylogenies of nine concatenated protein-coding regions. Bootstrap values above 0.50 are shown (Bayesian/Maximum likelihood). Scale bars in Figures 1-3 represents 10% nucleotide divergence in Bayesian analyses. See text for details of the methods.
Individual gene trees
With the exceptions of actin I and cct-γ, discussed below, all individual CDS phylogenies were congruent to each other and grouped S. nova with S. histriomuscorum, with strong bootstrap support in Bayesian analysis (≥94%). O. trifallax diverged earlier from this clade, and S. lemnae typically diverged earlier than this group. N. ovalis provided an outgroup. The ef-1 FNS and CDS gene trees had minor differences from each other, affecting only the branching order of S. lemnae and S. nova. Most other individual FNS and CDS gene trees were identical to each other, except as described below.
Actin I phylogeny
While the 5 taxon actin I FNS tree grouped O. trifallax with S. histriomuscorum, the 5 taxon CDS tree grouped S. lemnae and S. histriomuscorum (not shown). Actin I gene trees also disagreed with the concatenated tree on the placement of S. nova (Fig. 2). Therefore, we included additional strains of both S. lemnae and S. nova to improve the resolution of this tree (see Discussion).
Figure 2.
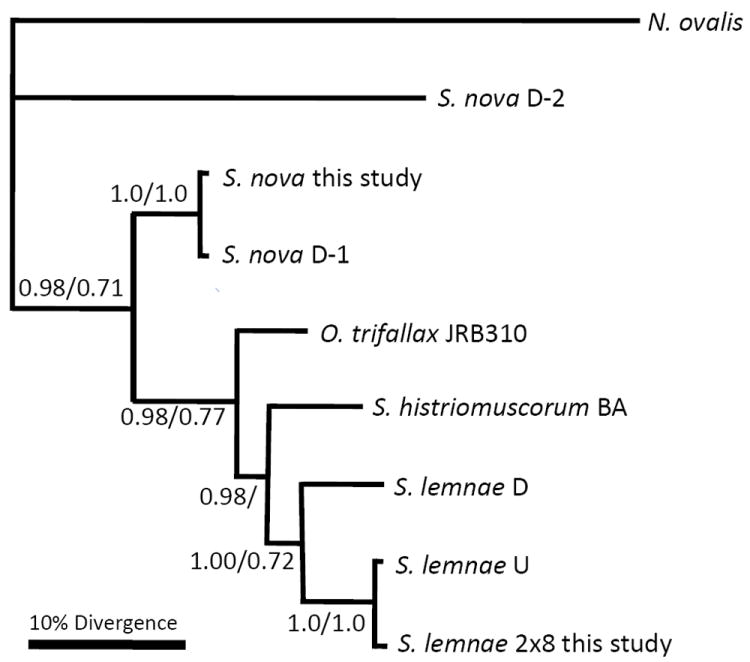
Bayesian and maximum likelihood nucleotide phylogeny of actin I protein-coding region. Bootstrap values above 0.50 are shown (Bayesian/maximum likelihood). S. lemnae U refers to accession AY046534 (Hogan et al. 2001). S. lemnae D refers to the consensus sequence of 4 very similar alleles from German strains D9 and B1, accession DQ108617 (Mollenbeck et al. 2006). S. nova D-1 refers to accession M22480 (Prescott and Greslin 1992), and S. nova D-2 refers to accession AF134156. See text for details.
Cct-γphylogeny
The cct-γ FNS and CDS gene trees were concordant with each other and suggest that S. histriomuscorum diverged earlier than O. trifallax, with S. lemnae and S. nova diverging later as a monophyletic lineage on this tree. This does not support the claim that S. nova is a member of the genus Sterkiella (Fig. 3).
Figure 3.
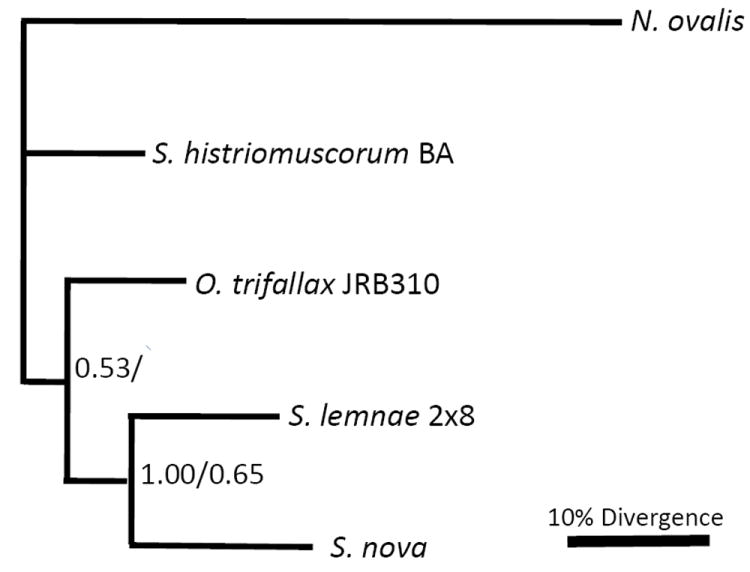
Bayesian and maximum likelihood phylogeny of cct-γprotein-coding region. Bootstrap values above 0.50 are shown (Bayesian/maximum likelihood). See text for details.
Small-subunit ribosomal RNA tree
We also constructed a tree from small-subunit (SSU) rDNA sequences for several stichotrichous ciliates (Fig. 4), rooted with two Uroleptus representatives that appear to be members of the earliest diverging lineage with scrambled genes (Chang et al. 2005; Hogan et al. 2001) and provide outgroups to the oxytrichids shown here. This analysis used the same locus and species that Schmidt et al. (2007) used to support the renaming of O. trifallax; hence this study essentially allowed us to place the bona fide S. histriomuscorum strain BA on their tree. We first note that the tree topology in Figure 4 is congruent with the concatenated CDS tree; hence we find no problems with the phylogenetic analysis in Schmidt et al. (2007) nor with the use of rDNA as a molecular marker for ciliate phylogeny. However, as in our concatenated protein tree, this reanalysis that includes both O. trifallax and S. histriomuscorum also supports the grouping of S. histriomuscorum with S. nova and suggests that O. trifallax and S. lemnae diverged earlier than these two species. Furthermore, S. histriomuscorum (strain BA) groups robustly with Pattersoniella vitiphila (99.4% identical) and is also 99.2% identical to G. steinii, whereas O. trifallax (strain JRB310) is clearly on a distinct lineage from the other species. Together, these phylogenetic analyses illustrate that O. trifallax and a bona fide S. histriomuscorum have accumulated significant evolutionary divergence from each other relative to other species.
Figure 4.
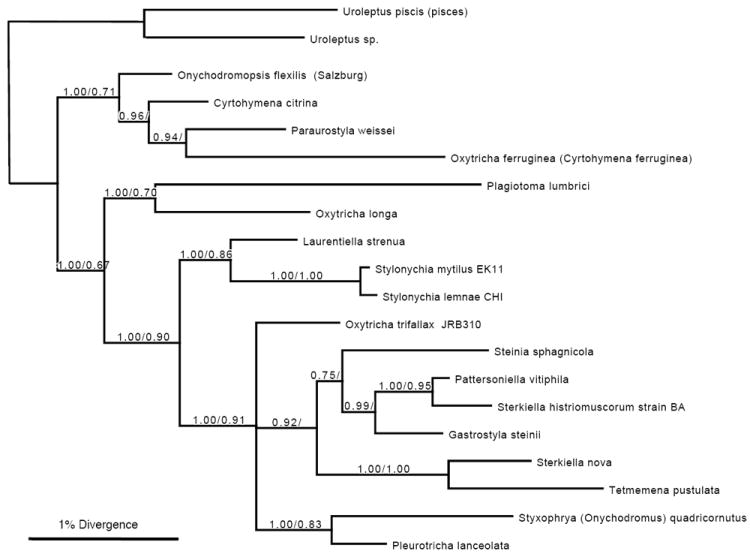
Bayesian and maximum likelihood phylogeny of small-subunit rRNA genes. Bootstrap values above 0.50 are shown (Bayesian/maximum likelihood). O. trifallax (JRB310), accession AF164121, is identical to O. trifallax AF508770; S. nova is accession X03948, S. lemnae CHI is AJ310496, S. mytilus EK11 is AJ310499, L. strenua AJ310487, S. sphagnicola AJ310494, P. vitiphila AJ310495, G. steinii AF508758, Tetmemena pustulata X03947 (formerly Stylonychia pustulata; identical to AF508775), Styxophya (also Onychodromus) quadricornutus X53485, P. lanceolata AF508768, O. longa AF508763, P. lumbrici AY547545, Oxytricha (also Cyrtohymena) ferruginea AF370027, P. weissei AJ310485, O. flexilis (Salzburg) AM412764, and C. citrina represented by AY498653. Uroleptus piscis (also pisces) AF508780, 98% identical to Uroleptus sp. AY294646 from in our lab (Chang et al. 2005), are both outgroups. S. histriomuscorum strain BA is new to this study with accession number HQ615720. The scale bar represents nucleotide divergence in Bayesian analysis.
Original isolation and characterization of O. trifallax
During 1985 and 1986 over 500 isolates of Oxytricha-like ciliates were prepared from 30 diverse freshwater sites in Indiana by selecting stichotrichs for the same general size, shape and lateral flexibility as the original O. fallax. These collection sites include a number of collections from the same stream where the original O. fallax was isolated. 268 cell lines that showed good to moderate growth and encystment were selected for further screening by dot and Southern hybridization of whole-cell DNAs to two cloned O. fallax macronuclear genes and to an O. fallax TBE1 transposon micronuclear probe (see Methods). 84 cell line DNAs cross-hybridized with the cloned macronuclear genes that had been selected for low conservation among strichotrichs, so that cross-hybridization would suggest close relatedness to O. fallax. For example, S. nova, which Berger moved to the S. histriomuscorum complex (Berger 1999), shows no cross hybridization under the conditions employed. The strongest hybridizing cell lines (like the O. fallax controls) and the moderately and weakly hybridizing classes were tentatively grouped and named unifallax, bifallax, and trifallax, respectively. All three groups are morphologically indistinguishable from the original O. fallax in protargol preparations (Fig. 5 compares the original O. fallax [unifallax] to O. trifallax) and demonstrated major lateral flexibility (Fig. 6), a criterion for the genus Oxytricha, in contrast to the rigidity of the Histriculus or Sterkiella genera (Berger 1999; Borror 1972). A morphometric analysis was not useful for identification because of overlapping size differences with large variation within a culture and in different protargol preparations (Supplementary Table S2), suggesting that such characters cannot distinguish these stichotrichs; however, we do note that size can distinguish S. histriomuscorum BA from the O. trifallax complex. All three groups also cross-hybridized in dot blots to the O. fallax TBE1 transposon probe (Herrick et al. 1985). Consistently, the cell lines that did not hybridize to the O. fallax macronuclear gene probes also did not hybridize to the TBE1 probe. The TBE1 probe also did not hybridize detectably to DNA from the Sterkiella histriomuscorum BA strain, identified by Foissner, nor to DNA of S. nova. Therefore, two features that can together define the O. fallax sibling species complex are hybridization to the TBE1 transposon probe (or TBE1-specific PCR intervals) and morphological match to established descriptions of O. fallax (Grimes 1972). These conclusions are consistent with the phylogenetic conclusions of this study (see Discussion).
Figure 5.
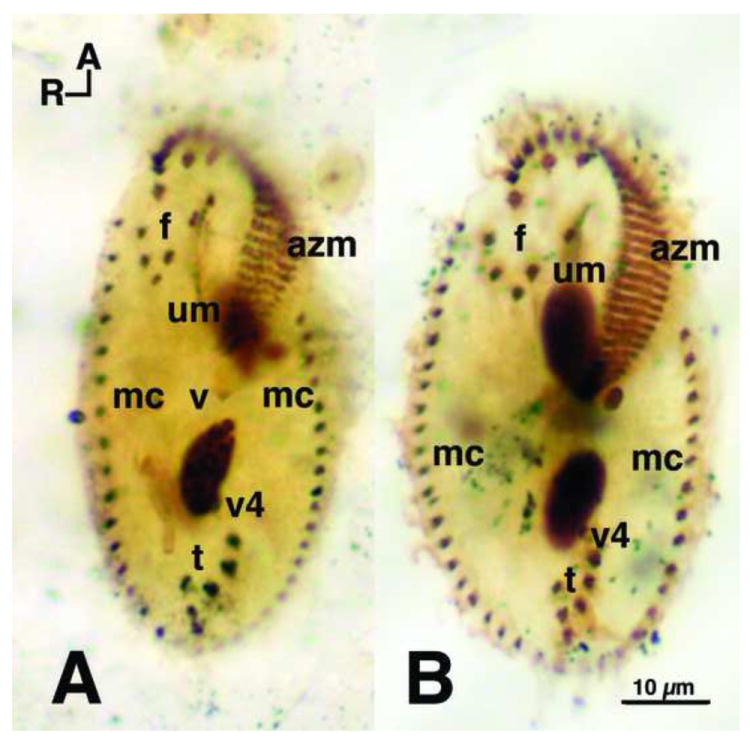
Protargol comparison of O. fallax and O. trifallax. Protargol stained cells, fixed with Perenyi’s fixative, showing the classic Oxytricha ventral surface ciliature. A. Oxytricha fallax stock 9D1 (the original Grimes stock) and B. Oxytricha trifallax JRB310, both showing the standard (Berger 1999) 18 fronto-ventral-transverse (fvt) cirri with 8 frontal cirri (f), 4 ventral cirri (v) (not all 4 shown because of focal level), and 6 transverse (t) cirri. Marginal cirri (mc) border the left and right sides, and extend to the posterior end of the cells. Caudal cirri (not shown) are also present and arise as described by Grimes (1972). The adoral zone of membranelles (azm) extend around the anterior end of the cells, down along the left side, and bend in toward the mid-ventral surface. Ventral cirri (v1 and v2) (not shown) lie below the posterior end of the azm. The undulating membranes (um), also called the endoral and paroral membranes, consist of rows of kinetosomes that appear to cross one another – a standard Oxytricha pattern (Berger 1999; Borror 1972). Dorsal ciliary rows arise as described by Grimes (1972). R and A indicate right and anterior axes, respectively.
Figure 6.
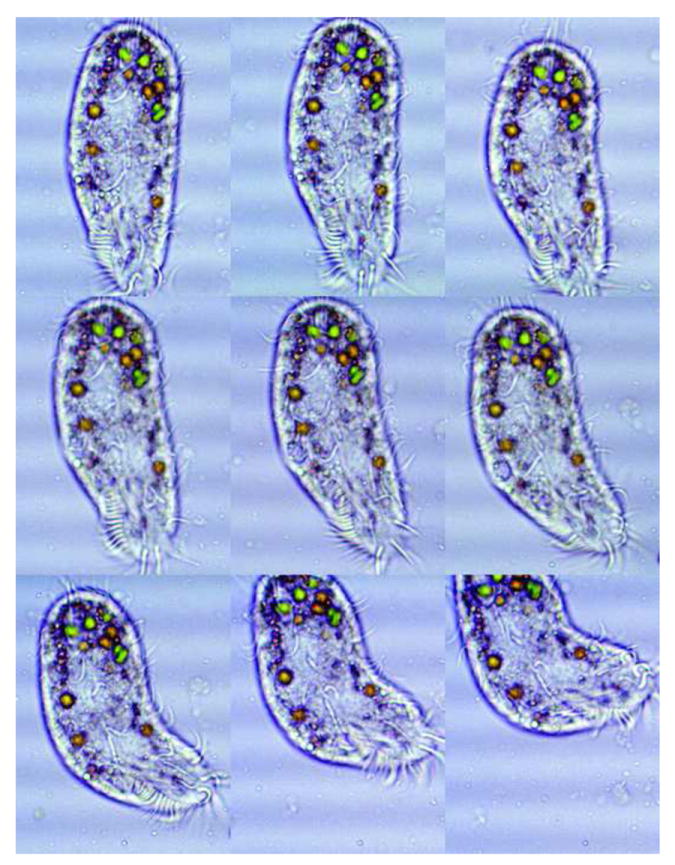
Lateral flexibility of O. trifallax. A series of nine time lapse micrographs of Oxytricha trifallax (JRB310) showing the lateral flexibility of this species. The green bodies are algae present in food vacuoles, resulting from standard growth conditions (Nowacki et al. 2008). In our experience, members of the Oxytricha genus show flexibility in a wide variety of growth conditions. Note that the adoral zone of membranelles is approximately one third total body length, while in the fixed specimen in Figure 5 it is closer to 40% and also varies with growth state, leading us to conclude that it is not a useful taxonomic character, although it was used by Foissner and Berger (1999). The ventral cirri are discernable in the middle of the body. Marginal cirri are observable on the right edges and the frontal cirri can be seen just below the anterior part of the azm. Cell movements were slowed using Protoslo®. Time lapse interval was 0.2 seconds between frames. 450X magnification.
Crosses of many of these lines in the sibling species complex to each other and to the original O. fallax revealed that O. unifallax and O. bifallax lines are either incapable of mating together, or only capable of selfing. However, the 41 O. trifallax lines fell into a single complex of eight mating groups (See Supplementary Material), reminiscent of the complex system suggested by Valbonesi, Ortenzi and Luporini (1992) for Euplotes crassus, and consistent with a mating type system defined by multiple alleles and co-dominance. Significantly, two cell lines (JRB27 and JRB51) were “promiscuous”, capable of mating with most other cell lines. These two lines can play a major role in the future identification of new O. trifallax cell lines and sibling species, because mating tests with them can detect over 80% (33 of 41) of all isolated O. trifallax tested thus far, and the addition of JRA52 (a mate-group VIII cell line) increases detection to 88% (36 of 41) of the O. trifallax cell lines, with survival through conjugation generally ranging from 65% to 95%. We also remark that O. trifallax is diploid, but rare triallelic patterns have been observed in some O. fallax strains (Herrick et al. 1987), perhaps, we propose here, due to the epigenetic transmission of somatic substitutions via RNA template-guided DNA proofreading (Nowacki et al. 2008).
Discussion
Resolving incongruent topologies
While support for the concatenated tree is strong (Fig. 1), and it is congruent with most single gene phylogenies in this study, actin I and cct-γ offer two notable exceptions (Figs 2, 3) that we discuss here.
Actin I
Because the actin I CDS gene tree was incongruent to the concatenated CDS gene tree (Figs 1, 2), we included within-species data (only CDS regions were available, but these are also more conservative) from multiple strains of S. lemnae and S. nova. The within-species data consistently group together and their presence does not alter the overall topology. All actin I CDS trees suggest that O. trifallax is divergent from S. histriomuscorum, but the placements of S. lemnae and S. nova differ from the concatenated tree. Contamination is an unlikely source of error, because DNA from different strains derived from separate experiments, often different labs in the case of intra-species data, and our own experiments displayed no evidence of PCR contamination in negative controls. However, Prescott et al. identified the presence of two actin paralogs in S. nova (Prescott and Greslin 1992). Additionally, a screen of actin I sequences against the Paramecium (Arnaiz et al. 2007) and Tetrahymena (Coyne et al. 2008) genome databases retrieved 22 and 8 copies, respectively. Thus, we cannot exclude the possibility of unrecognized paralogy (or potential loss in some lineages of the respective orthologs) confounding the actin I gene phylogeny and altering its topology (Martin and Burg 2002).
Cct-γ
The cct-γ CDS gene tree was also incongruent with the concatenated CDS tree (Figs 1, 3), although both still suggest that O. trifallax is not monophyletic with S. histriomuscorum. We recovered 4 and 1 additional cct-γ sequences from the Paramecium and Tetrahymena genome databases, respectively. Moreover, Drosophila melanogaster has two isoforms of cct (Tilley et al. 2008) that share only 60% nucleotide identity. It is therefore possible that the cct-γ phylogeny in this study is also undermined by hidden paralogy.
These two cases (actin I and cct-γ) both illustrate some common pitfalls of single gene phylogenies, suggesting that other factors may skew the tree topology (Philippe et al. 2004; Rokas et al. 2003). Likewise, it is possible that the trees presented in Schmidt et al. (2007), also based on the analysis of a single gene locus, may not be congruent to the species tree.
Hsp70
Because the hsp70 gene in most eukaryotes has distinct paralogs that target the cytoplasm, endoplasmic reticulum and mitochondria, respectively, we verified that the hsp70 sequences used in this study encode the cytoplasmic form, based on the presence of specific conserved amino-acids for the ciliate hsp70 orthologs (Budin and Philippe 1998); however, we cannot exclude the possibility of paralogs of the cytoplasmic form. The hsp70 phylogenies were concordant with the concatenated tree, however.
Congruent gene trees and concatenated CDS tree
S. histriomuscorum appears monophyletic with S. nova with 100% bootstrap support on the concatenated tree. O. trifallax diverged before this branch, also with high bootstrap support, and S. lemnae appears to have diverged earlier than O. trifallax. The long branch length to the N. ovalis outgroup could raise concern, but no earlier diverging spirotrich sequences were available from species that use the same genetic code as the main query taxa. The use of a concatenated gene tree minimizes anomalies in the data arising from lateral gene transfer, hidden paralogy, and other tree reconstruction artifacts (Philippe et al. 2004), suggesting that it would more closely approach the overall species tree (Rokas et al. 2003).
Ribosomal RNA (rRNA) comparison
The SSU ribosomal RNA tree (Figure 4) also supports the monophyly of S. histriomuscorum strain BA and S. nova, together with several other stichotrich species, and shows that Oxytricha trifallax (lab strain JRB310) diverged before this clade, with high bootstrap support for this conclusion. Hence this analysis is congruent with the concatenated protein-coding tree. This comparison notably revisited the same dataset that Schmidt et al. (2007) used to support the reclassification of S. histriomuscorum; however, their analysis did not include the S. histriomuscorum strain BA that Foissner identified (Villalobo et al. 2002) and that we sequenced in this study.
Conclusions from molecular phylogenetic analyses
Both Bayesian and maximum likelihood phylogenetic analyses of multiple data sets suggest that O. trifallax is as divergent from S. histriomuscorum as it is from other stichotrich ciliates. Our results are compatible with previous phylogenetic studies that examined subsets of these taxa (e.g., Croft et al. 2003; Hewitt et al. 2003; Hoffman and Prescott 1997a). Therefore, the 1999 morphological re-classification of O. trifallax (Foissner and Berger 1999), which lacked molecular support, should be revoked. We unequivocally identify O. trifallax (strain JRB310) as a separate and unique organism from S. histriomuscorum (strain BA). We also confirm the renaming of O. nova as Sterkiella nova, because it does group monophyletically with S. histriomuscorum.
Moreover, we are not aware of any published studies of O. trifallax that referred to it exclusively as S. histriomuscorum; however, from the time of introduction of that taxon, many authors have usually provided both names, and readers should note the strain name whenever provided. Papers where the name S. histriomuscorum has been used exclusively, such as elegant work on encystment from Baroin-Tourancheau and collaborators (e.g., Baroin-Tourancheau et al. 1999; Grisvard et al. 2008) have used the S. histriomuscorum stock BA isolated by Baroin-Tourancheau et al. (1999).
Furthermore, unless the strain name is provided, we cannot be confident that studies of O. trifallax from labs other than our own are from the species we describe in this paper. Adl and Berger obtained JRB310 and 510 from the Herrick lab to perform the seminal biological characterizations reported in 1997 and 2000, but refer to the species as “S. histriomuscorum”. The Prescott lab reported the original discovery of scrambled genes in the clonal strains of O. trifallax supplied by one of us (R.L.H.) (Greslin et al. 1989) and Prescott and Greslin (1992) reported data from JRB 310, one of those clones. However, in 1991 they isolated a new ciliate they named “O. trifallax WR” from a Colorado lake (DuBois and Prescott 1995). With the exception of Prescott and Greslin (1992), which used O. trifallax JRB310, studies of “O. trifallax” post-1991 often stated that they used DNA or cells provided by Prescott (e.g. Bryan et al. 1998; Inagaki and Doolittle 2000, 2001; Lingner et al. 1994) without strain designations, such as JRB 310, 510 or WR. Similarly, some publications from the Prescott lab after 1991 did not indicate the strain (e.g. Hewitt et al. 2003; Hoffman and Prescott 1997 a, b; Prescott et al. 1998).
Original isolation and identification of sibling species of O. fallax
The protocols used for isolation and screening of natural isolates ensured that they were all closely related to the O. fallax previously identified by Borror (Grimes 1972). While none of the isolates were identical to the original O. fallax, we conclude that all cross-hybridizing cell lines are closely related to and represent sibling species of the original O. fallax. This view is further supported by the identical cortical type (Fig. 5) and lateral flexibility (Fig. 6) in these isolates. Furthermore, hybridization to a TBE1 transposon probe (see Methods) is also limited to O. fallax and its sibling species, but notably absent from the BA strain and O. nova, which Foissner and Berger classify as S. histriomuscorum and S. nova, respectively. Therefore, hybridization to a TBE1 transposon probe can be used to screen and identify members of this sibling species complex. Future work could also use PCR to survey with conserved published TBE1 primer sets (e.g., Witherspoon et al. 1997 or Nowacki et al. 2009). Studies of within-species variation in the original O. fallax and O. trifallax support the closeness of these sibling species (e.g., Seegmiller et al. 1996).
The Oxytricha species complex
Following Sonneborn’s naming of the Paramecium aurelia sibling species (syngen) complex (Sonneborn 1975), we suggest a numerical system for the Oxytricha species complex based on genetic divergence from the original O. fallax described by Grimes. Consequently, the original O. fallax will be given the name Oxytricha unifallax, the next most closely related group of isolates will be called O. bifallax, and the third mostly closely related group is Oxytricha trifallax. Furthermore, we suggest that this classification system should not be applied to all Oxytricha fallax in the historical literature but restricted to the Oxytricha fallax identified by Borror and described by Grimes, because molecular data are unavailable for historical strains, and because the Grimes cell line, though 40 years old and incapable of successful mating, is cryogenically preserved, viable, and available for molecular comparison. While this system is not all-inclusive and new members of this species complex may yet be isolated, it provides a quantitative and general screen. The descriptions for organisms in the O. unifallax and O. bifallax categories are admittedly vague and much work would be necessary to characterize them completely. However, the description of the Oxytricha trifallax species is specific and meets all of the criteria for a biological species in the Ernst Mayr tradition. We also remark that the original Grimes’ Oxytricha fallax (Grimes 1972) was identified by A. Borror using procedures and a system of classification consistent with past practices extending into the 1800’s (Stein 1859; Stokes 1887). Because of this, the name Oxytricha fallax relates to a wealth of historical observations and experimental data from the mid 1880’s and early 1900’s that merits recognition. (Dawson 1920)
O. trifallax’s mating competency also readily permits identification of new isolates against a small number of tester lines, with JRB27, JRB51 and JRA52 able to detect approximately 90% of all known isolates of O. trifallax. These tester lines of O. trifallax, along with strains JRB310 and JRB510, which have been widely distributed to stichotrich researchers, will be deposited with ATCC to provide a reliable method for describing new isolates of O. trifallax. An additional 22 known isolates of O. trifallax will also be deposited with ATCC to provide a large population of genetically diverse clones of O. trifallax. Because these cell lines contain many allelic differences, the use of specific cell line labels (stock or strain names) is essential in any publications.
The following five criteria serve to identify future isolates of O. trifallax as part of the O. fallax sibling species complex. New isolates should:
Have a length between 60-80 microns, when feeding.
Show lateral flexibility (most easily observed in less well-fed cultures).
Demonstrate the general O. fallax ciliature pattern (see Fig. 5 legend).
Hybridize to a specific TBE1 transposon probe or demonstrate the presence of TBE1 transposons by PCR (Witherspoon et al. 1997), relative to a TBE1-lacking outgroup, such as S. nova; (this criterion alone demonstrates membership in the sibling species complex).
Show mating compatibility with one or more of the standard tester lines (JRB27, JRB51, and JRA52); this criterion confirms O. trifallax identity for all but 10% of the known lines.
The first four criteria are qualitative judgments to permit candidate screening; the final criterion is an unequivocal test of species identity, as defined for the Paramecium and Tetrahymena species complexes (Nanney and McCoy 1976; Sonneborn 1975). While a sterile isolate of O. trifallax might satisfy the first four criteria, we hope that most wild isolates would be capable of fertile matings with one or more tester lines (Haggard 1974).
We have examined S. histriomuscorum BA using these five criteria and found that its mean length and width are significantly greater than for the O. trifallax complex (Supplementary Table S2). S. histriomuscorum BA does not possess TBE1 transposons, nor can it mate with any of the standard tester lines, as well as 17 other O. trifallax cell lines. Our conclusion would be that S. histriomuscorum BA is most likely outside the O. trifallax complex, given existing data.
Therefore, in the spirit of Articles 67 and 69 of the International Code of Zoological Nomenclature (ICZN 1999) the name of Oxytricha trifallax should remain as stated to maintain nomenclature continuity and priority. O. trifallax should no longer be classified as a nomen nudum and should be recognized as an independent ciliate species, related to–but separate from–S. histriomuscorum. Our proposal follows that of Foissner and Berger (Foissner and Berger 1999).
Methods
Genomic DNA isolation
Macronuclear DNA for O. trifallax strain JRB310 was isolated from starved vegetative cells using DNAzol according to manufacturer’s instructions (Invitrogen Corp., Carlsbad, CA.). Macronuclear DNA for S. lemnae strain 2×8 was provided by Hans Lipps (University of Witten, Germany). Total DNA for S. nova was isolated from vegetative cells (gift from Martin Horvath, University of Utah, USA) as above, and suspended in 10 mM Tris and 1mM EDTA at 100ng/μL. Genomic DNA from Sterkiella histriomuscorum strain BA, identified by Foissner (Villalobo et al. 2002), was a gift from A. Baroin-Tourancheau (Université Paris-Sud). Nyctotherus ovalis cells were kindly provided by J. Hackstein (Radboud University) and DNA isolated by W.-J. Chang (Liang et al. 2005).
PCR amplification
The following nine protein-coding genes were chosen for comparative sequence analysis among the five organisms: elongation factor-1 (ef-1), 2-oxoglutarate dehydrogenase (sucA), turgor pressure sensor (turgor), ribosomal protein L10 (rL10), ubiquitin conjugating enzyme E2 (Ubc), heat shock protein 70 (hsp70), actin I, choline-phosphate cytidylyltransferase-γ (cct-γ), and tata box binding protein (tbp). The turgor, Ubc, and rL10 genes were selected using a bioinformatic screen as follows. Initially, a comprehensive search of S. histriomuscorum (strain BA) genes was conducted on GenBank. Fifteen potential macronuclear coding sequences were screened with tBLASTx against a locally-compiled database containing all current O. trifallax, S. lemnae, and N. ovalis DNA sequences (http://oxytricha.princeton.edu/genome/blast/blast.html). Genes with at least 35% nucleotide identity across all examined species were aligned with ClustalX v2.0.5 (Thompson et al. 1994). The four genes that showed specific regions of high conservation were selected for use, and PCR primers were designed from the conserved regions.
The ef-1, sucA, hsp70, actin I, cct-γ, and tbp genes were chosen due to their high conservation across eukaryotes and previous use in multi-gene phylogenies of microbial eukaryotes (e.g., Patron et al. 2007). All sequences were initially screened against GenBank to retrieve matches in ciliates. Genes that had macronuclear sequence available for ciliates related to O. trifallax were subjected to a tBLASTx search of all O. trifallax, S. lemnae, and N. ovalis sequences available at http://oxytricha.princeton.edu/genome/blast/blast.html. Genes with sequence data available from several species were selected, and primers were designed from conserved regions to recover any missing data by PCR as described below. This combined bioinformatic and experimental approach led to the production of a dataset of fifty conserved gene sequences for this study.
For initial sequencing, the polymerase chain reaction (PCR) was used with degenerate primers designed from conserved regions. Once several hundred nucleotides were sequenced from the middle of each gene from each organism, non-degenerate primers were designed for use in Telomere Suppression PCR (TSP, also telomere-specific PCR; Curtis and Landweber 1999; Siebert et al. 1995), performed as in (Chang et al. 2004) (Supplementary Table S1). This allowed recovery of complete 5’ and 3’ gene sequences all the way to the telomere, including the subtelomeric, non-coding regions, to provide complete nanochromosome sequences.
TSP primers were designed to meet the following specific conditions: Tm of 67-69 °C, at least 10 non-degenerate base pairs at the 3’ terminus, and a self-dimer ΔG value below -8.00 kcal/mol to ensure sufficient primer concentration in the reaction mixture. Tm and self-dimer values for primers were calculated using the Integrated DNA Technologies (Integrated DNA Technologies, Coralville, IA) Oligo Analyzer (www.idtdna.com/analyzer/applications/oligoanalyzer/). Consensus primers tolerated no more than 6 mismatches over a 30 base pair primer. TSP reactions used the following conditions: 100 ng template DNA, 0.2μM each oligonucleotide primer, 50mM NaCl, 1.5mM MgCl2, 0.05mM each dNTP in 50 μL containing 1x PCR buffer and 2.5 units Taq polymerase (Roche, Indianapolis, IN). All reactions included a no-template negative control to exclude the possibility of contamination. The second round of TSP (TSP2) used the first round TSP product (TSP1) diluted 1:400 as template. Each PCR used a standard TSP thermocycler program (A) adapted from Chang et al. (Chang et al. 2004). To reduce background, TSP2 sometimes used the following stringent conditions (program B): 94 °C hold; 5 cycles of (94 °C 30 sec; 73 °C 240 sec), followed by 20 cycles of (94 °C 30 sec; 69 °C 240 sec) and then 69 °C for an additional 240 sec. More relaxed conditions (program C) used 3 mM MgCl2 (Supplementary Table S1).
DNA sequencing
Each PCR was optimized to yield a specific product after one round of nested PCR, TSP2. Amplified products were directly sequenced (Genewiz, Inc. New Brunswick, NJ) after treatment with ExoSAP-IT (USB Corporation, Cleveland, OH). Occasionally products were cloned (Strataclone Cloning kit; Stratagene, Cedar Creek, TX) according to the manufacturer’s protocol, when multiple bands were present. For longer products, nested primers were used to “walk” to the end of the sequence.
Sequence alignments
We compiled full telomere-to-telomere sequence for each of the nine genes among the five ciliates. To restrict the analysis to the CDS, intron/exon structure was inferred by alignment against well-annotated orthologs when available. Where no published orthologs were available, we used the NCBI ORF Finder (http://www.ncbi.nlm.nih.gov/projects/gorf/) or Augustus (Stanke and Waack 2003) gene predictions (trained on manually curated Oxytricha trifallax gene predictions), to infer the correct CDS for each gene according to published criteria (Cavalcanti et al. 2004).
Protein sequences were initially aligned using Kalign (Lassmann and Sonnhammer 2005) with default parameters (gap opening penalty: 11.0; gap extension penalty: 0.85; terminal gap penalty: 0.45). The coding sequence multiple sequence alignment was produced by mapping aligned protein sequences back to nucleotide sequences by a custom, Genewise (Birney et al. 2004) wrapper written in Python. We then manually checked and adjusted these CDS alignments in Geneious (Drummond et al. 2009) by comparing their translated alignment to protein multiple sequences alignments generated by Clustalw2 (Larkin et al. 2007) (gap opening penalty of 10; gap extension penalty of 0.2). Poorly aligned nucleotide regions were deleted from the concatenated sequence alignment (600 nt out of 12,849 nt, or just under 5%). rRNA sequences were aligned with ClustalX (Thompson et al. 1994) (gap opening penalty of 10; gap extension penalty of 0.2).
The CDS alignments excluded 5’ and 3’ untranslated regions and introns that were inferred from our gene predictions and alignments. Most genes lacked introns, although Ubc and cct-γ each appeared to contain three. For each species we compiled a single concatenated sequence of the nine FNS or the nine CDS, with the genes ordered alphabetically by gene name.
Phylogenetic analysis
Maximum likelihood (Olsen et al. 1994) and Bayesian (Huelsenbeck and Ronquist 2001; Ronquist and Huelsenbeck 2003) phylogenetic analyses (Figs 1-4) were performed on individual gene CDS, and concatenated CDS alignments.
We used PAUP* v4.0b10 (Swofford 2002) with default parameters for maximum likelihood analysis. Unrooted trees were generated using the monophyletic outgroup method, with N. ovalis as the outgroup for all protein-coding genes. We set the number of substitution types to 2 from the Hasegawa-Kishino-Yano 85 model and the transition to transversion ratio to 2 (Hasegawa et al. 1985). The Rogers-Swofford approximation method was used to infer branch lengths (Swofford 2002). The branch and bound search method generated both scaled and unscaled trees, and a heuristic search yielded a consensus 50% majority rule tree with 1000 bootstrap replicates.
We generated Bayesian trees with MrBayes v3.1.2 (Huelsenbeck and Ronquist 2001, 2003) using a 4 by 4 nucleotide model and a transition to transversion ratio of 2 (Hasegawa et al. 1985). Branch lengths were unconstrained and all topologies were given equal probability. We ran the single gene analyses for 10,000 generations with a sampling frequency of 10 generations until the average standard deviation reached below 0.01. We ran additional generations if the average standard generation did not yet drop below this value. In all cases, the potential scale reduction factor (Gelman and Rubin 1992) approached 1.00, indicating the convergence of multiple runs upon a single topology. Again, we generated both scaled and unscaled trees, performing 1,500 bootstrap replicates to produce a consensus 50% majority rule tree. For the concatenated datasets, we ran analyses for 100,000 generations with a sampling frequency of 10 generations and 15,000 bootstrap replicates. We finally imported all sampled trees and the respective consensus trees into Mesquite v2.5 (Maddison and Maddison 2010) to generate high-resolution drawings.
For rDNA phylogenies, we used the same sequences as in Schmidt et al. (2007) to permit direct comparison of tree topologies and addition of the bona fide S. histriomuscorum to their tree. Also, we excluded cox-1, because the outgroup N. ovalis lacks it in its hydrogenosome genome (de Graaf et al. 2011), which has undergone extensive evolutionary divergence from other mitochondrial genomes, since it’s an anaerobe and doesn’t use oxygen as a terminal electron acceptor.
Protargol staining and photomicroscopy. Protargol stained specimens were prepared by a modification of the procedure of Jerka-Dziadosz and Frankel (1969). In this modification cells were preserved in Perenyi’s fixative (5 min), washed in 50% ethanol (5 min), attached to albuminized slides and then covered with a layer of flexible collodian prior to staining. Micrographs of slightly starved (because algae in food vacuoles obscure many of the ciliate’s features) and living Oxytricha trifallax (stock JRB310) were visualized at 450x using an Olympus BH-2 compound microscopic equipped with an Olympus U-PMTVC digital camera with images captured using Pixelink Capture® software (v.3.1). The software was set to capture up to nine consecutive images at 0.2 seconds per image.
Southern hybridization
Southern analysis of agarose gels and dot blots was performed as in Cartinhour and Herrick (1984). The three probes were prepared from two cloned O. fallax macronuclear genes pMA83 and pMA H1, which were from the same macronuclear DNA library (Cartinhour and Herrick 1984), and from the O. fallax transposon TBE1 (Herrick et al. 1985). Wash temperatures of 50 °C permitted strong hybridization of the O. fallax macronuclear clones to O. fallax DNA, and no detectable hybridization to S. nova DNA.
Culture and mating tests
Cells were grown and mated as described in Nowacki et al. (2008), except that small-scale mating tests were conducted in glass depression slides.
Supplementary Material
Acknowledgments
We thank Jingmei Wang for culturing O. trifallax, S. lemnae, and S. nova. Thanks to David J. Witherspoon and Kevin R. Williams for the hybridization screens with a TBE1 probe. We thank Anne Baroin-Tourancheau (S. histriomuscorum strain BA) and Wei-Jen Chang (N. ovalis) for providing DNA samples. We acknowledge funding from NSF (grant 0622112 to L.F.L.), NIH (grants GM59708 to L.F.L. and GM25203 to G.H.), Ball State University (to R.L.H.), and Princeton University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adl SM, Berger JD. Timing of life cycle morphogenesis in synchronous samples of Sterkiella histriomuscorum. I. The vegetative cell cycle. Eur J Protistol. 1997;33:99–109. doi: 10.1111/j.1550-7408.2000.tb00073.x. [DOI] [PubMed] [Google Scholar]
- Adl SM, Berger JD. Timing of life cycle morphogenesis in synchronous samples of Sterkiella histriomuscorum. II. The sexual pathway. J Eukaryot Microbiol. 2000;47:443–449. doi: 10.1111/j.1550-7408.2000.tb00073.x. [DOI] [PubMed] [Google Scholar]
- Arnaiz O, Cain S, Cohen J, Sperling L. Parameciumdb: A community resource that integrates the Paramecium tetraurelia genome sequence with genetic data. Nucleic Acids Res. 2007;35(Database issue):D439–444. doi: 10.1093/nar/gkl777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroin-Tourancheau A, Morin L, Yang T, Perasso R. Messenger RNA in dormant cells of Sterkiella histriomuscorum (Oxytrichidae): indentification of putative regulatory gene transcripts. Protist. 1999;150:137–147. doi: 10.1016/s1434-4610(99)70017-9. [DOI] [PubMed] [Google Scholar]
- Berger H. Monograph of the Oxytrichidae (Ciliophora, Hypotrichia) Monographiae Biologicae. 1999;78:i–xii. 1–1080. [Google Scholar]
- Bernhard D, Stechmann A, Foissner W, Ammermann D, Hehn M, Schlegel M. Phylogenetic relationships within the class Spirotrichea (Ciliophora) inferred from small subunit rRNA gene sequences. Mol Phylogenet Evol. 2001;21:86–92. doi: 10.1006/mpev.2001.0997. [DOI] [PubMed] [Google Scholar]
- Birney E, Clamp M, Durbin R. GeneWise and Genomewise. Genome Res. 2004;14:988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borror AC. Revision of the Order Hypotrichida (Ciliophora, Protozoa) J Protozool. 1972;19:1–23. doi: 10.1111/j.1550-7408.1972.tb03407.x. [DOI] [PubMed] [Google Scholar]
- Bryan TM, Sperger JM, Chapman KB, Cech TR. Telomerase reverse transcriptase genes identified in Tetrahymena thermophila and Oxytricha trifallax. Proc Natl Acad Sci USA. 1998;95:8479–8484. doi: 10.1073/pnas.95.15.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budin K, Philippe H. New insights into the phylogeny of eukaryotes based on ciliate hsp70 sequences. Mol Biol Evol. 1998;15:943–956. doi: 10.1093/oxfordjournals.molbev.a026010. [DOI] [PubMed] [Google Scholar]
- Cartinhour SW, Herrick GA. Three different macronuclear DNAs in Oxytricha fallax share a common sequence block. Mol Cell Biol. 1984;4:931–938. doi: 10.1128/mcb.4.5.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalcanti AR, Dunn DM, Weiss R, Herrick G, Landweber LF, Doak TG. Sequence features of Oxytricha trifallax (class Spirotrichea) macronuclear telomeric and subtelomeric sequences. Protist. 2004;155:311–322. doi: 10.1078/1434461041844196. [DOI] [PubMed] [Google Scholar]
- Chang W-J, Stover NA, Addis VM, Landweber LF. A micronuclear locus containing three protein-coding genes remains linked during macronuclear development in the spirotrichous ciliate Holosticha. Protist. 2004;155:245–255. doi: 10.1078/143446104774199628. [DOI] [PubMed] [Google Scholar]
- Chang W-J, Bryson PD, Liang H, Shin M-K, Landweber LF. The evolutionary origin of a complex scrambled gene. Proc Natl Acad Sci USA. 2005;102:15149–15154. doi: 10.1073/pnas.0507682102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne RS, Thiagarajan M, Jones KM, Wortman JR, Tallon LJ, Haas BJ, Cassidy-Hanley DM, Wiley EA, Smith JJ, Collins K, Lee SR, Couvillion MT, Liu Y, Garg J, Pearlman RE, Hamilton EP, Orias E, Eisen JA, Methe BA. Refined annotation and assembly of the Tetrahymena thermophila genome sequence through EST analysis, comparative genomic hybridization, and targeted gap closure. BMC Genomics. 2008;9:562. doi: 10.1186/1471-2164-9-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft KE, Dalby AB, Hogan DJ, Orr KE, Hewitt EA, Africa RJ, DuBois ML, Prescott DM. Macronuclear molecules encoding actins in spirotrichs. J Mol Evol. 2003;56:341–350. doi: 10.1007/s00239-002-2405-2. [DOI] [PubMed] [Google Scholar]
- Curtis EA, Landweber LF. Evolution of gene scrambling in ciliate micronuclear genes. Ann N Y Acad Sci. 1999;870:349–350. doi: 10.1111/j.1749-6632.1999.tb08900.x. [DOI] [PubMed] [Google Scholar]
- Dawson D, Herrick G. Telomeric properties of C4A4-homologous sequences in micronuclear DNA of Oxytricha fallax. Cell. 1984;36:171–177. doi: 10.1016/0092-8674(84)90086-2. [DOI] [PubMed] [Google Scholar]
- Dawson JA. An experimental study of an amicronucleate Oxytricha. II. The formation of doublet animals or ‘twins’. J Exp Zool. 1920;30:128–157. [Google Scholar]
- de Graaf RM, Ricard G, van Alen TA, Duarte I, Dutilh BE, Burgtorf C, Kuiper JW, van der Staay GW, Tielens AG, Huynen MA, Hackstein JH. The organellar genome and metabolic potential of the hydrogen-producing mitochondrion of Nyctotherus ovalis. Mol Biol Evol. 2011;28:2379–2391. doi: 10.1093/molbev/msr059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doak TG, Cavalcanti A, Stover NA, Dunn DM, Weiss R, Herrick G, Landweber LF. Sequencing the Oxytricha trifallax macronuclear genome: A pilot project. Trends Genet. 2003;19:603–607. doi: 10.1016/j.tig.2003.09.013. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Ashton B, Cheung M, Heled J, Kearse M, Moir R, Stones-Havas S, Thierer T, Wilson A. Geneious v4.8. 2009 http://www.Geneious.Com.
- DuBois M, Prescott DM. Scrambling of the actin I gene in two Oxytricha species. Proc Natl Acad Sci USA. 1995;92:3888–3892. doi: 10.1073/pnas.92.9.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foissner W, Berger H. Identification and ontogenesis of the nomen nudum hypotrichs (protozoa: Ciliophora) Oxytricha nova (= sterkiella nova sp. N.) and O. trifallax (= S. histriomuscorum) Acta Protozool. 1999;38:215–248. [Google Scholar]
- Foissner W, Blatterer H, Berger H, Kohmann F. Taxonomische und ökologische Revision der Ciliaten des Saprobiensystems - Band I: Cyrtophorida, Oligotrichida, Hypotrichia, Colpodea. 91. Vol. 1. Informationsberichte Bayer Landesamt für Wasserwirtschaft; München: 1991. pp. 1–478. [Google Scholar]
- Foissner W, Moon-van der Staay SY, van der Staay GWM, Hackstein JHP, Krautgartner W-D, Berger H. Reconciling classical and molecular phylogenies in the stichotrichines (Ciliophora, Spirotrichea), including new sequences from some rare species. Eur J Protistol. 2004;40:265–281. [Google Scholar]
- Fryd-Versavel G, Lemullois M, Aubusson-Fleury A. Maintaining cell polarity through vegetative cell pattern dedifferentiation: cytoskeleton and morphogenesis in the hypotrich ciliate Sterkiella histriomuscorum. Protist. 2010;161:222–236. doi: 10.1016/j.protis.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Gelman A, Rubin DB. Inference from iterative simulation using multiple sequences. Statistical Sci. 1992;7:457–472. [Google Scholar]
- Greslin AF, Prescott DM, Oka Y, Loukin SH, Chappell JC. Reordering of nine exons is necessary to form a functional actin gene in Oxytricha nova. Proc Natl Acad Sci USA. 1989;86:6264–6268. doi: 10.1073/pnas.86.16.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes GW. Cortical structure in nondividing and cortical morphogenesis in dividing Oxtricha fallax. J Protozool. 1972;19:428–445. [Google Scholar]
- Grimes GW, Aufderheide KJ. Cellular aspects of pattern formation: The problem of assembly. Monog Devel Biol. 1991;22:1–94. [PubMed] [Google Scholar]
- Grisvard J, Lemullois M, Morin L, Baroin-Tourancheau A. Differentially expressed genes during the encystment-excystment cycle of the ciliate Sterkiella histriomuscorum. Eur J Protistol. 2008;44:278–286. doi: 10.1016/j.ejop.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Haggard BW. Interspecies crosses in Paramecium aurelia (syngen 4 by syngen 8) J Protozool. 1974;21:152–159. doi: 10.1111/j.1550-7408.1974.tb03630.x. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Herrick G, Cartinhour SW, Williams KR, Kotter KP. Multiple sequence versions of the Oxytricha fallax 81-MAC alternate processing family. J Eukaryot Microbiol. 1987;34:429–434. doi: 10.1111/j.1550-7408.1987.tb03207.x. [DOI] [PubMed] [Google Scholar]
- Herrick G, Cartinhour S, Dawson D, Ang D, Sheets R, Lee A, Williams K. Mobile elements bounded by C4A4 telomeric repeats in Oxytricha fallax. Cell. 1985;43:759–768. doi: 10.1016/0092-8674(85)90249-1. [DOI] [PubMed] [Google Scholar]
- Hewitt EA, Müller KM, Cannone J, Hogan DJ, Gutell R, Prescott DM. Phylogenetic relationships among 28 spirotrichous ciliates documented by rDNA. Mol Phylogenet Evol. 2003;29:258–267. doi: 10.1016/s1055-7903(03)00097-6. [DOI] [PubMed] [Google Scholar]
- Hoffman DC, Prescott DM. Phylogenetic relationships among hypotrichous ciliates determined with the macronuclear gene encoding the large, catalytic subunit of DNA polymerase alpha. J Mol Evol 1997. 1997a;45:301–310. doi: 10.1007/pl00006234. [DOI] [PubMed] [Google Scholar]
- Hoffman DC, Prescott DM. Evolution of internal eliminated segments and scrambling in the micronuclear gene encoding DNA polymerase alpha in two Oxytricha species. Nucleic Acids Res. 1997b;25:1883–1889. doi: 10.1093/nar/25.10.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan DJ, Hewitt EA, Orr KE, Prescott DM, Müller KM. Evolution of IESs and scrambling in the actin I gene in hypotrichous ciliates. Proc Natl Acad Sci USA. 2001;98:15101–15106. doi: 10.1073/pnas.011578598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. Mrbayes: Bayesian inference of phylogeny. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- ICZN, International Commission on Zoological Nomenclature. International Code of Zoological Nomenclature. London, UK: The International Trust for Zoological Nomenclature; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki Y, Ford Doolittle W. Evolution of the eukaryotic translation termination system: origins of release factors. Mol Biol Evol. 2000;17:882–889. doi: 10.1093/oxfordjournals.molbev.a026368. [DOI] [PubMed] [Google Scholar]
- Inagaki Y, Doolittle WF. Class I release factors in ciliates with variant genetic codes. Nucleic Acids Res. 2001;29:921–927. doi: 10.1093/nar/29.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerka-Dziadosz M, Frankel J. An analysis of the formation of ciliary primordial in the hypotrich ciliate Urostyla weissei. J Protozool. 1969;16:612–637. doi: 10.1111/j.1550-7408.1969.tb02321.x. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lassmann T, Sonnhammer EL. Kalign--an accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics. 2005;6:298. doi: 10.1186/1471-2105-6-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, Wong J, Bao Q, Cavalcanti ARO, Landweber LF. Decoding the decoding region: analysis of eukaryotic release factor stop codon-binding residues. J Mol Evol. 2005;60:337–344. doi: 10.1007/s00239-004-0211-8. [DOI] [PubMed] [Google Scholar]
- Lingner J, Hendrick LL, Cech TR. Telomerase RNAs of different ciliates have a common secondary structure and a permuted template. Genes Dev. 1994;8:1984–1998. doi: 10.1101/gad.8.16.1984. [DOI] [PubMed] [Google Scholar]
- Lynn DH, Small EB. Phylum Ciliophora Doflein, 1901. In: Lee JJ, Leedale GF, Bradbury PC, editors. An Illustrated Guide to the Protozoa. 2. Society of Protozoologists; Lawrence, KS: 2002. pp. 371–656. [Google Scholar]
- Maddison WP, Maddison DR. Mesquite: A modular system for evolutionary analysis. Version 2.74. 2010 http://mesquiteproject.org.
- Martin AP, Burg TM. Perils of paralogy: Using hsp70 genes for inferring organismal phylogenies. Syst Biol. 2002;51:570–587. doi: 10.1080/10635150290069995. [DOI] [PubMed] [Google Scholar]
- Möllenbeck M, Cavalcanti AR, Jönsson F, Lipps HJ, Landweber LF. Interconversion of germline-limited and somatic DNA in a scrambled gene. J Mol Evol. 2006;3:69–73. doi: 10.1007/s00239-005-0166-4. [DOI] [PubMed] [Google Scholar]
- Nanney DL, Wynne McCoy J. Characterization of the species of the Tetrahymena pyriformis complex. Trans Am Microsc Soc. 1976;95:664–682. [PubMed] [Google Scholar]
- Narasimharao MV, Prescott DM. Micronuclear RNA synthesis in Paramecium caudatum. J Cell Biol. 1967;33:281–285. doi: 10.1083/jcb.33.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowacki M, Haye JE, Fang W, Vijayan V, Landweber LF. RNA-mediated epigenetic regulation of DNA copy number. Proc Natl Acad Sci USA. 2010;107:22140–22144. doi: 10.1073/pnas.1012236107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowacki M, Higgins BP, Maquilan G, Swart E, Doak TG, Landweber LF. A functional role for transposases in a large eukaryotic genome. Science. 2009;324:935–938. doi: 10.1126/science.1170023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowacki M, Vijayan V, Zhou Y, Schotanus K, Doak TG, Landweber LF. RNA-mediated epigenetic programming of a genome-rearrangement pathway. Nature. 2008;451:153–158. doi: 10.1038/nature06452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen G, Matsuda H, Hagstrom R, Overbeek R. FastDNAml: A tool for construction of phylogenetic trees of DNA sequences using maximum likelihood. Comput Appl Biosci. 1994;10:41–48. doi: 10.1093/bioinformatics/10.1.41. [DOI] [PubMed] [Google Scholar]
- Paiva TS, Borges BN, Harada ML, Silva-Neto ID. Comparative phylogenetic study of Stichotrichia (Alveolata: Ciliophora: Spirotrichea) based on 18S-rDNA sequences. Genet Mol Res. 2009;8:223–246. doi: 10.4238/vol8-1gmr529. [DOI] [PubMed] [Google Scholar]
- Patron NJ, Inagaki Y, Keeling PJ. Multiple gene phylogenies support the monophyly of cryptomonad and haptophyte host lineages. Curr Biol. 2007;17:887–891. doi: 10.1016/j.cub.2007.03.069. [DOI] [PubMed] [Google Scholar]
- Philippe H, Snell EA, Bapteste E, Lopez P, Holland PW, Casane D. Phylogenomics of eukaryotes: Impact of missing data on large alignments. Mol Biol Evol. 2004;21:1740–1752. doi: 10.1093/molbev/msh182. [DOI] [PubMed] [Google Scholar]
- Pluta AF, Kaine BP, Spear BB. The terminal organization of macronuclear DNA in Oxytricha fallax. Nucleic Acids Res. 1982;10:8145–8154. doi: 10.1093/nar/10.24.8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott JD, DuBois ML, Prescott DM. Evolution of the scrambled germline gene encoding alpha-telomere binding protein in three hypotrichous ciliates. Chromosoma. 1998;107:293–303. doi: 10.1007/s004120050311. [DOI] [PubMed] [Google Scholar]
- Prescott DM. Genome gymnastics: Unique modes of DNA evolution and processing in ciliates. Nat Rev Genet. 2000;1:191–198. doi: 10.1038/35042057. [DOI] [PubMed] [Google Scholar]
- Prescott DM, Greslin AF. Scrambled actin I gene in the micronucleus of Oxytricha nova. Dev Genet. 1992;13:66–74. doi: 10.1002/dvg.1020130111. [DOI] [PubMed] [Google Scholar]
- Rae PMM, Spear BB. Macronuclear DNA of the hypotrichous ciliate Oxytricha fallax. Proc Natl Acad Sci USA. 1978;75:4992–4996. doi: 10.1073/pnas.75.10.4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricard G, de Graaf RM, Dutilh BE, Duarte I, van Alen TA, van Hoek AH, Boxma B, van der Staay GW, Moon-van der Staay SY, Chang W-J, Landweber LF, Hackstein JH, Huynen MA. Macronuclear genome structure of the ciliate Nyctotherus ovalis: single-gene chromosomes and tiny introns. BMC Genomics. 2008;9:587. doi: 10.1186/1471-2164-9-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokas A, Williams BL, King N, Carroll SB. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature. 2003;425:798–804. doi: 10.1038/nature02053. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. Mrbayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Schmidt SL, Bernhard D, Schlegel M, Foissner W. Phylogeny of the Stichotrichia (Ciliophora; Spirotrichea) reconstructed with nuclear small subunit rRNA gene sequences: Discrepancies and accordances with morphological data. J Eukaryot Microbiol. 2007;54:201–209. doi: 10.1111/j.1550-7408.2007.00250.x. [DOI] [PubMed] [Google Scholar]
- Seegmiller A, Williams KR, Hammersmith RL, Doak TG, Witherspoon D, Messick T, Storjohann LL, Herrick G. Internal eliminated sequences interrupting the Oxytricha 81 locus: allelic divergence, conservation, conversions, and possible transposon origins. Mol Biol Evol. 1996;13:1351–1362. doi: 10.1093/oxfordjournals.molbev.a025581. [DOI] [PubMed] [Google Scholar]
- Siebert PD, Chenchik A, Kellogg DE, Lukyanov KA, Lukyanov SA. An improved pcr method for walking in uncloned genomic DNA. Nucleic Acids Res. 1995;23:1087–1088. doi: 10.1093/nar/23.6.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonneborn TM. The Paramecium aurelia complex of fourteen sibling species. Trans Am Micros Soc. 1975;94:155–178. [Google Scholar]
- Stanke M, Waack S. Gene prediction with a Hidden-Markov Model and a new intron submodel. Bioinformatics. 2003;19(suppl 2):ii215–ii225. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- Stein F. Der Organismus der Infusionsthiere nach eigenen Forschungen in systematischer Reihenfolge bearbeitet 1 Abth. Engelmann; Leipzig: 1859. p. 206. [Google Scholar]
- Stokes AC. Some new hypotrichous infusoria from American fresh waters. Ann Nat Hist. 1887;20:104–114. [Google Scholar]
- Swanton MT, Heumann JM, Prescott DM. Gene-sized DNA molecules of the macronuclei in three species of hypotrichs: Size distributions and absence of nicks. DNA of ciliated protozoa VIII Chromosoma. 1980;77:217–227. doi: 10.1007/BF00329546. [DOI] [PubMed] [Google Scholar]
- Swofford DL. PAUP*. Phylogenetic Analysis Using Parsimony (*and other methods) Sinauer Associates; Sunderland, MA: 2002. [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. Clustal w: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilley DM, Evans CR, Larson TM, Edwards KA, Friesen JA. Identification and characterization of the nuclear isoform of Drosophila melanogaster ctp: Phosphocholine cytidylyltransferase. Biochemistry. 2008;47:11838–11846. doi: 10.1021/bi801161s. [DOI] [PubMed] [Google Scholar]
- Valbonesi A, Ortenzi C, Luporini P. The species problem in a ciliate with a high multiple mating type system, Euplotes crassus. J Eukaryot Microbiol. 1992;39:45–54. [Google Scholar]
- Villalobo E, Morin L, Moch C, Lescasse R, Hanna M, Xiao W, Baroin-Tourancheau A. A homologue of croc-1 in a ciliated protist (Sterkiella histriomuscorum) testifies to the ancient origin of the ubiquitin-conjugating enzyme variant family. Mol Biol Evol. 2002;19:39–48. doi: 10.1093/oxfordjournals.molbev.a003980. [DOI] [PubMed] [Google Scholar]
- Witherspoon DJ, Doak TG, Williams KR, Seegmiller A, Seger J, Herrick G. Selection on the protein-coding genes of the TBE1 family of transposable elements in the ciliates Oxytricha fallax and O. trifallax. Mol Biol Evol. 1997;14:696–706. doi: 10.1093/oxfordjournals.molbev.a025809. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.