

Abstract
We have assembled a reliable phosphoproteomic data set for budding yeast Saccharomyces cerevisiae and have investigated its properties. Twelve publicly available phosphoproteome data sets were triaged to obtain a subset of high-confidence phosphorylation sites (p-sites), free of “noisy” phosphorylations. Analysis of this combined data set suggests that the inventory of phosphoproteins in yeast is close to completion, but that these proteins may have many undiscovered p-sites. Proteins involved in budding and protein kinase activity have high numbers of p-sites and are highly over-represented in the vast majority of the yeast phosphoproteome data sets. The yeast phosphoproteome is characterized by a few proteins with many p-sites and many proteins with a few p-sites. We confirm a tendency for p-sites to cluster together and find evidence that kinases may phosphorylate off-target amino acids that are within one or two residues of their cognate target. This suggests that the precise position of the phosphorylated amino acid is not a stringent requirement for regulatory fidelity. Compared with nonphosphorylated proteins, phosphoproteins are more ancient, more abundant, have longer unstructured regions, have more genetic interactions, more protein interactions, and are under tighter post-translational regulation. It appears that phosphoproteins constitute the raw material for pathway rewiring and adaptation at various evolutionary rates.
The application of mass spectrometry combined with affinity techniques that enrich for phosphopeptides has revolutionized the field of phosphoproteomics, such that hundreds, or even thousands, of phosphorylation sites (p-sites)1 may be identified in a single experiment. However, as with any high-throughput (HTP) technique, there are concerns about data quality and potential biases in the enrichment and identification procedures (1–3). Thus, there is a need for a stringent data evaluation to filter out possibly spurious p-sites before drawing any general conclusions about the structure and properties of a phosphoproteome.
There are several reasons for using yeast to benchmark these novel phosphoproteomics technologies. The most important of which is that a large number of phosphoproteomics experiments have been performed with Saccharomyces cerevisiae, under a reasonably wide range of conditions (4–14). Mass spectrometry-based proteomic methods sample the available proteome in a quasi-random manner (15). Moreover, a large fraction (∼80%) of the predicted yeast proteome has been found to be expressed under normal laboratory growth conditions, with high-throughput tagging or MS-based proteomics (16–18). In a specific example that highlights the power of HTP proteomics, tandem MS approaches have managed to identify ∼85–90% of all yeast mitochondrial proteins (19). Finally, there is a wealth of relevant functional genomic information available for the organism, including data on protein abundance, half-lives, and the number of kinases targeting a given protein (17, 20–22), among others. All of these factors should assist in an in-depth bioinformatics analysis of the yeast phosphoproteome.
EXPERIMENTAL PROCEDURES
Twelve high-throughput experiments between the years 2005 and 2009 were merged in the 12HQ data set (where HQ stands for high quality). Details of these data sets may be found in Table I and in the supplemental Files 1a, 1b, 1c and 2). These experiments were performed with yeast cells in a variety of physiological and developmental states, including mating, exponential growth, different phases of the cell cycle and also challenged by DNA-damaging agents, osmotic stress, rapamycin, or cycloheximide. Apart from two cell-cycle experiments, asynchronous populations were analyzed. There is substantial variation between these data sets in terms of both yeast strains used and the analytical protocols employed to identify p-sites.
Table I. The twelve publicly available phosphoproteomic data sets of the 12HQ compendium.
| Chron. order | Author/date | Conditions | p-sites | Phospho-proteins |
|---|---|---|---|---|
| 1 | Gruhler et al., 2005 | Alpha factor treated cells | 676 | 470 |
| 2 | Chi et al., 2007 | ? | 724 | 422 |
| 3 | Li et al., 2007 | Alpha factor arrested cells | 1433 | 755 |
| 4 | Albuquerque et al., 2008 | DNA-damage response (MMS) | 3155 | 1513 |
| 5 | Bodenmiller et al., 2008 | ? | 2274 | 1071 |
| 6 | Beltrao et al., 2009 | Exponential growth in rich media | 201 | 177 |
| 7 | Huber et al., 2009 | Rapamycin/ Cycloheximide | 311 | 160 |
| 8 | Holt et al., 2009 | Asynchronous population | 1939 | 857 |
| 9 | Holt et al., 2009 | Arrested in mitosis with the spindle poison nocodazole | 3348 | 1286 |
| 10 | Holt et al., 2009 | Arrested in late mitosis by overexpression of a non-degradable cyclin, Clb2-delta | 4321 | 1400 |
| 11 | Gnad et al., 2009 | Grown for 10 generations on YNB (i.e. minimal) ?+glucose until they reached log-phase | 1546 | 726 |
| 12 | Soufi et al., 2009 | Osmotic stress | 1155 | 682 |
To ensure the high quality of the combined phosphoproteome data set, we required that phosphopeptides were correctly identified with a probability of ≥99%, and that p-sites were correctly localized with a similar probability, in each experiment (see supplemental material for the thresholds applied to each public data set). These are more stringent criteria than those used in the original published studies and, therefore, our 12HQ represents a high-confidence subset of the original data, which should also address the potential problem of false positives in some of the data (by false positives, we mean inaccurate assignment of p-sites by the MS identification technology).
RESULTS AND DISCUSSION
Before analyzing the properties of the phosphoproteome, we needed to ensure that all experiments may be used and, therefore, a series of quality controls were performed.
No Single Data Set Dominates the Compendium
First, we determined whether analyses of 12HQ would be distorted by experiments with a relatively excessive number of p-sites dominating the combined data set. By removing each of the original data sets individually from 12HQ, resulted in a 0–16% reduction in the number of p-sites and a 0–11% reduction in the number of phosphoproteins. Therefore, no individual experiment dominates 12HQ, and the degree of overlap between experiments provides further assurance of the quality of the combined data set.
The Various Experiments Significantly Overlap With Each Other
Every published experiment identifies a number of p-sites that had been identified previously by other experiments. On average, it appears that, for any two experiments, ∼12% of p-sites and ∼28% of phosphoproteins are shared. The overlap observed between any two experiments is always statistically significant, whether it is for p-sites or the phosphoproteins identified (chi-square p < 0.05). Therefore, there was no need to exclude any of these twelve experiments from our study. Interestingly, two experiments from different groups that were performed in very similar conditions (alpha-factor treated cells) (9, 12) had a much lower overlap (11% of p-sites and 31% of phosphoproteins) between them than two experiments of the same group that were performed in two different phases of the cell cycle (28% and 54% respectively) (10) (see supplemental Tables 2 and 3). In terms of p-site identification, the implication is that the protocols used seem to be more important than the experimental conditions.
Saturation of the Current Phosphorylation Data Set Compendium for Yeast
Next, we investigated the likelihood that 12HQ contains the majority p-sites and phosphoproteins that make up the entire yeast phosphoproteome. Fig. 1 (below) shows the incremental increase in the total number of nonredundant p-sites and phosphoproteins identified, following each HTP experiment. It is evident that the current compendium has not reached saturation in terms of p-sites. Moreover, the manually curated data set from Phosphogrid (14), that contains only high-confidence functional p-sites from low-throughput (LTP) studies, supports this conclusion. Only 27% (131/480) of the PhosphoGrid p-sites have also been identified in any of the HTP phosphoproteomics experiments. Stark et al. (14) reached the same conclusions when they compared the PhosphoGrid data set with a more limited yeast phosphoproteomics compendium. Huber et al. (11) reported that many rapamycin-sensitive phosphorylation events that were known from the literature could not be found in their HTP experiment. Finally, Albuquerque et al. (4) reported that the extent of phosphorylation of two low-abundance proteins, Rad9p and Mrc1p was 50% less in the HTP experiment compared with another experiment in which these two proteins were purified.
Fig. 1.

Incremental increase, with time, of the compendium for nonredundant p-sites and phosphoproteins.
Regarding the identification of phosphoproteins, Fig. 1 suggests that the recorded phosphoproteome is slowly approaching saturation. Again, the PhosphoGrid data set supports this conclusion, because 85% (122/144) of the LTP-identified phosphoproteins in PhosphoGrid were also identified by the HTP phosphoproteomics experiments. Furthermore, in a study that compiled fewer experiments, Beltrao et al. (5) estimated that the detected yeast phosphoproteome from HTP studies is at the level of 81–92% saturation. Most probably, several phosphoproteins have yet to be detected, either because of their very low expression levels or because an insufficient number of biological conditions have been studied. Nevertheless, all the above facts converge on the notion that the majority of phosphoproteins has already been detected. The two trends in Fig. 1 are further supported by the fact that the average overlap of p-sites, observed between any two experiments, is 12% compared with one of 28% for phosphoproteins.
The lack of saturation in terms of p-sites could be attributed to an insufficient number of environmental conditions having been tested in the experiments, or due to biases and weaknesses of the current phosphoproteomics technologies and protocols employed. Indeed, 57% of p-sites in the compendium have been identified only once. Interestingly, the majority of detected p-sites in a given experiment do not seem to be regulated in that specific condition. Gruhler et al. (9) reported that only 18% of the detected phosphopeptides were regulated by alpha factor in their experiment, whereas Soufi et al. (13) reported that 15% of detected p-sites changed status after osmotic shock treatment. Similar conclusions were reached by Huber et al. (11) in another experiment with rapamycin treatment. In addition, Huber et al. observed that rapamycin-sensitive p-sites that had been rigorously defined by LTP experiments were not detected in their HTP experiment. This is a clear demonstration that these technologies and protocols require further development and refinement.
The Nonphosphoproteome
We wanted to investigate if there are any basic differences between phosphorylated and nonphosphorylated proteins that may affect the detection of p-sites. It is important to determine if any underlying differences have a biological basis or whether they stem from biases in the MS technologies or other experimental protocols used. Therefore, we identified a collection of yeast proteins for which there is no extant evidence of their being phosphorylated in any of the 12 HTP experiments, even if we do not apply any filters at all. We call this collection of proteins the nonphosphoproteome; it is composed of 2219 ORFs (see supplemental File S2).
It is conceivable that the nonphosphoproteome is an artifactual data set that merely contains proteins that are inherently undetectable by high-throughput (HTP) proteomic Mass-Spectrometry (MS) technologies. In order to account for this potential inherent undetectability, we also generated a subset (1418 out of the 2219 proteins) of the original nonphosphoproteome that was actually detectable by HTP-MS proteomics (designated as MS-detectable nonphosphoproteome). To this end, we used two HTP yeast proteomic data sets that were detectable by MS technology (16, 18) (see supplemental Material). These two HTP-MS experiments, when combined together, identified 4656 yeast proteins in total, where 86% of them are found in both data sets. This is a strong confirmation of the reproducibility of the MS technology for protein detection, even by different laboratories. In this study, any analyses performed with the nonphosphoproteome were also performed with the MS-detectable nonphosphoproteome, to control for protein detectability.
GO-slim analysis with Bingo (23) on the nonphosphoproteome revealed a statistically significant enrichment in proteins found in the mitochondria, membranes, cell wall, endoplasmic reticulum, and the extracellular space (the above conclusions, with the exception of those for the cell wall, are also supported by the MS-detectable nonphosphoproteome). Nevertheless, membrane proteins are considered more difficult to detect by mass spectrometry, than are cytosolic proteins. Gnad et al. (8) also reported that their data set was underrepresented in mitochondrial and endoplasmic reticulum proteins. We investigated a small data set from Reinders et al. (24) that was specifically designed to detect phosphorylation events in the mitochondrial fraction of the proteome. Previous analyses have detected ∼850 proteins in mitochondria, with a coverage of 85% of known mitochondrial proteins (19, 24, 25). From the 78 p-sites found in 46 proteins, 22 p-sites (28%) and 24 proteins (52%) were also detected in the 12HQ data set. Therefore, we believe that the under-representation of mitochondrial proteins in the HTP phosphoproteome generated by MS analyses has a biological basis. This finding may relate to the prokaryotic origin of mitochondria and the recent observations (26–34) that prokaryotic proteins are not as extensively phosphorylated as are those of eukaryotes.
The Impact of the Abundance and Half-life of Proteins
Next, we investigated if protein abundance is a confounding factor for the detection of a phosphoprotein in the MS experiments and, for this purpose, we used three comprehensive protein abundance data sets (17, 21) (see supplemental material and supplemental File S2). We compared protein abundances of the phosphoproteome (2781 proteins) against the nonphosphoproteome (2219 proteins) and observed that the phosphoproteome had, on average, a 2–3 times higher abundance than the nonphosphoproteome data set. This result was consistent for each of the three abundance data sets (Wilcoxon p < 3 e−7). We also determined whether, in each of the 12 experiments, the detected phosphoproteins were members of the higher abundance classes. In 78% of the cases (three abundance data sets for 12 MS experiments), we found the phosphoproteins to have significantly higher abundance (Wilcoxon p < 0.05). Beltrao et al. (5) also reported that phosphopeptides were three times more abundant than nonphosphorylated proteins, but regard this as a small difference, given the eight orders of magnitude span in protein abundances (5). Although a bias clearly exists, we also believe that it is not the determining factor for identifying a phosphoprotein because there is no clear distinction in the level of abundance of the phosphoproteome versus the nonphosphoproteome. The abundances of both groups of proteins span a similar range of orders of magnitude (see supplemental Material, supplemental Fig. S1). The above conclusions are also supported when accounting for HTP MS-detectability (see supplemental Material).
Another factor that might affect the detection of a phosphoprotein is its half-life, because rapid degradation could make a phosphoprotein more difficult to detect. The data indicate that, although protein turnover is a consideration, it is not a major one. When we analyzed a comprehensive yeast protein half-life data set (20) (see supplemental File S2), we found that the 12HQ phosphoproteins had, on average, a 50% lower half-life than nonphosphoproteins (Wilcoxon p < 0.0011). The above conclusions are also supported when accounting for HTP MS-detectability (see supplemental Material, Controlling for MS-detectability).
The Importance of Protein Structure
Protein kinases have a high preference for phosphorylating serine, threonine, and tyrosine (STY) amino acids that are embedded within intrinsically disordered regions (35). We wanted to investigate whether nonphosphorylated proteins lacked disordered regions and, for this purpose, the intrinsic disorder (ID) of yeast proteins was predicted (36) (see supplemental File S2). The 12HQ phosphoproteins had, on average, ID regions that were 182% longer than those of the nonphosphoproteins (an average ID length of 330 and 117 residues per protein for phosphoproteins and nonphosphoproteins respectively; this is statistically significant, Wilcoxon p = 0). Interestingly, the 12HQ phosphoproteins had, on average, nonID regions that were 38% longer than those of the nonphosphoproteins (an average non-ID length of 294 and 214 residues per protein for phosphoproteins and nonphosphoproteins, respectively; this is statistically significant, Wilcoxon p < 2e−16). The above conclusions are also supported when accounting for HTP MS-detectability (see supplemental Material, Controlling for MS-detectability). In each of the 12 experiments, proteins that were detected as phosphorylated (even if they were excluded from the 12HQ data set) always had longer ID regions than nondetected (i.e. nonphosphorylated) proteins (Wilcoxon p = 0). Thus a strong bias exists, and there is a substantial difference in the total length of disordered regions in phosphoproteins compared with nonphosphoproteins (see Fig. 2). In addition, we observed a moderate correlation (Pearson coefficient = 0.55) between the number of p-sites on a protein and the length of its ID region. It should be noted that ID regions are not only involved in interactions with kinases, but in transient protein interactions in general (37).
Fig. 2.

Boxplot of the length (in amino acids) of regions of intrinsic disorder, for the nonphosphoproteome (denoted with 0) and the 12HQ phosphoproteome (denoted with 1).
Peptide Analysis
We next investigated whether there were likely to be differences in the length or relative charge of the digested peptides generated for MS analysis between phosphorylated and nonphosphorylated proteins. It is conceivable that the current protocols can detect only a narrow spectrum of the phosphopeptides that is not present in the negative data set. We thus performed a theoretical trypsin digestion of the proteins in the two data sets with the proteogest tool (38) and calculated the length and relative charge of the theoretical peptides. We did not observe any substantial difference in the distribution of either variable between the two data sets (see supplemental Material, supplemental Fig. S2). The above conclusions are also supported when accounting for HTP MS-detectability (see supplemental Material, Controlling for MS-detectability).
Functionality of p-Sites and Biological Noise
Recently, concerns have been raised about the functionality of p-sites detected in analyses using MS and that the importance of biological noise has been underestimated in these HTP experiments (2, 3). Lienhard has raised the possibility that, due to the high sensitivity of these MS instruments, biologically noisy p-sites are being detected (3). “Biological noise,” in this case, represents phosphorylation events occurring in degenerate motifs by noncognate kinases; frequent (but low abundance) off-target phosphorylations, etc. Landry et al. exploited evolutionary information to estimate that up to 65% of p-sites in these HTP experiments could be nonfunctional, thus indicating that biological noise could be a significant problem (2).
The presence of such a high number of HTP MS experiments for yeast allows us to address this very important issue. First of all, a basic assumption is made, that a p-site identified in many experiments is probably not because of stochastic off-target kinase interactions but, rather, has a high probability of being functional. Several factors could possibly invalidate this basic assumption, such as the inherent detectability of certain proteins or p-sites, driven by protein abundance, modification stoichiometry, peptide properties etc. In addition, we cannot exclude the possibility that the functional effect of particular phosphorylation events is entirely neutral. Nevertheless, strong indications of the validity of this basic assumption come from five independent analyses, shown below:
First, the 12HQ compendium was compared with a list of proteins that might be phosphorylated in any of the 12 MS experiments but did not meet our stringent filtering criteria; the 12HQ set was found to be more enriched in PhosphoGrid proteins (5.1% versus 1.7% respectively; chi-squared p = 5e−7). Within the 12HQ compendium, we compared a list of proteins identified as phosphorylated in 3 or more experiments versus another list of proteins identified as phosphorylated in 1 or 2 experiments and found that the first list was more enriched in PhosphoGrid proteins (6.4% versus 3.4% respectively; chi-squared p = 0.0015). Therefore, as our filtering criteria become more stringent, the corresponding data sets are also becoming more enriched in proteins from the “gold-standard ” PhosphoGrid data set, which is compiled from low-throughput experiments and so is not affected by biases of the high-throughput experiments.
Second, we wanted to exclude the possibility that protein abundance, or the length or chemistry of a digested peptide, affected the number of times a protein was detected as phosphorylated in our 12HQ compendium. To examine this possibility, we binned proteins in 12 groups, depending on how many times they were found to be phosphorylated. These bins were compared for each of the above three properties (abundance, peptide length, peptide relative charge), but no significant differences were found between the different bins (see supplemental Material and supplemental Figs. S2, S4, and S5).
Third, we investigated whether p-sites identified in only a few experiments tend to be found within more degenerate motifs than p-sites identified in many experiments. For this analysis, we used the netphosyeast prediction algorithm that is considered the best performing algorithm for yeast motifs (39) (see supplemental File S1a). Netphosyeast makes predictions for serines and threonines, but not for tyrosines. As a negative comparator, we used the 195,109 ST amino acids in the 12HQ proteins, for which there is no evidence of phosphorylation, even if we do not apply any filters on the data (see supplemental File S1b). This collection of ST amino acids constitute the no-p-sites data set, symbolized with zero in Fig. 3. As a positive comparator, we also used the 473 ST amino acids in the PhosphoGrid data set; these are known to be phosphorylated and functional (see supplemental File S1c). This collection of ST amino acids is symbolized with 13 in Fig. 3. It is evident that p-sites with a coverage of 2 × or more have very similar median prediction scores to that of the PhosphoGrid set of known functional p-sites. Furthermore, the differences in netphosyeast scores were statistically different among the various adjacent bins of p-site coverage (negative versus 1×, 1x versus 2×; Wilcoxon test p < 2e-16 and p < 2e-16 respectively).
Fig. 3.

Boxplot of netphosyeast prediction scores for p-sites with a certain coverage (number of times identified). 0 = the 195,109 ST amino acids in the 12HQ proteins, for which there is no evidence that they are phosphorylated, even when no filters are applied. 1–12 = the number of experiments in the 12HQ set in which an ST amino acid has been detected as phosphorylated. 13 = the 473 ST amino acids known to be phosphorylated and functional according to the PhosphoGrid data set.
Fourth, the same conclusions about motif degeneracy are reached when we analyze the predictions supplied by Mok et al. (40) (see supplemental Files S1a, S1b, S1c). This group used a peptide library approach to determine consensus phosphorylation site motifs for almost half the yeast protein kinases (61/122). By integrating these sequence motifs together with other features, such as evolutionary conservation, disorder and protein surface accessibility, they used a Bayesian algorithm (MOTIPS) to predict phosphorylation sites for certain kinases. By applying a likelihood threshold of >0.5, we observed that MOTIPS could assign a kinase to 34% (165/480) p-sites in PhosphoGrid, 40% (1027/2566) p-sites in 12HQ_3x (p-sites that have been detected in 3 or more experiments), 34% (3359/9783) p-sites of 12HQ, and 15% (34916/239269) of the non-p-sites in the 12HQ data set. Thus the Mok et al. predictions, which are independent from those of netphosyeast, confirm that p-sites with higher coverage have both higher prediction scores from netphosyeast and more predicted phosphorylation motifs from MOTIPS.
Fifth, an additional indication that our assumption holds is the observation that, for disordered regions, p-sites detected in 3 or more experiments evolve 8% more slowly than p-sites detected in only 1 experiment (Wilcoxon p < 9e−6). In addition, for disordered regions, p-sites detected in 3 or more experiments evolve 10% more slowly than nonphosphorylated S/T/Ys from 12HQ proteins (Wilcoxon p < 3e−12). For this analysis, we used the evolutionary rates calculated by Landry et al. (2) (see supplemental File S1a and S1b).
An important question, then, is: in how many experiments should a p-site have been discovered in order to confidently designate it as functional? To address this, we simulated the 12 phosphoproteomic experiments by shuffling the positions of the p-sites. For the simulation, we took into account the structure of the proteins (order/disorder), the number of STY amino acids in each protein, and the number of phosphorylation events detected in each experiment. 1000 simulations were performed and the results with the highest (by chance) coverage were retained for comparison with the observed coverage in the real data set. Fig. 4 shows the cumulative distribution of the coverage of p-sites for both the 12 experiments and for the simulation. In essence, Fig. 4 investigates how many repeated observations of low-stoichiometry off-target p-sites we would expect to find by chance, if all of the phosphorylations were low-stoichiometry off-target events.
Fig. 4.

Cumulative distribution of coverage for p-sites of the 12HQ data set and for the simulation.
We observed that, if all phosphogroups were assigned in a totally random manner, then we would expect 1614 p-sites with a coverage 2 × or more, just by chance, whereas the observed number is 4204. The equivalent percentage for ≥3 × coverage would be 177 expected and 2566 observed, whereas for ≥4 × coverage it would be 27 expected and 1734 observed. Therefore, it seems reasonable to select ≥3 × coverage as a very stringent cut-off in order to filter out off-target phosphorylation events. This cut-off does not necessarily mean that any specific p-site that has been identified only once or twice represents an example of a low-stoichiometry off-target phosphorylation event. It only provides a very conservative and confident subset, that we designate 12HQ_3×.
The distribution in Fig. 4 reveals that a substantial number of p-sites are found in many experiments. According to Soufi et al. (13) this could be explained by the asynchronous state of the cell populations in most of the experiments. However, the relatively high overlap (28 and 54% for p-sites and phosphoproteins respectively) in the 2 Holt et al. experiments (10), which characterized the phosphoproteome at two different stages of the cell cycle, indicates that this cannot be a complete explanation. We would suggest that some p-sites are ubiquitously in an “ON ” state (phosphorylated). It may be that the cell keeps a small percentage of the expressed protein molecules of a gene in this phosphorylated state and that this percentage changes according to external stimuli.
Having excluded technical noise, and having validated the high quality of the 12HQ data set, we were in a position to investigate the properties of the yeast phosphoproteome.
General Characteristics of the Phosphoproteome
The compilation of the above 12 experiments leads to the 12HQ data set, with 9783 p-sites found in 2374 phosphoproteins and the 12HQ_3 × data set with 2566 p-sites in 1112 phosphoproteins (see supplemental Files S1a, S1b, S1c, S2). Recently, Yachie et al. (41) analyzed a compendium of ∼3500 phosphoproteins containing 26,000 p-sites. We accredit this discrepancy in the absolute numbers of p-sites and phosphoproteins mainly to the very rigorous protocol we applied to filter out technical false-positive and low-abundance off-target phosphorylations, which are a major concern (2, 3). 17% of 12HQ p-sites and 12% of 12HQ_3 × p-sites are found inside or in the vicinity (± 10 amino acids) of an annotated Pfam domain (we excluded Pfam-B domains, that are un-annotated and automatically generated). Serines, threonines, and tyrosines constitute, respectively, 81%, 17%, and 2% of the phosphorylated residues in yeast. This pattern of site preference is consistent across all of the 12 HTP experiments as well as the PhosphoGrid data set, albeit with some variation. Therefore, we do not consider it an artifact of the MS technologies. The very low percentage of phosphorylated tyrosines is explained by the lack of tyrosine kinases in yeast and the dual specificity of certain kinases that may phosphorylate some tyrosines (12, 42).
Previous analyses show that most p-sites are found in disordered regions (2). Indeed, we also observed that 91% of STY p-sites are found in disordered regions, compared with 54% of nonphosphorylated STY sites. In PhosphoGrid, the percentage is very similar, around 92%. The structural properties of the region around the p-site probably play an important functional role. It is considered that kinases tend to phosphorylate sites that are easy to access, thus it makes sense that p-sites should be embedded within unstructured regions. These conclusions are robust for the 12HQ_3 × subset as well.
Functional Analysis Using GO-Slim
Functional categories that are related to cell budding and kinase activity are highly over-represented in the vast majority of the HTP experiments. Furthermore, a GO-Slim enrichment heat-map (see supplemental material; supp_GOSlim_heatmap.pdf) reveals that the functional categories of proteins that are usually found to be phosphorylated are very similar among the various experiments. Our findings are in agreement with those of Beltrao et al. (5) using data for three different yeast species (S. cerevisiae, Candida albicans, Schizosaccharomyces pombe). Their analysis showed functional categories such as budding, cytokinesis, and signal transduction to be over-represented in the phosphoproteins of all three species. This consistency in terms of GO-Slim categories is in contrast to the statistically significant, but nevertheless rather moderate, overlap of p-sites between the 12 S. cerevisiae HTP experiments. Apparently, in every HTP experiment, different p-sites are found to be phosphorylated, but usually either on the same proteins or on proteins within the same functional categories. This conclusion may be biologically meaningful, but artifacts because of the range of physiological and developmental conditions studied, or the experimental protocols employed to identify phosphorylated proteins, cannot be excluded.
Distribution of p-Sites in Yeast Proteins
On average, we found four p-sites per phosphoprotein in the 12HQ data set. Proteins with functions involved in cell budding, cytoskeleton, and signal transduction have a higher than average number (six to eight) of p-sites per phosphoprotein. The distribution of p-sites in phosphoproteins is markedly skewed (Fig. 5). Most phosphoproteins have a small number of p-sites, whereas a very small number of phosphoproteins have many p-sites. For example, the top 10 most phosphorylated proteins have between 32–54 p-sites each. When we use the more stringent 12HQ_3 × data set, we observe the same skewed distribution, but the top 10 most phosphorylated proteins have between 10–23 p-sites. Due to the incompleteness of the phosphoproteomics data sets we expect the actual number of p-sites to be significantly higher. The most phosphorylated protein (with 54 p-sites) is Sec16p (YPL085W), which is a coat protein of the COPII vesicle, required for ER transport (43). A similar distribution has been reported for other species, such as human and mouse (44).
Fig. 5.

Distribution of p-sites in proteins, for the 12HQ and 12HQ_3× data sets.
Phosphoproteins are of More Ancient Origin than Nonphosphorylated Proteins
In a previous analysis with a smaller data set, Chi et al. (7) observed that phosphoproteins tend to be of more ancient evolutionary origin than randomly chosen proteins. By using a published data set of yeast orthologous groups (45) that was based on phylogenetic analysis and chromosomal synteny from YGOB (46), we identified orthologs in the genomes of nine other fungi and observed that a higher fraction of S. cerevisiae phosphoproteins have orthologs in other fungal genomes than do nonphosphorylated proteins (see Fig. 6). The above conclusions are also supported when analyzing the 12HQ_3 × data set and also when accounting for HTP MS-detectability (see supplemental Material, Controlling for MS-detectability).
Fig. 6.

Orthology ratio for various fungi (orthologs based on phylogeny and YGOB) for the 12HQ versus negative data set (blue color), for the 12HQ_3x versus negative data set (yellow color) and the MS-detectable 12HQ versus negative data set (red color). A ratio value >1 indicates that phosphoproteins have more yeast orthologs than the negative data set, in that particular fungus.
Phosphoproteins and Essentiality
For this analysis, we used a high-confidence list of 956 well-defined essential genes (47–49) (see supplemental File S2). We observed that 23% of phopshoproteins are products of these essential genes, compared with 10% of nonphosphoproteins (Chi-squared < 6e−23). Nevertheless, the relationship between phosphorylation and essentiality is not a simple one, because only 17% of proteins with many p-sites (≥10) are essential. Thus a high number of phosphorylation sites on a protein does not necessarily increase the likelihood of its being essential. Indeed, in a previous study, we have shown that genes encoding phosphoproteins are more likely to be maintained in duplicate following whole-genome duplication events (50). Our findings seem to contradict a previous analysis by Chi et al. (7), performed with a much smaller data set, where there was no difference in essentiality between phosphoproteins and randomly selected proteins. Interestingly, when accounting for HTP MS-detectability (based on peptide count) a higher fraction of phoshoproteins is essential, compared with nonphosphoproteins, but the difference is not statistically supported any more (see supplemental Material, Controlling for MS-detectability). Therefore, this issue of enrichment for essential genes/proteins in the phosphoproteome has yet to be clearly resolved.
Phosphoproteins are Under Tighter Regulatory Control than Nonphosphorylated Proteins
Gsponer et al. (37) have independently shown that unstructured proteins are under tight regulation at many levels of gene expression; therefore, we wanted to investigate if phosphoproteins are under tighter regulatory control than nonphosphorylated proteins. We analyzed functional data such as the number of TFs that bind to the promoters of genes (51–53), protein half-lives (20), protein ubiquitination (54), high-confidence genetic (55) or protein-protein (56) interactions and the number of different kinases targeting a protein (22) (see supplemental File S2). We observed no statistically significant difference in the number of TFs that bind the promoters of genes that encode phosphoproteins compared with those that encode nonphosphorylated proteins. Nevertheless, phosphoproteins have, on average, 43–50% shorter protein half-lives (Wilcoxon p < 0.0011) than nonphosphorylated proteins. In addition, a higher fraction of phosphoproteins are ubiquitinated, compared with the nonphosphoproteome (27 and 9% respectively; Chi-squared < 2e−16). Phosphoprotein genes have, on average, 39–40% more genetic interactions than the nonphosphorylated data set (Wilcoxon p < 1.6 e−15). Furthermore, phosphoproteins have 45–48% more protein-protein interactions (Wilcoxon p < 2e−13) than nonphosphoproteins, in accordance with a recent analysis on another yeast data set (41). Additionally, we observed a moderate correlation between the number of p-sites on a protein and the number of proteins interacting with it (Spearman coefficient = 0.3, for the 12HQ data set). All of the above conclusions hold for the 12HQ_3 × data set and even when accounting for HTP MS-detectability (see supplemental Material, Controlling for MS-detectability). Recently, Shou et al. (57), demonstrated that different types of molecular networks rewire at different rates with their order being (from fast to slow) transcriptional, phosphorylation, genetic interaction, miRNA, protein interaction and metabolic pathway networks. Apparently, phosphoproteins are enriched in those elements that act as the evolutionary raw material for adaption at various speeds.
Weak Correlation Between the Number of Phosphorylation Sites on a Protein and the Number of Different Kinases that Target It
For this analysis, we used the in vitro protein array experiment of Ptacek et al. (22). As expected, the phosphoproteins data set (2374 proteins) had over three times more kinase interactions than the nonphosphoprotein data set (2219 proteins); Wilcoxon p = 0. Interestingly, the number of kinases interacting with a phosphoprotein did not correlate strongly with the number of p-sites found in the protein (Pearson coefficient = 0.18). One potential explanation is that the specific experiment that measures which kinases target a protein is noisy because it is performed in vitro. Nevertheless, there exists a statistically significant overlap among phosphoproteins found by MS experiments and by the in vitro protein array experiments (687 proteins found in both data sets phosphorylated; chi-squared <2.2 e−16). Further support for the in vitro approach comes from the fact that, for each of the 12 experiments, the proteins identified as phosphorylated were found to be targeted by 2.2–2.9 times more kinases than the proteins that were not identified as phosphorylated (Wilcoxon p < 7e−10) by the HTP in vivo approach. A second, and more plausible, explanation is that there is no 1:1 relationship between kinases and phosphorylation sites. One kinase may phosphorylate many p-sites in a protein, or the same p-site may be phosphorylated by several closely related kinases. Indeed, Schweiger and Linial showed that groups of neighboring p-sites in a protein may be phosphorylated by the same kinase (58).
Clusters of p-Sites
For more than half (51%) of 12HQ p-sites, there exists another p-site within a distance of ≤8 amino acids; for more than a third (33.5%) of the 12HQ p-sites, the interval is even smaller, ≤3 amino acids. Schweiger and Linial (58) have demonstrated, for a different data set, that this clustering is statistically significant. We repeated their analysis with the 12HQ and 12HQ_3 × high quality data sets to ensure that this clustering is not an artifact resulting from the MS analysis mistakenly assigning the phosphogroup to a neighboring STY amino acid. Such mistakes would be expected to be more common for the neighboring amino acids of the most frequently phosphorylated p-sites. For our analysis, we used p-sites with a very high probability of correct localization (>99% for each p-site), therefore, our data set tackles this very important issue.
Simulations were performed where we shuffled the p-sites in a protein, but did not change either the overall amino acid sequence, the distribution of p-sites found between disordered and ordered regions, or the total number of p-sites on the protein. One thousand simulations with the 12HQ data set showed clearly that p-sites tend to cluster together more frequently than would be expected by chance (Fig. 7), as Linial and Schweiger originally reported (58). The simulations were repeated for the 12HQ_3 × data set, with the same conclusions.
Fig. 7.

Cumulative distribution of distance among neighboring p-sites.
It has been suggested that the clustering of p-sites allows a greater flexibility of control over a protein's activity, permitting variability in the sensitivity or rapidity of the response to different intra- or extra-cellular stimuli (58–60). A more mundane, but perfectly feasible, alternative is that this clustering may partly result from misphosphorylations made by the protein kinase. It is known that protein kinases recognize very degenerate target sequences and that the specificity inside the cell is determined by many other factors (e.g. co-expression, colocalization, scaffolding, etc) (61, 62). Thus, most of the time, a kinase would detect its cognate motif and phosphorylate the correct STY amino acid. However, if there were another STY amino acid close by, then the kinase might phosphorylate this second residue in error. The error will depend on the quality of the motif. The more degenerate the noncognate motif, the less frequently an off-target phosphorylation will occur.
We measured the distance between all adjacent pairs of p-sites for the 12HQ data set and, for each of these pairs, we determined the log2 ratio of netphosyeast score and the log2 ratio of 12HQ p-site coverage. We reasoned that, if some kinases phosphorylate a neighboring (off-target) ST amino acid, then the more degenerate the motif (lower netphosyeast score), the lower the probability its being the subject of an off-target phosphorylation. We tested the correlation for various distances (e.g. one residue, two residues, etc…) against a background correlation of distance ≥20 residues. We also calculated the 95% confidence intervals for all these correlations and found that, for the 12HQ data set and a distance of two residues, the correlation is quite high (Pearson coefficient = 0.48) and also significantly higher than the background correlation (Pearson coefficient = 0.22) (see Fig. 8). We infer that some kinases may be prone to making such neighboring off-target phosphorylations, especially for amino acids within two residues of the cognate site. Interestingly, Schweiger and Linial indicate that the most prevalent distances in the observed clustering were one to four amino acids. We believe that these inherent neighboring off-target phosphorylations of kinases are tolerated by the cell because the precise positioning of phosphorylation sites is not always required for proper regulation (63), thus highlighting the robustness of this molecular network.
Fig. 8.
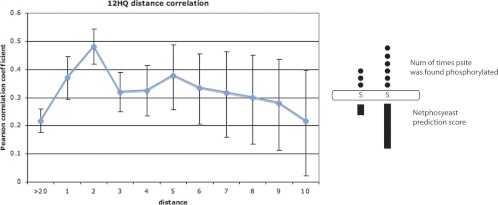
Plot of the Pearson coefficient values for log2 ratio of netphosyeast score and log2 ratio of coverage of p-sites. The correlation was calculated for neighboring p-sites with a certain distance. For example, for neighboring p-sites with a distance of two amino acids, we observe that the p-site with the higher netphosyeast score is also found in more experiments than the other p-site and that the correlation is quite high (0.48).
CONCLUSIONS
In this analysis, we integrated 12 HTP phosphoproteomic data sets from S. cerevisiae, published between the years 2005 and 2009, together with literature-curated LTP data from the PhosphoGrid database. We applied very stringent criteria to filter out both technical false-positives and low-stoichiometry off-target phosphorylations, which are a major concern (2, 3), and one that is not addressed properly in many analyses. We have thus provided a high quality data set of p-sites, which may be employed to study the general properties of the yeast phosphoproteome. Our quality controls demonstrated that every HTP experiment correctly captured a fraction of the yeast phosphoproteome, but there is still plenty of room for improvements in the technologies and protocols used. The compendium may well be approaching saturation in terms of identifying all yeast phosphoproteins, but it is far from complete in terms of identifying all the p-sites on those proteins.
The yeast phosphoproteome is characterized by a few proteins with many p-sites and many proteins with a few p-sites. Proteins involved especially in budding and protein kinase activity have high numbers of p-sites. We confirm a tendency for p-sites to cluster together and find evidence that kinases may be involved in low-stoichiometry off-target phosphorylations of amino acids that are within one or two residues of their cognate target. This suggests that the precise position of the phosphorylated amino acid is not a stringent requirement for regulatory fidelity. Compared with nonphosphorylated proteins, phosphoproteins are more ancient in evolutionary terms, have longer unstructured regions (that are fast evolving), are encoded by genes with more genetic interactions, have more protein interactions, and are under tighter post-translational regulation. Shou et al. (57) recently demonstrated that different types of molecular networks rewire at different rates with their order being (from fast to slow): transcriptional, phosphorylation, genetic interaction, miRNA, protein interaction and metabolic pathway networks. Therefore, it is conceivable that phosphoproteins act as the raw material for adaption at various evolutionary speeds.
Several of the properties that we observed in the current phosphoproteome were also observed correctly in previous and much smaller data sets, with less stringent filtering criteria. Therefore, despite the incompleteness of the current compendium, we suggest that this high-quality sample is sufficient to accurately reveal the major properties of the entire yeast phosphoproteome.
Footnotes
* This work was supported by awards to SGO from the Biotechnology and Biological Sciences Research Council (Grant BB/C505140/1) and the UNICELLSYS Collaborative Project (No. 201142) of the European Commission. GDA acknowledges additional support from EMBO (ALTF-930-2007) and the Research Commitee of the University of Thessaly, Greece (No. 4290.01.09). Y.V.d.P. acknowledges support from the Inter-University Network for Fundamental Research (P6/25) (BioMaGNet).
 This article contains supplemental Material, Files S1 and S2, Figs. S1 to S8, and Tables S1 to S6.
This article contains supplemental Material, Files S1 and S2, Figs. S1 to S8, and Tables S1 to S6.
1 The abbreviations used are:
- p-site
- phosphorylation site
- HTP
- high-throughput
- LTP
- low-throughput
- 12HQ
- high-quality compendium of the 12 data sets
- 12HQ_3X
- high-quality compendium of the 12 data sets with p-sites found in at least 3 or more experiments
- ID
- intrinsic disorder
- GO
- Gene ontology.
REFERENCES
- 1. Bodenmiller B., Mueller L. N., Mueller M., Domon B., Aebersold R. (2007) Reproducible isolation of distinct, overlapping segments of the phosphoproteome. Nat. Methods 4, 231–237 [DOI] [PubMed] [Google Scholar]
- 2. Landry C. R., Levy E. D., Michnick S. W. (2009) Weak functional constraints on phosphoproteomes. Trends Genet. 25, 193–197 [DOI] [PubMed] [Google Scholar]
- 3. Lienhard G. E. (2008) Non-functional phosphorylations? Trends Biochem. Sci. 33, 351–352 [DOI] [PubMed] [Google Scholar]
- 4. Albuquerque C. P., Smolka M. B., Payne S. H., Bafna V., Eng J., Zhou H. (2008) A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol. Cell. Proteomics 7, 1389–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beltrao P., Trinidad J. C., Fiedler D., Roguev A., Lim W. A., Shokat K. M., Burlingame A. L., Krogan N. J. (2009) Evolution of phosphoregulation: comparison of phosphorylation patterns across yeast species. PLoS Biol. 7, e1000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bodenmiller B., Campbell D., Gerrits B., Lam H., Jovanovic M., Picotti P., Schlapbach R., Aebersold R. (2008) PhosphoPep–a database of protein phosphorylation sites in model organisms. Nat. Biotechnol. 26, 1339–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chi A., Huttenhower C., Geer L. Y., Coon J. J., Syka J. E., Bai D. L., Shabanowitz J., Burke D. J., Troyanskaya O. G., Hunt D. F. (2007) Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc. Natl. Acad. Sci. USA 104, 2193–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gnad F., de Godoy L. M., Cox J., Neuhauser N., Ren S., Olsen J. V., Mann M. (2009) High-accuracy identification and bioinformatic analysis of in vivo protein phosphorylation sites in yeast. Proteomics 9, 4642–4652 [DOI] [PubMed] [Google Scholar]
- 9. Gruhler A., Olsen J. V., Mohammed S., Mortensen P., Faergeman N. J., Mann M., Jensen O. N. (2005) Quantitative phosphoproteomics applied to the yeast pheromone signaling pathway. Mol. Cell. Proteomics 4, 310–327 [DOI] [PubMed] [Google Scholar]
- 10. Holt L. J., Tuch B. B., Villén J., Johnson A. D., Gygi S. P., Morgan D. O. (2009) Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325, 1682–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huber A., Bodenmiller B., Uotila A., Stahl M., Wanka S., Gerrits B., Aebersold R., Loewith R. (2009) Characterization of the rapamycin-sensitive phosphoproteome reveals that Sch9 is a central coordinator of protein synthesis. Genes Dev. 23, 1929–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li X., Gerber S. A., Rudner A. D., Beausoleil S. A., Haas W., Villén J., Elias J. E., Gygi S. P. (2007) Large-scale phosphorylation analysis of alpha-factor-arrested Saccharomyces cerevisiae. J. Proteome Res. 6, 1190–1197 [DOI] [PubMed] [Google Scholar]
- 13. Soufi B., Kelstrup C. D., Stoehr G., Fröhlich F., Walther T. C., Olsen J. V. (2009) Global analysis of the yeast osmotic stress response by quantitative proteomics. Mol. Biosyst. 5, 1337–1346 [DOI] [PubMed] [Google Scholar]
- 14. Stark C., Su T. C., Breitkreutz A., Lourenco P., Dahabieh M., Breitkreutz B. J., Tyers M., Sadowski I. (2010) PhosphoGRID: a database of experimentally verified in vivo protein phosphorylation sites from the budding yeast Saccharomyces cerevisiae. Database 2010, bap026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 16. de Godoy L. M., Olsen J. V., Cox J., Nielsen M. L., Hubner N. C., Fröhlich F., Walther T. C., Mann M. (2008) Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 455, 1251–1254 [DOI] [PubMed] [Google Scholar]
- 17. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 18. Wu R., Dephoure N., Haas W., Huttlin E. L., Zhai B., Sowa M. E., Gygi S. P. (2011) Correct interpretation of comprehensive phosphorylation dynamics requires normalization by protein expression changes. Mol. Cell. Proteomics 10, M111.009654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sickmann A., Reinders J., Wagner Y., Joppich C., Zahedi R., Meyer H. E., Schönfisch B., Perschil I., Chacinska A., Guiard B., Rehling P., Pfanner N., Meisinger C. (2003) The proteome of Saccharomyces cerevisiae mitochondria. Proc. Natl. Acad. Sci. USA 100, 13207–13212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Belle A., Tanay A., Bitincka L., Shamir R., O'Shea E. K. (2006) Quantification of protein half-lives in the budding yeast proteome. Proc. Natl. Acad. Sci. USA 103, 13004–13009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Newman J. R., Ghaemmaghami S., Ihmels J., Breslow D. K., Noble M., DeRisi J. L., Weissman J. S. (2006) Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature 441, 840–846 [DOI] [PubMed] [Google Scholar]
- 22. Ptacek J., Devgan G., Michaud G., Zhu H., Zhu X., Fasolo J., Guo H., Jona G., Breitkreutz A., Sopko R., McCartney R. R., Schmidt M. C., Rachidi N., Lee S. J., Mah A. S., Meng L., Stark M. J., Stern D. F., De Virgilio C., Tyers M., Andrews B., Gerstein M., Schweitzer B., Predki P. F., Snyder M. (2005) Global analysis of protein phosphorylation in yeast. Nature 438, 679–684 [DOI] [PubMed] [Google Scholar]
- 23. Maere S., Heymans K., Kuiper M. (2005) BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449 [DOI] [PubMed] [Google Scholar]
- 24. Reinders J., Wagner K., Zahedi R. P., Stojanovski D., Eyrich B., van der Laan M., Rehling P., Sickmann A., Pfanner N., Meisinger C. (2007) Profiling phosphoproteins of yeast mitochondria reveals a role of phosphorylation in assembly of the ATP synthase. Mol. Cell. Proteomics 6, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 25. Reinders J., Zahedi R. P., Pfanner N., Meisinger C., Sickmann A. (2006) Toward the complete yeast mitochondrial proteome: multidimensional separation techniques for mitochondrial proteomics. J. Proteome Res. 5, 1543–1554 [DOI] [PubMed] [Google Scholar]
- 26. Lin M. H., Hsu T. L., Lin S. Y., Pan Y. J., Jan J. T., Wang J. T., Khoo K. H., Wu S. H. (2009) Phosphoproteomics of Klebsiella pneumoniae NTUH-K2044 reveals a tight link between tyrosine phosphorylation and virulence. Mol. Cell. Proteomics 8, 2613–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Macek B., Gnad F., Soufi B., Kumar C., Olsen J. V., Mijakovic I., Mann M. (2008) Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol. Cell. Proteomics 7, 299–307 [DOI] [PubMed] [Google Scholar]
- 28. Macek B., Mijakovic I., Olsen J. V., Gnad F., Kumar C., Jensen P. R., Mann M. (2007) The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol. Cell. Proteomics 6, 697–707 [DOI] [PubMed] [Google Scholar]
- 29. Parker J. L., Jones A. M., Serazetdinova L., Saalbach G., Bibb M. J., Naldrett M. J. (2010) Analysis of the phosphoproteome of the multicellular bacterium Streptomyces coelicolor A3(2) by protein/peptide fractionation, phosphopeptide enrichment and high-accuracy mass spectrometry. Proteomics 10, 2486–2497 [DOI] [PubMed] [Google Scholar]
- 30. Prisic S., Dankwa S., Schwartz D., Chou M. F., Locasale J. W., Kang C. M., Bemis G., Church G. M., Steen H., Husson R. N. (2010) Extensive phosphorylation with overlapping specificity by Mycobacterium tuberculosis serine/threonine protein kinases. Proc. Natl. Acad. Sci. USA 107, 7521–7526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ravichandran A., Sugiyama N., Tomita M., Swarup S., Ishihama Y. (2009) Ser/Thr/Tyr phosphoproteome analysis of pathogenic and non-pathogenic Pseudomonas species. Proteomics 9, 2764–2775 [DOI] [PubMed] [Google Scholar]
- 32. Schmidl S. R., Gronau K., Pietack N., Hecker M., Becher D., Stülke J. (2010) The phosphoproteome of the minimal bacterium Mycoplasma pneumoniae: analysis of the complete known Ser/Thr kinome suggests the existence of novel kinases. Mol. Cell. Proteomics 9, 1228–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Soufi B., Gnad F., Jensen P. R., Petranovic D., Mann M., Mijakovic I., Macek B. (2008) The Ser/Thr/Tyr phosphoproteome of Lactococcus lactis IL1403 reveals multiply phosphorylated proteins. Proteomics 8, 3486–3493 [DOI] [PubMed] [Google Scholar]
- 34. Sun X., Ge F., Xiao C. L., Yin X. F., Ge R., Zhang L. H., He Q. Y. (2010) Phosphoproteomic analysis reveals the multiple roles of phosphorylation in pathogenic bacterium Streptococcus pneumoniae. J. Proteome Res. 9, 275–282 [DOI] [PubMed] [Google Scholar]
- 35. Iakoucheva L. M., Radivojac P., Brown C. J., O'Connor T. R., Sikes J. G., Obradovic Z., Dunker A. K. (2004) The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 32, 1037–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peng K., Radivojac P., Vucetic S., Dunker A. K., Obradovic Z. (2006) Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics 7, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gsponer J., Futschik M. E., Teichmann S. A., Babu M. M. (2008) Tight regulation of unstructured proteins: from transcript synthesis to protein degradation. Science 322, 1365–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cagney G., Amiri S., Premawaradena T., Lindo M., Emili A. (2003) In silico proteome analysis to facilitate proteomics experiments using mass spectrometry. Proteome Sci. 1, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ingrell C. R., Miller M. L., Jensen O. N., Blom N. (2007) NetPhosYeast: prediction of protein phosphorylation sites in yeast. Bioinformatics 23, 895–897 [DOI] [PubMed] [Google Scholar]
- 40. Mok J., Kim P. M., Lam H. Y., Piccirillo S., Zhou X., Jeschke G. R., Sheridan D. L., Parker S. A., Desai V., Jwa M., Cameroni E., Niu H., Good M., Remenyi A., Ma J. L., Sheu Y. J., Sassi H. E., Sopko R., Chan C. S., De Virgilio C., Hollingsworth N. M., Lim W. A., Stern D. F., Stillman B., Andrews B. J., Gerstein M. B., Snyder M., Turk B. E. (2010) Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci. Signal. 3, ra12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yachie N., Saito R., Sugiyama N., Tomita M., Ishihama Y. (2011) Integrative features of the yeast phosphoproteome and protein-protein interaction map. PLoS Comput. Biol. 7, e1001064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Manning G., Plowman G. D., Hunter T., Sudarsanam S. (2002) Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 27, 514–520 [DOI] [PubMed] [Google Scholar]
- 43. Supek F., Madden D. T., Hamamoto S., Orci L., Schekman R. (2002) Sec16p potentiates the action of COPII proteins to bud transport vesicles. J. Cell Biol. 158, 1029–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yachie N., Saito R., Sugahara J., Tomita M., Ishihama Y. (2009) In silico analysis of phosphoproteome data suggests a rich-get-richer process of phosphosite accumulation over evolution. Mol. Cell. Proteomics 8, 1061–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wapinski I., Pfeffer A., Friedman N., Regev A. (2007) Natural history and evolutionary principles of gene duplication in fungi. Nature 449, 54–61 [DOI] [PubMed] [Google Scholar]
- 46. Byrne K. P., Wolfe K. H. (2005) The Yeast Gene Order Browser: combining curated homology and syntenic context reveals gene fate in polyploid species. Genome Res. 15, 1456–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Giaever G., Chu A. M., Ni L., Connelly C., Riles L., Véronneau S., Dow S., Lucau-Danila A., Anderson K., André B., Arkin A. P., Astromoff A., El-Bakkoury M., Bangham R., Benito R., Brachat S., Campanaro S., Curtiss M., Davis K., Deutschbauer A., Entian K. D., Flaherty P., Foury F., Garfinkel D. J., Gerstein M., Gotte D., Guldener U., Hegemann J. H., Hempel S., Herman Z., Jaramillo D. F., Kelly D. E., Kelly S. L., Kötter P., LaBonte D., Lamb D. C., Lan N., Liang H., Liao H., Liu L., Luo C., Lussier M., Mao R., Menard P., Ooi S. L., Revuelta J. L., Roberts C. J., Rose M., Ross-Macdonald P., Scherens B., Schimmack G., Shafer B., Shoemaker D. D., Sookhai-Mahadeo S., Storms R. K., Strathern J. N., Valle G., Voet M., Volckaert G., Wang C. Y., Ward T. R., Wilhelmy J., Winzeler E. A., Yang Y., Yen G., Youngman E., Yu K., Bussey H., Boeke J. D., Snyder M., Philippsen P., Davis R. W., Johnston M. (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391 [DOI] [PubMed] [Google Scholar]
- 48. Pache R. A., Babu M. M., Aloy P. (2009) Exploiting gene deletion fitness effects in yeast to understand the modular architecture of protein complexes under different growth conditions. BMC Syst. Biol. 3, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Steinmetz L. M., Scharfe C., Deutschbauer A. M., Mokranjac D., Herman Z. S., Jones T., Chu A. M., Giaever G., Prokisch H., Oefner P. J., Davis R. W. (2002) Systematic screen for human disease genes in yeast. Nat. Genet. 31, 400–404 [DOI] [PubMed] [Google Scholar]
- 50. Amoutzias G. D., He Y., Gordon J., Mossialos D., Oliver S. G., Van de Peer Y. (2010) Posttranslational regulation impacts the fate of duplicated genes. Proc. Natl. Acad. Sci. USA 107, 2967–2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Balaji S., Iyer L. M., Babu M. M., Aravind L. (2008) Comparison of transcription regulatory interactions inferred from high-throughput methods: what do they reveal? Trends Genet. 24, 319–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Harbison C. T., Gordon D. B., Lee T. I., Rinaldi N. J., Macisaac K. D., Danford T. W., Hannett N. M., Tagne J. B., Reynolds D. B., Yoo J., Jennings E. G., Zeitlinger J., Pokholok D. K., Kellis M., Rolfe P. A., Takusagawa K. T., Lander E. S., Gifford D. K., Fraenkel E., Young R. A. (2004) Transcriptional regulatory code of a eukaryotic genome. Nature 431, 99–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee T. I., Rinaldi N. J., Robert F., Odom D. T., Bar-Joseph Z., Gerber G. K., Hannett N. M., Harbison C. T., Thompson C. M., Simon I., Zeitlinger J., Jennings E. G., Murray H. L., Gordon D. B., Ren B., Wyrick J. J., Tagne J. B., Volkert T. L., Fraenkel E., Gifford D. K., Young R. A. (2002) Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298, 799–804 [DOI] [PubMed] [Google Scholar]
- 54. Peng J., Schwartz D., Elias J. E., Thoreen C. C., Cheng D., Marsischky G., Roelofs J., Finley D., Gygi S. P. (2003) A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 21, 921–926 [DOI] [PubMed] [Google Scholar]
- 55. Costanzo M., Baryshnikova A., Bellay J., Kim Y., Spear E. D., Sevier C. S., Ding H., Koh J. L., Toufighi K., Mostafavi S., Prinz J., St Onge R. P., VanderSluis B., Makhnevych T., Vizeacoumar F. J., Alizadeh S., Bahr S., Brost R. L., Chen Y., Cokol M., Deshpande R., Li Z., Lin Z. Y., Liang W., Marback M., Paw J., San Luis B. J., Shuteriqi E., Tong A. H., van Dyk N., Wallace I. M., Whitney J. A., Weirauch M. T., Zhong G., Zhu H., Houry W. A., Brudno M., Ragibizadeh S., Papp B., Pál C., Roth F. P., Giaever G., Nislow C., Troyanskaya O. G., Bussey H., Bader G. D., Gingras A. C., Morris Q. D., Kim P. M., Kaiser C. A., Myers C. L., Andrews B. J., Boone C. (2010) The genetic landscape of a cell. Science 327, 425–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Batada N. N., Reguly T., Breitkreutz A., Boucher L., Breitkreutz B. J., Hurst L. D., Tyers M. (2006) Stratus not altocumulus: a new view of the yeast protein interaction network. PLoS Biol. 4, e317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shou C., Bhardwaj N., Lam H. Y., Yan K. K., Kim P. M., Snyder M., Gerstein M. B. (2011) Measuring the evolutionary rewiring of biological networks. PLoS Comput. Biol. 7, e1001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schweiger R., Linial M. (2010) Cooperativity within proximal phosphorylation sites is revealed from large-scale proteomics data. Biol. Direct 5, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gunawardena J. (2005) Multisite protein phosphorylation makes a good threshold but can be a poor switch. Proc. Natl. Acad. Sci. USA 102, 14617–14622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nash P., Tang X., Orlicky S., Chen Q., Gertler F. B., Mendenhall M. D., Sicheri F., Pawson T., Tyers M. (2001) Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature 414, 514–521 [DOI] [PubMed] [Google Scholar]
- 61. Linding R., Jensen L. J., Ostheimer G. J., van Vugt M. A., Jørgensen C., Miron I. M., Diella F., Colwill K., Taylor L., Elder K., Metalnikov P., Nguyen V., Pasculescu A., Jin J., Park J. G., Samson L. D., Woodgett J. R., Russell R. B., Bork P., Yaffe M. B., Pawson T. (2007) Systematic discovery of in vivo phosphorylation networks. Cell 129, 1415–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Won A. P., Garbarino J. E., Lim W. A. (2011) Recruitment interactions can override catalytic interactions in determining the functional identity of a protein kinase. Proc. Natl. Acad. Sci. USA 108, 9809–9814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Moses A. M., Liku M. E., Li J. J., Durbin R. (2007) Regulatory evolution in proteins by turnover and lineage-specific changes of cyclin-dependent kinase consensus sites. Proc. Natl. Acad. Sci. USA 104, 17713–17718 [DOI] [PMC free article] [PubMed] [Google Scholar]