

Abstract
Data processing forms an integral part of biomarker discovery and contributes significantly to the ultimate result. To compare and evaluate various publicly available open source label-free data processing workflows, we developed msCompare, a modular framework that allows the arbitrary combination of different feature detection/quantification and alignment/matching algorithms in conjunction with a novel scoring method to evaluate their overall performance. We used msCompare to assess the performance of workflows built from modules of publicly available data processing packages such as SuperHirn, OpenMS, and MZmine and our in-house developed modules on peptide-spiked urine and trypsin-digested cerebrospinal fluid (CSF) samples. We found that the quality of results varied greatly among workflows, and interestingly, heterogeneous combinations of algorithms often performed better than the homogenous workflows. Our scoring method showed that the union of feature matrices of different workflows outperformed the original homogenous workflows in some cases. msCompare is open source software (https://trac.nbic.nl/mscompare), and we provide a web-based data processing service for our framework by integration into the Galaxy server of the Netherlands Bioinformatics Center (http://galaxy.nbic.nl/galaxy) to allow scientists to determine which combination of modules provides the most accurate processing for their particular LC-MS data sets.
LC-MS is a well established analysis technique in the field of proteomics and metabolomics (1–5). It is frequently used for comparative label-free profiling of preclassified sets of samples with the aim to identify a set of discriminating compounds, which are either further used to select biomarker candidates or to identify pathways involved in the studied biological processes (6–8). However, the highly complex and large data sets necessitate the use of elaborated data processing workflows to reliably identify discriminatory compounds (9–11).
The main aim in the quantitative processing of label-free LC-MS data is to obtain accurate quantitative information about the measured compounds, as well as proper matching of the same compounds across multiple samples. Quantification of compounds from raw mass spectrometry data can be performed in a number of ways. Spectral counting methods (11–14) are mainly used for proteomics samples and exploit the number of MS/MS spectra that are acquired per peptide ion(s) for protein quantification. These methods are easy to implement because they use the output of the peptide/protein identification tools but are less accurate than methods based on ion intensity for the determination of protein ratios (15, 16). Other widely used methods rely on single-stage MS information for compound quantification. In single-stage MS data, compounds (peptides, proteins, and metabolites) are detected and quantified in the raw mass spectrometry data, but they are not identified. Instead, algorithms locate and quantify features corresponding to compound peaks in the raw data (see definition of “feature” and “peak” on page 4 of the supplemental material), i.e. compound-related signals above a given noise level, and assign a metric to each feature in the form of an intensity, height, area, or three-dimensional volume, correlated to compound concentration.
Label-free LC-MS data are often used for the relative quantification of compounds in several samples (17). However, when appropriate, quantified stable isotope standards (peptides or proteins) at known concentrations are added to each sample (17, 18), or when protein identification can be obtained from MS/MS data and by using a standard protein of known quantity (19), it is possible to calculate the absolute amount of proteins.
Matching the same features across multiple samples requires several steps. First, shifts between chromatograms in mass-to-charge ratio (m/z) and retention time dimensions must be corrected, the most challenging being the correction for nonlinear retention time shifts (20–27). For data obtained with high resolution mass spectrometers, recalibration of the m/z axis, e.g. by using background ions from common contaminants (28) or continuously co-ionizing standards, generally improves mass accuracy and facilitates matching the same feature across multiple chromatograms. After correcting shifts in retention time and m/z ratio between chromatograms, features are clustered and reported in the form of a quantitative matrix, where rows (or columns) correspond to matched features and columns (or rows) to samples. Alternatively, some programs perform alignment and clustering in one step in the same programming module. In addition to the two main modules of feature quantification and matching, there are optional modules such as noise filtering to improve feature detection/quantification or to remove redundancy of related features in the final quantitative matched feature matrix through decharging and deisotoping (29, 30). Feature quantification needs to be linked to compound identification (31–35) to understand the relevance of changes in their biological context.
A number of research groups have developed programs, such as MZmine (36), OpenMS (37), SuperHirn (38), and others (39–41), to process label-free LC-MS data. Each workflow uses different algorithms for feature detection/quantification and feature alignment/matching. The accuracy of data processing has a large impact on the ultimate result of a proteomics or metabolomics experiment and can lead to false discoveries. Although validation of LC-MS procedures has made considerable progress in recent years, there is a lack of understanding of how the performance of the individual modules of data processing workflows affects the overall result. Zhang et al. (42) showed that considerable differences in performance exist between algorithms using isotope pattern matching for feature picking, such as msInspect, and approaches using feature shape filtering, such as mzMine. To detect performance differences, Zhang et al. used receiver operating characteristics curves on a sample containing a protein mixture of 48 proteins, which resulted in ∼800 identified peptides after digestion with trypsin. Lange et al. (43) showed that time alignment approaches differ in their accuracy to correct nonlinear retention time shifts between chromatograms, which affects the accuracy of clustering the same features across multiple chromatograms significantly. Both articles only evaluate a specific part of the data processing workflow; however, they do not take possible combinatorial effects between feature detection/quantification and alignment/matching methods into account. It is important to consider such effects when evaluating the performance of entire workflows.
Recently Zhang et al. (44) compared the quantification performance of two commercial workflows, Progenesis and Elucidator. The authors propose seven metrics, such as the mean and variance of feature intensities, the mean and variance of feature intensity correlation between all quality control sample pairs, or the manual inspection to assess the validity of features that were only found by one of the workflows, to evaluate the overall quantification performance of the workflows. However, the large number of chosen metrics complicates the accurate comparison and ranking of the different workflows. Furthermore, metrics requiring manual evaluation and the lack of a global score make evaluation of the performance of a large number of different workflows or the optimization of parameters to maximize the performance of a given workflow extremely laborious and arbitrary. Nonetheless, all three comparison studies indicate that there are differences in data processing performance at different levels: between different feature detection, feature quantification, and alignment methods, as well as between complete workflows. Because it is difficult and tedious for a scientist with common informatics knowledge to install and familiarize himself with many different programs to apply various evaluation methods on a particular LC-MS data set, attempts have been made to integrate different programs into a single framework.
Currently, Corra (45) is the only existing framework providing a simple and uniform system to perform quantitative LC-MS data processing for scientists with limited bioinformatics knowledge. This framework allows quantitative data processing of LC-MS data sets using either SpecArray (41), SuperHirn (38), msBID (46), or OpenMS (37) and includes modules for statistical analysis. The developers of Corra also implemented a new data format called annotated putative peptide markup language (APML),1 which has been proposed as a standard format to store intermediate and final results of different data processing tools. This complements the mzQuantML standard, which is currently under development at the European Bioinformatics Institute together with the HUPO/PSI (47). APML facilitates the addition of new tools to the Corra framework; however, at this time only a limited number of tools support this format. Once the choice of data analysis tools has been made in the Corra framework, it applies to the entire processing pipeline; this prevents the user from assessing whether more accurate quantification can be obtained through the combination of data processing modules from different workflows. Our work shows that, in fact, it may be beneficial to combine modules from different published workflows to improve the overall result.
To compare the performance of different combinations of modules, we developed a framework, msCompare, interconnecting the feature detection/quantification and featuring alignment/matching methods of three publicly available open source workflows (SuperHirn, MZmine, and OpenMS). In addition, we included stand-alone modules for feature detection/quantification (N-M rule algorithm) (48), and an in-house-developed time alignment algorithm Warp2D (27) combined with feature matching across multiple samples (Fig. 1). Within msCompare it is also possible to add new modules that perform a single step or multiple steps in the overall data processing workflow, making this a very flexible framework to optimize data processing with respect to a given data set. To facilitate the use of msCompare and to allow interfacing with other data processing frameworks, such as Corra, we developed an AMPL converter that allows export of either feature lists or the quantitative matched feature matrix in APML format.
Fig. 1.

msCompare computational framework combining modules from different open source data processing workflows. a, overview of different open source data processing workflows modularized in msCompare. b, overview of the computational framework, which allows execution of any combination of feature detection/quantification or feature alignment/matching modules of the original pipelines.
The msCompare framework can assess the performance of all combinations of the implemented modules based on a unified scoring method applied to the matched feature matrix of an arbitrary data processing workflow. The scoring function requires a set of LC-MS chromatograms obtained from one sample of interest as biological matrix, in which known molecules were spiked at minimally two concentration levels. In this article, we compare the performance of homogenous and heterogeneous combinations of modules for feature detection/quantification and feature alignment/matching for the analysis of LC-MS data from human urine and trypsin-digested porcine CSF. Furthermore, we provide easy-to-use processing services by integrating msCompare into the Galaxy framework (49, 50), freely accessible at http://galaxy.nbic.nl/galaxy.
MATERIALS AND METHODS
Modularization of Data Processing Tools
msCompare was designed to interconnect different modules of independent open source LC-MS data analysis programs. We first divided all programs into two main modules: feature detection/quantification and feature alignment/matching. These two modules were implemented by writing wrappers around the data processing tools of OpenMS (version 1.2 and 1.5 for feature detection/quantification and version 1.5 for alignment/matching) (37, 51–53) and SuperHirn (version 0.05) (38), which execute the processing steps of the program for either feature detection/quantification or feature alignment/matching. MZmine (version 0.6) (36, 54, 55) can be only accessed via a graphical user interface. For that reason we implemented MZmine in msCompare by writing a program that encapsulates MZmine as a Java library. Additionally, a wrapper was written around our in-house feature alignment/matching tool based on the Warp2D algorithm (27) and around a feature detection/quantification module (N-M rules) that we implemented based on the approach published by Radulovic et al. (48). Two XML-based data formats were developed: one is the FeatureLists format, which stores the feature detection/quantification results, and the other is the FeatureMatrix format, which stores the resulting quantitative matrix obtained after feature alignment/matching. The formats are described in detail in the following section and were developed to enable bidirectional conversion between the different internal data formats of the various tools.
We have implemented additional modules to extend the functionality of msCompare to perform filtering of the feature list by setting criteria for feature properties (e.g. deleting features with extreme width in the retention time and/or mass to charge ratio dimensions), to export feature lists or feature matrices into a tab-delimited or APML format, or to perform conversions between the different feature list formats. The latter module has the additional benefit of allowing the visualization of any feature list in the TOPPView program (53), which is part of the OpenMS TOPP framework, independently of the used feature detection/quantification module. msCompare uses mzXML (56) as input format, but mzData (57) and mzML (58) data formats are also supported through the use of the file converter of OpenMS.
Data Integration
Our data format for the feature list is generic and allows storage of the complete information of the feature lists provided by all programs integrated in msCompare (supplementary Section 1.1). The implementation does not provide a link between the corresponding feature attributes with different names (e.g. attribute names used for feature quantity may be feature intensity, feature area, or feature volume) or between the same attributes having different units, such as retention time attributes in minutes or seconds, nor does it provide a solution for absent feature attributes. Such links are, however, necessary for the conversion between different feature list formats. To solve the conversion problem, we developed a set of rules described in XML format called FeatureConversion XML, which are used at runtime to perform the conversion between the different feature attributes. Because we had four feature detection/quantification and four feature alignment/matching modules, we describe the conversion rules between all possible scenarios of format conversion (16 rules in total). This approach facilitates the integration of new modules, because, by defining a new set of conversion rules in XML, msCompare is able to convert data between one of the already integrated modules and the newly added module. We have implemented msCompare in a way that conversion to the FeatureList XML format from the internal format of the feature quantification module is performed as the last step. The first step of all feature alignment/matching modules is therefore the conversion from the FeatureList XML format to the feature list format used by the integrated program.
APML is the only currently available format for storing processed quantitative LC-MS proteomics data at the feature list and matched feature matrix levels. We have decided to design a different format, because it is not possible to store all information obtained with the different integrated modules in the APML format. APML format has a predefined set of feature properties, and it contains a number of feature properties that are optional. Accordingly, we do not provide an import module for feature list in APML format. However, to facilitate integration of msCompare with other programs supporting the APML format (for example the statistical modules in Corra), we designed two export functions: one to export feature lists and the other to export feature matrices in APML format.
Implementation of msCompare in Galaxy
To provide an easy-to-use web-based interface for scientists with limited bioinformatics expertise, we implemented msCompare in the Galaxy processing framework (49, 50). Galaxy offers relatively simple integration for command line tools. Command line tools require definition of a “tool XML config file,” which describes the command line usage, input and output formats, and input parameters. Implementation of various data processing modules of msCompare in Galaxy was therefore relatively simple because all modules can be run from the command line. One particularly useful property of Galaxy is that it keeps track of user histories including the data, parameter settings, and data processing tasks. The history stores the input, output, and parameter settings of the executed data processing tasks, which can be reused for future data processing. In addition, processing tasks stored in the history can be used to build data processing workflows. Workflows may also be built using an integrated visual workflow editor. Histories (data and parameters) and workflows can be shared with other users, facilitating collaboration between multiple users in large scientific projects. Galaxy is able to execute processing tasks either on a local computer or on a computer cluster, thus providing a high throughput data processing and analysis environment. All hardware-related implementation details are handled by the Galaxy framework and thus hidden from the user.
Integrating New Modules in msCompare
The current version of msCompare supports four different feature detection/quantification and four different feature alignment/matching modules. To add new modules to the msCompare framework, it is necessary to write parsers to make the output and/or input formats compatible with either the FeatureList and/or FeatureMatrix XML format of msCompare. The next step is to define the conversion rules between the feature attributes that are used by the new module and the feature attributes that are used by the already integrated modules. This can be a challenging task when the number of modules increases. To facilitate this task, we developed a stand-alone FeatureMatcher Java tool. Using FeatureMatcher, a user can define conversion rules for the new feature list with respect to an already existing feature list format. FeatureMatcher automatically adds all other conversions rules as needed for all other integrated feature list formats. The last step in module integration is adding the module to the galaxy framework by writing the tool XML config file as mentioned in the previous section.
Scoring Module
The accurate comparison and assessment of workflow performance requires knowledge of the “ground truth,” i.e. the exact molecular composition of the samples and the amount of each compound. In complex biological samples it is, however, not possible to know the exact quantitative and qualitative molecular composition. It is thus necessary to add (“spike”) known compounds at defined levels to biological samples to define the “ground truth” for the added compounds. To evaluate the capacity of a given workflow to detect and quantify correctly, spiking must be done at two different concentration levels or more and the concentration difference must be larger than the measurement error of the analytical system. It is also important that the biological matrix has the same molecular composition and concentration for the different spiking levels.
Because the number of detected and quantified features is not constant across workflows, classical binary statistical tests do not apply, leading us to develop a novel figure of merit to measure performance based on a score of the ranked spiked features rather than a match of measured feature strengths to “ground truth” values (Equation 1). This score is based on the number of detected features that correspond to spiked peptides and their relative rank among the most discriminating features. The scoring module requires a data set in which a sample with an unknown composition is spiked at different concentration levels with peptides (or other compounds) that can be assigned based on their known mass to charge ratios and retention times. The detected features are sorted according to decreasing t values in the matched feature matrix, and the score is calculated using Equation 1,
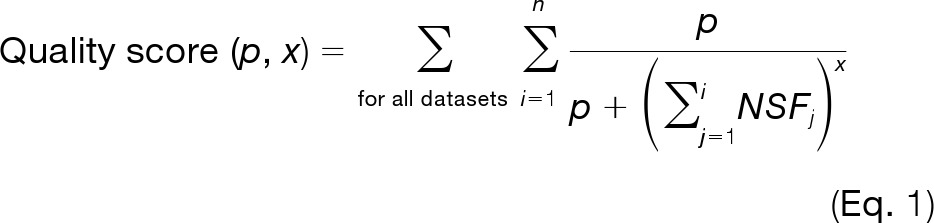 |
where n is the number of all features in a data set, NSF (number of non-spike-related features) is the number of features occurring between rank 1 and index i in the matched feature matrix that are not related to the spiked standard compounds, and p and x are constants determining the degree of score attenuation for non-spike-related features among the most discriminating features. A decoy approach is applied to calculate a score corresponding to random distribution of the detected spiked peptide-related features in the complete feature list, and the decoy score is subtracted from the score obtained for a particular pipeline. The random distribution of features related to spiked peptides in the feature lists is obtained by randomly reshuffling the order of all features in the quantitative matched feature matrix. This decoy score provides the background value for a given data processing result and corrects for differences in the number of all detected features (n) and in the number of identified spiked peptide-related features.
Fig. 2 gives two examples to demonstrate the scoring mechanism using several values for p and x. By decreasing the values for p, one increases the penalty after each non-spike-related feature that occurs in the rank list. In that case, our scoring method distinguishes clearly between feature lists containing few or many non-spike-related features among the most discriminating features. The value of x defines the degree to which non-spike-related features affect the further increase in score for the less discriminatory spike-related features. When x is large (x is close to 1), the score attenuation for each additional spike-related feature following an NSF is large, and for small x values (x is close to 0), this effect is small. Supplemental Fig. S1 summarizes the main steps of the scoring mechanism starting with sorting features according to their t value, followed by assigning features to spiked standard compounds and the final application of Equation 1.
Fig. 2.

Examples showing the operational mechanism of the scoring function on four different quantitative matched feature matrices per parameter choice. Black dots (●) represent spike-related features, whereas white dots (○) represent other features (NSF in Equation 1). Equation 1 contains two constants, p and x, which influence the final scores. When p and x have stringent settings (left panel), the presence of NSFs in high rank positions of the matched feature matrix leads to a rapid decrease of the score for subsequent spike-related features. The value of x defines the degree to which non-spike-related features affect the score increase for less discriminatory spike-related features and has weaker influence on the score than p (see scores values for various x and p in supplemental Fig. S6 in the supplementary material). Setting these parameters more leniently (right panel) allows for more NSFs with lower discriminatory ranks without penalizing subsequent standard features severely. For evaluation of different workflows, we used p = 5 and x = 1. To remove the dependence of the total number of detected features n from the score, we corrected the score using a decoy approach. The decoy approach includes subtraction of the score obtained for randomly reshuffled matched feature matrix/matrices from the score obtained with the real matched feature matrix/matrices sorted according to the t value.
LC-MS Data Acquisition and Analysis
Collection of Urine Samples
50 midstream morning urine samples from 15 healthy females and 35 healthy males aged 26.9–72.9 years were obtained from the Department of Pathology and Laboratory Medicine at the University Medical Centre Groningen (Groningen, The Netherlands). A pooled urine sample was prepared by combining 200 μl from each sample, which served as biological matrix for all LC-MS analyses. Sample preparation was performed as previously described. The amount of urine injected into the LC-MS system was normalized to 50 nmol of creatinine. All of the subjects that participated in this study gave their oral and/or written informed consent. The study protocol was in agreement with local ethical standards and the Helsinki declaration of 1964, as revised in 2004.
Preparation of Spiked Urine Samples
Urine samples were spiked with different volumes of a stock solution containing a tryptic digest of carbonic anhydrase (Sigma; C3934) plus seven synthetic peptides. 600 μl of carbonic anhydrase (CA) solution at 22 mg/ml in 50 mmol/liter NH4HCO3 buffer at pH 7.8 were divided into six equal aliquots. Ten μl of 100 mm DTT were added to each aliquot, and the solution was incubated at 50 °C for 30 min followed by the addition of 40 μl of 137.5 mm iodoacetamide and incubation at room temperature for another 60 min. Reduced and alkylated CA was digested by adding 40 μl of 0.5 μg/μl sequencing grade modified porcine trypsin (Promega, Madison, WI; V5111) and subsequent incubation at 37 °C overnight. The reaction was stopped by the addition of 10 μl of pure formic acid (FA). The excess of DTT and iodoacetamide was removed by solid phase extraction using a 100 mg Strata C-18 SPE column with the following protocol: the column was conditioned with 2 ml of methanol followed by one washing step with 2 ml of water. Each aliquot of digested CA was loaded on the SPE column, and the column was subsequently washed with 2 ml of 5% aq. methanol. Peptides were eluted with 1 ml of 80% aq. methanol. The eluate was dried in a vacuum centrifuge and redissolved in 200 μl of 30% ACN and 1% FA. Finally 500 μl of digested CA were mixed with 200 μl of a stock solution of the synthetic peptides resulting in a standard mixture stock solution with a calculated digested CA concentration of 240 μm and the following concentrations for the seven synthetic peptides: VYV, 83 μm; YGGFL, 57 μm; DRVYIHPF, 29 μm; YPFPGPI, 46 μm; YPFPG, 60 μm; GYYPT, 54 μm; and YGGWL, 57 μm.
Analysis of Spiked Urine Samples by Reversed Phase LC-MS
All of the LC-MS analyses were performed on an 1100 series capillary high performance liquid chromatography system equipped with a cooled autosampler (4 °C) and an SL ion trap mass spectrometer (Agilent Technologies, Santa Clara, CA). The samples were desalted on an Atlantis dC18 precolumn (Waters Corporation, Milford, MA; 2.1 × 20 mm, 3-μm particles, 10-nm pores) using 0.1% FA in 5% ACN at a flow rate of 50 μl/min for 16 min. Compounds were back-flushed from the precolumn onto a temperature-controlled (25 °C) Atlantis dC18 analytical column (1.0 × 150 mm, 3-μm particles, 30-nm pores) and separated over 90 min at a flow rate of 50 μl/min, during which the percentage of solvent B (0.1% FA in ACN) in solvent A (0.1% FA in ultrapure H2O) was increased from 5.0 to 43.6% (eluent gradient of 0.43%/min). Settings of the electrospray ionization interface and the mass spectrometer were as follows: nebulization gas, 40.0 psi N2; drying gas, 6.0 liters/min N2; capillary temperature, 325 °C; capillary voltage, 3250 V; skimmer voltage, 25 V; capillary exit voltage, 90 V; octapole 1 voltage, 8.5 V; octapole 2 voltage, 4.0 V; octapole RF voltage, 175 V; lens 1 voltage, −5 V; lens 2 voltage, −64.6 V; trap drive, 67; scan speed, 5500 m/z s−1; accumulation time, 50 ms (or 30,000 ions); and scan range, 100–1500 m/z. A Gaussian smoothing filter (width 0.15 m/z) was applied for each mass spectrum; rolling average was disabled, resulting in a rate of ∼70 mass spectra/min. The spectra were saved in profile mode.
Following the gradient, both columns were washed with 85% B for 5 min and equilibrated with 5% B for 10 min prior to the next injection. Different volumes of the standard mixture (CA digest plus peptides) were injected on the precolumn prior to injection of the pooled urine sample to obtain the desired final concentrations. Supplemental Table S1 provides the list of dilution factors with the corresponding concentrations expressed in terms of the practical lower limit of quantification (pLLOQ; supplemental Section 1.2) for the respective peptides. The injection system was cleaned with 70% ACN after each injection and filled with 0.1% FA in 5% ACN. Mass spectrometry settings were optimized for detection of singly and doubly charged ions of DRVYIHPF without provoking upfront fragmentation. Raw data converted to mzXML format are available at http://tinyurl.com/msCompareData.
Data Analysis of Spiked Urine Samples
After the LC-MS analysis, the raw LC-MS profile data were exported in mzXML format using CompassXport v1.3.6. These data were then analyzed by all different workflow combinations (see supplemental Section 1.3 for parameters and execution details), which lead to the construction of multiple matched feature matrices, each containing 10 LC-MS analyses at two spiking concentration levels (five LC-MS analyses for each level). We used the score module with the list of spike-related features (supplemental Section 1.4) to calculate the scores for one feature matrix. The final scores for the high, medium, and low categories were obtained by summing the individual scores of several quantitative matched feature matrices (see supplemental Table S2 for an overview of the feature matrices used to construct the three categories).
Collection of CSF Samples
Porcine CSF was obtained from the Animal Sciences Group of Wageningen University (Division of Infectious Diseases, Lelystad, The Netherlands). CSF was collected from the cerebromedullary cistern of the subarachnoid space in the cervical region directly after killing the animal (by intravenous injection of T61® pentobarbital followed by exsanguination). The sample was collected under mild suction using a syringe with a 22-gauge needle. The CSF sample was centrifuged immediately after sampling (10 min at 1500 × g). The total protein concentration was measured using the Micro BCATM Assay (Pierce), and the final concentration was 860 ng/μl.
Preparation of Spiked CSF Samples
Digestion with trypsin was performed according to the following procedure: 200 μl of CSF and 200 μl of 0.1% RapiGestTM (in 50 mm ammonium bicarbonate) (Waters, Milford, MA) were added to a sample tube (Greiner Bio-One, Alphen aan den Rijn, The Netherlands; part 623201). The sample was reduced by adding 4 μl of 0.5 m DTT followed by incubation at 60 °C for 30 min. After cooling to room temperature, the sample was alkylated with 20 μl of iodoacetamide (0.3 m) in the dark for 30 min at room temperature. Eight μl of sequencing grade modified porcine trypsin (1 μg/μl) was added to give a trypsin to protein ratio of ∼1:20 (w/w). The sample was incubated for ∼16 h at 37 °C under vortexing (450 rpm) in a thermomixer comfort (Eppendorf). Thereafter 40 μl of hydrochloric acid (0.5 m) were added to stop the digestion followed by incubation for 30 min at 37 °C. The sample was centrifuged at 13,250 × g for 10 min at 4 °C to remove the insoluble part of the hydrolyzed RapiGestTM. The spiking procedure was carried out according to the following protocol: 20 μl of CSF digest were mixed with 20 μl of a tryptic digest of horse hearth cytochrome c (Fluka; part 30396) at different concentrations (25, 5, 2.5, 0.5, 0.05, 0.025, and 0.005 fmol/μl). The samples at each spiking level were aliquoted in five tubes containing 8 μl each. Spiked, trypsin-digested CSF was injected five times at each spiking level (4 μl from individual vials) in a random order (amount of injected cytochrome c, 50, 10, 5, 1, 0.1, 0.05, and 0.01 fmol). Supplemental Table S3 provides the list of dilution factors with the corresponding concentrations expressed in terms of the practical lower limit of quantification (pLLOQ; see supplemental Section 1.5) for the respective peptides.
Analysis of Spiked CSF Samples by Reversed Phase Chip-LC-MS
Peptides were separated on a reverse phase chip-LC (Protein ID chip 3; G4240–63001 SPQ110: Agilent Technologies; separating column, 150 mm × 75 μm Zorbax 300SB-C18, 5 μm; trap column, 160 nl of Zorbax 300SB-C18, 5 μm) coupled to a nano LC system (Agilent 1200) with a 40-μl injection loop. Ions were generated by ESI and transmitted to a quadrupole time-of-flight mass spectrometer (Agilent 6510). Instrumentation was operated using the MassHunter data acquisition software (version B.01.03; Build 1.3.157.0; Agilent Technologies, Santa Clara, CA).
For LC separation the following eluents were used: eluent A, ultra-pure water with 0.1% FA, and eluent B, acetonitrile with 0.1% FA. The samples were injected on the trap column at a flow rate of 3 μl/min (3% B). After 10 min, the sample was back flushed from the trap column and transferred to the analytical column at a flow rate of 250 nl/min, and the peptides were eluted using the following gradient: 95-min linear gradient from 3 to 70% B; 2-min linear gradient from 70 to 3% B, which was maintained for 10 min before injecting the next sample. The samples were analyzed in a random order with blanks and quality control samples (200 fmol of trypsin-digested cytochrome c) injected after every fifth sample.
The MS analysis was done in the 2-GHz extended dynamic range mode under the following conditions: mass range, 100–2000 m/z; acquisition rate, 1 spectrum/s; data storage, profile and centroid mode; fragmentor, 175 V; skimmer, 65 V; OCT 1 RF Vpp, 750 V; spray voltage, ∼1800 V; drying gas temp, 325 °C; drying gas flow (N2), 6 liter/min. Mass correction was performed during analysis using internal standards with m/z of 371.31559 (originating from a ubiquitous background ion of dioctyl adipate (plasticizer)) and m/z of 1221.990637 (HP-1221 calibration standard) continuously evaporating from a wetted wick inside the spray chamber.
Data Analysis of Spiked CSF Samples
Raw LC-MS data were exported in mzData format using quantitative analysis (B.03.01) in the MassHunter software package in centroid mode to limit file size and analysis time. These data were processed by all different workflow combinations (see supplemental Section 1.3 for parameters and execution details), which lead to the construction of multiple matched feature matrices, each containing 10 LC-MS analyses at two spiking concentration levels (five LC-MS analysis for each level). We used the list of spike-related features (supplemental Section 1.6) to calculate the score for each feature matrix based on the score module with Equation 1. The final scores for the high, medium, and low categories were obtained by summing the individual scores of several feature matrices (see supplemental Table S4 for an overview of the feature matrices used to construct the three categories).
RESULTS
Comparison of Homogenous Workflows
For most LC-MS data analyses, one homogenous data processing workflow is used. We therefore first investigated the performance of the homogenous workflows of MZmine, OpenMS, and SuperHirn (Fig. 1a). The obtained results for spiked human urine samples presented in Fig. 3 (a–c) show that the OpenMS workflow results in scores exceeding those of MZmine and SuperHirn when the differences in spiking level are high (8- or 16-fold pLLOQ versus lower spiking levels; see supplemental Table S2 for details). The performances of the homogenous workflows are similar to each other for low spiking levels (0.5- or 1-fold pLLOQ versus lower levels). To get an insight into the basis for the difference between the workflows, we investigated the 10 most discriminatory features according to their t values, which are not related to the spiked in peptides, for the feature matrix with the largest concentration difference (blank versus samples spiked at 16-fold the pLLOQ level).
Fig. 3.

Comparison of the performance of the published, open source data processing workflows SuperHirn, OpenMS, and MZmine with LC-MS data derived from the analysis of human urine (a–c) and porcine CSF (d and e) samples spiked with a range of peptides. The scores were calculated with Equation 1. All of the workflows were compared with respect to high (a and d), medium (b), and low (c and e) concentration differences of the spiked peptides (see supplemental Tables S2 and S4). The OpenMS workflow outperforms the other two workflows at large (a) and medium (b) spiked concentration differences, whereas performances are approaching each other at the lowest (c) spiked concentration difference in human urine data sets. In porcine CSF, OpenMS performed best at both high and low spiked concentration differences.
The difference in performance of OpenMS and the other two homogenous workflows appears to be related to the higher ranks of discriminating features that are unrelated to the spiked in peptides (Fig. 4). Most of these features were only quantified in samples at one spike level, although they are visible in the raw data at other spike levels using extracted ion chromatograms (EICs). Majority of these errors were due to the splitting up of one peak at feature detection/quantification step, which is incorrectly matched at the feature alignment/matching step of the samples of the other spiking level (see supplemental Table S5). We also investigated the number of detected and quantified features that are related to the spiked peptides among the 100 most discriminating features and determined the number of overlapping features between homogenous workflows. MZmine and SuperHirn detected approximately 50 features, whereas OpenMS found 64 spike-related features among the 100 most discriminatory features. 78 unique features related to spiked peptides were found by the three workflows, with 28 features being found by all of them, and 23 features were found by OpenMS and one of the two other workflows. It is surprising that MZmine and SuperHirn detect two quite distinct sets of features, whereas OpenMS is capable of detecting most of those features. In total, 27 unique features related to spiked peptides were found exclusively by one of the workflows, of which approximately half (13) were found by OpenMS only (Fig. 5). The number of features only detected by one workflow is relatively high (35%), indicating that one part of spike-related features is difficult to detect and quantify.
Fig. 4.

Overview of the score evaluation function for the most discriminating features for three homogenous workflows (see Fig. 3) when comparing the 16-fold pLLOQ spiked samples with the blank (0.1-fold pLLOQ) obtained with the human urine data set. The bars at the bottom of the graph provide visual indication of the ranks at which features related to the spiked peptides were found for the respective workflow (blue, OpenMS; orange, SuperHirn; red, MZmine). Non-spike-related features are represented in this subplot as white squares. The OpenMS workflow found only one non-spiked-related feature up to rank 48, whereas the other two workflows showed a less consistent performance, leading to lower scores.
Fig. 5.
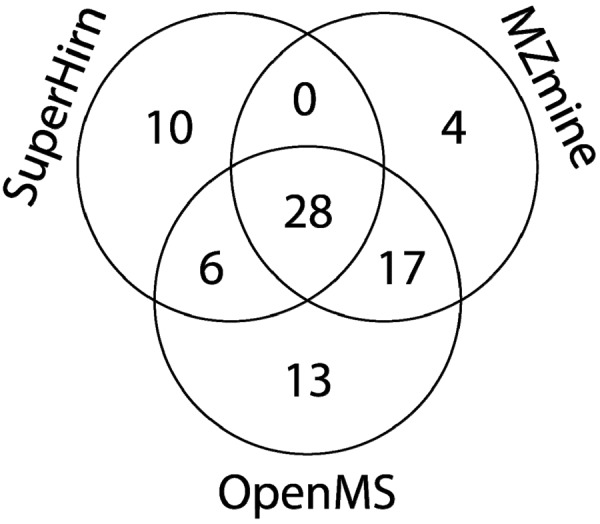
Venn diagram of spike-related features found among the 100 most discriminatory features by the three homogenous workflows (see Fig. 3) obtained with the spiked human urine data set. The data were obtained by comparing the 16-fold pLLOQ spike level with the blank (0.1-fold pLLOQ). OpenMS found 64 (82% of the total number of unique features found by all workflows) of all unique features related to the spiked peptides. It also identified the highest number (13) of unique features related to the spiked compounds not identified by any of the other workflows.
We have prepared one other set of spiked samples with a completely different composition than the spiked pooled human urine samples. This sample set consisted of one porcine CSF sample as biological matrix that was spiked with tryptic digest of horse hearth cytochrome c. The data were acquired with a quadrupole time-of-flight instrument at 12,000 resolution compared with the pooled human urine sample set, which was acquired with a three-dimensional quadrupole ion trap at 2000 resolution. Trypsin digest of horse heart cytochrome c containing 14 peptides was used at spiking levels ranging from 2.5× to 0.005× pLLOQ. Because of the lower spiking levels in the porcine CSF samples only high and low spiking level differences were analyzed. The results for the spiked porcine CSF samples show a similar pattern to the results obtained for the urine data set (Fig. 3, d and e), indicating that workflow performance is rather insensitive to the biological matrix, spiking level, or mass spectrometer used. The score of the homogenous workflow of OpenMS exceeds those of SuperHirn and MZmine for both spiking level differences. Scores measured for SuperHirn and MZmine are close to each other for both spiking difference levels, with the SuperHirn workflow having a slightly higher score for low spiking level differences (0.5 pLLOQ versus lower spiking levels [0.05, 0.025, and 0.005]).
Comparison of Heterogeneous Workflows
Because the msCompare analysis framework allows combination of modules from different workflows, we next investigated whether combining feature detection/quantification and feature alignment/matching modules from different workflows could improve the overall performance beyond that of the homogenous workflows. To this end we interconnected modules in msCompare in a total of 16 combinations (Fig. 1b). A number of the newly created heterogeneous workflows outperformed the original homogenous ones when tested with the spiked human urine data set, notably at high spiking level differences (8- or 16-fold pLLOQ versus lower levels for pooled human urine set and 0.5 versus lower levels for porcine CSF sample set; Fig. 6) for both human urine and porcine CSF data sets. Overall, the two best performing workflows at large spike level differences were the combination of the feature detection/quantification module of OpenMS with the in-house developed feature alignment/matching algorithm, the homogenous OpenMS workflow in the case of the human urine data set, and combination of OpenMS and SuperHirn in the case of the porcine CSF data set. Combination of the feature detection/quantification modules of SuperHirn and MZmine with the in-house or OpenMS feature alignment/matching modules resulted in clearly improved performance when compared with the original, homogenous workflows (Figs. 3 and 6). As the difference in spiking level decreases, the advantages of these workflow combinations are reduced, and a new combination based on the feature detection/quantification module “N-M rules” with the in-house developed feature alignment/matching module for the human urine data set.
Fig. 6.

Comparison of the performance of 16 and 12 different combinations of feature detection/quantification and feature alignment/matching modules at high, medium, and low concentration differences of spiked peptides (see supplemental Tables S6 and S7) using the spiked human urine data set (a) and the spiked porcine CSF data set (b), respectively. Labels of the hybrid workflows (x axis) start with the name of the feature detection/quantification module followed by the name of the feature alignment/matching module. The best performing workflows at each concentration level difference are highlighted in red. The homogeneous OpenMS workflow and combinations of the OpenMS feature detection/quantification module with the in-house developed feature alignment/matching module result in the highest scores when concentration differences are large or medium for the spiked human urine data set (a), whereas the respective combination of the OpenMS-SuperHirn heterogeneous workflow provides the best performance for the porcine CSF data set spiked with large concentration differences (b). The scores level out at medium concentration differences, although some combinations do not perform well at any level (e.g. SuperHirn to MZmine). The combination N-M rules feature detection/matching module with the in-house developed feature alignment/matching module (Inhouse D.) performs best for low spiked concentration differences for spiked human urine data set (M-N rule peak picking was not performed for porcine CSF data set because of the incompatibility of this approach with high resolution data), whereas the best performing combination of feature detection/quantification and feature alignment/matching modules for the low spiked concentration difference of the porcine spiked CSF data set is the respective OpenMS homogenous workflow.
The OpenMS homogeneous pipeline provides the best performance for low concentration differences of spiked peptides in the porcine CSF data set. M-N rules feature detection was not applied to analyze the porcine CSF data set due to its incompatibility with high resolution (quadrupole time-of-flight) data because of high memory requirements. The results of the remaining 12 workflow combinations (Fig. 6b) on the porcine CSF data set show a remarkably similar pattern to the results obtained for the human urine data set (Fig. 6a) for both high and low levels of spiked concentration differences. The combination of the OpenMS feature detection/quantification module with any feature alignment/matching module outperforms all combinations including MZmine or SuperHirn for feature detection/quantification. These results show that for both low and high resolution data, OpenMS homogenous pipeline and OpenMS peak detection/quantification combined with other peak alignment/matching modules give reasonably good quantification results for all spiked concentration level differences.
DISCUSSION
Quality assessment of LC-MS data processing workflows is difficult because different errors may occur at various stages during feature detection, feature quantification, the correction for retention time and mass shifts between chromatograms, and clustering of the same feature across multiple chromatograms. For example, the detection/quantification module may split large peaks because of peak tailing into multiple features, whereas small peaks may not be detected. Features may also not be detected because of an unexpected peak shape.
Binary statistics cannot be applied because the exact molecular composition of the biological matrix is unknown, and the different workflows detect different numbers of noise-, compound-, and spiked peptide-related features. In sets of spiked complex biological samples, where one sample is used to prepare the spiked sample set, the ground truth is known for the spiked peptides, whereas the biological matrix serves as constant background signal. Based on the information provided by the spiked peptides, we have developed a generic method to score the quantitative processing results taking the rank of spike-related features among the most discriminating features into account. We have successfully applied this method to capture the differences of various data processing workflows in a single value. There are significant differences in performance of workflows with respect to data processing accuracy when different modules are used for feature detection/quantification and feature alignment/matching, although our scoring method serves to compare the relative performance of the workflows for one particular type of data and to assure that a workflow provides optimal performance for a given data set. However, the scoring method cannot identify the data processing errors that underlie the observed performance differences.
Comparing quantification performance of homogenous workflows on the spiked human urine data set, we observed that the majority of the 10 most discriminating non-spike-related features were only detected/quantified at one of the two spiking levels by all three homogenous workflows (supplemental Table S5; the three most discriminating non-spike-related features are visualized by means of EICs). Moreover, the 10 most discriminating non-spike-related features were different for the three homogenous workflows. This indicates that this type of quantification error is due to random failure of the feature detection/quantification or feature alignment/matching modules to detect a particular feature across different samples and at different spiking levels. Random failure may be related to the large number of compounds that are not related to the spiked peptides relative to the low number of available samples per sample group, which increases the chance that this kind of random error occurs only in samples at one of two spiking levels. We also observed that the combinatorial effect between feature detection/quantification, and feature alignment/matching modules can result in data processing errors in the form of highly discriminatory features that are not related to any of the spiked peptides. We observed, for example, that an isotopic peak of a highly abundant non-spiked peptides was detected as two separate features (i.e. feature splitting) in one of 10 samples by the feature detection/quantification module of SuperHirn. The subsequent feature alignment/matching resulted in two matched features in the feature matrix, one of which was highly discriminatory between the two spiking levels (supplemental Fig. S2). Although the exact reasons for the generation of highly discriminatory non-spike-related features provided by the different data processing workflows are not always easy to determine, their negative effect on the overall performance is captured by our scoring method.
The Venn diagram in Fig. 5 shows that different homogenous workflows in the human urine data set detect different spike-related features. In fact, MZmine and SuperHirn detected quite different sets of spike-peptide related features (28 features were detected by both workflows, of which 16 were uniquely detected by SuperHirn and 21 by MZmine), although OpenMS detected significantly more of the spiked peptide-related features (64 of the 78 unique features found by all three workflows). Because the detected spiked peptide-related features of MZmine and SuperHirn have little overlap, merging them into a single feature matrix greatly increased the overall number of discriminatory features and thus the performance of data processing. Alternatively, taking the intersection of two or more feature matrices may be used to reduce the number of features (e.g. biomarker candidates) to be followed up as those features are in general easy to detect, therefore decreasing the probability that they are data processing artifacts. However, this comes at the risk of missing relevant features that were only detected by one of the workflows.
The probability to detect more spiked peptide-related features in the union of two or more feature matrices depends on the balance between the accumulation of spiked peptide-related features (true positives) and other features (false positives) among the most discriminatory features. We evaluated the union of all feature matrices obtained with the homogenous workflows (supplemental Fig. S3). The union of the feature matrices obtained with SuperHirn or MZmine for the largest concentration difference (blank and samples spiked at 16 times the pLLOQ level) resulted in a slightly increased score (supplemental Fig. S4). The union of the feature matrices of the other homogenous workflows did not result in improved scores, indicating that the union also contained a larger number of non-spike-related features among the most discriminating features. The decrease in performance when combining OpenMS with other workflows is due to the accumulation of non-spike-related features among the most discriminatory features, because OpenMS already detected most of the spiked peptide-related features when compared with the other workflows (Fig. 5). When calculating the score for the high spiking level difference by summing up the scores of the union feature matrices of the SuperHirn and MZmine workflows, we did not observe any improvement of the score (supplemental Fig. S3).
Visualization of selected peaks using EICs is an important quality check to assess the accuracy of quantitative LC-MS data processing. For example, EICs can be used to verify whether the outcome of the feature detection/quantification modules is supported by the raw LC-MS data. For this purpose we added a module to the msCompare framework, which allows the user to create EICs (supplemental Fig. S5) based on a list of features with their corresponding retention times and mass to charge ratios. It is also possible to export any feature list in our XML-based format to the feature list format of the original workflows. This can be used to export feature lists to the OpenMS format and to use the TOPPView application of OpenMS for visualization (supplemental Fig. S5).
Our scoring method has limitations in that it provides relative scores, which makes it impossible to compare the scores obtained by two different workflows on different data sets. This becomes apparent when comparing the scores of the CSF data set with those of the urine data set. The scores for the CSF data set regarding the high spiked in concentration difference category the best performing workflow only reaches 41, whereas the score of the worst performing workflow in the urine data set is 116. These differences can be explained mainly by the differences in number of spiked peptides in the two data sets. In fact, the porcine CSF data set used 14 peptides for spiking, whereas in the human urine data set 70 peptides were used for spiking. Even using the same set of spiked peptides at the same concentration levels would not result in similar scores, because of ionization differences resulting in differences in charge state distribution of the spiked peptides on different instrument (59). In addition, upfront fragmentation, unspecific cleavage, association with adduct ions (e.g. sodium), and peptide modifications such as methionine oxidation or acetylation further increase the number of spiked peptide-related features. The scoring method was solely designed to compare different data processing workflows for label-free LC-MS analyzing the same data set(s) and not to compare data sets with each other. A useful aspect of our scoring algorithm is that the score is largely independent of the parameters used, with similar ranks of workflow performances obtained within one data set for a wide range of parameters (supplemental Fig. S6).
Another important aspect of the scoring method is that it was initially designed to support the biomarker discovery studies. These studies are generally performed by comparing a control group with a group of interest. Our scoring method mimics this situation closely by using two groups of samples spiked with known compounds at different concentration levels. The scoring approach can, however, also be applied to time series analysis or other experimental designs because it provides a general performance assessment of the feature quantification and matching accuracy independently from the number of sample groups in an experiment.
Most “omics” studies use a representative, pooled sample to control the quality of the analytical profiling method. Because our scoring method requires a data set containing samples that are spiked with known compounds at different concentration levels, it is possible to extend this concept to include pooled samples that have been spiked with known compounds at different levels, for example, with a standardized peptide mixture that is currently available from various suppliers. Including such a set of samples would allow assessment of the performance of the analytical platform as well as that of the data processing workflow and to determine which workflow or combination of workflows provides the optimal performance in a particular case.
In summary, we have developed a generic framework that harmonizes the various formats used by modules of different quantitative LC-MS data processing workflows, thus allowing their integration and the user-defined combination of distinct modules. We expect that integration will be greatly facilitated by acceptance of standard data formats for feature lists and matched, quantitative feature matrices based on the APML standard used in the Corra framework (45) or the currently ongoing development of mzquantML (47). The msCompare framework supports the most common standard data formats to read raw LC-MS data such as mzXML, mzML, and mzData (the latter two are supported by modules in the OpenMS framework).
The msCompare framework facilitates future incorporation of additional modules from other workflows, thereby increasing the possibilities for creating “custom-made” data processing workflows. Additionally, the framework enables the use of existing modules in combination with new modules, allowing rapid evaluation of new data processing tools. By creating a modular, computational framework, we open the possibility of combining feature detection/quantification and feature alignment/matching modules from different open source workflows into “hybrid” heterogeneous workflows that may outperform their original predecessors. Data processing workflows such as OpenMS, SuperHirn, or MZmine are continuously improving their algorithms, and new developments from other research groups enter the field. Our framework forms the basis for integrating these new developments and comparing their relative performance based on well designed data sets. The testing ground can be further extended based on spiked sample sets in data repositories such as PRIDE (http://www.ebi.ac.uk/pride/) and TRANCHE (https://proteomecommons.org/tranche/), covering the effect of sample complexity, the biological matrix, variable retention time shifts, and the influence of mass analyzers of different resolution and mass accuracy on the final result. To enable easy use of msCompare for the wider community of life scientists, we have implemented msCompare in the Galaxy framework and provide a web-based online processing service at the Galaxy server of the Netherlands Bioinformatics Center accessible at http://galaxy.nbic.nl/galaxy.
In conclusion, we show that data processing has a crucial effect on the outcome of comprehensive proteomics profiling experiments. Our framework (which is available for download at https://trac.nbic.nl/mscompare/, including the LC-MS data, information on the parameters used for this article and a Galaxy installation manual) demonstrates that existing workflows contain modules that, when properly combined, result in optimal individual or combined heterogeneous workflows that may outperform the originals. Differences between the best and the worst performing workflows can be surprisingly large, and the choice of algorithm can strongly affect further statistical analysis and the biological interpretation of the results.
Footnotes
* The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- APML
- annotated putative peptide markup language
- CSF
- cerebrospinal fluid
- CA
- carbonic anhydrase
- FA
- formic acid
- EIC
- extracted ion chromatogram
- RF
- radio frequency.
REFERENCES
- 1. Chen G., Pramanik B. N. (2009) Application of LC/MS to proteomics studies: Current status and future prospects. Drug Discov. Today 14, 465–471 [DOI] [PubMed] [Google Scholar]
- 2. Nilsson T., Mann M., Aebersold R., Yates J. R., 3rd, Bairoch A., Bergeron J. J. (2010) Mass spectrometry in high-throughput proteomics: Ready for the big time. Nat. Methods 7, 681–685 [DOI] [PubMed] [Google Scholar]
- 3. Domon B., Aebersold R. (2010) Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 28, 710–721 [DOI] [PubMed] [Google Scholar]
- 4. Allwood J. W., Goodacre R. (2010) An introduction to liquid chromatography-mass spectrometry instrumentation applied in plant metabolomic analyses. Phytochem. Anal. 21, 33–47 [DOI] [PubMed] [Google Scholar]
- 5. Griffiths W. J., Wang Y. (2009) Mass spectrometry: From proteomics to metabolomics and lipidomics. Chem. Soc. Rev. 38, 1882–1896 [DOI] [PubMed] [Google Scholar]
- 6. Choudhary C., Mann M. (2010) Decoding signalling networks by mass spectrometry-based proteomics. Nat. Rev. Mol. Cell Biol. 11, 427–439 [DOI] [PubMed] [Google Scholar]
- 7. Wepf A., Glatter T., Schmidt A., Aebersold R., Gstaiger M. (2009) Quantitative interaction proteomics using mass spectrometry. Nat. Methods 6, 203–205 [DOI] [PubMed] [Google Scholar]
- 8. Yang W., Cai Q., Lui V. W., Everley P. A., Kim J., Bhola N., Quesnelle K. M., Zetter B. R., Steen H., Freeman M. R., Grandis J. R. (2010) Quantitative proteomics analysis reveals molecular networks regulated by epidermal growth factor receptor level in head and neck cancer. J. Proteome Res. 9, 3073–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. America A. H., Cordewener J. H. (2008) Comparative LC-MS: A landscape of peaks and valleys. Proteomics 8, 731–749 [DOI] [PubMed] [Google Scholar]
- 10. Katajamaa M., Oresic M. (2007) Data processing for mass spectrometry-based metabolomics. J. Chromatogr. A 1158, 318–328 [DOI] [PubMed] [Google Scholar]
- 11. Mueller L. N., Brusniak M. Y., Mani D. R., Aebersold R. (2008) An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J. Proteome Res. 7, 51–61 [DOI] [PubMed] [Google Scholar]
- 12. Shinoda K., Tomita M., Ishihama Y. (2010) emPAI Calc: For the estimation of protein abundance from large-scale identification data by liquid chromatography-tandem mass spectrometry. Bioinformatics 26, 576–577 [DOI] [PubMed] [Google Scholar]
- 13. Ishihama Y., Oda Y., Tabata T., Sato T., Nagasu T., Rappsilber J., Mann M. (2005) Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics 4, 1265–1272 [DOI] [PubMed] [Google Scholar]
- 14. Lundgren D. H., Hwang S. I., Wu L., Han D. K. (2010) Role of spectral counting in quantitative proteomics. Expert Rev. Proteomics 7, 39–53 [DOI] [PubMed] [Google Scholar]
- 15. Schulze W. X., Usadel B. (2010) Quantitation in mass-spectrometry-based proteomics. Annu. Rev. Plant Biol. 61, 491–516 [DOI] [PubMed] [Google Scholar]
- 16. Grossmann J., Roschitzki B., Panse C., Fortes C., Barkow-Oesterreicher S., Rutishauser D., Schlapbach R. (2010) Implementation and evaluation of relative and absolute quantification in shotgun proteomics with label-free methods. J. Proteomics 73, 1740–1746 [DOI] [PubMed] [Google Scholar]
- 17. Bantscheff M., Schirle M., Sweetman G., Rick J., Kuster B. (2007) Quantitative mass spectrometry in proteomics: A critical review. Anal. Bioanal. Chem. 389, 1017–1031 [DOI] [PubMed] [Google Scholar]
- 18. Brun V., Masselon C., Garin J., Dupuis A. (2009) Isotope dilution strategies for absolute quantitative proteomics. J. Proteomics 72, 740–749 [DOI] [PubMed] [Google Scholar]
- 19. Silva J. C., Gorenstein M. V., Li G. Z., Vissers J. P., Geromanos S. J. (2006) Absolute quantification of proteins by LCMSE: A virtue of parallel MS acquisition. Mol. Cell. Proteomics 5, 144–156 [DOI] [PubMed] [Google Scholar]
- 20. Christin C., Hoefsloot H. C., Smilde A. K., Suits F., Bischoff R., Horvatovich P. L. (2010) Time alignment algorithms based on selected mass traces for complex LC-MS data. J. Proteome Res. 9, 1483–1495 [DOI] [PubMed] [Google Scholar]
- 21. Christin C., Smilde A. K., Hoefsloot H. C., Suits F., Bischoff R., Horvatovich P. L. (2008) Optimized time alignment algorithm for LC-MS data: Correlation optimized warping using component detection algorithm-selected mass chromatograms. Anal. Chem. 80, 7012–7021 [DOI] [PubMed] [Google Scholar]
- 22. Clifford D., Stone G., Montoliu I., Rezzi S., Martin F. P., Guy P., Bruce S., Kochhar S. (2009) Alignment using variable penalty dynamic time warping. Anal. Chem. 81, 1000–1007 [DOI] [PubMed] [Google Scholar]
- 23. Lange E., Gröpl C., Schulz-Trieglaff O., Leinenbach A., Huber C., Reinert K. (2007) A geometric approach for the alignment of liquid chromatography-mass spectrometry data. Bioinformatics 23, 273–281 [DOI] [PubMed] [Google Scholar]
- 24. Lommen A. (2009) MetAlign: Interface-driven, versatile metabolomics tool for hyphenated full-scan mass spectrometry data preprocessing. Anal. Chem. 81, 3079–3086 [DOI] [PubMed] [Google Scholar]
- 25. Prakash A., Mallick P., Whiteaker J., Zhang H., Paulovich A., Flory M., Lee H., Aebersold R., Schwikowski B. (2006) Signal maps for mass spectrometry-based comparative proteomics. Mol. Cell. Proteomics 5, 423–432 [DOI] [PubMed] [Google Scholar]
- 26. Sadygov R. G., Maroto F. M., Hühmer A. F. (2006) ChromAlign: A two-step algorithmic procedure for time alignment of three-dimensional LC-MS chromatographic surfaces. Anal. Chem. 78, 8207–8217 [DOI] [PubMed] [Google Scholar]
- 27. Suits F., Lepre J., Du P., Bischoff R., Horvatovich P. (2008) Two-dimensional method for time aligning liquid chromatography-mass spectrometry data. Anal. Chem. 80, 3095–3104 [DOI] [PubMed] [Google Scholar]
- 28. Scheltema R. A., Kamleh A., Wildridge D., Ebikeme C., Watson D. G., Barrett M. P., Jansen R. C., Breitling R. (2008) Increasing the mass accuracy of high-resolution LC-MS data using background ions: A case study on the LTQ-Orbitrap. Proteomics 8, 4647–4656 [DOI] [PubMed] [Google Scholar]
- 29. Jaitly N., Mayampurath A., Littlefield K., Adkins J. N., Anderson G. A., Smith R. D. (2009) Decon2LS: An open-source software package for automated processing and visualization of high resolution mass spectrometry data. BMC Bioinformatics 10, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Strohalm M., Kavan D., Novák P., Volný M., Havlícek V. (2010) mMass 3: A cross-platform software environment for precise analysis of mass spectrometric data. Anal. Chem. 82, 4648–4651 [DOI] [PubMed] [Google Scholar]
- 31. Bowen B. P., Northen T. R. (2010) Dealing with the unknown: Metabolomics and metabolite atlases. J. Am. Soc. Mass. Spectrom. 21, 1471–1476 [DOI] [PubMed] [Google Scholar]
- 32. Neumann S., Bocker S. (2010) Computational mass spectrometry for metabolomics: Identification of metabolites and small molecules. Anal. Bioanal Chem. 398, 2779–2788 [DOI] [PubMed] [Google Scholar]
- 33. Li M., Zhou Z., Nie H., Bai Y., Liu H. (2010) Recent advances of chromatography and mass spectrometry in lipidomics. Anal. Bioanal. Chem. 399, 243–249 [DOI] [PubMed] [Google Scholar]
- 34. Menschaert G., Vandekerckhove T. T., Baggerman G., Schoofs L., Luyten W., Van Criekinge W. (2010) Peptidomics coming of age: A review of contributions from a bioinformatics angle. J. Proteome Res. 9, 2051–2061 [DOI] [PubMed] [Google Scholar]
- 35. Duncan M. W., Aebersold R., Caprioli R. M. (2010) The pros and cons of peptide-centric proteomics. Nat. Biotechnol. 28, 659–664 [DOI] [PubMed] [Google Scholar]
- 36. Katajamaa M., Miettinen J., Oresic M. (2006) MZmine: Toolbox for processing and visualization of mass spectrometry based molecular profile data. Bioinformatics 22, 634–636 [DOI] [PubMed] [Google Scholar]
- 37. Kohlbacher O., Reinert K., Gröpl C., Lange E., Pfeifer N., Schulz-Trieglaff O., Sturm M. (2007) TOPP: The OpenMS proteomics pipeline. Bioinformatics 23, e191–197 [DOI] [PubMed] [Google Scholar]
- 38. Mueller L. N., Rinner O., Schmidt A., Letarte S., Bodenmiller B., Brusniak M. Y., Vitek O., Aebersold R., Müller M. (2007) SuperHirn: A novel tool for high resolution LC-MS-based peptide/protein profiling. Proteomics 7, 3470–3480 [DOI] [PubMed] [Google Scholar]
- 39. Leptos K. C., Sarracino D. A., Jaffe J. D., Krastins B., Church G. M. (2006) MapQuant: Open-source software for large-scale protein quantification. Proteomics 6, 1770–1782 [DOI] [PubMed] [Google Scholar]
- 40. Yu T., Park Y., Johnson J. M., Jones D. P. (2009) apLCMS: Adaptive processing of high-resolution LC/MS data. Bioinformatics 25, 1930–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li X. J., Yi E. C., Kemp C. J., Zhang H., Aebersold R. (2005) A software suite for the generation and comparison of peptide arrays from sets of data collected by liquid chromatography-mass spectrometry. Mol. Cell. Proteomics 4, 1328–1340 [DOI] [PubMed] [Google Scholar]
- 42. Zhang J., Gonzalez E., Hestilow T., Haskins W., Huang Y. (2009) Review of peak detection algorithms in liquid-chromatography-mass spectrometry. Curr. Genomics 10, 388–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lange E., Tautenhahn R., Neumann S., Gröpl C. (2008) Critical assessment of alignment procedures for LC-MS proteomics and metabolomics measurements. BMC Bioinformatics 9, 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang R., Barton A., Brittenden J., Huang J., Crowther D. (2010) Evaluation of computational platforms for LC-MS based label-free quantitative proteomics: A global view. J. Proteomics Bioinformatics 3, 260–265 [Google Scholar]
- 45. Brusniak M. Y., Bodenmiller B., Campbell D., Cooke K., Eddes J., Garbutt A., Lau H., Letarte S., Mueller L. N., Sharma V., Vitek O., Zhang N., Aebersold R., Watts J. D. (2008) Corra: Computational framework and tools for LC-MS discovery and targeted mass spectrometry-based proteomics. BMC Bioinformatics 9, 542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hwang D., Zhang N., Lee H., Yi E., Zhang H., Lee I. Y., Hood L., Aebersold R. (2008) MS-BID: A Java package for label-free LC-MS-based comparative proteomic analysis. Bioinformatics 24, 2641–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Orchard S., Jones A., Albar J. P., Cho S. Y., Kwon K. H., Lee C., Hermjakob H. (2010) Tackling quantitation: A report on the annual Spring Workshop of the HUPO-PSI 28–30 March 2010, Seoul, South Korea. Proteomics 10, 3062–3066 [DOI] [PubMed] [Google Scholar]
- 48. Radulovic D., Jelveh S., Ryu S., Hamilton T. G., Foss E., Mao Y., Emili A. (2004) Informatics platform for global proteomic profiling and biomarker discovery using liquid chromatography-tandem mass spectrometry. Mol. Cell. Proteomics 3, 984–997 [DOI] [PubMed] [Google Scholar]
- 49. Blankenberg D., Von Kuster G., Coraor N., Ananda G., Lazarus R., Mangan M., Nekrutenko A., Taylor J. (2010) Galaxy: A web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. Chapter 19, Unit 19.10, 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Giardine B., Riemer C., Hardison R. C., Burhans R., Elnitski L., Shah P., Zhang Y., Blankenberg D., Albert I., Taylor J., Miller W., Kent W. J., Nekrutenko A. (2005) Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 15, 1451–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sturm M., Bertsch A., Gröpl C., Hildebrandt A., Hussong R., Lange E., Pfeifer N., Schulz-Trieglaff O., Zerck A., Reinert K., Kohlbacher O. (2008) OpenMS: An open-source software framework for mass spectrometry. BMC Bioinformatics 9, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Reinert K., Kohlbacher O. (2010) OpenMS and TOPP: Open source software for LC-MS data analysis. Methods Mol. Biol. 604, 201–211 [DOI] [PubMed] [Google Scholar]
- 53. Sturm M., Kohlbacher O. (2009) TOPPView: An open-source viewer for mass spectrometry data. J. Proteome Res. 8, 3760–3763 [DOI] [PubMed] [Google Scholar]
- 54. Pluskal T., Castillo S., Villar-Briones A., Oresic M. (2010) MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Katajamaa M., Oresic M. (2005) Processing methods for differential analysis of LC/MS profile data. BMC Bioinformatics 6, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pedrioli P. G., Eng J. K., Hubley R., Vogelzang M., Deutsch E. W., Raught B., Pratt B., Nilsson E., Angeletti R. H., Apweiler R., Cheung K., Costello C. E., Hermjakob H., Huang S., Julian R. K., Kapp E., McComb M. E., Oliver S. G., Omenn G., Paton N. W., Simpson R., Smith R., Taylor C. F., Zhu W., Aebersold R. (2004) A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 22, 1459–1466 [DOI] [PubMed] [Google Scholar]
- 57. Orchard S., Montechi-Palazzi L., Deutsch E. W., Binz P. A., Jones A. R., Paton N., Pizarro A., Creasy D. M., Wojcik J., Hermjakob H. (2007) Five years of progress in the Standardization of Proteomics Data 4th Annual Spring Workshop of the HUPO-Proteomics Standards Initiative April 23–25, 2007 Ecole Nationale Superieure (ENS), Lyon, France. Proteomics 7, 3436–3440 [DOI] [PubMed] [Google Scholar]
- 58. Deutsch E. (2008) mzML: A single, unifying data format for mass spectrometer output. Proteomics 8, 2776–2777 [DOI] [PubMed] [Google Scholar]
- 59. Horvatovich P., Govorukhina N. I., Reijmers T. H., van der Zee A. G., Suits F., Bischoff R. (2007) Chip-LC-MS for label-free profiling of human serum. Electrophoresis 28, 4493–4505 [DOI] [PubMed] [Google Scholar]