

Abstract
Skeletal muscle tissue contains slow as well as fast twitch muscle fibers that possess different metabolic and contractile properties. Although the distribution of individual proteins in fast and slow fibers has been investigated extensively, a comprehensive proteomic analysis, which is key for any systems biology approach to muscle tissues, is missing. Here, we compared the global protein levels and gene expression profiles of the predominantly slow soleus and fast extensor digitorum longus muscles using the principle of in vivo stable isotope labeling with amino acids based on a fully lysine-6 labeled SILAC-mouse. We identified 551 proteins with significant quantitative differences between slow soleus and fast extensor digitorum longus fibers out of >2000 quantified proteins, which greatly extends the repertoire of proteins differentially regulated between both muscle types. Most of the differentially regulated proteins mediate cellular contraction, ion homeostasis, glycolysis, and oxidation, which reflect the major functional differences between both muscle types. Comparison of proteomics and transcriptomics data uncovered the existence of fiber-type specific posttranscriptional regulatory mechanisms resulting in differential accumulation of Myosin-8 and α-protein kinase 3 proteins and mRNAs among others. Phosphoproteome analysis of soleus and extensor digitorum longus muscles identified 2573 phosphosites on 973 proteins including 1040 novel phosphosites. The in vivo stable isotope labeling with amino acids-mouse approach used in our study provides a comprehensive view into the protein networks that direct fiber-type specific functions and allows a detailed dissection of the molecular composition of slow and fast muscle tissues with unprecedented resolution.
Skeletal muscles contain different types of fibers, which are responsible for specific biological properties and functions of individual muscles. Muscle fibers have been classified into slow type I and fast type II fibers mainly based on myofibrillar ATP staining and immunohistochemistry using specific antibodies (1).
Slow type I fibers show a red tint, contain high numbers of mitochondria, and their energy supply is mainly based on oxidative metabolism. These features enable slow fibers to execute long lasting contractions, which are essential for the maintenance of body posture. The primary function of type II fibers is the rapid contraction of muscles. Fast fibers are divided into three additional subclasses: Type IIb and IIx (also known as IId) are glycolytic fibers, whereas type IIa fibers are more comparable to oxidative type I fibers (2). Type II fibers, which mainly derive their energy from glycolysis, are thus more susceptible to fatigue compared with Type I fibers.
Muscle fibers have also been classified based on the expression of different isoforms of myosin heavy chain (MyHC) proteins. Myosins are the major contractile proteins and their activation by ATP and Ca2+ ions results in shortening of muscle fibers. For example, slow type I fibers express MyHCIβ and the three fast fiber types express MyHCIIa, MyHCIIb, and MyHCIIx (3), respectively. Although the MyHC-based classification is used most often, several other marker proteins for slow and fast muscles have been described. For example cardiac troponin C (Tnnc1), a regulatory Ca2+ binding protein, is expressed in slow and cardiac muscle tissue, whereas Troponin C/STNC (Tnnc2) is predominant in fast type II fibers (4). Likewise, the calcium-ATPase pumps SERCA1 and 2, which are required for re-uptake of calcium into the sarcoplasmatic reticulum after contraction and for subsequent muscle relaxation (5) are also expressed differentially in slow and fast twitch fibers. SERCA1 is more abundant in fast twitch fibers, whereas SERCA2 is overrepresented in slow muscle fibers (6). Another example of an unequal protein distribution between fast and slow fibers is the catalytic A-subunit of the calcium-dependent serine-threonine phosphatase calcineurin, which shows predominant expression in fast fibers (7). Activation of calcineurin-dependent pathways plays an important role for muscle hypertrophy and fiber transition (8) in response to physical exercise, aging, or metabolic diseases such as diabetes (9, 10).
Our current knowledge of fiber type specific protein expression has been established primarily by traditional biochemical and immunohistochemical techniques. More recently, several cDNA-microarray based studies providing a more systematic and unbiased characterization of muscle tissues has helped to improve our understanding of muscle physiology and the mechanisms of fiber transition (11, 12). However, transcriptional profiles do not necessarily correlate with the steady-state levels of corresponding proteins and fail to give insight into signaling pathways such as are relayed e.g. by protein kinase cascades.
So far, most studies aiming at in-depth characterization of the skeletal or heart muscle proteomes have chosen an approach based on two-dimensional gel electrophoresis (13, 14, 15), which yielded a limited number of reliably identified proteins. Recent advances in mass spectrometry based proteomics, however, offer new options allowing identification and quantification of thousands of proteins (16, 17, 18). These new techniques have already been exploited in a few studies to characterize differences in protein abundance between different muscle tissues, including limb and extraocular muscles using one dimensional-gel or isoelectric focusing fractionation in combination with high mass accuracy liquid chromatography-tandem MS (LC-MS/MS) analysis (19, 20).
Here we report a large-scale quantitative analysis to compare the proteomes and transcriptomes of slow soleus and fast extensor digitorum longus (EDL)1 muscles of the mouse. Our analysis is based on the use of 13C6-lysine-labeled mice (“SILAC-mouse”), which provides an internal protein standard. We quantified >2100 proteins in slow and fast muscles, of which ∼25% showed a differential expression pattern. Comparison of proteomics data to mRNA expression profiles obtained from the same samples revealed the existence of specific post-transcriptional regulatory mechanisms in slow and fast muscle fibers although the majority of proteins and mRNA molecules showed a similar distribution. Furthermore, we identified 1040 novel class 1 phosphorylation sites indicating a widespread phosphorylation of sarcomeric proteins in skeletal muscle cells. All data were implemented into the newly developed Quantimus database and are readily accessible.
EXPERIMENTAL PROCEDURES
Materials and Reagents
Mouse diet substituted for 13C6-lysine was obtained from Silantes (Martinsried, Germany). Mice were generated in house from a C57Bl/6 colony. All chemicals used for tissue extraction, digests, and liquid chromatography were purchased from Sigma - Aldrich and Carl Roth GmbH (Karlsruhe, Germany), except for Lysyl endopeptidase (R), which was obtained from WAKO GmbH (Neuss, Germany).
Generation of 13C6-Lysine Labeled Mice
Mice fully labeled with 13C6-Lysine were generated as described (21), except that food pellets containing the heavy lysine were purchased from a commercial source (Silantes, Martinsried, Germany). Labeling efficiency was >96% in the F2 generation of mice maintained on a heavy lysine substituted diet as described previously. Ten-week-old individuals of the F2 generation were used for all experiments. The incorporation rate and labeling efficiency of heavy SILAC labeled mice was monitored in each experiment by assessing the average SILAC ratio on a subset of proteins from muscle tissue.
Sample Preparation
Mice were sacrificed by ketanest injection. The thorax was opened immediately after respiratory arrest before the heart was perfused with PBS via the left ventricle and the right ventricle was cut open for drainage. Successful perfusion and blood cell removal was assessed by observing the color change from red to grayish-white of blood rich organs such as lung and liver. Soleus and Extensor digitorum muscles were dissected from the hindlimbs of the animals, washed in PBS, and snap frozen in liquid nitrogen (nheavy = 2, nlight = 2). Muscles from heavy and light SILAC-labeled animals (8.5–13 mg wet weight) were mechanically homogenized in 250 μl of ice cold modified RIPA buffer containing 1% Nonidet P-40, 0.1% sodium deoxycholate, 50 mm Tris-HCl pH 7.5, 150 mm NaCl, and 1 mm EDTA supplemented with complete protease inhibitor mixture (Roche) using an Ultraturrax disperser (IKA, Staufen, Germany) and incubated for 5 min on ice to extract proteins. Alternatively, we used SDS lysis buffer (4% SDS, 100 mm Tris/HCl pH 7.6) to extract muscle fibers. To remove debris lysates were centrifuged at 19,000 × g for 10 min. Supernatants were collected and protein content was determined using detergent compatible Lowry protein assay (Bio-Rad DC). Fifty to 80 μg of a 1:1 mixture of heavy and light muscle RIPA extracts were then separated by gel electrophoresis on precast 4–12% NuPAGE gradient gels (Invitrogen, Carlsbad, CA) and stained with the Colloidal Blue Staining Kit (Invitrogen). Each lane was cut into 15 evenly sized gel pieces and processed for GeLC-MS/MS. Briefly, proteins within gel pieces were subjected to reduction and alkylation, followed by endopeptidase Lys C cleavage. Peptides were then extracted as described (22), desalted, and concentrated by Stage Tips (23). SDS lysates were subjected to the FASP protocol followed by Offgel separation as described in (24). For phosphopeptide enrichment we used a combination of (FASP) and strong-cation chromatography followed by titanium dioxide enrichment (25).
LC-MS/MS Analysis
Each sample, representing the peptide content of one gel piece was analyzed by nano-Reversed Phase Chromatography using an Agilent 1100 nanoflow system that was online coupled via in house packed fused silica capillary column emitters (length 15 cm; ID 75 μm; resin ReproSil-Pur C18-AQ, 3 μm) and a nanoelectrospray source (Proxeon) to an LTQ Orbitrap XL mass spectrometer (Thermo Scientific). Peptides were eluted from the C18 column by applying a linear gradient from 5–35% buffer B (80% acetonitrile, 0.5% acetic acid) over 150 min. The mass spectrometer was operated in the data-dependent mode, collecting collision induced MS/MS spectra from the five most intense peaks in the MS (LTQ-FT full scans from m/z 300 to m/z 1800; resolution r = 60,000; LTQ isolation and fragmentation at a target value of 10000). For the identification of phosphopeptides, an LTQ-Orbitrap Velos mass spectrometer was used and MS/MS spectra were generated by higher C-trap dissociation. Briefly, 30,000 ions were accumulated in the c-trap and MS/MS spectra were detected in the orbitrap at a resolution of 7500 (26). Raw data files were then processed by MaxQuant software (Version 1.0.014.10) in conjunction with Mascot database searches (Version 2.2) (27). All data were searched against the International Protein Index sequence database (mouse IPI, version 3.54) with 56,149 entries. The database was extended with commonly observed contaminants and concatenated with reversed versions of all sequences. The parameter settings were: LysC as digesting enzyme, a maximum of two missed cleavages, a minimum of six amino acids, carbamidomethylation at cysteine residues as fixed and oxidation at methionine residues as variable modifications. For the phosphopeptide analysis we set phosphorylation of serine, threonine, and tyrosine as variable modification. The false discovery rate was set to 1% at the peptide and protein level. All peptides and phosphopeptides with their SILAC ratios were uploaded to the Quantimus database. The database is based on an Apache Server and MySQL (Version 5). SILAC labeling with 13C6-lysine was specified and accounted for within MaxQuant. The maximum allowed mass deviation was 10ppm for MS and 0.5 Da for MS/MS scans. Only proteins identified with one uniquely assigned peptide to the corresponding protein were included.
Protein Classification with Gene Ontology
Detected proteins from IPI 3.54 mouse database and Affymetrix transcript IDs were used for a Gene Ontology (GO) term analysis. Term annotations were derived from uniprot. GO annotatable proteins were used for a hypergeometric test to reveal overexpressed terms. We defined GO terms as overexpressed, having a Benjamini and Hochberg corrected p value smaller than 0.05. Calculations were done by the Cytoscape plugin Bingo. We tested each data set (Soleus and EDL) on mRNA and protein level for overexpression at GO domains cellular component, molecular function, and biological process. Separated lists are provided in an additional table (supplemental Table S1).
Microarrays
Tissues were dissected from PBS-perfused animals, and total RNA was isolated using the TRIzol method (Invitrogen). RNA quality was checked on the Agilent 2100 Bioanalyzer using the RNA 6000 Nano Kit. For mRNA expression analysis, the Affymetrix GeneChip Mouse Genome 1.0 Array was employed with the respective 1-cycle target labeling protocol. Data were analyzed by the RMA algorithm using the Affymetrix Expression Console. An unpaired t test was performed with log2-transformed data to identify significantly differentially expressed transcripts, and FC was calculated using DNAStar ArrayStar 3.0 software.
RESULTS
Large-scale Quantification of Proteins in Slow and Fast Muscles Using SILAC-assisted Mass Spectrometry
For accurate protein quantification we used the stable isotope labeling of amino acid in cell culture approach (SILAC), which is based on the metabolic incorporation of non-radioactive amino acids in which carbon and/or nitrogen atoms are substituted by heavy isotopes (i.e. 13C6-lysine) (28). Recently, we extended this technique to in vivo conditions in which a mouse is labeled with the 13C6-lysine isotope (21). By feeding mice over two generations with a 13C6 lysine diet, virtually all natural 12C6 containing lysine amino acids are replaced by the heavy amino acid.
To identify and quantify proteins that are either predominantly found in fast and slow twitching muscle fibers or common to both muscle types we isolated fast (EDL muscle) and slow (soleus muscle) fibers from the lower leg of the mouse hind limb. In contrast to rats, where the soleus muscle consists mainly of type I fibers, soleus muscles in mice contain an equal amount of type I and type IIa/IIx fibers (29). Despite these restrictions we choose the soleus muscle as the slow muscle reference for our proteomic analysis because it contains the highest amount of type I fibers and the remaining type II fibers are mostly oxidative. Based on peak intensities (see methods), equal amounts of labeled soleus proteins were mixed with unlabeled soleus and EDL muscle extracts, respectively. As a control we performed a crossover experiment by mixing labeled EDL with unlabeled EDL and Soleus extracts (Fig. 1). Forward and crossover experiments were performed with two biological replicates each. To decrease sample complexity, proteins were separated by one-dimensional SDS-PAGE. Each lane was separated into 15 slices following Coomassie-blue staining and subjected to in-gel digest with LysC to obtain lysine only containing peptides. We used the filter aided sample preparation (FASP) protocol in combination with isoelectric focusing of peptides (Offgel, Agilent) to increase our identification rate for proteins with reduced solubility. In total, we analyzed 123 fractions by LC-MS/MS using an LTQ-Orbitrap XL or an LTQ-Orbitrap XL Velos (Thermo-Fisher Scientific) mass spectrometer. Using a false discovery rate <1% we identified 537,282 MS/MS spectra, which resulted in the identification of 28,924 peptides corresponding to 3447 proteins with at least one unique peptide (supplemental Table S1). Quantifications (e.g. SILAC ratios) were obtained for 2163 proteins between Soleus and EDL muscle tissue.
Fig. 1.

Design of forward and reverse in vivo SILAC experiments. For relative quantification of proteins in slow versus fast twitch muscle fibers fully lysine-6 labeled tissues (heavy) were used as internal standard. Ratios between unlabeled slow and unlabeled fast muscle fibers were calculated as indicated.
To facilitate data analysis and to provide public access to our skeletal muscle data set, an online proteomic database called QuantiMus was developed. This database contains all information obtained by using the MaxQuant software tool, including the number of identified peptides, unique peptides, Silac-ratios, PTMs and Mascot-scores (27). Both unregulated and regulated proteins are grouped into ratio bins. Data base searches create graphical overviews, which display localization of all identified peptides within a protein sequence (http://quantimus.mpi-bn.mpg.de). For the comparison of soleus and EDL muscles we selected only proteins that were quantified with at least one unique peptide in all Sol versus EDL and respective crossover measurements. In addition, by taking advantage of the QuantiMus database, we also included several proteins that were identified in soleus(h) versus EDL(l) or EDL(h) versus soleus(l) experiments but which lacked a corresponding ratio in control (soleus(h)/soleus(l) or EDL(h)/EDL(l)) experiments. This correction was necessary because a complete absence of proteins in either soleus or EDL generates misleading ratios. For example, slow troponin T was detected with a 25.5-fold change in Sol (h)/EDL (l) and with the inverse ratio of 0.02 in EDL (h)/Sol (l), which indicated low levels or even complete absence of slow troponins in fast muscle tissue.
One challenge for mass spectrometry is the simultaneous detection of low and high abundant peptides within a complex protein sample such as extracts of total muscle. To test whether the peak intensity of detected SILAC-pairs would influence the overall ratio distribution and thus accuracy of our quantitative determinations we plotted the sum of the light and heavy peak intensities against the corresponding log2 ratios. As shown in (Fig. 2A), the ratio distribution of labeled versus non-labeled soleus sample was close to 1, indicating accurate quantification within the whole range of peak intensities. Of note, the overall ratio distribution is more scattered between soleus and EDL compared with the control soleus and soleus ratio (Fig. 2A) illustrating quantitative differences in the proteome composition between both muscle fiber types. A similar observation is made when binned log ratios of all quantified proteins are displayed in a frequency distribution plot: The distributions centered more closely on a ratio of 1 for the comparison between light and heavy muscle fibers of the same type, although they are clearly more dispersed when different fiber types were compared (Fig. 2C). The reproducibility of our quantitative measurements appeared very high as indicated by the Pearson correlation coefficient between both experiments (0.86) that was obtained by plotting all log ratios obtained from the soleus (h)/EDL (l) experiment against the respective crossover ratios (EDL (h)/soleus (l)) (Fig. 2B).
Fig. 2.

Comparison of slow and fast muscle proteins. A, Distribution of ratios for peptides identified from labeled versus non-labeled extracts. The log ratio of all peptides were plotted against the sum of heavy and light peak intensities for both the comparison of the same fiber type (Sol/Sol; upper panel) and different fiber types (Sol/EDL; lower panel). Note that some proteins (red) do not show any significant changes during fiber comparison, whereas other proteins e.g. calsequestrin (green) are differentially regulated. B, Histogram representation of the binned log ratios of all quantified proteins. Distributions center more closely around a ratio of 1 in comparisons of light and heavy muscle fibers of the same type while they are more dispersed in comparisons of different fiber types. C, Correlation plot between forward (Sol/EDL) and reverse experiment (EDL/Sol), representative examples of proteins overrepresented in soleus fibers are marked in green, examples of proteins overrepresented in EDL muscle are marked in blue. D, Employing SILAC-labeled control mice as internal standard simplifies the identification of different protein abundances in both fiber types by determining the direct ratios between unlabeled soleus and unlabeled EDL muscle. A ratio of 1 indicates equal distribution.
Because extracts from the SILAC-labeled mouse served as an internal protein standard in our experiments we were able to calculate protein ratios between unlabeled soleus and unlabeled EDL simply by dividing ratio 2 through ratio 1 (see Fig. 1), which yields quantitative differences between both muscle types. Most detected proteins showed a ratio close to 1:1, which indicates equal amounts of proteins in both muscles. Myopodin, f.e., was detected with a Sol(h)/Sol(l) ratio of 1 and Sol(h)/EDL(l) ratio of 0.8, which finally resulted in a Sol(l)/EDL(l) ratio of 1.2. The crossover experiment showed for Myopodin also an EDL(l)/Sol(l) 1 ratio (Fig. 2D). Some proteins, such as the ATP citrate lyase were measured with a ratio of 4.3 between Sol(h)/Sol(l). However, a similar ratio of 4.9 was also determined comparing Sol(h)/EDL(l), resulting finally in a direct SOL(l)/EDL(l) ratio of 1.2. Similarly, Glutathione peroxidase 3 was detected with a Sol(h)/Sol(l) ratio of 0.4 and Sol(h)/EDL(l) ratio of 0.4, respectively, which again resulted in a SOL(l)/EDL(l) ratio of 1:1.1 (Fig. 2D). To estimate the total number of proteins that were differentially regulated between slow and fast muscle we calculated the geometric mean between the forward and crossover experiments and set the cutoff value to 1.5 or 2 (30, 31). Using this definition we identified 252 proteins that were enriched in the soleus and 299 proteins enriched in the EDL muscle (supplemental Table S1).
SILAC-assisted Mass Spectrometry Greatly Increases the Number of Known Slow and Fast Muscle Enriched Proteins
To confirm the overall quality of our in vivo-SILAC approach we first focused on proteins that are well described components of either slow or fast muscle fibers, for example, the sarcoplasmic/endoplasmic reticulum calcium associated ATPases 1 and 2 (6), which are important for the regulation of Ca2+ homeostasis. SERCA1 has been shown to be more abundant in fast twitch fibers, whereas SERCA2 is the predominant form in slow muscle fibers. Our analysis yielded very similar results indicating the validity of the mass spectrometry approach. Fig. 3 depicts mass spectra of SILAC-peptide pairs for SERCA1 (A) and 2 (B) as representative examples. The corresponding ratios of both proteins are listed in (Table IA, B). The identification of specific isoforms of myosin heavy chains (MyHC) by mass spectrometry presents a more challenging problem because extensive sequence homologies exist between different MyHC proteins (32). We took a conservative approach and used only unique peptides for protein quantification to avoid any misinterpretation of our data set (Fig. 4A), which reduced the number of quantifiable peptides considerably. For example, MyHCs-Iβ, -IIa, -IIb, Myosin-8, and Myosin-13 share the peptide HADSVAELGEQIDNLQRVK (Fig. 4C), which therefore was not used for quantification. Only ∼24 unique peptides out of ∼90 peptides which were measured from the MyHCIβ protein group were used for quantification because of sequence identities. The measured ratio of 11:1 between Soleus versus EDL indicated greatly increased MyHCIβ (gene: myh7) levels in the slow soleus compared with the fast EDL muscle (Fig. 4B, upper left two panels). We also identified increased levels of MyHC-IIa in soleus muscle compared with EDL (Fig. 4B, lower left two panels). In contrast, MyHC-IIb and MyHC-13 were more abundant in the EDL muscle (Fig. 4B, right hand panels), which is consistent with previous reports about the distribution of MyHC proteins (33). Similar results were obtained for several slow myosin light chains (MyLC). In conclusion, the in vivo SILAC approach was able to generate a rapid and accurate estimation of the distribution of proteins in slow and fast muscle fibers even for molecules that are relatively difficult to distinguish by traditional biochemical means.
Fig. 3.
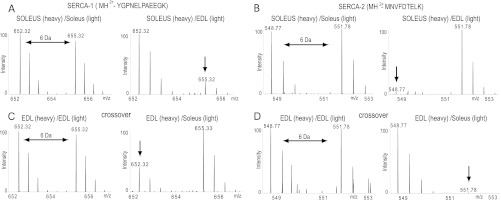
MS spectra of heavy and light peptide pairs derived from SERCA1 and SERCA2. The SERCA1 peptide MH2+-YGPNELPAEEGK is less abundant in soleus muscle fibers (A; arrow), whereas the SERCA2 derived peptide MH2+-MNVFDTELK is overrepresented in EDL fibers (B; arrow). The inverse patterns are seen in the corresponding crossover experiments (C, D).
Table I. (A) List of selected proteins enriched in the Soleus. The table is divided into different cellular compartments and fold changes are shown for the forward (soleus/EDL, S/E) and crossover (EDL/soleus, E/S) experiments. The p values of the mRNA fold changes are indicated in green (p value < 0.001), in yellow (p value 0.05–0.001), and in red (p value > 0.05). (B) List of selected proteins enriched in the EDL muscle.
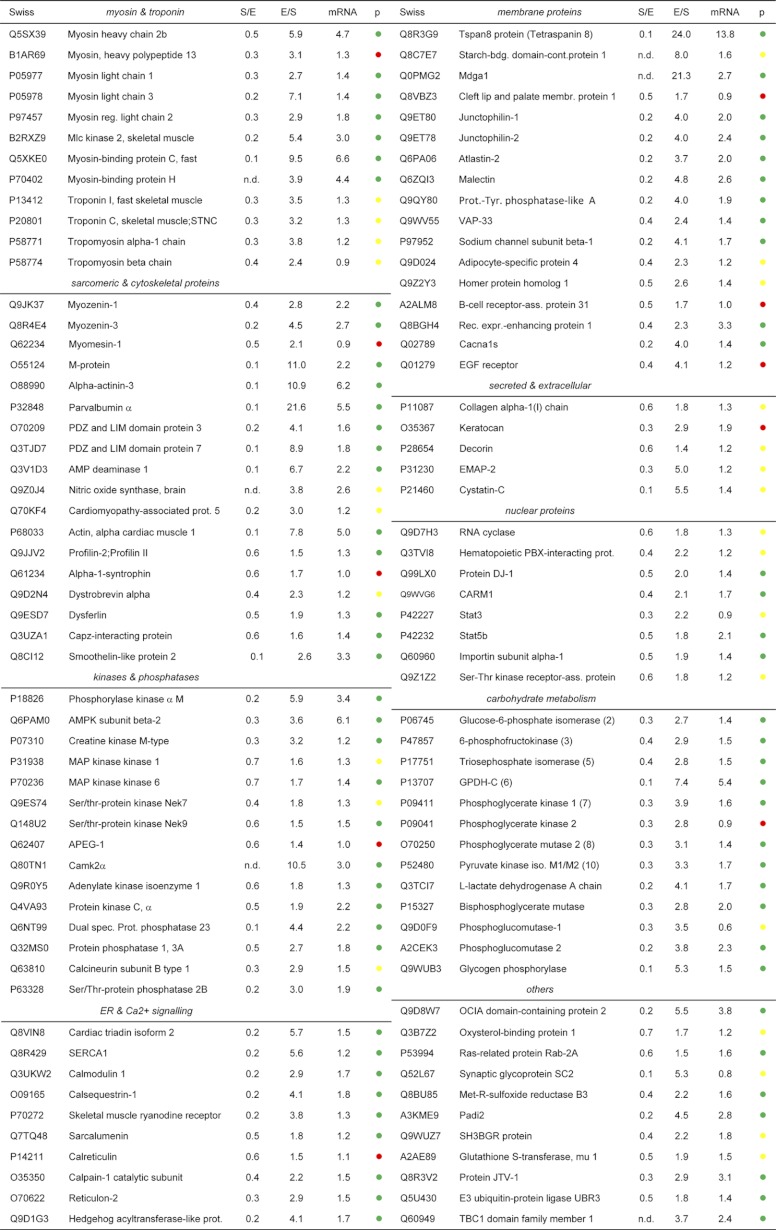
Fig. 4.

Schematic alignment of myosin heavy chains. A, Mapping of quantifiable myosin heavy chain (MyHC) peptides to their corresponding gene locus. Unique peptides used for quantification are indicated in black, non-unique peptides (not used for quantification) are marked in gray. Localization of phoshorylation sites are shown in red. B, MS spectra of representative SILAC peptide pairs derived from MyHC-1β, -IIa, -IIb, or MyHC-13 that show over- or underrepresentation in either soleus or EDL muscle. C, Example of a quantifiable peptide that is shared by various members of the MyHC1β family and that was therefore excluded from quantification.
Pronounced quantitative differences between slow and fast muscle fibers were also found for several other proteins located in or near the Z-disc: the ankyrin repeat domain containing protein 2 (also known as mArpp) (34), which was described as a stretch sensor specifically expressed in type I muscle fibers, showed a top-ranking SILAC-ratio of 39:1 between soleus and EDL. FATZ-1 (also known as calsarcin-2/Myozenin-1) and FATZ-3 (calsarcin-3/Myozenin-3) (35) showed a strong enrichment in fast fibers whereas FATZ-2 (calsarcin-1/Myozenin-2) was more abundant in the soleus muscle corroborating previous reports. Differential accumulation in slow versus fast muscles was also found for M-band proteins that are required for the regular packing and organization of thick filaments. So far, three Myomesins, namely Myomesin-1 (also known as Skelemin or EH-myomesin) (36), Myomesin-2 (also known as M-protein) (37), and Myomesin-3 (38) have been localized to the M-band. Our results supported previous findings that Myomesin-2 is predominantly located in fast (EDL) and Myomesin-3 in slow muscles (soleus). In contrast, increased levels of Myomesin-1 were detected in the EDL, which corrects the view that Myomesin-1 is uniformly distributed in all skeletal muscle tissues.
So far, ∼60 proteins have been described to be localized within sarcomeric structures mostly based on immunohistochemistry and two dimensional gel electrophoresis experiments (39, 40). In our single large-scale study we determined the relative distribution between slow and fast muscles for virtually all known sarcomeric proteins. Moreover, we were able to measure quantitative differences of a large group of additional muscle proteins in slow and fast muscles (Fig. 5A), which might help to resolve the molecular composition of the contractile apparatus and its specific properties in different muscle types.
Fig. 5.

Schematic representation of skeletal muscle fiber structure and composition. A, Selected muscle proteins identified and quantified in our screen are depicted according to their subcellular localization (green = overrepresented in EDL; red = overrepresented in Soleus, black = no quantitative difference between EDL and Soleus). B, Venn diagram summarizing the results of a gene ontology analysis of proteins differentially expressed in EDL and soleus. Significantly overrepresented GO terms are provided for EDL (left) and soleus (right).
Bioinformatical Analysis Reveals Differential Enrichment of GO-terms in Slow and Fast Muscles
To obtain a better insight into differences between slow and fast muscles, which are reflected by different protein concentrations we performed a comprehensive search for specific GO-terms, which were overrepresented among proteins differentially expressed in soleus and EDL. First, we used a GO-term analysis to identify enriched terms for “biological process,” “cellular localization,” and “molecular function” in soleus and EDL tissue. The soleus muscle showed a strong overrepresentation of categories for oxidative metabolism, fatty acid metabolic processes, and mitochondrial proteins, which mirrors its dependence on oxidative phosphorylation required for efficient generation of ATP (Fig. 5B). Furthermore, we detected several marker proteins typical for high oxygen consumption, such as Myoglobin (41) and carbonic anhydrase III (42). In contrast, analysis of EDL proteins revealed a clear overrepresentation of GO-terms for carbohydrate metabolic processes, epitomized by the detection of almost all glycolytic enzymes. Overrepresentation of sarcoplasmatic structures (GO: 0016529) emphasized the need of enhanced Ca2+ release and re-uptake in EDL muscles.
Although the differential distribution of the above-mentioned processes were not surprising and in line with previous reports we also detected a number of unexpected differences between both fiber types. Interestingly, several proteins of the extracellular matrix (ECM), including biglycan, a secreted proteoglycan, asporin, and periostin-2, which are secreted by muscle fibers, were all increased in the soleus muscle. Other proteoglycans, such as keratocan, and Glypican, were enriched in the EDL. Several other ECM proteins such as lumican fibromodulin, fibronectin, decorin, and several collagens did not show any significant changes.
Another class of enzymes that was markedly different between both muscle types were Glutathione S-transferases (GSTs), which are detoxifying enzymes (43) also involved in cell signaling (44). Four different GST enzymes, including Mu-7, Omega-1, kappa-1, and the microsomal GST-I were enriched in the soleus muscle, emphasizing their role in the prevention of oxidative damage. In contrast, GST Mu1, 2 and 5 showed increased levels in EDL. The GST-Mu2 isoform was described as a regulator of the ryanodine receptor (RyR) (45). Similar protein concentrations in soleus and EDL muscles were determined for other GST′s including A4, P1, and theta1. The presence of specific subsets of GST enzymes in slow and fast muscle fibers reflects potential differences in detoxification and GST-mediated signaling processes that may impinge on the overall physiology of both fiber types. We also observed enriched GO-terms for amino acid metabolism in EDL samples. The enrichment of 9 amino acid tRNA synthetases in fast muscle tissue might indicate a higher metabolic activity in the fast EDL muscle because of the increased mechanical “wear and tear” and a demand for higher protein turnover in these fibers.
Slow and Fast Muscles Show Different Protein Kinase and Phosphatase Expression Levels
Muscle contraction and relaxation requires phosphorylation and dephosphorylation of numerous proteins. Therefore, we were particularly interested to determine potential differences in fiber type specific expression of protein kinases and phosphatases. In total, we identified and quantified more than 122 kinases and 86 phosphatases. Apart from nearly all glycolytic kinases, we found a 10-fold higher expression of the calcium/calmodulin-dependent protein kinase type IIα (CaM) in the EDL compared with the soleus muscle supporting the importance of Ca2+ signaling for fast muscle fibers. The 5′-AMP-activated protein kinase also showed increased levels in the EDL. This enzyme is one of the immediate downstream targets of CaM kinase, which acts as a metabolic master switch and regulates several metabolic pathways including glucose uptake and glycolysis. Similarly, phosphorylase kinase, an important regulator of glycogen homeostasis (46), was also detected at higher levels in the EDL underscoring the requirement of sufficient glucose supply in the EDL. Furthermore, we detected enriched levels of the ERK activator kinase 1, 2, 4, and 6 in the EDL, which belong to mitogen-activated protein kinases (MAPK). An important substrate of the MAPK signaling is the 90 kDa ribosomal S6 kinase, which was also enriched in the EDL muscle. The S6 kinase phosphorylates the ribosomal protein S6 and acts as an activator of protein biosynthesis indicating increased protein turnover in fast muscles.
Five dual specific phosphatases (DUSP), enzymes that are able to dephosphorylate both tyrosine and serine/threonine residues, were detected in our samples. Three of them were enriched in the EDL including DUPD1, dual specificity phosphatase 3 (Dusp3), and dual specificity protein phosphatase 23 (Dusp23). In contrast, the protein phosphatase regulatory subunit 1A (Ppp1r1a), the phosphatase 1B (Ppm1b), the Inositol polyphosphate 4-phosphatase type IIβ (Inpp4b) and the catalytic subunit of the Myo-inositol monophosphatase A2 (Impa2) were more abundant in the soleus muscle.
The distinct expression pattern of kinases and phosphatases, which distinguishes soleus from EDL, suggests significant differences of corresponding target proteins and the kinetics of phosphorylation and dephosphorylation. It seems reasonable to assume that such differential phosphorylation events are instrumental for different physiological processes in fast and slow muscles.
Toward a Skeletal Muscle Phosphoproteome
As a first step to establish a comprehensive skeletal muscle phosphoproteome we isolated phosphopeptides from EDL and soleus muscles by cation exchange chromatography, further enriched the phosphopeptides by TiO2 affinity purification, and subjected them to mass spectrometry. In total, we identified 2573 phosphorylation sites on 973 proteins with high accuracy and a localization probability > 0.75 (median localization probability 0.99) (supplemental Table S2). Comparison with entries from other phosphopeptide databases, such as www.Phosida.org (∼25,000 phosphopeptides), L6 rat cell culture line (2230 phosphopeptides) (47), mouse atlas (48), and www.PhosphoSitePlus.org (∼ 94,000 phosphopeptides) revealed that 1040 candidate phosphorylation sites were novel. For example, we identified unknown phosphosites for several sarcomeric proteins, including sites for tropomyosins, for muscle creatine kinase, and for myosin heavy chain proteins. Interestingly, the overlap to other phospho-databases that feature different tissues was rather low suggesting the existence of tissue specific phosphorylation patterns. We found that the overall distribution of serine (∼78%) and threonine (∼18%) phosphorylation sites was similar to datasets obtained in other tissues (25, 47) (supplemental Fig. S1A). However, we monitored a clear increase of tyrosine phosphorylation sites of ∼4% compared with 1–2% usually found in other cells and tissues (25, 47). For example, 42 novel tyrosine phosphorylation sites were detected on the giant protein titin. Mapping of these sites revealed a preferential accumulation of newly mapped tyrosine phosphorylation sites to functional domains, including the fibronectin type 3 like domain (FN3-like) and the PEVK domain (supplemental Fig. S2). Analysis of phosphorylation motifs by MaxQuant indicated that proline dependent CK1/2 and PKA motifs were more abundant compared with other motifs (supplemental Fig. S1B). We would like to emphasize that the repertoire of phosphorylation sites obtained in this study represents a profound repository and starting point for future studies focusing on the regulation of cell signaling events in skeletal muscle. All identified phosphorylation sites were implemented in the Quantimus database.
Comparison of RNA and Protein Expression Levels in Slow and Fast Muscles Indicates Multiple Levels of Regulation
cDNA hybridization to microarrays is an important technology to obtain global views of transcript levels within cells or tissues. To investigate the correlation between mRNA transcript levels and protein abundances we performed hybridizations with Affymetrix Chip sets (mouse gene 1.0 ST Array; n = 3 for each muscle type) using RNA isolated from soleus and EDL muscles. To allow comparison of microarray probes to proteins we matched international protein index (IPI 3.54) identifiers to microarray probes characterized by common Entrez gene identifiers.
In total, we profiled 22,360 Entrez genes in skeletal muscle. Statistical significant quantification was possible for 16,985 genes after background subtraction (Fig. 6A, supplemental Table S3). A Pearson correlation coefficient of 0.81 (Fig. 6B) indicated a significant overlap of all detected transcripts and proteins, which fits to results obtained in other experimental systems including yeast (49) and mouse (50). Supplemental Table S1 shows the numbers of regulated genes between soleus and EDL dependent on different p values and ratio cut offs. Next, we compared all transcript intensities with mRNA intensities from our protein dataset (supplemental Fig. S3). Not surprisingly, most of the identified proteins were detected from transcripts with higher abundance. This shows that several orders of magnitudes in sensitivity and dynamic range are needed to detect lower abundant transcripts from skeletal muscle tissue.
Fig. 6.

Correlation between proteomic and genomic data sets. A, Hybridization of cDNAs isolated from both muscle fiber types revealed enrichment for 1442 (p < 0.05) transcripts in either the EDL or the Soleus muscle. All transcripts with a fold change > 1.5 (p value <0.001) are indicated in green. Transcripts with fold change >1.5 and p values of 0.001–0.05 are indicated as yellow dots. Transcripts with fold changes <1.5 are shown in black. B, SILAC ratios of all quantified proteins plotted versus the fold change of mRNA abundance of the corresponding proteins. Pearson correlation coefficient = 0.81. C, D, Correlation of protein abundances (SILAC ratios) with mRNA expression levels (fold change mRNA expression) for proteins up-regulated either in EDL fibers (C) or soleus fibers (D). Selected proteins are encircled.
For the soleus muscle we observed 700 genes with a ratio >1.5 and a p value <0.05. Similarly, 688 genes were enriched in the EDL muscle using the same statistical cutoff. 112 proteins that were enriched in the EDL (37%) also showed enrichment at the mRNA level (p value <0.05). Further 124 candidates (41%) showed changes in the same direction (fold change >1) with a p value <0.05, which results in a Pearson correlation of 0.55 (Fig. 6C).
In the Soleus, 151 of the enriched proteins (59%) were also enriched on the mRNA level and a further 63 proteins and genes (21%) showed changes in the same direction generating a Pearson correlation of 0.78 (Fig. 6D).
To assess the biological function of transcriptionally regulated genes a GO-term analysis was performed in a similar manner to the analysis of the proteomics data. Using a 1.5-fold change as cutoff we observed an enrichment for GO-terms for muscle contraction (GOBP: 0006936), for lipid metabolic process (GOBP: 0006629, and for fatty acid metabolic process (GOBP: 0006631) in the soleus. GO-analysis of EDL samples showed a similar enrichment of GO-terms as in our proteomics dataset including terms for carbohydrate metabolism and fast muscle contraction. Taken together the enrichment for similar GO-terms in our proteomics and mRNA expression analysis reflects similar regulations of mRNA and protein abundances for most of the analyzed genes (supplemental Fig. S4).
Interestingly, we found striking differences in protein and mRNA expression levels for several molecules indicating that the identity of slow and fast muscles is in part achieved by post-transcriptional regulation. For example, 10 genes showed higher mRNA levels in Soleus (five genes) and EDL (five genes), which was not matched by changes at the protein level (supplemental Table S5). In particular, we noted increased mRNA levels for two complement proteins in the Soleus, which was surprising given the fact that these proteins are secreted serum proteins. Similarly, α-protein kinase 3 also known as Myocytic induction/differentiation originator (Midori) (51) was enriched at the mRNA level in soleus but showed no concentration differences at the protein level.
Another group of genes showed elevated protein levels in either soleus or EDL muscles but had similar mRNA levels in both fiber types (fold change <1.5, p value >0.05). Most of the proteins with increased levels in the soleus but identical mRNA levels were localized to mitochondria (16 proteins) or within the membrane fraction (eight proteins). In addition, Myosin-8 protein was increased in the soleus but showed matching mRNA concentrations in either muscle (supplemental Fig. S5). Forty-three proteins were enriched in the EDL but did not show significant changes on the mRNA level. For example, five tRNA synthetases, four secreted proteins, three kinases, and four membrane proteins including the EGF-receptor showed similar mRNA expression level but increased concentrations at the protein level. Furthermore, the M-band protein Myomesin-1 (Skelemin) and the extraocular myosin heavy chain EO-MyHC (Myosin, heavy polypeptide 13) were more abundant in the EDL as proteins compared with their mRNAs suggesting fiber-specific regulatory mechanisms for these sarcomeric proteins (supplemental Fig. S6). Our findings reveal the existence of regulatory events at the posttranscriptional or posttranslational level in fast and slow muscles and emphasize the complex relationship between gene expression and protein abundances, which help to shape the distinct phenotype of different muscle fiber types.
DISCUSSION
Numerous studies have described the ultrastructure and molecular diversity of skeletal muscle tissues after the initial recognition of differences between white and red muscle tissue by Ranvier in the year 1873. The general structure of muscle fibers is rather similar despite the presence of different fiber types and the ability of skeletal muscle fibers to adapt to different physiological demands. So far, the composition of skeletal muscle or individual muscle fibers was mostly assessed in a qualitative manner Only limited numbers of proteins were quantified. Very few proteomics studies attempted to quantify proteins in muscle tissues (52–54) and to this end only one two-dimensional-gel electrophoresis-based compared soleus and EDL muscle in rats detecting a limited number of proteins (15). Most studies were devoted to rats, guinea pigs, and rabbits whereas considerably less is known for smaller mammalia such as mice (1, 39). The in vivo SILAC approach used in this study allowed a comprehensive assessment of proteins in slow and fast muscle fibers in mice. Comparison of protein and mRNA data extended the analysis and provided insights into the regulatory events that shape the identity of fast and slow muscles at the post-transcriptional level.
The SILAC method employs the metabolic incorporation of stable isotopes of essential amino acids into cellular proteomes and has been successfully applied for in vitro-cell culture experiments (28). Here, we used metabolic labeling in the SILAC-mouse, in which all tissues are completely labeled with the heavy 13C6 lysine isotope (21, 55) to allow global protein quantification. This method, which is based on mixing control and experimental samples with the heavy protein standard, avoids additional post-extraction modification steps of proteins or peptides and therefore significantly reduces potential methodological errors. Analysis of SILAC-data sets is further facilitated by the use of several specialized software tools such as MSQuant, MaxQuant and Proteome Discoverer (Thermo Scientific), which can be readily used together with LysC digests generating peptide pairs with 6 Da mass differences.
Our approach allowed us to quantify more than 2100 proteins in fast and slow muscle fibers, which to the best of our knowledge, is the largest quantitative data set for proteins in skeletal muscle tissues in vivo. The identification of ∼550 proteins, which we found to be differentially expressed in slow or fast muscles, greatly extends the number of fiber type-enriched proteins. Because virtually all already known fiber type-enriched proteins were contained in our data set we assume a fairly good coverage of the fast and slow skeletal proteomes. The Quantimus database, in which these data are stored, represents an easy and convenient tool to identify proteins of interest via a simple search function. Further links provide additional information such as associated peptides, which are described by their position in the protein, Mascot-scores, phosphorylation status, and SILAC ratios between soleus and EDL. The database will be updated continuously as new entries with corresponding parameters will become available in further experiments. Refined sample preparation protocols and improved resolution and sensitivity of mass spectrometric instrumentation might help to further extend the coverage of muscle proteomes.
One of the most common ways to discriminate different muscle fiber types is based on the content of different MyHC proteins. However, as already indicated earlier, the amino acid sequences of individual members of this protein family are rather similar and peptide assignments after mass spectrometric analysis is often complicated by high sequence homologies (56). For exact identification and quantification of fiber type specific MyHC isoforms it is essential to select only peptides, which are unique for a given isoform. Using these criteria we were able to determine the abundance of MyHC′s in the soleus in the following order: MyHCIβ > MyHCIIa > MyHC-8, MyHC-10 in slow muscles. In contrast, we observed the following order for the EDL muscle: MyHC-IIb > MyHC-13, which is in good correlation to published datasets. Several studies confirmed the presence of the slow type II MyHC IIa in the soleus. Our finding reflects the mixed type I and type II fiber composition of the mouse soleus muscle, which is more heterogeneous compared with the soleus of other animals such as rats and rabbits (57). It has been suggested that the MyHC diversity of skeletal muscle fibers in different species depends mainly on their body size (3). “Light weight” animals such as mice might need only a small proportion of type I fibers to maintain their body posture and use the soleus mainly to support (fast) movements.
Two other important protein groups which have a strong impact on the phenotype and physiological properties of different muscle fiber types are kinases and phosphatases. We detected more than 100 kinases indicating good coverage of this protein class. Interestingly, marked differences in the expression profiles of several kinases were found. One of the kinases, which showed strong enrichment in fast compared with slow muscle was calcium/calmodulin dependent protein kinase II (CaMKII) (58). This kinase has been linked to pathological conditions including heart failure and hypertrophy but also to physiological processes such as mitochondrial biogenesis and fatty acid uptake (59). Although those processes are more prominent in slow muscles and cardiomyocytes the expression pattern of CamKIIα discloses an important role in fast muscles. Several other kinases such as AMP kinase and several ERK-activator kinases were also selectively enriched in the EDL. Activation of the mitogen-activated protein kinases (MAPK) are involved in fiber type switching during exercise and regeneration (60). The cGKI dependent kinase α was one of the most abundant kinases in the soleus, which was surprising because cGKI dependent kinase has been mainly described in smooth muscles where it is involved in various different processes including muscle relaxation. In principle, it is possible that cGKI dependent kinase was derived from smooth muscle cells of arteries although the lack of enrichment of other smooth muscle proteins in soleus samples argues against such a possibility.
The main feature of muscle cells is the generation of contractile force mediated by myofibrillar proteins within the sarcomer. Force transmission occurs via connective tissue that is attached to tendons and bones. The extracellular matrix deposited by muscle fibers plays an important functional role in this process and has attracted some interest because of its involvement in several musculoskeletal diseases (61). Formation of the extracellular matrix is based mainly on secretion and deposition of collagen fibers and collagen associated proteins such as proteoglycans and fascilin proteins, which are the main components that determine mechanical properties of the ECM (62, 63). Our study revealed similar concentrations for some ECM proteins in fast and slow muscle including laminins, dystrophin, tenascin, and perlecan. Other proteins implicated in the formation of interstitial connective tissue and the basement membrane showed a differential distribution (Table IA, B), which might in part account for distinct fiber strength between slow and fast muscles. The increased presence of fibrillin-1, periostin, and other ECM proteins in slow muscles seems to contribute to optimal mechanical stability of the connection between slow muscle fibers and associated tendons/bones. We propose that fast movements in the EDL require a more flexible ECM organization whereas the slow soleus muscle needs more stability and integrity.
The correlation of transcript and protein levels added another level of resolution to our study. No comprehensive comparison between protein concentrations and mRNA levels has been available for skeletal muscles preventing an unbiased view on posttranscriptional regulatory mechanisms. Although the bulk of proteins and mRNAs showed a good correlation in our study as indicated by a Pearson coefficient of 0.81, we identified a large group of proteins that was characterized by significant differences to corresponding mRNA levels. Because more molecules were differentially distributed between slow and fast muscles on the protein compared with the mRNA level we concluded that post-transcriptional mechanisms based on miRNAs and protein stability play an important role in establishing the specific molecular make-up of slow and fast muscle fibers. Although the correlation of mRNA and protein levels that we achieved is similar to other studies (64) one has to be aware of potential technical bias. For example, we were unable to measure large numbers of proteins expressed at low levels only in contrast to DNA microarray techniques that permit detection of low-abundance mRNA′s. Nevertheless, we believe that a combined analysis of microRNA expression profiles of soleus and EDL muscles together with the analysis of complementary sequences in 3′-untranslated regions of mRNAs of differentially regulated proteins will help to identify new targets for microRNA′s. A similar approach might be taken to identify processes that regulate the half-live of proteins in different muscles. Our results revealed that the number of proteins, which are present in different concentrations in slow and fast muscles, is higher than anticipated. The major extension of the catalogue of proteins with structural or regulatory functions in fast and slow muscles allows a more holistic view of the molecular networks responsible for distinct muscle functions.
Data Availability
All RAW files and sequence spectra can be downloaded from the Proteome Commons.org tranche (https://proteomecommons.org/tranche) server. Data set 1 (protein) contains all RAW data obtained from the Soleus/EDL SILAC quantification experiments. Data set 2 (phospho) includes all RAW files for the phosphopeptide identification. The PNG documents “Phosphopeptides” contain MS/MS spectra of identified phosphopeptides in this study.
Data sets are available via the following hash code: zoHoIJcZJXHZjxCP6p+y6L/GLoieoPvPCy2JFUGxyeHEHbOeW3mIdBaeRhcPxYTmWHLQEMEdgzS3DRhpy0PpKNAT1C4AAAAAAAACGA==. The content of the data sets are described in the preceeding paragraph.
Acknowledgments
We thank Sylvia Jeratsch and Sylvia Thomas for excellent technical assistance.
Footnotes
* This work was supported by the Max-Planck-Society, the Excellence Initiative “Cardiopulmonary System”, the University of Giessen-Marburg Lung Center (UGMLC) and the Cell and Gene Therapy Center (CGT) at the University of Frankfurt.
 This article contains supplemental Figs. and Tables.
This article contains supplemental Figs. and Tables.
1 The abbreviations used are:
- MyHC
- myosin heavy chain
- GO
- Gene Ontology
REFERENCES
- 1. Schiaffino S., Reggiani C. (1996) Molecular diversity of myofibrillar proteins: gene regulation and functional significance. Physiol. Rev. 76, 371–423 [DOI] [PubMed] [Google Scholar]
- 2. Booth F. W., Thomason D. B. (1991) Molecular and cellular adaptation of muscle in response to exercise: perspectives of various models. Physiol. Rev. 71, 541–585 [DOI] [PubMed] [Google Scholar]
- 3. Pette D., Staron R. S. (2000) Myosin isoforms, muscle fiber types, and transitions. Microsc. Res. Tech. 50, 500–509 [DOI] [PubMed] [Google Scholar]
- 4. Dhoot G. K., Perry S. V. (1979) Distribution of polymorphic forms of troponin components and tropomyosin in skeletal muscle. Nature 278, 714–718 [DOI] [PubMed] [Google Scholar]
- 5. MacLennan D. H., Toyofuku T., Lytton J. (1992) Structure-function relationships in sarcoplasmic or endoplasmic reticulum type Ca2+ pumps. Ann. N.Y. Acad. Sci. 671, 1–10 [DOI] [PubMed] [Google Scholar]
- 6. Delbono O., Meissner G. (1996) Sarcoplasmic reticulum Ca2+ release in rat slow- and fast-twitch muscles. J. Membr. Biol. 151, 123–130 [DOI] [PubMed] [Google Scholar]
- 7. Swoap S. J., Hunter R. B., Stevenson E. J., Felton H. M., Kansagra N. V., Lang J. M., Esser K. A., Kandarian S. C. (2000) The calcineurin-NFAT pathway and muscle fiber-type gene expression. Am. J. Physiol. Cell Physiol 279, C915–924 [DOI] [PubMed] [Google Scholar]
- 8. Chin E. R., Olson E. N., Richardson J. A., Yang Q., Humphries C., Shelton J. M., Wu H., Zhu W., Bassel-Duby R., Williams R. S. (1998) A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev. 12, 2499–2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Teran-Garcia M., Rankinen T., Koza R. A., Rao D. C., Bouchard C. (2005) Endurance training-induced changes in insulin sensitivity and gene expression. Am. J. Physiol. Endocrinol Metab 288, E1168–1178 [DOI] [PubMed] [Google Scholar]
- 10. Handschin C., Chin S., Li P., Liu F., Maratos-Flier E., Lebrasseur N. K., Yan Z., Spiegelman B. M. (2007) Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J. Biol. Chem. 282, 30014–30021 [DOI] [PubMed] [Google Scholar]
- 11. Noguchi S., Tsukahara T., Fujita M., Kurokawa R., Tachikawa M., Toda T., Tsujimoto A., Arahata K., Nishino I. (2003) cDNA microarray analysis of individual Duchenne muscular dystrophy patients. Hum. Mol. Genet. 12, 595–600 [PubMed] [Google Scholar]
- 12. Stevenson E. J., Giresi P. G., Koncarevic A., Kandarian S. C. (2003) Global analysis of gene expression patterns during disuse atrophy in rat skeletal muscle. J. Physiol. 551, 33–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Isfort R. J., Wang F., Greis K. D., Sun Y., Keough T. W., Bodine S. C., Anderson N. L. (2002) Proteomic analysis of rat soleus and tibialis anterior muscle following immobilization. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 769, 323–332 [DOI] [PubMed] [Google Scholar]
- 14. Gelfi C., Viganò A., De Palma S., Ripamonti M., Begum S., Cerretelli P., Wait R. (2006) 2-D protein maps of rat gastrocnemius and soleus muscles: a tool for muscle plasticity assessment. Proteomics 6, 321–340 [DOI] [PubMed] [Google Scholar]
- 15. Okumura N., Hashida-Okumura A., Kita K., Matsubae M., Matsubara T., Takao T., Nagai K. (2005) Proteomic analysis of slow- and fast-twitch skeletal muscles. Proteomics 5, 2896–2906 [DOI] [PubMed] [Google Scholar]
- 16. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 17. de Godoy L. M., Olsen J. V., Cox J., Nielsen M. L., Hubner N. C., Fröhlich F., Walther T. C., Mann M. (2008) Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 455, 1251–1254 [DOI] [PubMed] [Google Scholar]
- 18. Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fraterman S., Zeiger U., Khurana T. S., Rubinstein N. A., Wilm M. (2007) Combination of peptide OFFGEL fractionation and label-free quantitation facilitated proteomics profiling of extraocular muscle. Proteomics 7, 3404–3416 [DOI] [PubMed] [Google Scholar]
- 20. Kislinger T., Gramolini A. O., Pan Y., Rahman K., MacLennan D. H., Emili A. (2005) Proteome dynamics during C2C12 myoblast differentiation. Mol. Cell. Proteomics 4, 887–901 [DOI] [PubMed] [Google Scholar]
- 21. Krüger M., Moser M., Ussar S., Thievessen I., Luber C. A., Forner F., Schmidt S., Zanivan S., Fässler R., Mann M. (2008) SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell 134, 353–364 [DOI] [PubMed] [Google Scholar]
- 22. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 23. Rappsilber J., Mann M., Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 [DOI] [PubMed] [Google Scholar]
- 24. Wiśniewski J. R., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods. 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 25. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 26. Olsen J. V., Schwartz J. C., Griep-Raming J., Nielsen M. L., Damoc E., Denisov E., Lange O., Remes P., Taylor D., Splendore M., Wouters E. R., Senko M., Makarov A., Mann M., Horning S. (2009) A dual pressure linear ion trap Orbitrap instrument with very high sequencing speed. Mol. Cell. Proteomics. 8, 2759–2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 28. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 29. Matsuura T., Li Y., Giacobino J. P., Fu F. H., Huard J. (2007) Skeletal muscle fiber type conversion during the repair of mouse soleus: potential implications for muscle healing after injury. J. Orthop. Res. 25, 1534–1540 [DOI] [PubMed] [Google Scholar]
- 30. Blagoev B., Ong S. E., Kratchmarova I., Mann M. (2004) Temporal analysis of phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat. Biotechnol. 22, 1139–1145 [DOI] [PubMed] [Google Scholar]
- 31. Mann M., Kelleher N. L. (2008) Precision proteomics: the case for high resolution and high mass accuracy. Proc. Natl. Acad. Sci. U.S.A. 105, 18132–18138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fraterman S., Zeiger U., Khurana T. S., Wilm M., Rubinstein N. A. (2007) Quantitative proteomics profiling of sarcomere associated proteins in limb and extraocular muscle allotypes. Mol. Cell. Proteomics 6, 728–737 [DOI] [PubMed] [Google Scholar]
- 33. Schiaffino S., Reggiani C. (1994) Myosin isoforms in mammalian skeletal muscle. J. Appl. Physiol. 77, 493–501 [DOI] [PubMed] [Google Scholar]
- 34. Kemp T. J., Sadusky T. J., Saltisi F., Carey N., Moss J., Yang S. Y., Sassoon D. A., Goldspink G., Coulton G. R. (2000) Identification of Ankrd2, a novel skeletal muscle gene coding for a stretch-responsive ankyrin-repeat protein. Genomics 66, 229–241 [DOI] [PubMed] [Google Scholar]
- 35. Frey N., Richardson J. A., Olson E. N. (2000) Calsarcins, a novel family of sarcomeric calcineurin-binding proteins. Proc. Natl. Acad. Sci. U.S.A. 97, 14632–14637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Steiner F., Weber K., Fürst D. O. (1999) M band proteins myomesin and skelemin are encoded by the same gene: analysis of its organization and expression. Genomics 56, 78–89 [DOI] [PubMed] [Google Scholar]
- 37. Masaki T., Takaiti O. (1974) M-protein. J. Biochem. 75, 367–380 [DOI] [PubMed] [Google Scholar]
- 38. Schoenauer R., Lange S., Hirschy A., Ehler E., Perriard J. C., Agarkova I. (2008) Myomesin 3, a novel structural component of the M-band in striated muscle. J. Mol. Biol. 376, 338–351 [DOI] [PubMed] [Google Scholar]
- 39. Frank D., Kuhn C., Katus H. A., Frey N. (2006) The sarcomeric Z-disc: a nodal point in signalling and disease. J. Mol. Med. 84, 446–468 [DOI] [PubMed] [Google Scholar]
- 40. Lange S., Agarkova I., Perriard J. C., Ehler E. (2005) The sarcomeric M-band during development and in disease. J. Muscle Res. Cell. Motil. 26, 375–379 [DOI] [PubMed] [Google Scholar]
- 41. Wittenberg B. A., Wittenberg J. B., Caldwell P. R. (1975) Role of myoglobin in the oxygen supply to red skeletal muscle. J. Biol. Chem. 250, 9038–9043 [PubMed] [Google Scholar]
- 42. Riley D. A., Ellis S., Bain J. (1982) Carbonic anhydrase activity in skeletal muscle fiber types, axons, spindles, and capillaries of rat soleus and extensor digitorum longus muscles. J. Histochem. Cytochem. 30, 1275–1288 [DOI] [PubMed] [Google Scholar]
- 43. Townsend D. M. (2007) S-glutathionylation: indicator of cell stress and regulator of the unfolded protein response. Mol. Interv 7, 313–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Adler V., Yin Z., Fuchs S. Y., Benezra M., Rosario L., Tew K. D., Pincus M. R., Sardana M., Henderson C. J., Wolf C. R., Davis R. J., Ronai Z. (1999) Regulation of JNK signaling by GSTp. EMBO J. 18, 1321–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Abdellatif Y., Liu D., Gallant E. M., Gage P. W., Board P. G., Dulhunty A. F. (2007) The Mu class glutathione transferase is abundant in striated muscle and is an isoform-specific regulator of ryanodine receptor calcium channels. Cell Calcium 41, 429–440 [DOI] [PubMed] [Google Scholar]
- 46. Krebs E. G., Fischer E. H. (1955) Phosphorylase activity of skeletal muscle extracts. J. Biol. Chem. 216, 113–120 [PubMed] [Google Scholar]
- 47. Hou J., Cui Z., Xie Z., Xue P., Wu P., Chen X., Li J., Cai T., Yang F. (2010) Phosphoproteome analysis of rat L6 myotubes using reversed-phase C18 prefractionation and titanium dioxide enrichment. J. Proteome Res. 9, 777–788 [DOI] [PubMed] [Google Scholar]
- 48. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villen J., Haas W., Sowa M. E., Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Futcher B., Latter G. I., Monardo P., McLaughlin C. S., Garrels J. I. (1999) A sampling of the yeast proteome. Mol. Cell. Biol. 19, 7357–7368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tian Q., Stepaniants S. B., Mao M., Weng L., Feetham M. C., Doyle M. J., Yi E. C., Dai H., Thorsson V., Eng J., Goodlett D., Berger J. P., Gunter B., Linseley P. S., Stoughton R. B., Aebersold R., Collins S. J., Hanlon W. A., Hood L. E. (2004) Integrated genomic and proteomic analyses of gene expression in Mammalian cells. Mol. Cell. Proteomics 3, 960–969 [DOI] [PubMed] [Google Scholar]
- 51. Hosoda T., Monzen K., Hiroi Y., Oka T., Takimoto E., Yazaki Y., Nagai R., Komuro I. (2001) A novel myocyte-specific gene Midori promotes the differentiation of P19CL6 cells into cardiomyocytes. J. Biol. Chem. 276, 35978–35989 [DOI] [PubMed] [Google Scholar]
- 52. Hakimov H. A., Walters S., Wright T. C., Meidinger R. G., Verschoor C. P., Gadish M., Chiu D. K., Strömvik M. V., Forsberg C. W., Golovan S. P. (2009) Application of iTRAQ to catalogue the skeletal muscle proteome in pigs and assessment of effects of gender and diet dephytinization. Proteomics 9, 4000–4016 [DOI] [PubMed] [Google Scholar]
- 53. Feng J., Xie H., Meany D. L., Thompson L. V., Arriaga E. A., Griffin T. J. (2008) Quantitative proteomic profiling of muscle type-dependent and age-dependent protein carbonylation in rat skeletal muscle mitochondria. J. Gerontol. A Biol. Sci. Med. Sci. 63, 1137–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Duan X., Berthiaume F., Yarmush D., Yarmush M. L. (2006) Proteomic analysis of altered protein expression in skeletal muscle of rats in a hypermetabolic state induced by burn sepsis. Biochem. J. 397, 149–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Boettger T., Beetz N., Kostin S., Schneider J., Krüger M., Hein L., Braun T. (2009) Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J. Clin. Invest. 119, 2634–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nesvizhskii A. I., Aebersold R. (2005) Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics 4, 1419–1440 [DOI] [PubMed] [Google Scholar]
- 57. Soukup T., Zacharová G., Smerdu V. (2002) Fibre type composition of soleus and extensor digitorum longus muscles in normal female inbred Lewis rats. Acta Histochem. 104, 399–405 [DOI] [PubMed] [Google Scholar]
- 58. Soderling T. R., Chang B., Brickey D. (2001) Cellular signaling through multifunctional Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 276, 3719–3722 [DOI] [PubMed] [Google Scholar]
- 59. Raney M. A., Turcotte L. P. (2008) Evidence for the involvement of CaMKII and AMPK in Ca2+-dependent signaling pathways regulating FA uptake and oxidation in contracting rodent muscle. J. Appl. Physiol. 104, 1366–1373 [DOI] [PubMed] [Google Scholar]
- 60. Kramer H. F., Goodyear L. J. (2007) Exercise, MAPK, and NF-kappaB signaling in skeletal muscle. J. Appl. Physiol. 103, 388–395 [DOI] [PubMed] [Google Scholar]
- 61. Kjaer M. (2004) Role of extracellular matrix in adaptation of tendon and skeletal muscle to mechanical loading. Physiol. Rev. 84, 649–698 [DOI] [PubMed] [Google Scholar]
- 62. Zhang G., Ezura Y., Chervoneva I., Robinson P. S., Beason D. P., Carine E. T., Soslowsky L. J., Iozzo R. V., Birk D. E. (2006) Decorin regulates assembly of collagen fibrils and acquisition of biomechanical properties during tendon development. J. Cell. Biochem. 98, 1436–1449 [DOI] [PubMed] [Google Scholar]
- 63. Norris R. A., Damon B., Mironov V., Kasyanov V., Ramamurthi A., Moreno-Rodriguez R., Trusk T., Potts J. D., Goodwin R. L., Davis J., Hoffman S., Wen X., Sugi Y., Kern C. B., Mjaatvedt C. H., Turner D. K., Oka T., Conway S. J., Molkentin J. D., Forgacs G., Markwald R. R. (2007) Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J. Cell. Biochem. 101, 695–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Maier T., Güell M., Serrano L. (2009) Correlation of mRNA and protein in complex biological samples. FEBS Lett. 583, 3966–3973 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All RAW files and sequence spectra can be downloaded from the Proteome Commons.org tranche (https://proteomecommons.org/tranche) server. Data set 1 (protein) contains all RAW data obtained from the Soleus/EDL SILAC quantification experiments. Data set 2 (phospho) includes all RAW files for the phosphopeptide identification. The PNG documents “Phosphopeptides” contain MS/MS spectra of identified phosphopeptides in this study.
Data sets are available via the following hash code: zoHoIJcZJXHZjxCP6p+y6L/GLoieoPvPCy2JFUGxyeHEHbOeW3mIdBaeRhcPxYTmWHLQEMEdgzS3DRhpy0PpKNAT1C4AAAAAAAACGA==. The content of the data sets are described in the preceeding paragraph.