

Abstract
The hexameric purine nucleoside phosphorylase from Bacillus subtilis (BsPNP233) displays great potential to produce nucleoside analogues in industry and can be exploited in the development of new anti-tumor gene therapies. In order to provide structural basis for enzyme and substrates rational optimization, aiming at those applications, the present work shows a thorough and detailed structural description of the binding mode of substrates and nucleoside analogues to the active site of the hexameric BsPNP233. Here we report the crystal structure of BsPNP233 in the apo form and in complex with 11 ligands, including clinically relevant compounds. The crystal structure of six ligands (adenine, 2′deoxyguanosine, aciclovir, ganciclovir, 8-bromoguanosine, 6-chloroguanosine) in complex with a hexameric PNP are presented for the first time. Our data showed that free bases adopt alternative conformations in the BsPNP233 active site and indicated that binding of the co-substrate (2′deoxy)ribose 1-phosphate might contribute for stabilizing the bases in a favorable orientation for catalysis. The BsPNP233-adenosine complex revealed that a hydrogen bond between the 5′ hydroxyl group of adenosine and Arg43* side chain contributes for the ribosyl radical to adopt an unusual C3’-endo conformation. The structures with 6-chloroguanosine and 8-bromoguanosine pointed out that the Cl6 and Br8 substrate modifications seem to be detrimental for catalysis and can be explored in the design of inhibitors for hexameric PNPs from pathogens. Our data also corroborated the competitive inhibition mechanism of hexameric PNPs by tubercidin and suggested that the acyclic nucleoside ganciclovir is a better inhibitor for hexameric PNPs than aciclovir. Furthermore, comparative structural analyses indicated that the replacement of Ser90 by a threonine in the B. cereus hexameric adenosine phosphorylase (Thr91) is responsible for the lack of negative cooperativity of phosphate binding in this enzyme.
Introduction
Purine nucleoside phosphorylases (PNPs; EC 2.4.2.1) are versatile enzymes that catalyze the reversible phosphorolysis of purine (2′deoxy)ribonucleosides producing bases and (2′deoxy)ribose 1-phosphate [1]. Their key role in the purine salvage pathway made PNPs attractive targets for drug design against several pathogens, such as Mycobacterium tuberculosis [2], [3], Plasmodium falciparum [4]–[7], Trichomonas vaginalis [8]–[10] and Schistosoma mansoni [11], [12], which lacks the de novo pathway for purine nucleotides synthesis. Due to their catalytic function, PNPs have also been investigated for the synthesis of nucleoside analogues (NAs) [13] and the activation of prodrugs in anti-cancer gene therapies [14].
NAs can be used in the treatment of a range of human viral infections, such as those caused by HIV, herpesvirus and hepatitis B/C virus [15]–[19]. They are among the first cytotoxic molecules to be used in the treatment of cancer [20] and have been studied as potential drugs against tuberculosis [21], [22], malaria [7], [23], trichomoniasis [24] and schistosomiasis [25]. The chemical synthesis of these compounds is generally a costly multistep process that includes several protection and deprotection stages [13], [26]. This has encouraged the development of new methods for the synthesis of NAs using PNPs and other enzymes as biocatalysts [13], [27], [28]. The main advantages of this approach are the higher stereospecificity, regioselectivity and efficiency of enzymes, whose employment usually dispenses group protection and purification steps, optimizing the process [13].
The differences in substrate specificity regarding trimeric and hexameric PNPs have allowed the development of suicide gene therapies strategies against solid tumors [14], [29]. Trimeric PNPs are mainly found in mammalian species and are specific for guanine and hypoxanthine (2′-deoxy)ribonucleosides whereas hexameric PNPs are prevalent in bacteria and accept adenine as well as guanine and hypoxanthine (2′-deoxy)ribonucleosides as substrates [1]. Thus, nontoxic adenosine analogues, which are poor substrates for human PNP, can be cleaved to cytotoxic bases specifically in tumor cells transfected with the bacterial hexameric PNP gene [14]. Main advances in this field have been achieved with the E. coli PNP [30]–[33].
In this context, the aim of the present work was to shed light on how a diverse set of substrate modifications affects its binding and catalysis by hexameric PNPs using a structural approach. For this purpose, we choose the hexameric PNP (BsPNP233) from the model specie Bacillus subtilis, which displays great biotechnological potential to produce NAs, including the antiviral drug ribavirin [34]. We have solved the crystal structure of BsPNP233 in the apo form and in complex with 11 ligands comprising sulfate, bases, natural nucleosides and NAs, including clinically relevant compounds. The crystal structure of six ligands (adenine, 2′deoxyguanosine, aciclovir, ganciclovir, 8-bromoguanosine, 6-chloroguanosine) in complex with a hexameric PNP are presented for the first time.
Besides providing a broad structural basis for studies aiming at the rational design of BsPNP233 and its homologues for biotechnological applications, this work also bring new insights into the distinct kinetic models for phosphate binding in hexameric PNPs. Furthermore, the structural information showed here may also be instrumental for the development of new inhibitors against hexameric PNPs from pathogens such as Plasmodium falciparum and Trichomonas vaginalis [5], [6], [8], [9] and for the combined design of both hexameric PNPs and prodrugs to improve specificity and efficiency of anti-cancer PNP gene therapies [14].
Materials and Methods
Chemicals
Adenine (Ade), adenosine (Ado), 2-fluoradenosine (F-Ado), tubercidin (TBN), 2′-deoxyguanosine (dGuo), hypoxanthine (Hyp), ganciclovir (GCV), aciclovir (ACV), 8-bromoguanosine (Br-Guo) and 6-chloroguanosine (Cl-Guo) were all purchased from Sigma-Aldrich.
Expression and Purification of Recombinant BsPNP233
BsPNP233 was expressed in E. coli cells and purified by immobilized metal affinity and size-exclusion chromatographies as described in [35]. The protein concentration was determined by absorption spectroscopy at 280 nm using the theoretical molar extinction coefficient of 16 515 M−1cm−1 calculated by the program ProtParam [36].
Crystallization
BsPNP233 at 11 mg/ml in 20 mM Tris–HCl pH 7.0, 50 mM NaCl and 1 mM DTT was crystallized by sitting-drop vapor-diffusion technique according to conditions previously described [35]. The crystals belong to the space groups P321, P6322, P212121 and H32 with one, two or six monomers per asymmetric unit depending on symmetry and cell dimensions.
Preparation of BsPNP233-ligand Complexes
The protein-ligand complexes were prepared by adding 0.1 µl of 50 mM ligand, dissolved in DMSO, to 1 µl crystallization drops at least 12 h prior to data collection. The ligands used were nucleosides, purine bases and NAs (Table S1). This procedure was performed in drops containing BsPNP233 crystals grown in 0.1 M sodium acetate pH 4.6, 3.2 M sodium chloride, 5% (v/v) glycerol at 291 K.
X-ray Data Collection and Processing
X-ray diffraction experiments were performed on the W01B-MX2 beamline at the Brazilian Synchrotron Light Laboratory (Campinas, Brazil). The data collection was carried out using crystals soaked in a cryoprotectant solution composed by the mother liquor and 20% (v/v) glycerol and flash-cooled in a nitrogen-gas stream at 100 K. The radiation wavelength was set to 1.458 Å and a MAR Mosaic 225 mm CCD detector was used to record the X-ray diffraction data. Data were indexed, integrated and scaled using the HKL-2000 suite [37] or the programs MOSFLM [38] and SCALA [39] from the CCP4 package [40]. Data processing statistics are summarized in Table S1.
Structure Determination and Refinement
The structures were solved by molecular replacement using the programs MOLREP [41] or PHASER [42], both from the CCP4 suite [40]. The first BsPNP233 structure was determined using the atomic coordinates of B. anthracis PNP (PDB code 1XE3) [43] as a search model. The subsequent BsPNP233 structures were solved using the atomic coordinates of BsPNP233 solved at 1.7 Å resolution (BsPNP233-GCV dataset, Table S1) as template. Refinement was carried out using the programs REFMAC5 [44] and COOT [45]. After 20 cycles of rigid body refinement in REFMAC5 [44], the models were refined alternating cycles of restrained isotropic refinement in REFMAC5 [44] and manual rebuilding and real space refinement in COOT [45]. Water molecules were added after refinement of the protein model at chemically reasonable places using COOT [45]. Subsequently, the ligands were added to the model and refined as described above using library descriptions generated by the program SKETCHER from the CCP4 suite [40]. The intensity based twin refinement of REFMAC5 was applied to refine the structures of BsPNP233 in complex with adenosine, 2-fluoradenosine and adenine. The majority of models for the BsPNP233 protein included all but the first and last residues (1 and 233). In the electron density map of the crystal structure solved in the space group P212121 the residue 1 and additional eight residues from the N-terminal his-tag were clearly defined and added to the model. Ramachandran analysis carried out by Molprobity [46] showed that all residues from all models are found in allowed regions (except Gly121 of the BsPNP233-Ade structure, chain B). Refinement statistics are detailed in Table S1. Weighted 2Fo-Fc maps (2mFo-DFcalc) of ligands as well as a table of interactions between ligands and protein residues are presented in the supplementary material (Figure S1, Table S2). The atomic coordinates and structure factors of form I (4D8V), form II (4D8X), form III (4D8Y), form IV (4D98) and the complexes of BsPNP233 with Hyp (4DAB), Ade (4DAO), Ado (4D9H), dGuo (4DA0), F-Ado (4DAN), Cl-Guo(4DAE), Br-Guo (4DA8), TBN (4DAR), GCV (4DA6) and ACV (4DA7) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
Figure Preparation
The figures of structures were prepared using PyMOL [47].
Structural Alignment
All structural comparisons were performed using the SSM algorithm [48] available at the program COOT [45] or at the PDBeFold server [49].
Results and Discussion
BsPNP233 Conserves the Quaternary Structure and Topology of Hexameric PNPs
The crystal structure of BsPNP233 confirmed that it is a homohexamer with D3 symmetry as observed for other hexameric PNPs (Figure 1A) [50], [51]. It was solved by X-ray crystallography in four distinct space groups (P321, P212121, P6322 and H32). The crystal contacts are similar in the crystal structures solved in P321, P212121 and P6322 but differ in the H32 space group. In the later, we observed additional crystallographic interfaces, resulting from a more compact crystal packing with a lower solvent content (41%) than crystals belonging to other space groups (∼56%) (Figure S2) [35].
Figure 1. Overall structure of BsPNP233.
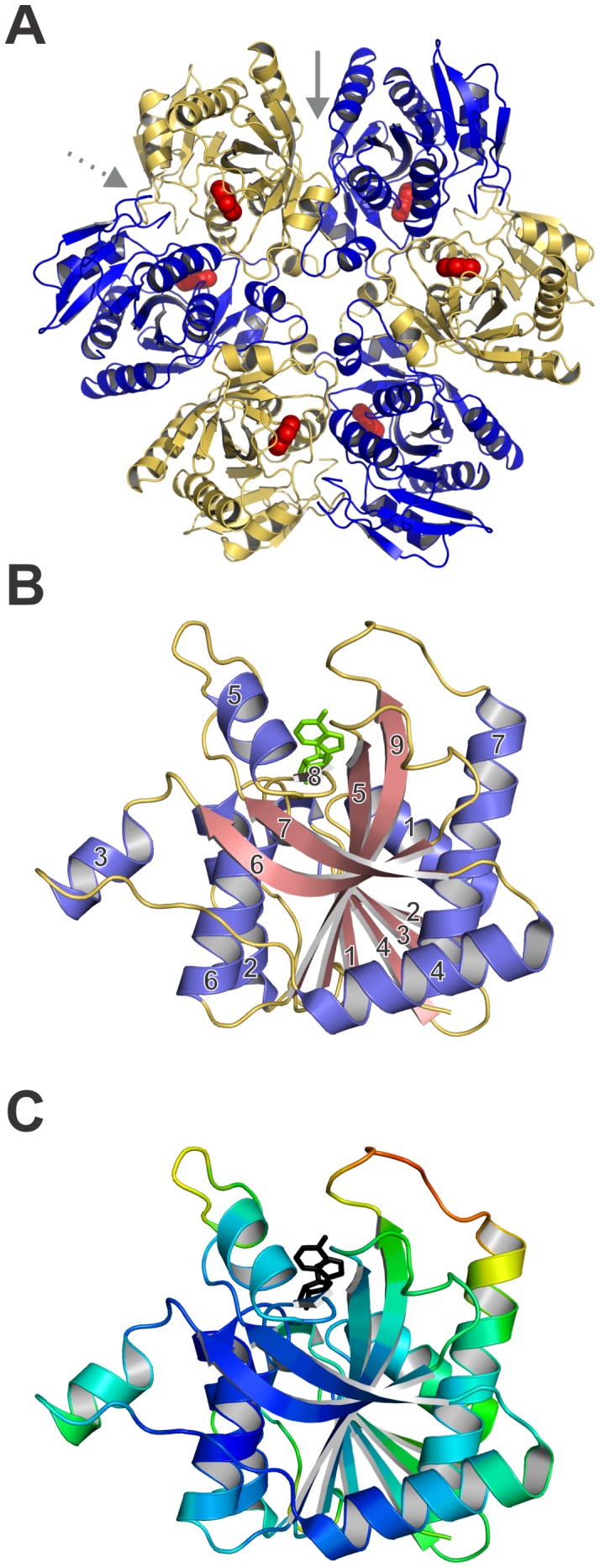
A. Cartoon representation of the hexamer BsPNP233 with adenine (red spheres) bound in the active site. Solid and dashed grey arrows indicate the inter-dimeric and catalytic interfaces, respectively. B. Cartoon representation of BsPNP233 protomer in complex with adenosine (green stick). Loops, α-helices and β-strands are shown in yellow, blue and pink. The α-helices and β-strands were numbered according to the Mao and colleagues notation [50]. C. BsPNP233 protomer colored by B-factors from dark blue (lowest) to red (highest). Adenosine is represented by a black stick.
The BsPNP233 subunits surround a central axis and alternate in an up/down fashion forming a disc-shaped structure with six active sites: three located at the top face and other three at the bottom face. Analogously to other hexameric PNPs, BsPNP233 is a trimer of dimers where each subunit interacts with the adjacent subunits forming two interfaces: the catalytic, which contains the active site, and the inter-dimeric, involved in hexamer stabilization (Figure 1A). The inter-dimeric interface is larger than the catalytic interface and both are mainly maintained by hydrophobic interactions. In the ligand-free crystal structure (form II), the inter-dimeric and the catalytic interface areas are 1711 and 1554 Å2, respectively.
The BsPNP233 subunit conserves the E. coli hexameric PNP (EcPNP) subunit topology with few exceptions. Its central mixed β-sheet lacks the short β10 strand observed in EcPNP [50] and is surrounded by eight (instead of seven) α-helices (Figure 1B). The extra 5-residues α-helix connects the strands β2 and β3 and is not labeled to preserves the Mao and colleagues notation [50]. BsPNP233 and EcPNP subunits share sequence identity of 58% (PDB code 1ECP, [50]) and superpose with a r.m.s.d of 0.93 Å for 231 Cα atoms aligned (Figure S3). Structural alignment of BsPNP233 subunit with hexameric PNPs subunits from other Bacillus species resulted in a r.m.s.d of 0.80 Å - 0.94 Å for 231 Cα atoms aligned and an average sequence identity of 71% (Figure S3).
Analysis of the B-factor distribution in the apo BsPNP233 crystal structure shows that the loop connecting β9 and α7 as well as the N-terminal portion of α7 present the highest B-factor values, highlighting its intrinsic flexibility. As this region surround the active site, its flexibility may be important for catalysis (Figure 1C).
Free Purine Bases Adopt Alternative Conformations in the Active Site
The crystal structures of BsPNP233 in complex with hypoxanthine (Hyp) and adenine (Ade) showed that the purine-binding site consists of residues Cys91, Gly92, Phe159, Val177 and Met179. Hydrophobic interactions are predominant in the stabilization of both ligands (Figure 2A).
Figure 2. Comparison of free bases bound to the BsPNP233 active site.

A. Structural comparison of a representative BsPNP233-Ade complex (purple carbon atoms) with the BsPNP233-Hyp complex (grey carbon atoms). B. The structure of the four BsPNP233-Ade complexes solved independently are superimposed. The sulfate-free Ade-complexes are colored in purple (chain A) and pink (chain B) whereas the two independent complexes solved with sulfate bound (dataset I) are colored in orange (chain A) and blue (chain B). C. The structure of the Ade-complex where Ade presents an alternative conformation (carbon atoms in orange) is superimposed in the structure of Hyp-complex (carbon atoms in grey). The surface of the glycerol molecule present at the Hyp-complex is shown to evidence the influence of this molecule in the position and orientation of Hyp in the active site. The hydrogen bonds are shown as dashed lines.
The BsPNP233-Ade binary complex was solved with (BsPNP233-Ade-SO4) or without (BsPNP233-Ade) sulfate ion and represent the first of their kind to be reported for hexameric PNPs. Two subunits were observed in the asymmetric unit of both crystal structures and all of them exhibited clear density for the ligand in the active site (Figure S1).
Superposition of BsPNP233-Ade and BsPNP233-Ade-SO4 complexes showed a preferential orientation of Ade in the base-binding site, except in one case where it is rotated by 49° around an axis perpendicular to the base plane (Figure 2B). This alternative orientation is not followed by significant conformational changes in the active-site residues (Figure 2B); however, it alters the solvation of the active-site pocket. In the alternative orientation, a crystallographic water molecule in the ribose-binding site is absent. This solvent molecule mediates a hydrogen bond between the AdeN9 atom and the carbonyl group of Ser90 in the presence of sulfate ion (Figure 2B).
Interestingly, the Hyp adopts an orientation similar to the alternative conformation of Ade (Figure 2C). In this case, a glycerol molecule is located in the ribose-binding site and seems to induce the displacement of Hyp, avoiding a steric clash with the HypN9 atom. This observation, along with those described above, suggests that binding of the co-substrate ribose-1-phoshate might contribute for stabilizing the base in the favorable orientation for catalysis.
The Hydrogen Bond between the 5′ Hydroxyl Group of Ado and Arg43* Side Chain Contributes for a Ribosyl C3′-endo Conformation
The base moiety of adenosine (Ado) binds to the BsPNP233 active site in a very similar fashion to that seen in homologous PNPs (Figure 3). However, the ribosyl group adopts a C3′-endo form instead of the C4′-endo or O4′-exo conformations usually observed in Ado complexes with hexameric PNPs (PDB codes 3UAW, [52]; 1ODI, [53]; 1PK7, [54]; 3U40, [55]; 1Z37, [51]; 1VHW, [56]) (Figure 3). This unusual conformation may be explained by a hydrogen bond between the 5′ hydroxyl group of Ado and Arg43* side chain (residues from the adjacent subunit are designated by an asterisk) not observed in other Ado complexes (Figure 3). Typically, the 5′-OH group of Ado is found interacting with one or two water molecules not observed in BsPNP233-Ado complex, suggesting that the hydration of the active site may influence the ribosyl conformation.
Figure 3. The different conformations of Ado ribosyl radical.
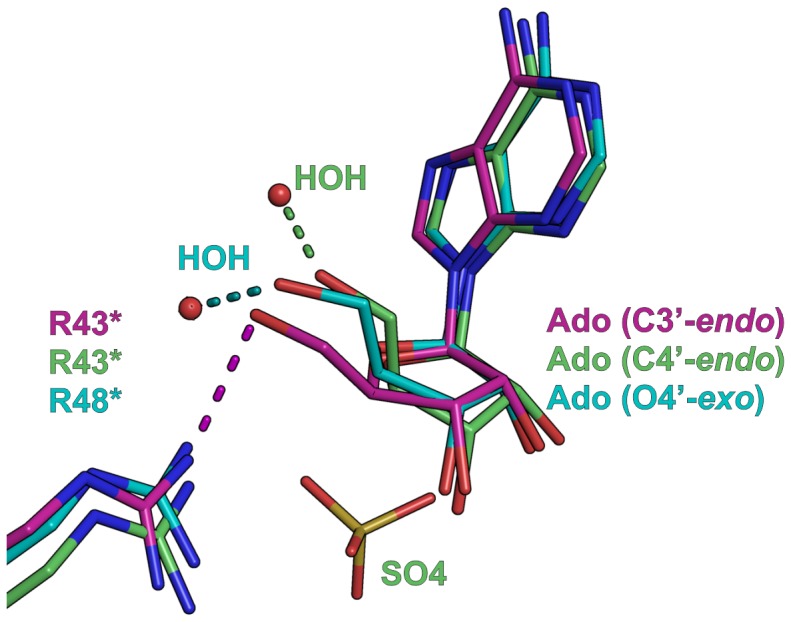
Structural superposition of BsPNP233-Ado (magenta carbon atoms), B. cereus adenosine phosphorylase (BcAdoP)-Ado-SO4 (green carbon atoms, PDB code 3UAW [52]) and Entamoeba histolytica PNP-Ado (cyan carbon atoms, PDB code 3U40 [55]) complexes. The different puckers adopted by the ribose moiety of adenosine are labeled and the hydrogen bonds involving the 5′-OH group of Ado in each complex are represented by dashed lines. The sugar puckers were assigned by the pucker.py script of PyMOL [47].
In sulfate/phosphate free complexes of hexameric PNPs with Ado, an O4′–exo conformation is usually found. However, in all complexes where the phosphate-binding site is occupied by a sulfate or phosphate, the ribosyl group shows a C4′–endo conformation, except for the Thermus thermophilus (TtPNP)-Ado complex (PDB code 1ODI, [53]). Since the side chain of Arg43* participates in phosphate binding, the presence of this co-substrate and the hydration of the active site probably prevent the interaction between Ado 5′-OH group and Arg43* side chain observed in BsPNP233-Ado complex favoring the ribose to adopt a C4′–endo conformation.
2′-deoxyguanosine Binding Mode Resembles to that Observed for Adenosine
In the BsPNP233-(2′-deoxyguanosine) complex structure, the base of 2′-deoxyguanosine (dGuo) binds to the active site in a similar manner to that observed for Ado (Figure 4). Neither the extra amino group at position 2 nor the carbonyl group at position 6 was observed making hydrogen bonds with the protein residues. A hydrogen bond between dGuoN7 and Ser202Oγ atoms slightly rotates the base and brings the residue Ser202 closer to the substrate (Figure 4B). The lack of the 2′-OH group in dGuo is counterbalanced by extra hydrophobic interactions between dGuoC2′ and Glu178 carbon atoms (Figure 4A). Comparisons between BsPNP233-dGuo and T. vaginalis PNP (TvPNP)-(2′-deoxyinosine) complexes showed that the deoxyribosyl group of both ligands conserves the binding mode, whereas the base assumes a little different orientation induced by the dGuoN7-Ser202Oγ hydrogen bond exclusively observed in BsPNP233-dGuo complex (Figure 4C).
Figure 4. The binding mode of 2′ deoxyguanosine and 2-fluoradenosine.

A. Representation of the BsPNP233 residues that interact with dGuo showing as spheres the atoms involved in hydrophobic interactions with dGuo C2′ atom. B. Structural alignment between the dGuo-complex (carbon atoms in orange) and the Ado-complex (carbon atoms in grey). C The structure of dGuo-complex and TvPNP-(2′-deoxyinosine) complex (carbon atoms in green, PDB code 1Z39, [51]) are superimposed. D. Representation of F-Ado complex showing the residues involved in van der Waals interactions with F2 atom (light blue sphere). E. Structural comparison of F-Ado complex (carbon atoms in yellow) with the Ado-complex (carbon atoms in grey). F. The structures of F-Ado complex, EcPNP-F-Ado-PO4 complex (carbon atoms in pink, PDB code 1PK9 [54]) and TvPNP-F-Ado complex (carbon atoms in green, PDB code 1Z35, [51]) are superimposed. In all panels hydrogen bonds are represented by dashed lines and color coded according to their respective structures.
BsPNP233 can be Explored as an Alternative in Gene Therapy Approaches using 2-Fluoradenosine as Prodrug
The compound 2-fluoradenosine (F-Ado) is an adenosine analogue which liberates the toxic metabolite 2-fluoradenine when cleaved. Its deoxy form has been studied as a prodrug in an anti-tumor gene therapy approach based on a modified human PNP [57]. The crystal structure of BsPNP233-(F-Ado) complex had two BsPNP233 subunits per asymmetric unit and both presented clear electronic density for the ligand (Figure S1). In the two independent active sites, F-Ado was found in the same orientation, similar to that of Ado (Figure 4E). The extra fluorine atom at position 2 is allocated in a hydrophobic micro-environment consisting of Ala156, Phe159, Val177 and Met179 (Figure 4D). This motif is fully conserved in EcPNP, which has been tested in anti-tumor gene therapy by activating produgs like F-ado [30].
Two hydrogen bonds (N6-Asp203Oδ and N7-Ser202Oγ) observed in BsPNP233-F-Ado complex, but not in BsPNP233-Ado complex, contribute for subtle changes in nucleoside position and base orientation (Figure 4E). The F-Ado ribosyl moiety adopts the catalytically favorable C4′-endo conformation supported by tight hydrogen bonds of nucleoside sugar hydroxyl groups with His4*, Arg87 and Glu180 side chains (Figure 4E). Structural comparisons among BsPNP233-F-Ado, TvPNP-F-Ado (PDB code 1Z35) and EcPNP-F-Ado (PDB code 1PK9) complexes showed a similar binding mode. However, the presence of phosphate in the EcPNP-(F-Ado) complex displaces by about 0.5 Å the ribosyl moiety, disrupts the N7-Ser202Oγ hydrogen bond and leads the N7 atom closer to the Asp203 side chain, favoring the catalysis (Figure 4F).
Since F-Ado binds to the BsPNP233 active site in a manner similar to that of the natural substrate Ado, placing the F2 atom in a hydrophobic pocket conserved in EcPNP, our structural data indicate that, as well as EcPNP [54], BsPNP233 is able to convert 2-fluoradenosine in the cytotoxic 2-fluoradenine. Thus, we concluded that BsPNP233 can be explored as an alternative in the development of anti-tumors gene therapy approaches using this prodrug or the less toxic 2-fluoro-2′-deoxyadenosine [57].
The Cl6 Substituent of 6-chloroguanosine Induces a Ribose C3′-exo Conformation and May Prevent Catalysis
The NA 6-chloroguanosine (Cl-Guo) can be used for the synthesis of 2-amino-6-chloro-9-(2,3-dideoxy-3-fluoro-beta-D-erythro-pentofuranosyl)purine, a compound with anti-HBV effects [58]. In addition, the free base 6-chloroguanine is an inhibitor of the trimeric PNP from Schistosoma mansoni [59]. Here we report the first crystal structure of a PNP in complex with Cl-Guo.
The molecule Cl-Guo displays a similar binding mode to that observed for dGuo (Figure 5A). However, as the chlorine van der Waals radius is larger than that of oxygen, the Cl6 substituent pushes the base in the direction of the ribosyl moiety to avoid steric clashes with Gly92Cα, Val205Cγ2, and Asp203Oδ1 atoms. This base displacement induces the ribosyl group to adopt an unusual C3′-exo conformation.
Figure 5. The influence of Cl6 and Br8 modifications in catalysis and nucleoside binding.

A. Structural alignment of Cl-Guo complex (pink carbon atoms) and dGuo complex (cyan carbon atoms). Spheres represent the van der Waals radius of Cl6, Gly92Cα, Asp203Oδ1 and Val205Cγ2 atoms. B. Superposition of Cl-Guo complex and EcPNP-Ado-PO4 complex (yellow carbon atoms, PDB code 1PK7 [54]). Spheres represent the van der Waals radius of Cl6 and EcPNP Asp204Oδ1 atoms to highlight the steric conflict imposed by the Cl6 atom. C. The Br-Guo complex (carbon atoms in green), dGuo complex (carbon atoms in orange) and sulfate complex (carbon atoms in magenta, form IV, chain B) structures are superimposed. The sphere represents the van der Waals radius of Br8 and the dashed lines represent hydrogen bonds colored according to the respective structures. D. Structural comparison of Br-Guo complex and the trimeric HsPNP-Guo-SO4 complex (purple carbon atoms, PDB code 1RFG, [63]). The spheres represent the van der Waals radius of Br8 and HsPNP Thr242Oγ1 atoms. The dashed circle has the same radius of Br8 and indicates the steric clash that would occur if BrGuo was placed at the Guo position in the HsPNP active site.
The C3′-exo pucker was already observed in the nucleoside 9-β-D-xylofuranosyladenine bound to EcPNP (PDB code 1PR6, [54]) and it is considered incompatible with the sugar conformation required for PNP catalysis [54]. Moreover, structural comparisons with the EcPNP-Ado-PO4 complex (PDB code 1PK7, [54]) showed that the chlorine atom may prevent the Asp203 side chain to approach to the N7 atom to donate a proton during catalysis (Figure 5B). Thus, these findings suggest that Cl-Guo as well as other NAs with 6-substituents heavier than chlorine cannot be cleaved by BsPNP233 and other hexameric PNPs.
The Br8 Substituent Displaces Ser90 Away from the Phosphate Binding Site and Might be Detrimental for Catalysis
The 8-bromoguanosine (Br-Guo) is a “poor substrate” of the trimeric PNP from calf spleen [60]. Its first crystallographic portrayal in complex with a protein is described here. The addition of a bromine radical at the C8 atom of guanosine results in the formation of a halogen bond between Br8 and Ser90Oγ atoms, which implicates in both positional and rotational displacement of the base by 0.3 Å and 7°, respectively (Figure 5C). The ribosyl moiety of Br-Guo presents a typical C4′–endo conformation and binds to the active site in a very similar fashion to that seen for 2′deoxyguanosine (Figure 5C).
In BsPNP233-(Br-Guo) complex, the side chain of the catalytic residue Asp203 is facing the O6 and N7 atoms of Br-Guo and the Ser90 side chain is pushed away from the active site in order to accommodate the bromine atom (Figure 5C). In hexameric PNPs, the hydroxyl group of Ser90 participates in the coordination of phosphate [61]. The position that it assumes in BsPNP233-sulfate and EcPNP-Ado-PO4 complexes (PDB code 1PK7 [54]) is incompatible with the presence of Br8 atom because of steric hindrance (Figure 5C). Thus, the bromine radical probably prevents the phosphate-Ser90 interaction being detrimental for binding and correct orientation of phosphate.
Site-directed mutations of human PNP phosphate binding site leads to a decrease in catalytic efficiency ranging from 25- to 185-fold [62]. Likewise, impairment of any phosphate interaction in the hexameric PNP active site may reduce the catalytic activity. As Br-Guo probably prevents the phosphate-Ser90 interaction it might be a “poor substrate” or even an inhibitor of BsPNP233 and other hexameric PNPs as well.
This interpretation cannot be applied for trimeric PNPs because Ser90 is not structurally conserved in trimeric PNPs. However, structural comparisons between BsPNP233-(Br-Guo) and human PNP in complex with guanosine and sulfate (PDB code 1RFG, [63]) indicate that Br-Guo is probably a “poor substrate” for trimeric PNPs because of steric hindrance involving the bromine and the side chain of Thr242, which would hinder the displacement of the N7 atom towards Asn243 side chain for stabilization of the transition state [64] (Figure 5D).
Corroboration of the Competitive Inhibition Mechanism of Hexameric PNP by Tubercidin
Tubercidin (7-deazaadenosine) is an adenosine analogue which presents antiviral, antischistosomal and antifungal properties as well as antitumor activity [65]–[68]. Furthermore, tubercidin and other 7-deazapurine nucleosides are inhibitors of EcPNP [69], [70].
TBN presented an interaction mode very similar to that seen for the natural substrate adenosine (Figure 6). Slightly differences were observed in its ribosyl moiety that assumed an O4′–exo pucker instead of the C3′-exo conformation of Ado in complex with BsPNP233 (Figure 6). The C7 substituent in TBN makes hydrophobic and van der Waals interactions with residues Cys91 and Ser202. The ribosyl moiety is stabilized by a conserved network of hydrogen bonds involving His4*, Glu180 and Arg87 side chains and by hydrophobic interactions with Glu178 (Cα and Cβ atoms) and Met179Cγ atom (Figure 6). Our structural data corroborate the competitive inhibition mechanism of hexameric PNP by TBN defined by in vitro studies [69]. The substitution of N7 by a carbon prevents the protonation step of the N7 atom required for catalysis [70], making TBN a non-cleavable adenosine analogue by EcPNP and probably by other PNPs.
Figure 6. The binding mode of tubercidin.
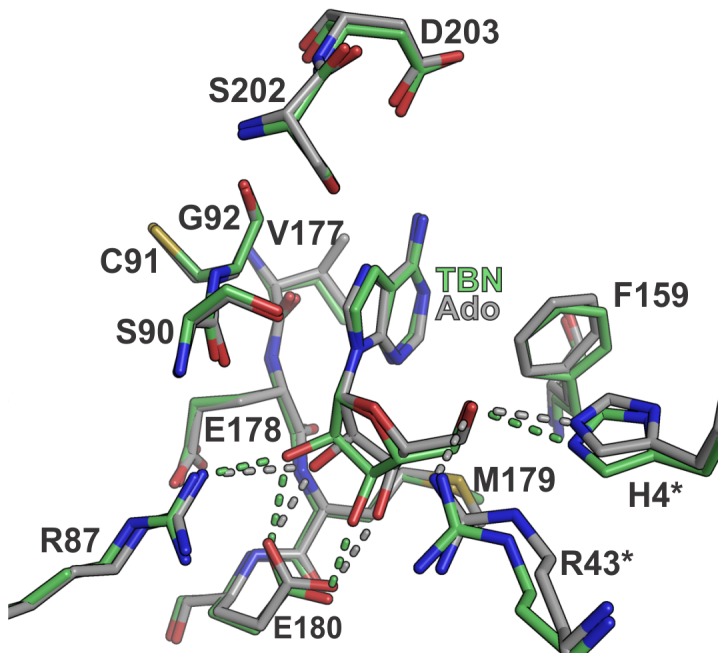
Structural comparison between the BsPNP233-TBN (carbon atoms in green) and BsPNP233-Ado (carbon atoms in grey) complexes. Dashed lines indicate hydrogen bonds and are colored according to their respective complexes.
Ganciclovir Inhibits Both Trimeric and Hexameric PNPs
Ganciclovir (GCV) is an acyclic NA used to treat cytomegalovirus infections [17]. It is also used together with herpes simplex virus thymidine kinase in a suicide gene therapy system that has been studied for the treatment of hepatocellular carcinoma [71]. GCV is an inhibitor of the human PNP (trimeric) [72] and probably has inhibitory effects on hexameric PNPs as well. Our structural data support this hypothesis revealing that GCV binds to the nucleoside binding site of BsPNP233 (Figure 7).
Figure 7. The binding mode of acyclic nucleosides.

A. Stick representation of GCV bound in the BsPNP233 active site. B. Structural comparison of GCV-complex (blue carbon atoms) with dGuo-complex (orange carbon atoms). C and D show the stick representation of the two conformations of ACV (ACV1 and ACV2) bound to the BsPNP233 active site. E. The structures of GCV-complex (grey) and ACV1,2-complex (green carbon atoms) are superimposed. F. Structural alignment of ACV1,2-complex with HsPNP-ACV complex (pink carbon atoms, PDB code 1PWY [74]). In all panels dashed lines indicate hydrogen bonds and are color coded according to their respective complexes.
The guanine moiety of GCV conserves the position observed for the 2′-deoxyguanosine base but it is rotated by about 10° to accommodate the acyclic chain in the ribose-binding site (Figure 7A–B). A water molecule mediates hydrogen bonds between the ligand O6 atom and the side chains of Ser202 and Asp203. The N7 atom interacts with Ser202Oγ and Gly92N atoms, and the base is stabilized by hydrophobic contacts with Ser90, Cys91, Ser202 and Phe159 (Figure 7A).
Interestingly, the three oxygens of the acyclic radical occupy similar positions to those observed for the three oxygens of dGuo ribosyl group, mimetizing its binding mode (Figure 7B). From the three hydrogen bonds observed for dGuo ribosyl moiety, the GCV acyclic radical conserves two, involving the His4* and Glu180 side chains. Moreover, the C4′ atom of the GCV acyclic moiety preserves the hydrophobic interactions with Met179Cβ and Met179Cγ atoms performed by the dGuo C3′ atom (Figure 7B). Therefore our data indicate that GCV is also a competitive inhibitor for hexameric PNPs.
Aciclovir Acyclic Chain Adopts Two Conformations in the BsPNP233 Ribosyl Binding Site
Aciclovir (ACV) is an antiviral drug used to treat herpes virus infections [73] and has modest inhibitory effects on human PNP [74]. Here, we present for the first time the crystal structure of a hexameric PNP with ACV. This structure revealed differences in the aciclovir binding mode, which can be explored for drug design targeting hexameric PNPs from pathogens such as P. falciparum [6] and T. vaginalis [8].
Aciclovir binds to the BsPNP233 nucleoside binding site and is stabilized by hydrophobic interactions and a hydrogen-bonding network mediated by solvent molecules (Figure 7C–D). Interestingly, the acyclic tail assumes two alternative conformations that, seen simultaneously, resemble the conformation observed for the ganciclovir acyclic radical (Figure 7C–E). In one of these conformations, the 3′ hydroxyl group of ACV is attached to the carboxyl group of Glu180 side chain while the carbon atoms make hydrophobic contacts with the main chain of Glu178 and with the Met179Cβ and Met179Cγ atoms (Figure 7C). A phosphate ion, modeled with half occupancy based on difference maps, also makes a hydrogen bond with the ligand 3′ hydroxyl group (Figure 7C). The other conformation is stabilized by a hydrogen bond between the 3′-OH group of ACV and the His4* side chain (Figure 7D).
The ACV guanine moiety assumes a different position and orientation from that observed for GCV (Figure 7E), getting closer to the Phe159 side chain. The main chain of Cys91 and the side chain of Val177 also contribute with hydrophobic interactions to the base (Figure 7C–D). The O6 atom makes water mediated hydrogen bonds with the Asp203 side chain and with the Phe159 carbonyl oxygen (Figure 7C and D). The same is observed for N1 and N2 atoms, which interact through a water molecule with the Gln158 carbonyl oxygen; for N7 atom, which makes water mediated hydrogen bonds with Asp203 side chain, and; for N9 atom, whose interaction with both Ser90 and Ser202 hydroxyl groups is also mediated by a solvent molecule (Figure 7C–D).
Structural comparison between BsPNP233-ACV and human PNP (HsPNP)-ACV (PDB code 1PWY, [74]) complexes showed differences in the binding mode. In the HsPNP-ACV complex, the base N1, N2, N7 and O6 atoms interact directly with active-site residues through hydrogen bonds. In addition, the acyclic chain adopts a different conformation, which is stabilized by hydrophobic interactions with Phe200 side chain and Ala116/Ala117 main chains (Figure 7F). To investigate if differences in the interaction mode of aciclovir with BsPNP233 and HsPNP may result in different binding affinities, we estimated the strength of protein–ligand interactions using the rerank score function of MOLEGRO [75]. According to this analysis, ACV presented similar predicted binding affinities in both complexes, which was slightly higher (lower rerank score value) for the BsPNP233 complex (Table S3). The same analysis was performed for GCV whose predicted binding affinity was considerable higher than that observed for ACV (Table S3). This result indicates that GCV is a better inhibitor for hexameric PNPs than ACV.
Structural Basis of Distinct Kinetic Models for Phosphate Binding in Hexameric PNPs
The asymmetric unit of the BsPNP233 crystal structure belonging to the H32 space group presented a catalytic dimer whose protomers adopt an open and a closed conformation, respectively (Figure 8A). The electron density map clearly showed a tetrahedral molecule in the active site of both subunits (Figure S1). As the crystallization condition was phosphate free and contained high concentrations of ammonium sulfate, we modeled sulfate ions in both sites.
Figure 8. Structural basis of distinct kinetic models for phosphate binding in hexameric PNPs.

A. Structural superposition of BsPNP233-sulfate open (green) and closed (pink) conformations with the BcAdoP-Ado complex (yellow, PDB code 3UAW, [52]). The cartoon representation highlights the conformational differences observed in the main chain of the β9-α7 loop and the N-terminal portion of helix α7 in the three structures. Dashed lines represent hydrogen bonds and follow the color code of their respective structures. B. The surface representation of BsPNP233 Phe220 in the closed conformation (pink) and of the BcAdoP Thr91 evidence the steric hindrance imposed by the Thr91Cγ2 atom to that Phe220 rotamer. C. The surface representation of BsPNP233 Phe220 and Ser90 in the closed conformation shows that the Ser90 side chain allows the Phe220 side chain to perform the conformational change needed for the closed conformation takes place.
The open and closed conformations of BsPNP233-sulfate complex were already observed in EcPNP-sulfate/phosphate structures and have been associated with two dissociation constants that characterize phosphate binding to EcPNP [61], [76]. The closed conformation is defined by a disruption of helix α7 and subsequent displacement of its N-terminal portion and the precedent loop towards the active site (Figure 8A). This conformation seems to be triggered by the interaction of Arg24 side chain with phosphate and results in an approximation of Arg216 to the catalytic residue Asp203 (Figure 8A) [61]. As BsPNP233 protomers are able to adopt open and closed conformations like EcPNP subunits, this suggests that the negative cooperativity of phosphate binding demonstrated for EcPNP [61] is also applied for BsPNP233.
Comparison between BsPNP233-sulfate and Bacillus cereus adenosine phosphorylase (BcAdoP)-sulfate complexes (PDB codes 3UAV, 3UAW, 3UAX, 3UAY, 3UAZ, [52]) showed that BcAdoP assumes an intermediate conformation where only the first turn of helix α7 is disrupted (Figure 8A). In the BcAdoP-sulfate complex structure (PDB code 3UAW, [52]), Arg217 (corresponding to BsPNP233-Arg216) points to the active site but it is not able to approach Asp204 (BsPNP233-Asp203) such as BsPNP233-Arg216 (Figure 8A).
The apparent inability of BcAdoP to adopt the closed conformation seems to be caused by a steric hindrance imposed by Thr91 to the conformational change that Phe221 (BsPNP233-Phe220) undergoes for the closed conformation being achieved (Figure 8B). In BsPNP233 and EcPNP this threonine residue is replaced by a serine, which allows Phe220 side chain to adopt the rotamer observed in the closed conformation (Figure 8C). These analyses suggest that the negative cooperativity model of phosphate binding displayed by EcPNP cannot be applied for BcAdoP, as BcAdoP apparently presents only one conformational state. This hypothesis is supported by functional studies, which showed that BcAdoP obeys Michaelis–Menten kinetics [77].
A previous work reported that BsPNP233 is specific for 6-aminopurine nucleosides [78]. However, Xie and coworkers [34] recently showed that BsPNP233 (named PNP702) exhibits a broad substrate specificity and present comparable activity towards both guanosine (6-oxopurine nucleoside) and adenosine (6-aminopurine nucleoside). Our structural data is in agreement with Xie and coworkers data indicating that BsPNP233 conserves the same catalytic mechanism proposed for EcPNP [76], where catalysis occurs in the closed conformation (Figure 8A).
Conclusion
This report provided a broad description of how the hexameric PNP from B. subtilis interacts with natural substrates and the impact of modifications in such substrates on binding and catalysis. The structural analysis reported here can be instrumental for studies aiming to optimize BsPNP233 or other hexameric PNPs for biotechnological applications such as industrial synthesis of nucleoside analogues or gene therapy against solid tumors. An initiative of this sort has been taken for E. coli PNP to optimize the cleavage of the prodrug Me(talo)-MeP-R with great success [29].
The crystal structure of six ligands (adenine, 2′deoxyguanosine, aciclovir, ganciclovir, 8-bromoguanosine and 6-chloroguanosine) in complex with a hexameric PNP are presented for the first time. The information extracted from these structures can be extended to homologous hexameric PNPs to help the development of new inhibitors against pathogens such as T. vaginalis [8] and P. falciparum [6] as well as new prodrugs for gene therapies against tumors [30], [79].
In addition, our results and comparative analyses shed light on distinct kinetic models for phosphate binding in hexameric PNPs. According to our model the substitution of the conserved residue Ser90 by a threonine disrupts the open/close mechanism of hexameric PNPs subunits, which results in the loss of the negative cooperativity of phosphate binding.
Supporting Information
Weighted 2Fo-Fc map (2mFo-DFcalc) of the ligands ( ball and stick ) bound to the BsPNP233 active site. A. Ade-complex (chain A). B. Ade-SO4 complex, evidencing only Ade (form I, chain A). C. Hyp-complex. D. Ado-complex (chain A). E. F-Ado complex (chain A). F. dGuo complex. G. Cl-Guo complex. H. Br-Guo complex. I. TBN complex. J. GCV complex. K. ACV complex. L. SO4 complex (form IV, chain A).
(TIF)
Crystallographic interfaces ( dark grey ) observed at the crystal structures solved at space groups P 321, P 212121, P 6322 (A) and at H 32 space group (B).
(TIF)
Structural alignment of BsPNP233 subunit with homologous hexameric PNPs protomers. The regions with the highest r.m.s.d. values are colored: BsPNP233 (green), BaPNP (blue - PDB 1XE3/F), BcPNP (yellow - PDB 2AC7/B), EcPNP (red - PDB 1ECP/A).
(TIF)
Data collection and refinement statistics.
(DOC)
Distances (Å) between the ligand atoms and interacting BsPNP233 atoms. Potential hydrogen bonds are highlighted by grey boxes. In the case of crystal structures containing more than one complex per asymmetric unit, only one of them is shown in the table. For the ligand adenine a representative structure of the preferential§ (BsPNP233-Ade complex, chain A) and alternative¥ (form I, chain A) conformations are presented.
(DOC)
In silico prediction of ligand binding affinity using the rerank score function of MOLEGRO [75] . ¥ The two values of BsPNP233-ACV complex correspond to the ACV1 and ACV2 alternative conformations, respectively. § HsPNP-ACV (PDB CODE: 1PWY).
(DOC)
Acknowledgments
We gratefully acknowledge the Brazilian Biosciences National Laboratory (CNPEM, Campinas, Brazil) and Brazilian Synchrotron Light Laboratory (CNPEM, Campinas, Brazil) for the use of the crystallization (RoboLab) and X-ray diffraction (MX2 beamline) facilities.
Funding Statement
This work was supported by the following research funding agencies: FAPESP [www.fapesp.br/] (grants numbers 2007/00194-9, 2010/51890-8) and CNPq [www.cnpq.br/]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Pugmire MJ, Ealick SE (2002) Structural analyses reveal two distinct families of nucleoside phosphorylases. Biochem J 361: 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Basso LA, Santos DS, Shi W, Furneaux RH, Tyler PC, et al. (2001) Purine nucleoside phosphorylase from Mycobacterium tuberculosis. Analysis of inhibition by a transition-state analogue and dissection by parts. Biochemistry 40: 8196–8203. [DOI] [PubMed] [Google Scholar]
- 3. Caceres RA, Timmers LFSM, Ducati RG, da Silva DON, Basso LA, et al. (2012) Crystal structure and molecular dynamics studies of purine nucleoside phosphorylase from Mycobacterium tuberculosis associated with acyclovir. Biochimie 94: 155–165 doi:10.1016/j.biochi.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 4. Lewandowicz A, Schramm VL (2004) Transition State Analysis for Human and Plasmodium falciparum Purine Nucleoside Phosphorylases†. Biochemistry 43: 1458–1468 doi:10.1021/bi0359123. [DOI] [PubMed] [Google Scholar]
- 5. Shi W, Ting L-M, Kicska GA, Lewandowicz A, Tyler PC, et al. (2004) Plasmodium falciparum purine nucleoside phosphorylase: crystal structures, immucillin inhibitors, and dual catalytic function. J Biol Chem 279: 18103–18106 doi:10.1074/jbc.C400068200. [DOI] [PubMed] [Google Scholar]
- 6. Madrid DC, Ting L-M, Waller KL, Schramm VL, Kim K (2008) Plasmodium falciparum purine nucleoside phosphorylase is critical for viability of malaria parasites. J Biol Chem 283: 35899–35907 doi:10.1074/jbc.M807218200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cassera MB, Hazleton KZ, Merino EF, Obaldia N 3rd, Ho M-C, et al (2011) Plasmodium falciparum parasites are killed by a transition state analogue of purine nucleoside phosphorylase in a primate animal model. PLoS ONE 6: e26916 doi:10.1371/journal.pone.0026916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Munagala N, Wang CC (2002) The purine nucleoside phosphorylase from Trichomonas vaginalis is a homologue of the bacterial enzyme. Biochemistry 41: 10382–10389. [DOI] [PubMed] [Google Scholar]
- 9. Munagala NR, Wang CC (2003) Adenosine is the primary precursor of all purine nucleotides in Trichomonas vaginalis. Mol Biochem Parasitol 127: 143–149. [DOI] [PubMed] [Google Scholar]
- 10. Rinaldo-Matthis A, Wing C, Ghanem M, Deng H, Wu P, et al. (2007) Inhibition and structure of Trichomonas vaginalis purine nucleoside phosphorylase with picomolar transition state analogues. Biochemistry 46: 659–668 doi:10.1021/bi061515r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pereira HD, Franco GR, Cleasby A, Garratt RC (2005) Structures for the potential drug target purine nucleoside phosphorylase from Schistosoma mansoni causal agent of schistosomiasis. J Mol Biol 353: 584–599 doi:10.1016/j.jmb.2005.08.045. [DOI] [PubMed] [Google Scholar]
- 12. Castilho MS, Postigo MP, Pereira HM, Oliva G, Andricopulo AD (2010) Structural basis for selective inhibition of purine nucleoside phosphorylase from Schistosoma mansoni: kinetic and structural studies. Bioorg Med Chem 18: 1421–1427 doi:10.1016/j.bmc.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 13.Patel RN (2006) Biocatalysis in the pharmaceutical and biotechnology industries. CRC Press. 924 p.
- 14. Zhang Y, Parker WB, Sorscher EJ, Ealick SE (2005) PNP anticancer gene therapy. Curr Top Med Chem 5: 1259–1274. [DOI] [PubMed] [Google Scholar]
- 15. De Clercq E (2007) Acyclic nucleoside phosphonates: past, present and future. Bridging chemistry to HIV, HBV, HCV, HPV, adeno-, herpes-, and poxvirus infections: the phosphonate bridge. Biochem Pharmacol 73: 911–922 doi:10.1016/j.bcp.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 16. De Clercq E (2009) Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. International Journal of Antimicrobial Agents 33: 307–320 doi:10.1016/j.ijantimicag.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 17. Faulds D, Heel RC (1990) Ganciclovir. A review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy in cytomegalovirus infections. Drugs 39: 597–638. [DOI] [PubMed] [Google Scholar]
- 18. Morfin F, Thouvenot D (2003) Herpes simplex virus resistance to antiviral drugs. J Clin Virol 26: 29–37. [DOI] [PubMed] [Google Scholar]
- 19. Paeshuyse J, Dallmeier K, Neyts J (2011) Ribavirin for the treatment of chronic hepatitis C virus infection: a review of the proposed mechanisms of action. Curr Opin Virol 1: 590–598 doi:10.1016/j.coviro.2011.10.030. [DOI] [PubMed] [Google Scholar]
- 20. Galmarini CM, Mackey JR, Dumontet C (2002) Nucleoside analogues and nucleobases in cancer treatment. The Lancet Oncology 3: 415–424 doi:10.1016/S1470–2045(02)00788-X. [DOI] [PubMed] [Google Scholar]
- 21. Long MC, Allan PW, Luo M-Z, Liu M-C, Sartorelli AC, et al. (2007) Evaluation of 3-deaza-adenosine analogues as ligands for adenosine kinase and inhibitors of Mycobacterium tuberculosis growth. J Antimicrob Chemother 59: 118–121 doi:10.1093/jac/dkl448. [DOI] [PubMed] [Google Scholar]
- 22. Van Calenbergh S, Pochet S, Munier-Lehmann H (2012) Drug design and identification of potent leads against mycobacterium tuberculosis thymidine monophosphate kinase. Curr Top Med Chem 12: 694–705. [DOI] [PubMed] [Google Scholar]
- 23.Evans GB, Furneaux RH, Kelly PM, Schramm VL, Tyler PC (2007) Transition state analogue inhibitors of N-ribosyltransferases: new drugs by targeting nucleoside processing enzymes. Nucleic Acids Symp Ser (Oxf): 63–64. doi:10.1093/nass/nrm032. [DOI] [PubMed]
- 24. Wright JM, Dunn LA, Kazimierczuk Z, Burgess AG, Krauer KG, et al. (2010) Susceptibility in vitro of clinically metronidazole-resistant Trichomonas vaginalis to nitazoxanide, toyocamycin, and 2-fluoro-2′-deoxyadenosine. Parasitol Res 107: 847–853 doi:10.1007/s00436-010-1938-3. [DOI] [PubMed] [Google Scholar]
- 25. el Kouni MH, Messier NJ, Cha S (1987) Treatment of schistosomiasis by purine nucleoside analogues in combination with nucleoside transport inhibitors. Biochemical Pharmacology 36: 3815–3821 doi:10.1016/0006–2952(87)90443-6. [DOI] [PubMed] [Google Scholar]
- 26. Pinheiro E, Vasan A, Kim JY, Lee E, Guimier JM, et al. (2006) Examining the production costs of antiretroviral drugs. AIDS 20: 1745–1752 doi:10.1097/01.aids.0000242821.67001.65. [DOI] [PubMed] [Google Scholar]
- 27. Ubiali D, Rocchietti S, Scaramozzino F, Terreni M, Albertini AM, et al. (2004) Synthesis of 2′-Deoxynucleosides by Transglycosylation with New Immobilized and Stabilized Uridine Phosphorylase and Purine Nucleoside Phosphorylase. Advanced Synthesis & Catalysis 346: 1361–1366 doi:10.1002/adsc.200404019. [Google Scholar]
- 28. Rocchietti S, Ubiali D, Terreni M, Albertini AM, Fernández-Lafuente R, et al. (2004) Immobilization and stabilization of recombinant multimeric uridine and purine nucleoside phosphorylases from Bacillus subtilis. Biomacromolecules 5: 2195–2200 doi:10.1021/bm049765f. [DOI] [PubMed] [Google Scholar]
- 29. Bennett EM, Anand R, Allan PW, Hassan AEA, Hong JS, et al. (2003) Designer gene therapy using an Escherichia coli purine nucleoside phosphorylase/prodrug system. Chem Biol 10: 1173–1181. [DOI] [PubMed] [Google Scholar]
- 30. Parker WB, Allan PW, Hassan AEA, Secrist JA 3rd, Sorscher EJ, et al (2003) Antitumor activity of 2-fluoro-2′-deoxyadenosine against tumors that express Escherichia coli purine nucleoside phosphorylase. Cancer Gene Ther 10: 23–29 doi:10.1038/sj.cgt.7700520. [DOI] [PubMed] [Google Scholar]
- 31. Martiniello-Wilks R, Dane A, Voeks DJ, Jeyakumar G, Mortensen E, et al. (2004) Gene-directed enzyme prodrug therapy for prostate cancer in a mouse model that imitates the development of human disease. J Gene Med 6: 43–54 doi:10.1002/jgm.474. [DOI] [PubMed] [Google Scholar]
- 32. Parker WB, Allan PW, Ealick SE, Sorscher EJ, Hassan AEA, et al. (2005) DESIGN AND EVALUATION OF 5′-MODIFIED NUCLEOSIDE ANALOGS AS PRODRUGS FOR AN E. COLI PURINE NUCLEOSIDE PHOSPHORYLASE MUTANT. Nucleosides, Nucleotides and Nucleic Acids 24: 387–392 doi:10.1081/NCN-200059807. [DOI] [PubMed] [Google Scholar]
- 33. Tai C-K, Wang W, Lai Y-H, Logg CR, Parker WB, et al. (2010) Enhanced efficiency of prodrug activation therapy by tumor-selective replicating retrovirus vectors armed with the Escherichia coli purine nucleoside phosphorylase gene. Cancer Gene Therapy 17: 614–623 doi:10.1038/cgt.2010.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie X, Xia J, He K, Lu L, Xu Q, et al.. (2011) Low-molecular-mass purine nucleoside phosphorylase: characterization and application in enzymatic synthesis of nucleoside antiviral drugs. Biotechnol Lett: 1107–1112. doi:10.1007/s10529-011-0535-6. [DOI] [PubMed]
- 35. Martins NH, Meza AN, Santos CR, de Giuseppe PO, Murakami MT (2011) Molecular cloning, overexpression, purification, crystallization and preliminary X-ray diffraction analysis of a purine nucleoside phosphorylase from Bacillus subtilis strain 168. Acta Crystallogr Sect F Struct Biol Cryst Commun 67: 618–622 doi:10.1107/S1744309111010414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, et al.. (2005) Protein Identification and Analysis Tools on the ExPASy Server. The Proteomics Protocols Handbook. Humana Press. 571–607.
- 37. Otwinowski Z, Minor W (1997) [20] Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography Part A. Academic Press, Vol. Volume 276: 307–326. [DOI] [PubMed] [Google Scholar]
- 38. Leslie AGW (2006) The integration of macromolecular diffraction data. Acta Crystallogr D Biol Crystallogr 62: 48–57 doi:10.1107/S0907444905039107. [DOI] [PubMed] [Google Scholar]
- 39. Kabsch W (1988) Evaluation of single-crystal X-ray diffraction data from a position-sensitive detector. Journal of Applied Crystallography 21: 916–924 doi:10.1107/S0021889888007903. [Google Scholar]
- 40. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallographica Section D Biological Crystallography 50: 760–763 doi:10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 41. Vagin A, Teplyakov A (1997) MOLREP: an Automated Program for Molecular Replacement. Journal of Applied Crystallography 30: 1022–1025 doi:10.1107/S0021889897006766. [Google Scholar]
- 42. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, et al. (2007) Phaser crystallographic software. J Appl Crystallogr 40: 658–674 doi:10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grenha R, Levdikov VM, Fogg MJ, Blagova EV, Brannigan JA, et al. (2005) Structure of purine nucleoside phosphorylase (DeoD) from Bacillus anthracis. Acta Crystallogr Sect F Struct Biol Cryst Commun 61: 459–462 doi:10.1107/S174430910501095X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53: 240–255 doi:10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 45. Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 doi:10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 46. Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, et al. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66: 12–21 doi:10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeLano WL (2002) The PyMOL Molecular Graphics System. Available:http://www.pymol.org.
- 48. Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774–797 doi:10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 49. Krissinel E, Henrick K (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60: 2256–2268. [DOI] [PubMed] [Google Scholar]
- 50. Mao C, Cook WJ, Zhou M, Koszalka GW, Krenitsky TA, et al. (1997) The crystal structure of Escherichia coli purine nucleoside phosphorylase: a comparison with the human enzyme reveals a conserved topology. Structure 5: 1373–1383. [DOI] [PubMed] [Google Scholar]
- 51. Zang Y, Wang W-H, Wu S-W, Ealick SE, Wang CC (2005) Identification of a subversive substrate of Trichomonas vaginalis purine nucleoside phosphorylase and the crystal structure of the enzyme-substrate complex. J Biol Chem 280: 22318–22325 doi:10.1074/jbc.M501843200. [DOI] [PubMed] [Google Scholar]
- 52. Dessanti P, Zhang Y, Allegrini S, Tozzi MG, Sgarrella F, et al. (2012) Structural basis of the substrate specificity of Bacillus cereus adenosine phosphorylase. Acta Crystallogr D Biol Crystallogr 68: 239–248 doi:10.1107/S090744491200073X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tahirov TH, Inagaki E, Ohshima N, Kitao T, Kuroishi C, et al. (2004) Crystal structure of purine nucleoside phosphorylase from Thermus thermophilus. J Mol Biol 337: 1149–1160 doi:10.1016/j.jmb.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 54. Bennett EM, Li C, Allan PW, Parker WB, Ealick SE (2003) Structural basis for substrate specificity of Escherichia coli purine nucleoside phosphorylase. J Biol Chem 278: 47110–47118 doi:10.1074/jbc.M304622200. [DOI] [PubMed] [Google Scholar]
- 55. Hewitt SN, Choi R, Kelley A, Crowther GJ, Napuli AJ, et al. (2011) Expression of proteins in Escherichia coli as fusions with maltose-binding protein to rescue non-expressed targets in a high-throughput protein-expression and purification pipeline. Acta Crystallogr Sect F Struct Biol Cryst Commun 67: 1006–1009 doi:10.1107/S1744309111022159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Badger J, Sauder JM, Adams JM, Antonysamy S, Bain K, et al. (2005) Structural analysis of a set of proteins resulting from a bacterial genomics project. Proteins 60: 787–796 doi:10.1002/prot.20541. [DOI] [PubMed] [Google Scholar]
- 57. Afshar S, Sawaya MR, Morrison SL (2009) Structure of a mutant human purine nucleoside phosphorylase with the prodrug, 2-fluoro-2′-deoxyadenosine and the cytotoxic drug, 2-fluoroadenine. Protein Sci 18: 1107–1114 doi:10.1002/pro.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Torii T, Onishi T, Izawa K, Maruyama T, Demizu Y, et al. (2006) Synthesis of 6-arylthio analogs of 2′,3′-dideoxy-3′-fluoroguanosine and their effect against hepatitis B virus replication. Nucleosides Nucleotides Nucleic Acids 25: 655–665 doi:10.1080/15257770600686394. [DOI] [PubMed] [Google Scholar]
- 59. Postigo MP, Guido RVC, Oliva G, Castilho MS, da R Pitta I, et al. (2010) Discovery of New Inhibitors of Schistosoma mansoni PNP by Pharmacophore-Based Virtual Screening. J Chem Inf Model 50: 1693–1705 doi:10.1021/ci100128k. [DOI] [PubMed] [Google Scholar]
- 60. Bzowska A, Kulikowska E, Darzynkiewicz E, Shugar D (1988) Purine nucleoside phosphorylase. Structure-activity relationships for substrate and inhibitor properties of N-1-, N-7-, and C-8-substituted analogues; differentiation of mammalian and bacterial enzymes with N-1-methylinosine and guanosine. J Biol Chem 263: 9212–9217. [PubMed] [Google Scholar]
- 61. Mikleušević G, Stefanić Z, Narczyk M, Wielgus-Kutrowska B, Bzowska A, et al. (2011) Validation of the catalytic mechanism of Escherichia coli purine nucleoside phosphorylase by structural and kinetic studies. Biochimie 93: 1610–1622 doi:10.1016/j.biochi.2011.05.030. [DOI] [PubMed] [Google Scholar]
- 62. Erion MD, Takabayashi K, Smith HB, Kessi J, Wagner S, et al. (1997) Purine nucleoside phosphorylase. 1. Structure-function studies. Biochemistry 36: 11725–11734 doi:10.1021/bi961969w. [DOI] [PubMed] [Google Scholar]
- 63. Canduri F, Silva RG, dos Santos DM, Palma MS, Basso LA, et al. (2005) Structure of human PNP complexed with ligands. Acta Crystallogr D Biol Crystallogr 61: 856–862 doi:10.1107/S0907444905005421. [DOI] [PubMed] [Google Scholar]
- 64. Ho M-C, Shi W, Rinaldo-Matthis A, Tyler PC, Evans GB, et al. (2010) Four generations of transition-state analogues for human purine nucleoside phosphorylase. Proc Natl Acad Sci USA 107: 4805–4812 doi:10.1073/pnas.0913439107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Acs G, Reich E, Mori M (1964) BIOLOGICAL AND BIOCHEMICAL PROPERTIES OF THE ANALOGUE ANTIBIOTIC TUBERCIDIN. Proc Natl Acad Sci U S A 52: 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. el Kouni MH, Diop D, Cha S (1983) Combination therapy of schistosomiasis by tubercidin and nitrobenzylthioinosine 5′-monophosphate. Proc Natl Acad Sci U S A 80: 6667–6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hwang BK, Ahn SJ, Moon SS (1994) Production, Purification, and Antifungal Activity of the Antibiotic Nucleoside, Tubercidin, Produced by Streptomyces-Violaceoniger. Canadian Journal of BotanyRevue Canadienne De Botanique 72: 480–485. [Google Scholar]
- 68. OWEN SP, SMITH CG (1964) CYTOTOXICITY AND ANTITUMOR PROPERTIES OF THE ABNORMAL NUCLEOSIDE TUBERCIDIN (NSC-56408). Cancer Chemother Rep 36: 19–22. [PubMed] [Google Scholar]
- 69. Perlman ME, Davis DG, Koszalka GW, Tuttle JV, London RE (1994) Studies of inhibitor binding to Escherichia coli purine nucleoside phosphorylase using the transferred nuclear Overhauser effect and rotating-frame nuclear Overhauser enhancement. Biochemistry 33: 7547–7559. [DOI] [PubMed] [Google Scholar]
- 70. A Bzowska ZK (1998) 7-Deazapurine 2′-deoxyribofuranosides are noncleavable competitive inhibitors of Escherichia coli purine nucleoside phosphorylase (PNP). Acta biochimica Polonica 45: 755–768. [PubMed] [Google Scholar]
- 71. Krohne TU, Shankara S, Geissler M, Roberts BL, Wands JR, et al. (2001) Mechanisms of cell death induced by suicide genes encoding purine nucleoside phosphorylase and thymidine kinase in human hepatocellular carcinoma cells in vitro. Hepatology 34: 511–518 doi:10.1053/jhep.2001.26749. [DOI] [PubMed] [Google Scholar]
- 72. Ray AS, Olson L, Fridland A (2004) Role of Purine Nucleoside Phosphorylase in Interactions between 2′,3′-Dideoxyinosine and Allopurinol, Ganciclovir, or Tenofovir. Antimicrob Agents Chemother 48: 1089–1095 doi:10.1128/AAC.48.4.1089–1095.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Thiers BH (1990) Acyclovir in the treatment of herpesvirus infections. Dermatol Clin 8: 583–587. [PubMed] [Google Scholar]
- 74. dos Santos DM, Canduri F, Pereira JH, Vinicius Bertacine Dias M, Silva RG, et al. (2003) Crystal structure of human purine nucleoside phosphorylase complexed with acyclovir. Biochem Biophys Res Commun 308: 553–559. [DOI] [PubMed] [Google Scholar]
- 75. Thomsen R, Christensen MH (2006) MolDock: a new technique for high-accuracy molecular docking. J Med Chem 49: 3315–3321 doi:10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- 76. Koellner G, Bzowska A, Wielgus-Kutrowska B, Luić M, Steiner T, et al. (2002) Open and closed conformation of the E. coli purine nucleoside phosphorylase active center and implications for the catalytic mechanism. J Mol Biol 315: 351–371 doi:10.1006/jmbi.2001.5211. [DOI] [PubMed] [Google Scholar]
- 77. Sgarrella F, Frassetto L, Allegrini S, Camici M, Carta MC, et al. (2007) Characterization of the adenine nucleoside specific phosphorylase of Bacillus cereus. Biochim Biophys Acta 1770: 1498–1505 doi:10.1016/j.bbagen.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 78. Jensen KF (1978) Two purine nucleoside phosphorylases in Bacillus subtilis. Purification and some properties of the adenosine-specific phosphorylase. Biochim Biophys Acta 525: 346–356. [DOI] [PubMed] [Google Scholar]
- 79. Sorscher EJ, Peng S, Bebok Z, Allan PW, Bennett LL Jr, et al. (1994) Tumor cell bystander killing in colonic carcinoma utilizing the Escherichia coli DeoD gene to generate toxic purines. Gene Ther 1: 233–238. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Weighted 2Fo-Fc map (2mFo-DFcalc) of the ligands ( ball and stick ) bound to the BsPNP233 active site. A. Ade-complex (chain A). B. Ade-SO4 complex, evidencing only Ade (form I, chain A). C. Hyp-complex. D. Ado-complex (chain A). E. F-Ado complex (chain A). F. dGuo complex. G. Cl-Guo complex. H. Br-Guo complex. I. TBN complex. J. GCV complex. K. ACV complex. L. SO4 complex (form IV, chain A).
(TIF)
Crystallographic interfaces ( dark grey ) observed at the crystal structures solved at space groups P 321, P 212121, P 6322 (A) and at H 32 space group (B).
(TIF)
Structural alignment of BsPNP233 subunit with homologous hexameric PNPs protomers. The regions with the highest r.m.s.d. values are colored: BsPNP233 (green), BaPNP (blue - PDB 1XE3/F), BcPNP (yellow - PDB 2AC7/B), EcPNP (red - PDB 1ECP/A).
(TIF)
Data collection and refinement statistics.
(DOC)
Distances (Å) between the ligand atoms and interacting BsPNP233 atoms. Potential hydrogen bonds are highlighted by grey boxes. In the case of crystal structures containing more than one complex per asymmetric unit, only one of them is shown in the table. For the ligand adenine a representative structure of the preferential§ (BsPNP233-Ade complex, chain A) and alternative¥ (form I, chain A) conformations are presented.
(DOC)
In silico prediction of ligand binding affinity using the rerank score function of MOLEGRO [75] . ¥ The two values of BsPNP233-ACV complex correspond to the ACV1 and ACV2 alternative conformations, respectively. § HsPNP-ACV (PDB CODE: 1PWY).
(DOC)