

Abstract
Using data from a genome-wide association study of 907 individuals with childhood acute lymphoblastic leukemia (cases) and 2,398 controls and with validation in samples totaling 2,386 cases and 2,419 controls, we have shown that common variation at 9p21.3 (rs3731217, intron 1 of CDKN2A) influences acute lymphoblastic leukemia risk (odds ratio = 0.71, P = 3.01 × 10−11), irrespective of cell lineage.
Acute lymphoblastic leukemia (ALL) is the major pediatric cancer in western countries, with B-cell precursor (BCP) ALL accounting for ~70% of cases. Although there is little evidence for a strong familial basis to ALL, evidence for an inherited susceptibility has recently been provided by genome-wide association (GWA) studies showing that IKZF1 (7p12.2), ARID5B (10q21.2) and CEBPE (14q11.2) variants confer a modest but significant risk1,2.
Our previously reported genome-wide association (GWA) studies of ALL were based on pooling data from two case-control series comprising a total of 907 cases and 2,398 controls successfully genotyped for 291,371 tagging SNPs1. To search for additional new variants influencing the risk of ALL, we have performed replication of 34 SNPs selected on the basis of statistical significance (P < 0.0001) coupled with considerations of minor allele frequency > 0.05, Hardy-Weinberg equilibrium (P > 0.05 in controls) and potential candidacy of nearby genes (on the basis of a role in B-cell and/or cancer biology) (Supplementary Table 1) in additional case-control series. The 34 SNPs were genotyped in an independent series of 1,428 ALL cases ascertained through the German Berlin-Frankfurt-Munster (BFM) group childhood ALL trials and 1,516 population controls. Thirty-three SNPs showed no significant evidence of an association at Ptrend < 0.01 in this series and were not further evaluated (Supplementary Table 1). rs3731217, mapping to 9p21.3, provided strong evidence for an association in the German series (P = 1.15 × 10−7) and was genotyped in case-control series from Spain (148 cases and 187 controls), Hungary (550 cases and 450 controls) and Canada (260 cases and 266 controls) (details in Supplementary Methods). Jointly, these four replication series provided strong evidence for association (combined odds ratio (OR) = 0.68, P = 1.13 × 10−8). Pooling genotype data for all of the studies provided unequivocal evidence for a relationship between rs3731217 and ALL risk (statistically significant after adjustment for multiple testing, assuming a conservative Bonferroni correction for the 291,371 tests in the original GWA study; OR for the T allele = 0.71, 95% CI 0.64–0.78, P = 3.01 × 10−11) (Fig. 1). This association was consistent in each of the replication series with nonsignificant statistics of between-study heterogeneity (P = 0.76, I2 = 0.0%).
Figure 1.

Forest plots of effect size and direction for 9p21.3 (rs3731217) association. (a–c) Association between all cases of ALL (a), BCP-ALL cases (b) and T-ALL cases and controls (c). Boxes denote OR point estimates, with their areas being proportional to the inverse variance weight of the estimate. Horizontal lines represent 95% CIs. The diamond and dashed line represent the summary OR computed under a fixed effects model, with the 95% CI indicated by the width of the diamond. The unbroken vertical line is at the null value (OR = 1.0).
rs3731217 localizes to intron 1 of CDKN2A (located at 21,974,661 base pairs; Fig. 2 and Supplementary Fig. 1) within a 174-kb region of linkage disequilibrium (LD) at 9p21.3. This region encompasses the CDKN2A and CDKN2B tumor-suppressor genes and the noncoding antisense RNA encoded by CDKN2BAS. CDKN2A encodes both p16 (INK4A), a negative regulator of cyclin-dependant kinases, and p14 (ARF1), an activator of p53. CDKN2A and CDKN2B are frequently inactivated in multiple hematological malignancies, and mono- or biallelic deletion of CDKN2A is one of the most frequent genetic events in childhood B- and T-lineage ALL3. CDKN2A deletions arise as secondary genetic events in cases of ALL initiated by ETV6-RUNX1 gene fusions4 and increase in frequency in cases of ALL relapse. Recent GWA studies have identified variation at 9p21.3 to be associated with nevus density5, melanoma6, basal cell carcinoma7, glioma8, type 2 diabetes9 and coronary heart disease10. It is possible that these associations are independent, but given the extent of LD across the region, it is also plausible that these multiple diseases have a common causal basis.
Figure 2.
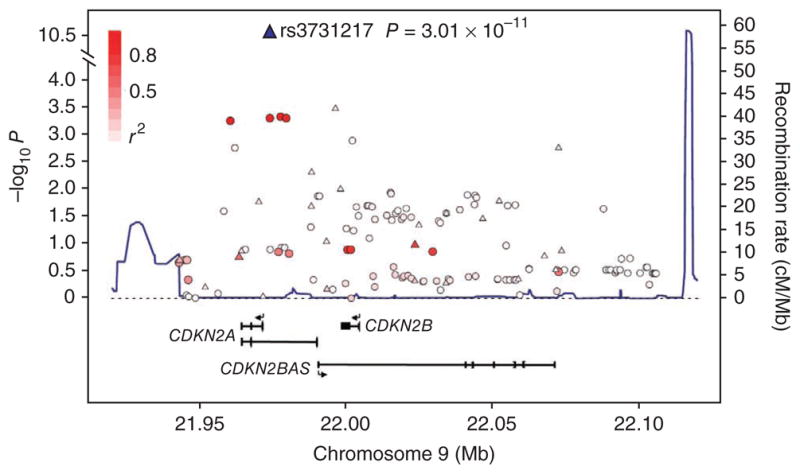
LD structure and association results for the 9p21.3 ALL locus. The local recombination rate is plotted in dark blue over this 174-kb chromosomal segment. Armitage trend test P values (as −log10 P values along the left y axis; note the broken axis between 4.0 and 10.5) are shown for the SNPs analyzed. Each triangle represents a SNP genotyped in the GWA study, circles represent imputed SNPs, and the most associated SNP in the combined analysis, rs3731217, is marked by a triangle (blue in combined analysis). The color intensity of each □ reflects the extent of LD with rs3731217, indicated by red (r2 > 0.8) going to white (r2 < 0.2). Physical positions are based on NCBI build 36 of the human genome. Also shown are the relative positions of genes mapping to each region of association. Exons of genes have been redrawn to show the relative positions in the gene, and therefore maps are not to physical scale.
Given the biological heterogeneity of ALL, we analyzed the association between the major subtypes of ALL and rs3731217 genotype. The association remained highly significant when the analysis was confined to BCP-ALL (combined OR = 0.72, 95% CI 0.64–0.80, P = 5.29 × 10−10, P of heterogeneity (Phet) = 0.41, I2 = 1.8%) and also for T-cell ALL (combined OR = 0.68, 95% CI 0.58–0.79, P = 1.88 × 10−7, Phet = 0.46, I2 = 0.0%) (Fig. 1). Subtype analysis of BCP-ALL in the UK GWA studies and the German case-control series provided no strong evidence that variation at 9p21.3 is associated with risk of a specific cytogenetic type of BCP-ALL (P = 0.22). Analysis of data from 378 UK-GWA2 study and 1,302 German cases provided no evidence that rs3731217 genotype was associated with event-free survival (P = 0.8 and P = 0.15 respectively; Supplementary Fig. 2). This is perhaps not unexpected given that CDKN2A-CDKN2B deletion status itself is not consistently associated with outcome in childhood ALL11.
Although CDKN2A is a strong candidate ALL-susceptibility gene on the basis of its biology, we cannot exclude a role of the other genes in 9p21.3 region as the basis of the association. To explore the entire genetic interval, we imputed 320 SNP genotypes within the 174-kb LD block containing rs3731217, using both HapMap and 1000 Genomes data in our GWA series (at 21,942,000–22,116,000 base pairs). Five SNPs showed a Ptrend < 10−3, and all were located within 15 kb of rs3731217 (Supplementary Table 2). None of these SNPs provided substantially better evidence for an association than rs3731217, and all were in strong LD with rs3731217 (r2 > 0.80, D′ > 0.97). Results from haplotype analysis provided evidence for a single risk variant defined by rs3731239-rs2811709-rs4074785-rs3731217-rs2811712-rs3218018 (Supplementary Table 3). To gain further insight into the causal basis of the association, we performed logistic regression using SNPs mapping to the 174-kb region of LD at 9p21.3. Analyses conditional on rs3731217 provided no support for a secondary association; however, a single disease-causing haplotype as inferred by ancestral recombination graph analysis could not be fully defined (Supplementary Table 3).
Inactivation of CDKN2A and CDKN2B is primarily a consequence of mono- or biallelic 9p21.3 deletion rather than promoter methylation11,12. To explore the possibility that the risk allele for rs3731217 is favored by somatic tumor evolution, we examined the relationship between genotype and CDKN2A and CDKN2B deletion in leukemic clones in 66 UK-GWA2 and 387 German cases. Although it is mechanistically plausible that somatic loss of the wild-type allele preferentially occurs in the leukemic clones of carriers, the frequency of CDKN2A deletion (mono- or biallelic) in leukemic clones was not different in carriers of the risk allele compared with non-carriers (P = 0.07 and P = 0.93 in the two datasets, respectively; Supplementary Table 4).
HapMap and 1000 Genomes data provided no evidence that rs3731217 is strongly correlated with a coding SNP in CDKN2A, CDKN2B or CDKN2BAS (r2< 0.001, D′ < 0.001; Supplementary Table 5). To explore the possibility that the association to disease might be mediated through differential CDKN2A or CDKN2B expression, we investigated the relationship between rs3731217 genotype and mRNA expression in 90 Epstein-Barr virus lymphoblastoid cells13. Although we found no association between rs3731217 genotype and expression of either mRNA transcript (Supplementary Fig. 3), steady-state levels of RNA at a single time point and in cycling mature B-cells may not adequately capture the impact of differential expression in leukemogenesis.
Elucidation of the causal basis of the 9p21.3 association will be contingent on fine-mapping and functional studies. To examine if any directly typed or imputed SNPs annotate a predicted transcription factor binding or enhancer elements, we conducted a bioinformatic search of the 174-kb region using TRANSFAC matrix database, PReMod and EEL software (see URLs). None of the SNPs associated with ALL risk (that is, having, P < 0.001) mapped within predicted regulatory elements (Supplementary Table 6 and Supplementary Fig. 1).
Irrespective of the causal basis of the 9p21.3 association, the potential impact of common alleles on gene expression will be modest and could occur at any time before disease diagnosis. Moreover, expression differences may only be relevant to a subpopulation of cells that provide ‘targets’ for leukemogenic mutations. There is some evidence that CDKN2A and CDKN2B deletions might be initiated by ‘off-target’ effects of the lymphoid mutagenic enzymes recombination activating proteins 1 and 2 (RAG1 and RAG2)14 or activation-induced cytidine deaminase (AID)15. Therefore, another possibility is that the association between increased risk of ALL and inherited variation in CDKN2A and CDKN2B reflects its structural or sequence-based vulnerability as a substrate.
There was no evidence of significant interaction between rs3731217 and the previously identified1,2 risk loci at 7p12.2 (IKZF1, rs4132601), 10q21.2 (ARID5B, rs7089424) and 14q11.2 (CEBPE, rs2239633), an observation compatible with each locus having an independent effect on ALL risk. Although the risk of ALL associated with these loci is modest, collectively they play a substantial role in the development of ALL, jointly accounting for as much as ~80% of the attributable risk in European populations. It will be intriguing to examine whether our new findings translate to nonwestern populations that have a different prevalence of ALL.
In summary, we have identified a new ALL risk locus at 9p21.3, and these findings provide additional insight into the development of ALL. Further studies are required to identify the causal variant(s) and to elucidate the biological basis of the association between genetic variation at CDKN2A loci and ALL pathogenesis.
URLs
R suite, http://www.r-project.org/ detailed information on the tag SNP panel, http://www.illumina.com/ dbSNP, http://www. ncbi.nlm.nih.gov/projects/SNP/ HapMap, http://www.hapmap.org/ 1958 Birth Cohort, http://www.cls.ioe.ac.uk/studies.asp?section=000100020003; MRC ALL 97 (Protocol 97PRT/14), http://www. thelancet.com/protocol-reviews/97PRT-14 United Kingdom Childhood Cancer Study, http://www.ukccs.org/ KBioscience, http://kbioscience.co.uk/ WGAViewer, http://www.genome.duke.edu/centers/pg2/downloads/wgaviewer.php; IMPUTE, https://mathgen.stats.ox.ac.uk/impute/impute.html SNPTEST, http://www.stats.ox.ac.uk/~marchini/software/gwas/snptest.html Margarita, http://www.sanger.ac.uk/resources/software/margarita/ EEL, http://www.cs.helsinki.fi/u/kpalin/EEL/ PReMod, http://genomequebec.mcgill.ca/PReMod/welcome.do TRANSFAC matrix database, http://www.biobase-international.com/pages/index.php?id=transfac; 1000 Genomes, http://www.1000genomes.org JASPAR2 database, http://jaspar.cgb.ki.se/ collaborators to the 1958 Birth Cohort dataset, http://www.wtccc.org.uk/.
Supplementary Material
Acknowledgments
The Kay Kendall Leukemia Fund and Leukemia Research (UK) provided principal funding for the study. Support from Cancer Research UK (C1298/A8362, supported by the Bobby Moore Fund) is also acknowledged. The study made use of genotyping data from the 1958 Birth Cohort which was generated and generously supplied to us by Panagiotis Deloukas of the Wellcome Trust Sanger Institute. For a full list of the investigators who contributed to the generation of the 1958 data, see URL section. We are grateful to S. Richards and J. Burrett (Clinical Trials Service Unit, Oxford) and L. Chilton (Leukaemia Research Cytogenetics Group, Northern Institute of Cancer Research, Newcastle Univ.), J. Simpson (Univ. York), P. Thomson and A. Hussain (Cancer Immunogenetics, School of Cancer Sciences, Univ. Manchester) for assistance with data harmonization. We thank the Leukaemia Research (LR) Childhood Cancer Leukaemia Group (CCLG) Childhood Leukaemia Cell Bank for access to the Medical Research Council ALL trial samples. We are grateful to the UK Cancer Cytogenetics Group (UKCCG) for data collection and provision of samples. P. Thomson (Cancer Immunogenetics, School of Cancer Sciences, University of Manchester) is funded by Children with Leukemia and we acknowledge their support. Genotyping of German cases and controls was partly covered by funding from Tumorzentrum Heidelberg-Mannheim, EU (Food-CT-2005-016320 and Health-F3-2007-2007-200767) and Deutsche Krebshilfe. The Hungarian sample collection was financially supported by grants from the Hungarian Scientific Research Fund (T042500), the Economic Competitiveness Operational Programme, Hungary (GVOP 3.1.1-2004-05-0022/3.0) and NKTH (National Research and Technology) TECH_08-A1/2-2008-0120. M.K. is a scholar of the Fonds de la Recherche en Santé du Québec. D.S. holds the François-Karl Viau Chair in Pediatric Oncogenomics and is also a scholar of the Fonds de la Recherche en Santé du Québec. J.H. is the recipient of a National Sciences and Engineering Research Council Canada Graduate’s scholarship. P. Garcia-Miguel and A. Sastre from Hospital Infantil La Paz, Madrid, and J. Luis Vivanco Martínez from Hospital 12 de Octubre, Madrid, were involved in the sample collection of the Spanish samples, and R. Alonso from Centro Nacional de Investigaciones Oncológicas was involved in the genotyping.
We are grateful to all the study subjects and individuals for their participation and, finally, we would also like to thank the clinicians, other hospital staff and study staff who contributed to the blood sample and data collection for this study.
Footnotes
AUTHOR CONTRIBUTIONS
R.S.H. and M.G. obtained financial support. R.S.H. designed and provided overall project management. R.S.H. drafted the manuscript with contributions from F.J.H., A.L.S. and M.G.; A.L.S. performed overall project management, development, database development and oversaw laboratory analyses; F.J.H. performed statistical analyses; F.J.H. and A.L.S. performed bioinformatics analyses; J.V. and E.P. performed UK sample preparation and genotyping; E.S. and S.E.K. performed curation and sample preparation of the Medical Research Council ALL 97 trial samples; T.L. and E.R. managed and maintained UKCCS sample data; M.T. performed curation and sample preparation of United Kingdom Childhood Cancer Study samples; J.M.A. and J.A.E.I. performed ascertainment, curation and sample preparation of the Northern Institute for Cancer Research case series. I.P.T. generated and managed UK colorectal cancer control genotypes. A.V.M. and C.J.H. performed UK CDKN2A deletion analysis; N.K., S.O. and H.P.K. carried out German CDKN2A deletion analysis; S.E.D. and Y.M. carried out HapMap and 1000 Genomes imputation; S.R. carried out survival analysis of UK data; M.Z. carried out survival analysis of German data; K.H. oversaw analysis of the German cohort; R.B.P., A.G. and R.K. conducted genotyping of German samples; R. Koehler, M. Stanulla, M. Schrappe and C.R.B. provided German DNA for analysis; D.J.E. and C.S. coordinated the data and sample collection of the Hungarian ALL cohort; A.F.S. genotyped the Hungarian samples; M.K., D.S. and J.H. performed curation and preparation of Canadian samples; A.G.N. was responsible for curation, management and genotyping of Spanish samples. All authors contributed to the final paper.
Note: Supplementary information is available on the Nature Genetics website.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
References
- 1.Papaemmanuil E, et al. Nat Genet. 2009;41:1006–1010. doi: 10.1038/ng.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Treviño LR, et al. Nat Genet. 2009;41:1001–1005. doi: 10.1038/ng.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mullighan CG, Downing JR. Leukemia. 2009;23:1209–1218. doi: 10.1038/leu.2009.18. [DOI] [PubMed] [Google Scholar]
- 4.Bateman C, et al. Blood. 2010 Jan 8; doi: 10.1182/blood-2009-10-25143. published online. [DOI] [Google Scholar]
- 5.Falchi M, et al. Nat Genet. 2009;41:915–919. doi: 10.1038/ng.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bishop DT, et al. Nat Genet. 2009;41:920–925. doi: 10.1038/ng.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stacey SN, et al. Nat Genet. 2009;41:909–914. doi: 10.1038/ng.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shete S, et al. Nat Genet. 2009;41:899–904. doi: 10.1038/ng.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scott LJ, et al. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McPherson R, et al. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sulong S, et al. Blood. 2009;113:100–107. doi: 10.1182/blood-2008-07-166801. [DOI] [PubMed] [Google Scholar]
- 12.Mirebeau D, et al. Haematologica. 2006;91:881–885. [PubMed] [Google Scholar]
- 13.Stranger BE, et al. PLoS Genet. 2005;1:e78. doi: 10.1371/journal.pgen.0010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitagawa Y, et al. J Biol Chem. 2002;277:46289–46297. doi: 10.1074/jbc.M208353200. [DOI] [PubMed] [Google Scholar]
- 15.Iacobucci I, et al. Leukemia. 2010;24:66–73. doi: 10.1038/leu.2009.197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.