

Abstract

The total synthesis of (±)-sorocenol B has been accomplished featuring key steps including silver nanoparticle (AgNP)-catalyzed Diels-Alder cycloaddition and late-stage Pd(II)-catalyzed oxidative cyclization. The synthetic natural product exhibited low micromolar cytotoxic activity against a number of human cancer cell lines.
Nanoparticle catalysis has emerged as a rapidly growing, multidisciplinary research area leading to increasing applications in organic synthesis.1 The development of nanoparticle-mediated methodologies to access complex molecules, especially natural products, is particularly attractive but has been achieved with limited success thus far.2 Herein, we report the first synthesis of the natural product sorocenol B in racemic form employing silver nanoparticle (AgNP)-catalyzed Diels-Alder cycloadditions of 2′-hydroxychalcones.3
Sorocenol B (1) was isolated from the root bark of Sorocea bonplandii with an overall yield of 3 milligrams per kilogram of dried bark.4 Due to the sparse distribution of its natural plant source, there has been no further report on this natural product since its first isolation in 1995.5 Structurally, compound 1 is characterized by an intriguing bicyclo[3.3.1] core which is postulated to be biosynthetically derived from oxidative cyclization of 2′-hydroxychalcone-derived Diels-Alder cycloadduct 2 (Scheme 1).4 Natural products containing similar bicyclic core structures include mulberrofuran I (3),6 australisin B (4),7 and mongolicin C (5)8 which are structurally related to chalcomoracin (6)9 and mulberrofuran C (7),10 respectively.
Scheme 1.
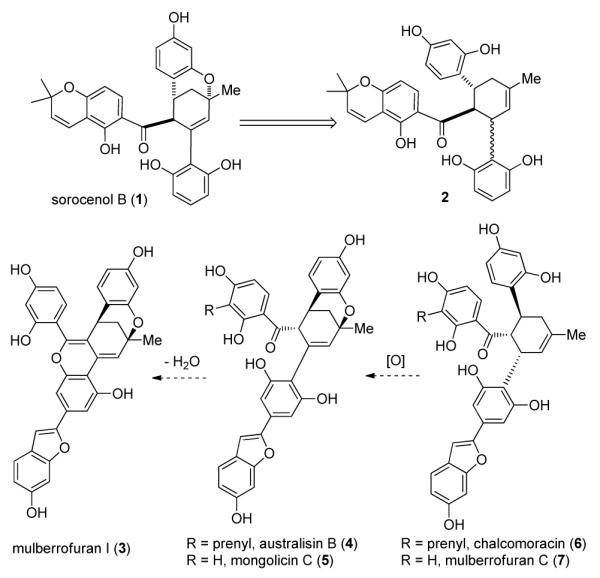
Biosynthesis of Sorocenol B and Related Natural Products
In our retrosynthetic analysis (Scheme 2), (±)-1 may be derived from MOM acetal precursor 8 which may be prepared through biomimetic, late-stage oxidative cyclization of cycloadducts 9 and/or 10. We envisioned that the synthesis of 9/10 could be achieved employing AgNP-catalyzed Diels-Alder cycloaddition3 between 2′-hydroxychalcone 11 and diene 12 which should be derived from commercially available chromene 13 and resorcinol (14), respectively.
Scheme 2.
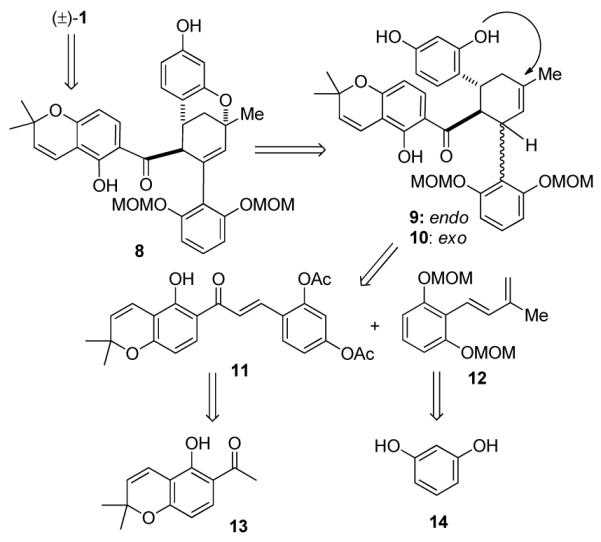
Retrosynthetic Analysis for Sorocenol B
The synthesis of the acetylated chalcone 11 commenced with Claisen-Schmidt condensation between chromene 13 and benzaldehyde 15 which smoothly generated chalcone 16 in 96% yield (Scheme 3). The use of NaH as a base in THF represents an improved protocol for chalcone formation which generally affords higher yields in shorter reaction times compared to conventional KOH/MeOH conditions.11 MOM hydrolysis of 16 using 3 M aqueous HCl in refluxing methanol, followed by acetylation, provided chalcone 11 in 79% yield (two steps).12
Scheme 3.
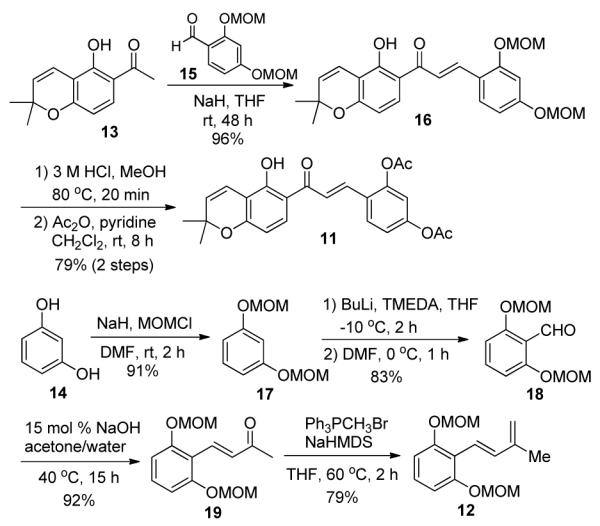
Syntheses of Chalcone 11 and Diene 12
The requisite diene 12 was prepared in four steps from resorcinol (14). Protection of 14 with MOMCl and NaH in DMF afforded MOM ether 17 in 91% yield. Regioselective formylation of 15 was carried out employing nBuLi in the presence of TMEDA followed by quenching the resulting aryl lithium intermediate with DMF which provided benzaldehyde 18 in 83% yield.13 Sequential base-catalyzed aldol condensation between 18 and acetone (92% yield)14 and Wittig olefination of the resulting α,β-unsaturated ketone 19 (79% yield) afforded the desired diene 12.
With both chalcone 11 and diene 12 in hand, we investigated the key Diels-Alder cycloaddition utilizing silica-supported silver nanoparticles (AgNP’s), a highly efficient and user-friendly catalyst recently developed in our laboratory.3a With 0.1 mol % catalyst loading at mild reaction temperature, 11 and 12 reacted cleanly in air to afford the desired cycloadducts in 90% combined yield with a 2:1 ratio of separable endo/exo diastereomers (20 and 21, Scheme 4). Notably, the endo-cycloadduct 20 was chromatographically inseparable from chalcone 11, therefore complete conversion of the starting material 11 was necessary for smooth access to the cycloadducts. In comparison, the uncatalyzed, thermal Diels-Alder cycloaddition of 11 and 12 did not proceed to completion to afford cycloadducts 20/21 (53% combined NMR yield, 1:2 ratio) even in the presence of large excess (11 equiv relative to 11) of diene 12.15
Scheme 4.
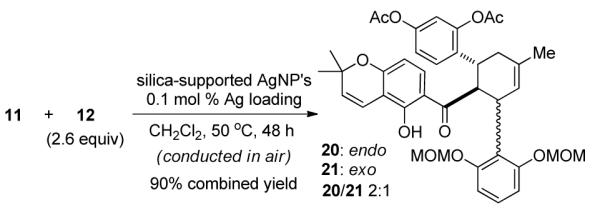
AgNP-Catalyzed Diels-Alder Cycloaddition of 11 and 12
In order to construct the bicyclo[3,3,1] framework of (±)-1, the next step entailed unmasking the acetyl-protected phenols of 20/21 followed by treatment with oxidants to accomplish oxidative cyclization, a process involving formal allylic C-H bond activation.16 After exploring a number of unproductive oxidative conditions (e.g. DDQ,17 CAN,18 and Pd(OAc)2/1,4-benzoquinone19), we found that substrate 9, prepared from endo diastereomer 20, reacted employing Stoltz’s conditions for oxidative Wacker cyclization (catalytic Pd(OAc)2/pyridine in toluene under an oxygen atmosphere)20 providing the desired bicyclic product 8 and its C-4 epimer 22 (2:1 ratio) in 50% combined yield (Scheme 5a). The relative stereochemistry of both 8 and 22 were unambiguously determined by key NOE signals (Scheme 6, e.g. H-4 and H-26 for 8, H-4 and H-6 for 22). Interestingly, deacylation of the exo diastereomer 21 to 10 followed by Pd-catalyzed oxidative cyclization did not afford the cyclized product (Scheme 5b).
Scheme 5.

Pd(II)-Catalyzed Oxidative Cyclization
Scheme 6.
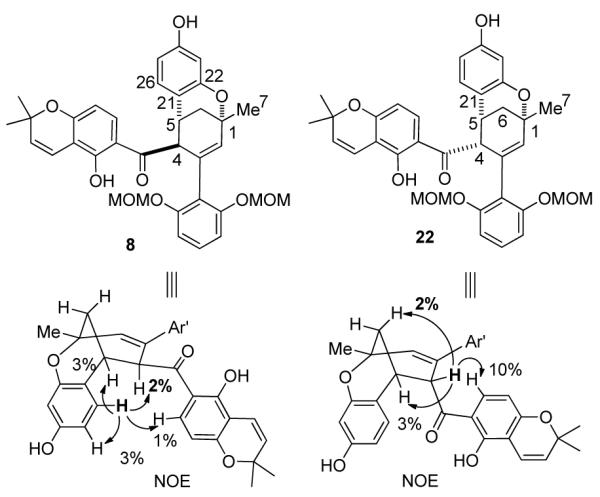
Key NOE’s Leading to Relative Stereochemistry Assignments of 8 and 22
The distinctive reactivities of 9 and 10 may be rationalized by the proposed mechanism for the Pd(II)-mediated oxidative cyclization.20 As shown in Scheme 5a, complexation of the Pd(II) catalyst with the unprotected phenol of 9 followed by intramolecular alkene insertion should generate intermediate 24. Subsequent syn-β-hydride elimination of 24 provides the desired bicyclo [3.3.1] product 8. In the case of 10, syn-β-hydride elimination is not feasible due to stereochemical restrictions. In addition, the presence of pyridine may lead to the formation of the minor product 22 through epimerization at the C-4 stereocenter of β,γ-unsaturated ketone 8.
Finally, MOM hydrolysis of 8 using aqueous HCl in refluxing MeOH completed the total synthesis of natural product (±)-1 in 74% yield without epimerization at the C-4 position. The relative stereochemistry of synthetic (±)-1 was confirmed by NOE analysis (Scheme 7, key signal: H-4 and H-26). Spectroscopic data for synthetic (±)-1 were identical to those reported in the original isolation paper.4
Scheme 7.
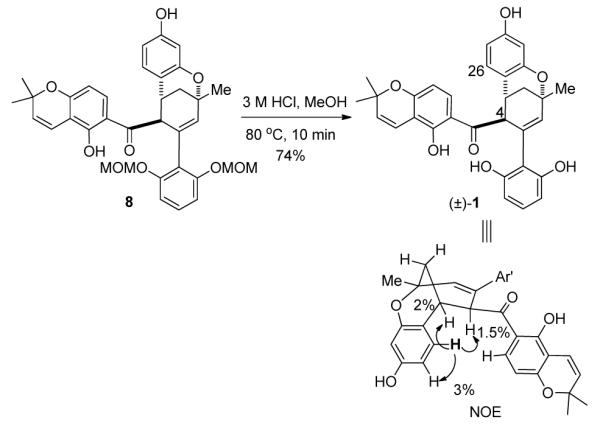
Synthesis of (±)-Sorocenol B
In preliminary studies, the biological activity of synthetic (±)-1 was evaluated in the National Cancer Institute (NCI) 60 human cancer cell screen.15 Single-dose assays at 10−5 M showed that (±)-1 exhibited antitumor activity with a mean cell growth of 26% of control. Subsequent full-dose response assays of (±)-1 exhibited low micromolar-level cytotoxic activities (mean GI50 values of 3.3 μM) and moderate selectivity. Tumor cell lines that are most sensitive to (±)-1 include prostate cancer PC-3 (GI50 = 1.1 μM), melanoma LOX IMVI (GI50 = 1.4 μM), leukemia MOLT-4 (GI50 = 1.4 μM), and colon cancer HCC-2998 (GI50 = 1.4 μM).
In summary, a concise total synthesis of (±)-sorocenol B has been accomplished employing a silver nanoparticle (AgNP)-catalyzed Diels-Alder cycloaddition. A late-stage Pd(II)-catalyzed oxidative cyclization of the derived endo cycloadduct has been used to access the bicyclo[3.3.1] framework of sorocenol B. Further studies on the applications of metal nanoparticle catalysts in complex molecule synthesis are ongoing and will be reported in due course.
Supplementary Material
Acknowledgment
Financial support from the National Institutes of Health (GM-073855 and GM-099920) is gratefully acknowledged. The authors thank Dr. John Beutler (Molecular Targets Laboratory, National Cancer Institute) for cytotoxicity testing against human cancer cell lines and Mr. Chao Qi (Boston University) for helpful discussions. H.C. thanks AstraZeneca for a graduate fellowship and the NIST and ACS for sponsorship of 2011 Kenneth G. Hancock Memorial Award. NMR (CHE-0619339) and MS (CHE-0443618) facilities at BU are supported by the U. S. NSF.
Footnotes
Supporting Information Available Expertimental procedures and characterization data for all new compounds are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1(a).Astruc D, editor. Nanoparticles and Catalysis. Wiley-VCH; Weinheim, Germany: 2007. [Google Scholar]; (b) Myers VS, Weir MG, Carino EV, Yancey DF, Pande S, Crooks RM. Chem. Sci. 2011;2:1632. [Google Scholar]; (c) Patil NT. ChemCatChem. 2011;3:1121. [Google Scholar]
- 2.Cong H, Porco JA. ACS Catal. 2012;2:65. doi: 10.1021/cs200495s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3(a).Cong H, Becker CF, Elliott SJ, Grinstaff MW, Porco JA. J. Am. Chem. Soc. 2010;132:7514. doi: 10.1021/ja102482b. For a recent application of the AgNP catalyst to natural product synthesis, see: Chee CF, Lee YK, Buckle MJC, Rahman NA. Tetrahedron Lett. 2011;52:1797.
- 4.Hano Y, Yamanaka J, Nomura T, Momose Y. Heterocycles. 1995;41:1035. [Google Scholar]
- 5.Based on a Scifinder search as of March 26, 2012.
- 6.Hano Y, Fukai T, Nomura T, Uzawa J, Fukushima K. Chem. Pharm. Bull. 1984;32:1260. doi: 10.1248/cpb.33.3195. [DOI] [PubMed] [Google Scholar]
- 7.Zhang QJ, Tang YB, Chen RY, Yu DQ. Chem. Biodivers. 2007;4:1533. doi: 10.1002/cbdv.200790133. [DOI] [PubMed] [Google Scholar]
- 8.Kang J, Chen RY, Yu DQ. Planta Med. 2006;72:52. doi: 10.1055/s-2005-873190. [DOI] [PubMed] [Google Scholar]
- 9.Takasugi M, Nagao S, Masamune T, Shirata A, Takahashi K. Chem. Lett. 1980;9:1573. [Google Scholar]
- 10(a).Hano Y, Suzuki S, Nomura T, Iitaka Y. Heterocycles. 1988;27:2315. For synthetic studies, see: Gunawan C, Rizzacasa MA. Org. Lett. 2010;12:1388. doi: 10.1021/ol9026705. Boonsri S, Gunawan C, Krenske EH, Rizzacasa MA. Org. Biomol. Chem. 2012 doi: 10.1039/c2ob25115a. DOI: 10.1039/c2ob25115a.
- 11.Nishida J, Kawabata J. Biosci. Biotechnol. Biochem. 2006;70:193. doi: 10.1271/bbb.70.193. [DOI] [PubMed] [Google Scholar]
- 12.Cong H, Ledbetter D, Rowe GT, Caradonna JP, Porco JA. J. Am. Chem. Soc. 2008;130:9214. doi: 10.1021/ja803094u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ballerini E, Minuti L, Piermatti O, Pizzo F. J. Org. Chem. 2009;74:4311. doi: 10.1021/jo9005365. [DOI] [PubMed] [Google Scholar]
- 14.Ballerini E, Minuti L, Piermatti O. J. Org. Chem. 2010;75:4251. doi: 10.1021/jo100785w. [DOI] [PubMed] [Google Scholar]
- 15.See Supporting Information for complete experimental details.
- 16(a).Gutekunst WR, Baran PS. Chem. Soc. Rev. 2011;40:1976. doi: 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]; (b) McMurray L, O’Hara F, Gaunt MJ. Chem. Soc. Rev. 2011;40:1885. doi: 10.1039/c1cs15013h. [DOI] [PubMed] [Google Scholar]
- 17(a).Floreancig PE. Synlett. 2007:191. [Google Scholar]; (b) Li CJ. Acc. Chem. Res. 2009;42:335. doi: 10.1021/ar800164n. [DOI] [PubMed] [Google Scholar]
- 18(a).Porco JA, Schoenen FJ, Stout TJ, Clardy J, Schreiber SL. J. Am. Chem. Soc. 1990;112:7410. [Google Scholar]; (b) Wood JL, Porco JA, Taunton J, Lee AY, Clardy J, Schreiber SL. J. Am. Chem. Soc. 1992;114:5898. [Google Scholar]
- 19.Wang DH, Engle KM, Shi BF, Yu JQ. Science. 2010;327:315. doi: 10.1126/science.1182512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20(a).Trend RM, Ramtohul YK, Ferreira EM, Stoltz BM. Angew. Chem. Int. Ed. 2003;42:2892. doi: 10.1002/anie.200351196. [DOI] [PubMed] [Google Scholar]; (b) Stoltz BM. Chem. Lett. 2004;33:362. [Google Scholar]; (c) Trend RM, Ramtohul YK, Stoltz BM. J. Am. Chem. Soc. 2005;127:17778. doi: 10.1021/ja055534k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.