

Abstract
Ventricular myocytes deficient in endothelial nitric oxide synthase (NOS3−/−) exhibit prolonged action potential (AP) duration and enhanced spontaneous activity (early and delayed afterdepolarizations) during β-adrenergic (β-AR) stimulation. Studies have shown that nitric oxide is able to regulate various K+ channels. Our objective was to examine if NOS3−/− myocytes had altered K+ currents. APs, transient outward (I to), sustained (I Ksus), and inward rectifier (I K1) K+ currents were measured in NOS3−/− and wild-type (WT) myocytes. During β-AR stimulation, AP duration (measured as 90% repolarization-APD90) was prolonged in NOS3−/− compared to WT myocytes. Nevertheless, we did not observe differences in I to, I Ksus, or I K1 between WT and NOS3−/− myocytes. Our previous work showed that NOS3−/− myocytes had a greater Ca2+ influx via L-type Ca2+ channels with β-AR stimulation. Thus, we measured β-AR-stimulated SR Ca2+ load and found a greater increase in NOS3−/− versus WT myocytes. Hence, our data suggest that the prolonged AP in NOS3−/− myocytes is not due to changes in I to, I Ksus, or I K1. Furthermore, the increase in spontaneous activity in NOS3−/− myocytes may be due to a greater increase in SR Ca2+ load. This may have important implications for heart failure patients, where arrhythmias are increased and NOS3 expression is decreased.
1. Introduction
Cardiac myocytes endogenously produce nitric oxide (NO) via two constitutively expressed NO synthase isoforms: endothelial NO synthase (NOS3) and neuronal NO synthase (NOS1). Both NOS1 and NOS3 play important roles in modulating cardiac function [1]. However, NOS1 and NOS3 signalings lead to different functional effects [2, 3]. NOS1 is localized to the sarcoplasmic reticulum (SR) and enhances cardiac contraction [2, 4], while NOS3 is localized to the caveolae and blunts the response to β-adrenergic (β-AR) stimulation due to a decreased L-type Ca2+ current (I Ca) [5, 6]. We also observed that NOS3 knockout (NOS3−/−) myocytes have prolonged action potential (AP) duration [5].
In addition to Ca2+ channels in the venitrcular myocyte, the AP waveform is also determined by potassium (K+) channels, which are essential for proper electrical activity of the heart as they are responsible for the resting membrane potential (RMP), the plateau phase, and repolarization [7]. K+ channels are also the most variable channels with multiple components, such as transient outward K+ current (I to), sustained outward K currents (I Ksus), inward rectifier K+ current (I K1), delayed rectifier K+ current (I Ks), and so forth [7]. In addition, there are different expression patterns of the various K+ channels in different species [8].
A previous study has shown that sodium nitroprusside, a NO donor, can enhance I Ks in guinea-pig cardiac myocytes [9]. Another study found that NOS3, but not NOS1, is responsible for the Ca2+-induced I Ks enhancement [10]. I Ks, which activates very slowly during depolarization and deactivates very slowly during repolarization, contributes to the repolarization during the late phase of the AP. However, I Ks channels are not expressed in adult murine ventricular myocytes [8, 11]. Thus, our observed changes in AP waveform in NOS3−/− mouse myocytes may result from alterations of other K+ channels, such as I Ksus, I to, and/or I K1. In mouse hearts, I Ksus is an important modulator of the plateau phase and repolarizing phase 3 of the AP. I to contributes to the repolarizing phase 1 and the plateau phase of AP. Previous data [12] showed that NO and NO donors inhibited human atrial I to. I K1 is responsible for setting the RMP and shaping the late repolarizing phase 3 of the AP. Gómez et al. [13] found that NO and NO donors can increase I K1 measured in human atrial cells. However, the role of NOS3 signaling on modulation of I to, I Ksus, and/or I K1 in ventricular myocytes is unclear. In addition to contributing to prolonged AP duration, altered K+ channel function can contribute to the generation of arrhythmias [14, 15]. Indeed, there is an increase in the incidence of arrhythmias in NOS3−/− mice [16]. Furthermore, our and others' data have shown increased spontaneous activity (i.e., early and delayed afterdepolarizations) in NOS3−/− myocytes [5, 17]. Thus, the purpose of this study is to determine if NOS3 signaling modulates K+ currents. In order to investigate the effects of NOS3 signaling on K+ currents, we used ventricular myocytes isolated from NOS3−/− and corresponding WT mice to investigate if alterations in one or all of K+ channels (I to, I Ksus, I K1) play a role in the prolongation of the AP, an effect expected to increase the propensity for spontaneous activity in NOS3−/− myocytes.
2. Materials and Methods
2.1. Isolation of Ventricular Myocytes
Age-matched, male and female NOS3−/− mice [18] and their corresponding control (C57Bl/6J) were obtained from Jackson Laboratories (Bar Harbor, ME, USA). Ventricular myocytes were isolated as previously described [5]. Briefly, the heart was mounted on a Langendorff apparatus and perfused with Ca2+ free normal Tyrode solution for 4 min. Blendzyme Type IV (0.077 mg/mL) (Roche Applied Science, Indianapolis, IN, USA) was then added to the perfusate. After 2–5 minutes, the heart was taken down, the right and left ventricles minced, and myocytes dissociated by trituration. Subsequently the myocytes were filtered, centrifuged, and resuspended in normal Tyrode solution containing 200 μmol/L Ca2+. We randomly selected our myocytes and the region of the heart (epicardium, endocardium, midmyocardium, etc.) could not be determined. Myocytes were used within 6 hours of isolation. All the animal protocols and procedures were performed in accordance with National Institutes of Health guidelines and approved by the Institutional Laboratory Animal Care and Use Committee at The Ohio State University.
2.2. Measurement of Action Potential
Action potentials (AP) were recorded with an Axopatch-200B amplifier and pClamp 8.1 software (Axon Instrument, Foster City, CA, USA) using the current-clamp mode. The pipette was filled with (in mM): NaCl (8), KCl (10), K-Aspartate (140), HEPES (5), MgATP (2), and pH 7.2 adjusted with KOH or HCl, with a resistance of 9–11 MΩ. Myocytes were perfused with normal Tyrode solution, which consisted of (in mM): NaCl (140), KCl (4), MgCl2 (1), CaCl2 (1), Glucose (10), HEPES (5), L-arginine (1), and pH 7.4 adjusted with NaOH or HCl. Isoproterenol (ISO, 1 μM, Sigma), a nonselective β-AR agonist, was prepared fresh each experimental day. A Grass S48 stimulator gated the amplifier for current injection to activate the AP, triggered by a 1.5 ms, 2 nA current injection. Measurements were performed at 37 ± 1°C.
2.3. Measurement of Potassium Currents
The amphotericin-B-perforated patch clamp technique was used to measure the various K+ currents. Myocytes were placed in a laminin-coated cell chamber (Cell Microcontrols, Norfolk, VA, USA) and superfused with bath solution containing (in mM): 135 NaCl, 5 MgCl2, 5 KCl, 10 glucose, 1.8 CaCl2, and 5 HEPES with pH adjusted to 7.4 with NaOH at temperature of 36 ± 0.5°C. Nifedipine (2 μM) was added to the superfusate to block the L-type calcium current. Borosilicate glass micropipettes with tip resistance of 1.5–3 MΩ were filled with pipette solution containing the following (in mM): 100 K-aspartate, 40 KCl, 5 MgCl, 5 EGTA, 5 HEPES, and pH adjusted to 7.2 with KOH.
Inward rectifier K+ current (I K1) was elicited by voltage steps from −140 to +40 mV from a holding potential of −40 mV (which will inactivate the sodium current). The current was measured at the end of each 100-ms test pulse. I K1 inward conductance (mS/cm2) was determined by calculating the slope of the linear portion of the current density-voltage relationship from −140 to −100 mV. Peak outward I K1 density was measured as the current at −60 mV (I −60) [19]. All currents (in picoamperes (pA)) were normalized to the cell capacitance (measured in picofarads (pF)) and expressed as pA/pF.
Outward K+ currents were elicited by a series of 300 ms test potentials from −50 to +50 mV from a holding potential of −60 mV (which will inactivate the sodium current). The sustained K+ current (I Ksus) was measured at the end of the 300 ms test pulse. The transient outward K+ current (I to) was determined by subtracting the sustained outward current from the peak outward current [19].
2.4. SR Ca2+ Load
SR Ca2+ load was measured at room temperature (22°C) by rapid application of 10 mmol/L caffeine for 10 sec. The amplitude of the caffeine-induced Ca2+ transient was used as an index of SR Ca2+ load [20].
2.5. Statistics
Myocyte data were presented as mean ± SEM. Differences between multiple groups were evaluated for statistical significance using an ANOVA (followed by Neuman-Keuls test) or unpaired Student's t-test for two groups. Statistical significance was accepted at the level of P < 0.05.
3. Results
3.1. NOS3−/− Myocytes Have Prolonged β-AR Stimulated APD but No Change in RMP
We have previously shown that during β-AR stimulation, NOS3−/− ventricular myocytes had a prolonged AP (measured as time to 90% repolarization-APD90) when measured at room temperature [5]. We repeated these experiments to examine if the phenomenon occurred at body temperature. Thus, we measured AP waveform in control (wildtype, WT) and NOS3−/− myocytes at 37°C. Representative AP traces measured in a WT and NOS3−/− myocyte (±β-AR stimulation with ISO) are shown in Figure 1(a). As shown in Figure 1(b), there was no difference between WT and NOS3−/− resting membrane potential (basal or with β-AR stimulation). However, we found that during β-AR stimulation, NOS3−/− myocytes (compared to WT) had a significant increase in APD90, consistent with our previous finding (Basal, WT: 75 ± 17 ms, NOS3−/−: 77 ± 10 ms; ISO, WT: 93 ± 24 ms, NOS3−/−: 117 ± 17 ms). The increase in APD90 with β-AR stimulation in NOS3−/− myocytes is shown in Figure 1(c). These data suggest that NOS3−/− myocytes have a prolonged AP with no change in the RMP during β-AR stimulation.
Figure 1.
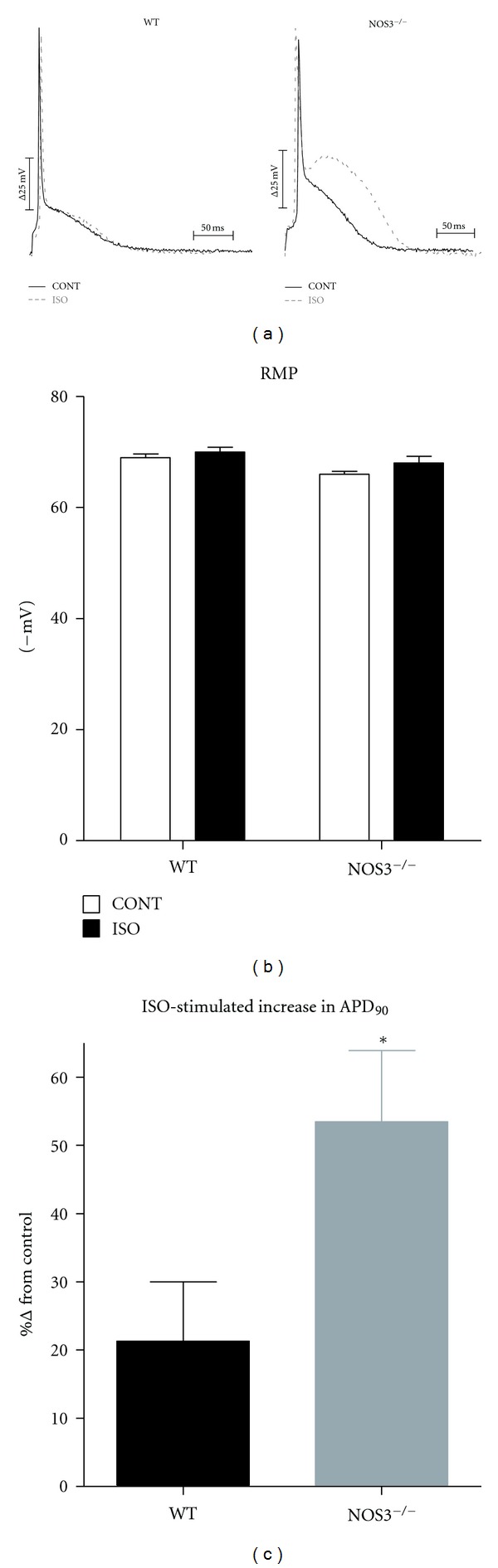
Prolonged APD90 in NOS3−/− myocytes after β-AR stimulation. (a) Representative AP traces from WT and NOS3−/− myocytes (control-CONT; isoproterenol-ISO). (b) Summary data (mean ± S.E.M.) of RMP in WT and NOS3−/− myocytes. (c) Summary data (mean ± S.E.M.) of β-AR stimulated increase of APD90 in WT and NOS3−/− myocytes. *P < 0.05, n = 4, 7 cells.
3.2. NOS3−/− Myocytes Do Not Have Altered Inward Rectifier K+ Currents
An important determinant of RMP and APD90 is the inward rectifier K+ current (I K1). Since we had prolonged APD90 with β-AR stimulation in NOS3−/− myocytes, we investigated if these myocytes had altered I K1. Representative I K1 currents measured in a WT and NOS3−/− myocyte are shown in Figure 2(a). As shown in Figures 2(b)–2(d), knockout of NOS3 did not alter the I K1 I-V relationship, the peak outward I K1, or the I K1 slope conductance. These data suggest that NOS3 signaling does not modulate I K1.
Figure 2.
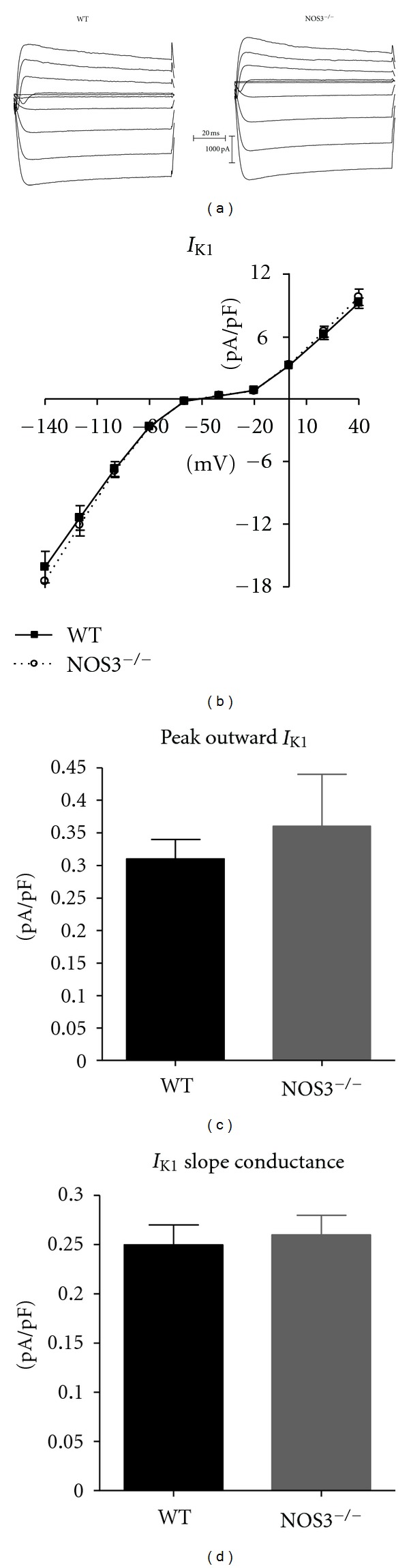
I K1 current was not changed in NOS3−/− myocytes. (a) Representative traces from WT and NOS3−/− myocytes. (b) I-V curves of I K1 in WT and NOS3−/− myocytes. (c) Summary data (mean ± S.E.M.) of peak outward I K1 in WT and NOS3−/− myocytes. (d) Summary data (mean ± S.E.M.) of I K1 slope conductance in WT and NOS3−/− myocytes. n = 6, 15 cells.
3.3. NOS3−/− Myocytes Do Not Have Altered Transient Outward or Sustained K+ Currents
Since other K+ currents are also involved in repolarization to determine the APD, we investigated if NOS3−/− myocytes had alterations in other K+ currents by measuring I to and I Ksus. Representative currents measured in a WT and NOS3−/− myocyte are shown in Figure 3(a). Our data show no difference in the I to or I Ksus I-V relationship in NOS3−/− compared to WT myocytes (Figure 3). With our voltage protocol, I Ksus is composed of various K+ currents, including I K,slow. However, since we did not observe a difference in composite I Ksus, we did not further investigate the contributing currents. These data indicate that NOS3 signaling does not modulate I to or I Ksus.
Figure 3.
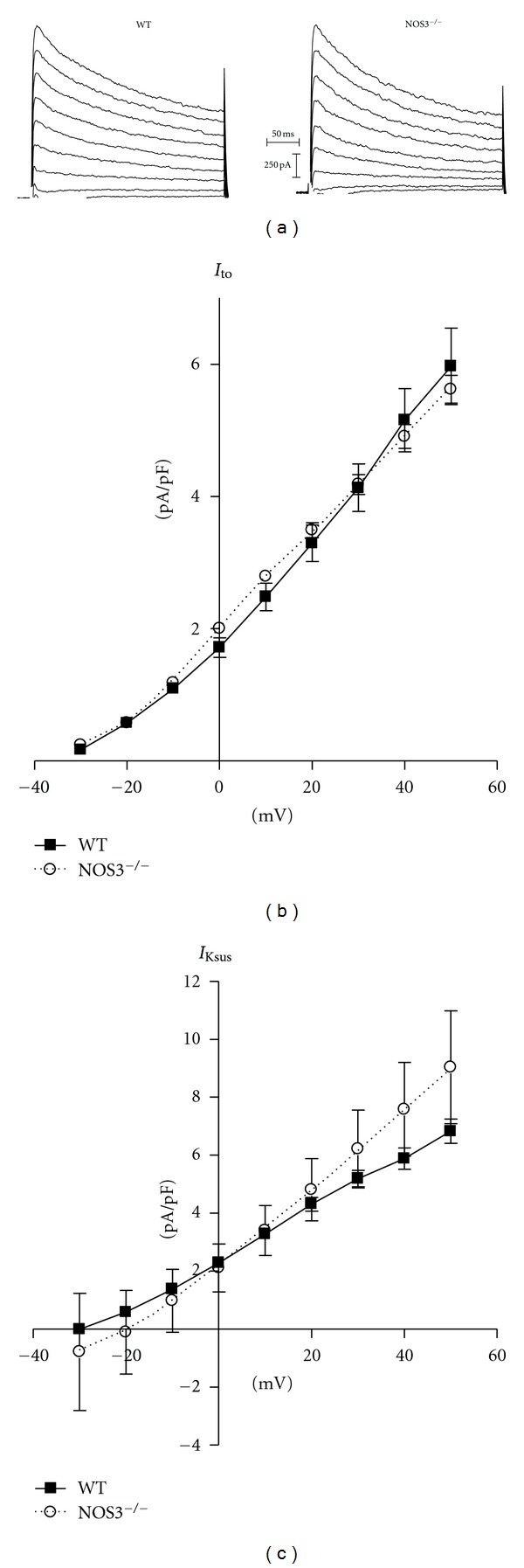
NOS3 knockout did not alter I to or I Ksus. (a) Representative current traces from WT and NOS3−/− myocytes. (b) Summary data (mean ± S.E.M.) of I to I-V curves in WT and NOS3−/− myocytes. (c) Summary data (mean ± S.E.M.) of I Ksus I-V curves in WT and NOS3−/− myocytes. n = 7, 13 cells.
3.4. β-AR Stimulated SR-Ca2+ Load Was Higher in NOS3−/− Myocytes
Altered K+ currents resulting in prolonged APD can contribute to the generation of afterdepolarizations [21, 22]. However, since we did not observe any differences in I to, I Ksus, or I K1, we performed additional experiments to measure SR Ca2+ load, which is also known to contribute to afterdepolarizations [22, 23]. We observed no difference in basal SR Ca2+ loads between NOS3−/− and WT myocytes (1.8 ± 0.2 versus 1.9 ± 0.2 ΔF/F 0). However, our data (Figure 4) show that NOS3−/− myocytes (compared to WT) have a larger increase in their SR Ca2+ load with β-AR stimulation. These data suggest that with β-AR stimulation there is a larger increase in SR Ca2+ load in NOS3−/− myocytes.
Figure 4.
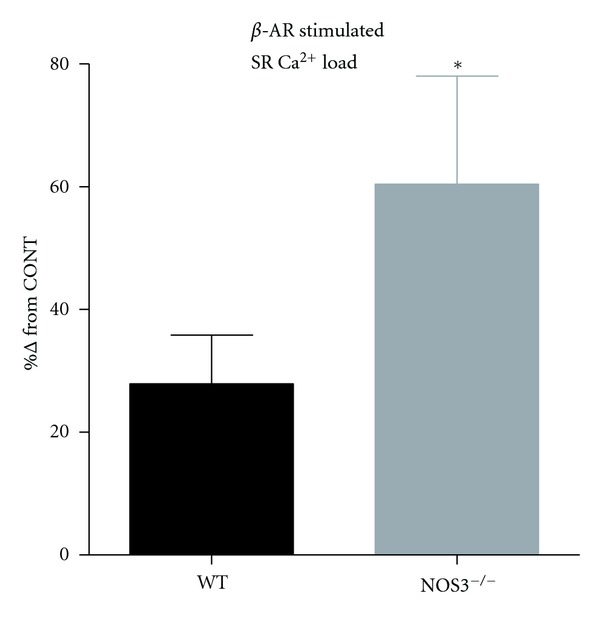
Larger increase in β-AR stimulated SR Ca2+ load in NOS3−/− myocytes. Summary data (mean ± S.E.M.) of the increase in SR Ca2+ load with β-AR stimulation in WT and NOS3−/− myocytes (control- CONT). *P < 0.05 versus WT, n = 11, 27 cells.
4. Discussion
The purpose of this study was to investigate the role of NOS3 signaling on the modulation of I to, I Ksus, and I K1. Our data show that I to, I Ksus, and I K1 are not altered in NOS3−/− ventricular myocytes, which suggests that NOS3 does not modulate I to, I Ksus, and I K1. Furthermore, the increased afterdepolarizations with β-AR stimulation in NOS3−/− myocytes may be due to enhanced spontaneous Ca2+ waves resulting from increased β-AR stimulated SR Ca2+ load.
4.1. Nitric Oxide Modulation of the β-AR Pathway
Stimulation of β-AR pathway is an essential regulator of cardiac contractility, leading to positive inotropic and lusitropic effects [24]. Our and other previous studies showed that NOS3 signaling blunts the functional response to β-AR stimulation [2, 5, 25–28]. That is, NOS3−/− myocytes have increased Ca2+ transient amplitudes and shortening amplitudes compared to WT, which is due to an enhanced β-AR stimulated I Ca. Our data also demonstrated that NOS3−/− myocytes had prolonged APD during β-AR stimulation [5]. This current study examined if alterations in K+ currents contributed to the prolonged APD in NOS3−/− myocytes.
4.2. Prolonged AP in NOS3−/− Myocytes Is Not due to Altered I to, I Ksus, and I K1
Previous studies have shown that alterations in I K1, which is an important K+ channel responsible for setting the RMP and shaping the late phase 3 of AP, can contribute to prolonging the AP [14]. However, our data (Figure 2) shows that I K1 is unaffected with knockout of NOS3. Furthermore, we observed similar RMP between WT and NOS3−/− myocytes (Figure 1), consistent with no difference in I K1. Thus, I K1 is not the cause for prolonged APD in NOS3−/− myocytes.
Besides I K1, many other K+ currents are key channels in shaping the AP waveform. I to is a transient outward current that contributes to the plateau phase of AP. Inhibition of I to has the ability to prolong the APD. In mouse myocytes, I Ksus is the sustained outward component of the repolarizing K+ current, and it plays an important role during the plateau and phase 3 of the AP. Interestingly, we also did not observe any differences in I to and I Ksus between NOS3−/− and WT myocytes (Figure 3). These data indicate that I to or I Ksus is not involved in the prolonged APD in NOS3−/− myocytes. Using our voltage protocol to elicit outward K+ channels, our composite I Ksus current will encompass I K,slow [29–32]. I K,slow is also an important K+ current in mice responsible for ~30% of the repolarization [29]. Since we did not observe a difference in I Ksus, these data suggest that I K,slow is also not modulated by NOS3 signaling. It should be noted that I to and I Ksus (including I K,slow) are not the only operating currents during the plateau phase. I Ca is an inward Ca2+ current that is counterbalanced by I to and I Ksus, which creates the plateau phase of the AP. Hence, the duration of the plateau is determined by the balance between I Ca and I to and I Ksus. We previously observed an increase in the β-AR stimulated I Ca in NOS3−/− myocytes compared to WT myocytes [5]. Thus, we suggest that in NOS3−/− myocytes an increased I Ca will overcome the unaltered I to and I Ksus, leading to more inward current resulting in prolonging the APD. This is consistent with previous studies which demonstrated that changes in I Ca will affect APD [33–35].
Previous studies demonstrated that NO donors are able to modulate K+ currents. The discrepancy between our findings and other groups may be due to the use of different species (human, guinea pig, and mouse). Another distinction between our current study and the previous studies is the use of endogenous NO (i.e., NOS3) or exogenous NO (i.e., NO donors). Our and others', work has shown the endogenous NO signaling via NOS1 and NOS3 is compartmentalized [1, 2]. However, when using NO donors, the entire myocyte will be exposed to NO, which is in contrast to the localized signaling observed with endogenous NO. In this study, we used isolated myocytes from NOS3−/− and WT hearts to directly investigate the effects of NOS3 signaling on K+ currents. While Gómez et al. [12] did observe effects of NO donors on I to, they also did not observe any differences in I to between WT and NOS3−/− ventricular myocytes (consistent with our observations). Thus, we are confident in our findings that NOS3 signaling does not modulate I to, I Ksus, or I K1.
4.3. Increased SR Ca2+ Load in NOS3−/− Myocytes with β-AR Stimulation
β-AR stimulation can result in triggered arrhythmias [36], which are observed in NOS3−/− mice [16]. These premature beats are analogous to afterdepolarizations in isolated myocytes, which we and others have demonstrated to occur in NOS3−/− myocytes [5, 17]. While alterations in K+ currents can contribute to the increased incidence of these triggered arrhythmias, we did not observe any difference in K+ currents in NOS3−/− myocytes. Another contributing factor to the generation of afterdepolarizations is in increase in the SR Ca2+ load [23, 37–39]. Studies have shown that early afterdepolarizations are due to an increase in I Ca [40] and delayed afterdepolarizations are due to spontaneous release of Ca2+ from the SR (i.e., Ca2+ wave) [37]. However, recent data now suggests that early afterdepolarizations caused by β-AR stimulation may also be due to spontaneous SR Ca2+ release [41, 42]. Thus, an important finding in the present study is the increased SR Ca2+ load with β-AR stimulation in NOS3−/− myocytes (Figure 4). It is known that enhanced Ca2+ influx via ICa will result in increased SR Ca2+ load [33, 43]. Thus, our higher SR Ca2+ load in NOS3−/− myocytes is consistent with the observed greater Ca2+ influx via I Ca. In fact, it has been suggested that Ca2+ waves and increased I Ca act synergistically to produce early and delayed afterdepolarizations [42]. Thus, NOS3−/− myocytes are tailored for afterdepolarizations since these myocytes exhibit an increased I Ca and SR Ca2+ load during β-AR stimulation. In fact, our previous study showed that the vast majority of NOS3−/− myocytes have afterpolarizations [5].
In addition to I Ca and the various K+ currents, it is known that other currents, most notably the sodium current [44], can not only affect APD but also contribute to the generation of afterdepolarizations. Since the purpose of this study was to examine if NOS3 signaling modulates K+ currents, we did not measure sodium current. Thus, the effect of NOS3 on sodium current and its corresponding effect on APD and afterdepolarizations cannot be excluded. Future studies are needed to investigate if NOS3 signaling can modulate the sodium current.
In conclusion, in NOS3−/− mouse ventricular myocytes, I to, I Ksus, and I K1 are normal and do not contribute to the prolonged APD and increased incidence of afterdepolarizations. We suggest that in NOS3−/− myocytes the increase in β-AR stimulated I Ca is the reason for the prolonged APD. Furthermore, the increased afterdepolarizations in NOS3−/− myocytes during β-AR stimulation are caused by the increased I Ca along with the increased SR Ca2+ load. This may have important implication for arrhythmias and sudden cardiac death in heart failure, where NOS3 expression is decreased [45, 46] and β-AR tone is increased [47].
References
- 1.Ziolo MT, Bers DM. The real estate of NOS signaling location, location, location. Circulation Research. 2003;92(12):1279–1281. doi: 10.1161/01.RES.0000080783.34092.AF. [DOI] [PubMed] [Google Scholar]
- 2.Barouch LA, Harrison RW, Skaf MW, et al. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416(6878):337–339. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- 3.Ziolo MT, Kohr MJ, Wang H. Nitric oxide signaling and the regulation of myocardial function. Journal of Molecular and Cellular Cardiology. 2008;45(5):625–632. doi: 10.1016/j.yjmcc.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang H, Kohr MJ, Traynham CJ, Wheeler DG, Janssen PML, Ziolo MT. Neuronal nitric oxide synthase signaling within cardiac myocytes targets phospholamban. American Journal of Physiology. 2008;294(6):C1566–C1575. doi: 10.1152/ajpcell.00367.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang H, Kohr MJ, Wheeler DG, Ziolo MT. Endothelial nitric oxide synthase decreases β-adrenergic responsiveness via inhibition of the L-type Ca2+ current. American Journal of Physiology. 2008;294(3):H1473–H1480. doi: 10.1152/ajpheart.01249.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H, Kohr MJ, Traynham CJ, Ziolo MT. Phosphodiesterase 5 restricts NOS3/Soluble guanylate cyclase signaling to L-type Ca2+ current in cardiac myocytes. Journal of Molecular and Cellular Cardiology. 2009;47(2):304–314. doi: 10.1016/j.yjmcc.2009.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiological Reviews. 2005;85(4):1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 8.Nerbonne JM. Molecular basis of functional voltage-gated K+ channel diversity in the mammalian myocardium. Journal of Physiology. 2000;525(2):285–298. doi: 10.1111/j.1469-7793.2000.t01-1-00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bai CX, Takahashi C, Masumiya H, Sawanobori T, Furukawa T. Nitric oxide-dependent modulation of the delayed rectifier K+ current and the L-type Ca2+ current by ginsenoside Re, an ingredient of Panax ginseng, in guinea-pig cardiomyocytes. British Journal of Pharmacology. 2004;142(3):567–575. doi: 10.1038/sj.bjp.0705814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bai CX, Namekata I, Kurokawa J, Tanaka H, Shigenobu K, Furukawa T. Role of nitric oxide in Ca2+ sensitivity of the slowly activating delayed rectifier K+ current in cardiac myocytes. Circulation Research. 2005;96(1):64–72. doi: 10.1161/01.RES.0000151846.19788.E0. [DOI] [PubMed] [Google Scholar]
- 11.Marx SO, Kurokawa J, Reiken S, et al. Requirement of a macromolecular signaling complex for β adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295(5554):496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 12.Gómez R, Núñez L, Vaquero M, et al. Nitric oxide inhibits Kv4.3 and human cardiac transient outward potassium current (I to1) Cardiovascular Research. 2008;80(3):375–384. doi: 10.1093/cvr/cvn205. [DOI] [PubMed] [Google Scholar]
- 13.Gómez R, Caballero R, Barana A, et al. Nitric oxide increases cardiac I K1 by nitrosylation of cysteine 76 of Kir2.1 Channels. Circulation Research. 2009;105(4):383–392. doi: 10.1161/CIRCRESAHA.109.197558. [DOI] [PubMed] [Google Scholar]
- 14.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current, and residual β-adrenergic responsiveness. Circulation Research. 2001;88(11):1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- 15.Maruyama M, Lin SF, Xie Y, et al. Genesis of phase 3 early afterdepolarizations and triggered activity in acquired long-QT syndrome. Circulation. 2011;4(1):103–111. doi: 10.1161/CIRCEP.110.959064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rakhit A, Maguire CT, Wakimoto H, et al. In vivo electrophysiologic studies in endothelial nitric oxide synthase (eNOS)-deficient mice. Journal of Cardiovascular Electrophysiology. 2001;12(11):1295–1301. doi: 10.1046/j.1540-8167.2001.01295.x. [DOI] [PubMed] [Google Scholar]
- 17.Kubota I, Han X, Opel DJ, et al. Increased susceptibility to development of triggered activity in myocytes from mice with targeted disruption of endothelial nitric oxide synthase. Journal of Molecular and Cellular Cardiology. 2000;32(7):1239–1248. doi: 10.1006/jmcc.2000.1158. [DOI] [PubMed] [Google Scholar]
- 18.Shesely EG, Maeda N, Kim HS, et al. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(23):13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sridhar A, Dech SJ, Lacombe VA, et al. Abnormal diastolic currents in ventricular myocytes from spontaneous hypertensive heart failure rats. American Journal of Physiology. 2006;291(5):H2192–H2198. doi: 10.1152/ajpheart.01146.2005. [DOI] [PubMed] [Google Scholar]
- 20.Trafford AW, Díaz ME, Eisner DA. A novel, rapid and reversible method to measure Ca buffering and time-course of total sarcoplasmic reticulum Ca content in cardiac ventricular myocytes. Pflugers Archiv European Journal of Physiology. 1999;437(3):501–503. doi: 10.1007/s004240050808. [DOI] [PubMed] [Google Scholar]
- 21.Choi BR, Burton F, Salama G. Cytosolic Ca2+ triggers early afterdepolarizations and Torsade de Pointes in rabbit hearts with type 2 long QT syndrome. Journal of Physiology. 2002;543(2):615–631. doi: 10.1113/jphysiol.2002.024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annual Review of Physiology. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 23.Venetucci LA, Trafford AW, O’Neill SC, Eisner DA. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovascular Research. 2008;77(2):285–292. doi: 10.1093/cvr/cvm009. [DOI] [PubMed] [Google Scholar]
- 24.Ziolo MT, Katoh H, Bers DM. Positive and negative effects of nitric oxide on Ca2+ sparks: influence of β-adrenergic stimulation. American Journal of Physiology. 2001;281(6):H2295–H2303. doi: 10.1152/ajpheart.2001.281.6.H2295. [DOI] [PubMed] [Google Scholar]
- 25.Champion HC, Georgakopoulos D, Takimoto E, Isoda T, Wang Y, Kass DA. Modulation of in vivo cardiac function by myocyte-specific nitric oxide synthase-3. Circulation Research. 2004;94(5):657–663. doi: 10.1161/01.RES.0000119323.79644.20. [DOI] [PubMed] [Google Scholar]
- 26.Gödecke A, Heinicke T, Kamkin A, et al. Inotropic response to β-adrenergic receptor stimulation and anti-adrenergic effect of ACh in endothelial NO synthase-deficient mouse hearts. Journal of Physiology. 2001;532(1):195–204. doi: 10.1111/j.1469-7793.2001.0195g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gyurko R, Kuhlencordt P, Fishman MC, Huang PL. Modulation of mouse cardiac function in vivo by eNOS and ANP. American Journal of Physiology. 2000;278(3):H971–H981. doi: 10.1152/ajpheart.2000.278.3.H971. [DOI] [PubMed] [Google Scholar]
- 28.Varghese P, Harrison RW, Lofthouse RA, Georgakopoulos D, Berkowitz DE, Hare JM. β3-adrenoceptor deficiency blocks nitric oxide-dependent inhibition of myocardial contractility. Journal of Clinical Investigation. 2000;106(5):697–703. doi: 10.1172/JCI9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo W, Xu H, London B, Nerbonne JM. Molecular basis of transient outward K+ current diversity in mouse ventricular myocytes. Journal of Physiology. 1999;521(3):587–599. doi: 10.1111/j.1469-7793.1999.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu H, Barry DM, Li H, Brunet S, Guo W, Nerbonne JM. Attenuation of the slow component of delayed rectification, action potential prolongation, and triggered activity in mice expressing a dominant-negative Kv2 α subunit. Circulation Research. 1999;85(7):623–633. doi: 10.1161/01.res.85.7.623. [DOI] [PubMed] [Google Scholar]
- 31.Xu H, Guo W, Nerbonne JM. Four kinetically distinct depolarization-activated K+ currents in adult mouse ventricular myocytes. Journal of General Physiology. 1999;113(5):661–678. doi: 10.1085/jgp.113.5.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou J, Jeron A, London B, Han X, Koren G. Characterization of a slowly inactivating outward current in adult mouse ventricular myocytes. Circulation Research. 1998;83(8):806–814. doi: 10.1161/01.res.83.8.806. [DOI] [PubMed] [Google Scholar]
- 33.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Dordrecht, The Netherlands: Kluwer Academic; 2001. [Google Scholar]
- 34.Pott C, Philipson KD, Goldhaber JI. Excitation-contraction coupling in Na+-Ca2+ exchanger knockout mice: reduced transsarcolemmal Ca2+ flux. Circulation Research. 2005;97(12):1288–1295. doi: 10.1161/01.RES.0000196563.84231.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saegusa N, Moorhouse E, Vaughan-Jones RD, Spitzer KW. Influence of pH on Ca(2)(+) current and its control of electrical and Ca(2)(+) signaling in ventricular myocytes. The Journal of General Physiology. 2011;138:537–559. doi: 10.1085/jgp.201110658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pogwizd SM, Bers DM. Cellular basis of triggered arrhythmias in heart failure. Trends in Cardiovascular Medicine. 2004;14(2):61–66. doi: 10.1016/j.tcm.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Marban E, Robinson SW, Wier WG. Mechanisms of arrhythmogenic delayed and early afterdepolarizations in ferret ventricular muscle. Journal of Clinical Investigation. 1986;78(5):1185–1192. doi: 10.1172/JCI112701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Díaz ME, Trafford AW, O’Neill SC, Eisner DA. Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. Journal of Physiology. 1997;501:3–16. doi: 10.1111/j.1469-7793.1997.003bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tweedie D, Harding SE, MacLeod KT. Sarcoplasmic reticulum Ca content, sarcolemmal Ca influx and the genesis of arrhythmias in isolated guinea-pig cardiomyocytes. Journal of Molecular and Cellular Cardiology. 2000;32(2):261–272. doi: 10.1006/jmcc.1999.1070. [DOI] [PubMed] [Google Scholar]
- 40.January CT, Riddle JM. Early afterdepolarizations: mechanism of induction and block. A role for L-type Ca2+ current. Circulation Research. 1989;64(5):977–990. doi: 10.1161/01.res.64.5.977. [DOI] [PubMed] [Google Scholar]
- 41.Volders PGA, Kulcsár A, Vos MA, et al. Similarities between early and delayed afterdepolarizations induced by isoproterenol in canine ventricular myocytes. Cardiovascular Research. 1997;34(2):348–359. doi: 10.1016/s0008-6363(96)00270-2. [DOI] [PubMed] [Google Scholar]
- 42.Zhao Z, Wen H, Fefelova N, et al. Revisiting the ionic mechanisms of early afterdepolarizations in cardiomyocytes: predominant by Ca waves or Ca currents? American Journal of Physiology. 2012;302(8):1636–1644. doi: 10.1152/ajpheart.00742.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang M, Zhang X, Li Y, et al. Enhanced basal contractility but reduced excitation-contraction coupling efficiency and β-adrenergic reserve of hearts with increased Cav1.2 activity. American Journal of Physiology. 2010;299(2):H519–H528. doi: 10.1152/ajpheart.00265.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wagner S, Dybkova N, Rasenack ECL, et al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. Journal of Clinical Investigation. 2006;116(12):3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Champion HC, Skaf MW, Hare JM. Role of nitric oxide in the pathophysiology of heart failure. Heart Failure Reviews. 2003;8(1):35–46. doi: 10.1023/a:1022142904202. [DOI] [PubMed] [Google Scholar]
- 46.Damy T, Ratajczak P, Shah AM, et al. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. The Lancet. 2004;363(9418):1365–1367. doi: 10.1016/S0140-6736(04)16048-0. [DOI] [PubMed] [Google Scholar]
- 47.Tsukamoto T, Morita K, Naya M, et al. Decreased myocardial β-adrenergic receptor density in relation to increased sympathetic tone in patients with nonischemic cardiomyopathy. Journal of Nuclear Medicine. 2007;48(11):1777–1782. doi: 10.2967/jnumed.107.043794. [DOI] [PubMed] [Google Scholar]