

Abstract
The biallelic expression of the imprinted gene ZAC1/PLAGL1 underlies ∼60% of all cases of transient neonatal diabetes mellitus (TNDM) that present with low perinatal insulin secretion. Molecular targets of ZAC1 misexpression in pancreatic β cells are unknown. Here, we identified the guanine nucleotide exchange factor Rasgrf1 as a direct Zac1/Plagl1 target gene in murine β cells. Doubling Zac1 expression reduced Rasgrf1 expression, the stimulus-induced activation of mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) pathways, and, ultimately, insulin secretion. Normalizing Rasgrf1 expression reversed this phenotype. Moreover, the transplantation of Zac1-overexpressing β cells failed to reinstate euglycemia in experimental diabetic mice. In contrast, Zac1 expression did not interfere with the signaling of the glucagon-like peptide 1 receptor (GLP-1R), and the GLP-1 analog liraglutide improved hyperglycemia in transplanted experimental diabetic mice. This study unravels a mechanism contributing to insufficient perinatal insulin secretion in TNDM and raises new prospects for therapy.
INTRODUCTION
Transient neonatal diabetes mellitus (TNDM) due to chromosome 6q anomalies is a rare cause of early-onset hyperglycemia in term newborn infants (1). Neonates typically present with low birth weight and high blood glucose values, features of low pancreatic insulin secretion in utero and after delivery. Initially insulin treatment is required, whereas by 3 months insulin secretion has improved to allow its discontinuation. Patients remain in apparent remission until adolescence, when half of them relapse with a loss of first-phase insulin secretion similar to type 2 diabetes (T2D) (47, 52).
Three genetic anomalies have been identified in TNDM: paternal uniparental isodisomy of chromosome 6, unbalanced paternal duplication of 6q24 (the TNDM locus), and maternal methylation anomalies (1, 13, 38, 53). Two overlapping imprinted genes with the silencing of the maternal allele have been discovered at the TNDM locus, ZAC1 (zinc finger [ZF] protein regulating apoptosis and cell cycle arrest; alias PLAGL1, pleomorphic adenoma gene-like 1) and HYMAI (hydatidiform mole-associated and imprinted transcript) (3, 13, 23).
While the function of the nontranslated HYMAI RNA remains unknown, ZAC1 encodes a zinc finger protein regulating cell cycle arrest and apoptosis under forced expression (50, 54). In addition to the context-dependent coregulation of nuclear receptors (19), p53, and p73 (17, 18), Zac1 binds to different classes of DNA elements that determine transcriptional activator versus repressor activities (15–17).
Zac1 null mice are growth retarded and suffer from cardiac malformations and high perinatal lethality due to lung failure (55, 61). In contrast, transgenic mice overexpressing ZAC1 present with neonatal hyperglycemia and impaired glucose tolerance in later life (37). Embryonic pancreata show a reduction of β cells followed by overcompensated proliferation in early postnatal life. Despite this, early neonates remain hyperglycemic because of inadequate insulin secretion. The increase in β-cell number disappears in adults, and glucose tolerance deteriorates with signs of reduced insulin secretion.
Pancreatic targets of ZAC1 misexpression in TNDM are unknown. In the present study, we identified the guanine nucleotide exchange factor Rasgrf1 as a direct Zac1 target gene in β cells. Rasgrf1 is activated by Ca2+-calmodulin and serves as a regulator and effector of Ras pathways by integrating Ca2+ signals elicited by Ca2+ influx and G-protein-coupled receptors (60). We further investigated the consequences of Zac1-dependent Rasgrf1 regulation for insulin secretion. Our results raise new prospects for the pharmacotherapy of TNDM patients.
MATERIALS AND METHODS
Cell culture and transfection experiments.
INS-1 and Min6 β cells were cultured as described previously (39) or in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, 40 mM sodium bicarbonate, and 70 μM 2-mercaptoethanol. R7T1 β cells were grown in DMEM supplemented with 15% horse serum, 2.5% fetal calf serum, and 2 μg/ml tetracycline (Tc). Transient and stable transfections were performed with Turbofect transfection reagent (Fermentas, St. Leon-Roth, Germany).
Following the transfection of a Zac1 expression vector (4 μg of pRK.SV40-Hygromycin-CMV.Flag-Zac1), R7T1 β cells were selected (150 μg/ml hygromycin B; Calbiochem, Merck KGaA, Darmstadt, Germany) and pooled (R7-Z). R7-Z cells were transfected with a Rasgrf1 expression vector (4 μg pRK.SV40-Blasticidin-CMV.HA-Rasgrf1), selected (2 μg/ml blasticidin; Calbiochem), and pooled (R7-Z-R). Primary pools (Zac1, n = 8; Rasgrf1, n = 6) were amplified separately for 1 month to prepare a batch of master stocks. Transgene expression was determined by quantitative reverse transcription-PCR (qRT-PCR) to identify suitable populations either mimicking biallelic Zac1 (n = 3) or reconstituting Rasgrf1 (n = 3) expression. Thereafter, cultures were initiated on demand from master stocks and cultivated for up to 2 months. Stock cultures were grown in medium supplemented with antibiotics used for selection throughout to sustain proper transgene expression.
Proliferation rates were measured by a Coulter Counter (Beckman Coulter, Krefeld, Germany); briefly, 5 × 103 cells were seeded into 12-well plates and maintained in the absence or presence of Tc (2 μg/ml) for the indicated time. Medium was replenished every third day.
Rasgrf1 promoter regions were cloned in the pGL3-basic vector (Promega, Mannheim, Germany). Gaussia luciferase values were normalized to firefly luciferase activity from a cotransfected expression vector (pRK7Luc; 0.1 μg). Amounts of plasmids transfected are described in the respective figure legends.
Small interfering RNA (siRNA) oligonucleotides (Zac1, CGTGGGTTTCTTTGAGGAA; nonsilencing control, AGGTAGTGTACGCCTTGTT) were transfected by Turbofect siRNA transfection reagent (Fermentas).
qRT-PCR and RNA extractions.
Primer sequences are listed in Table S1 in the supplemental material. The housekeeping gene for mitochondrial ATP synthase-coupling factor 6 (ATP5J) served for normalization. RNA extraction and qRT-PCR analysis were performed as described previously (17).
Immunoblotting, immunohistochemistry, and antibodies.
Whole-cell extracts were fractionated by SDS-PAGE. To determine phospho-Erk1/2 immunoreactivity, cells were kept for 5 days in the appropriate medium at 80% confluence if not indicated otherwise. Medium was replenished for 12 h. Cells were washed twice with glucose-free modified Krebs-Ringer buffer (KRBH; 125 mM NaCl, 4.74 mM KCl, 1 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 5 mM NaHCO3, 25 mM HEPES, pH 7.4, 0.1% bovine serum albumin [BSA]) and kept for 1 h in the same buffer. Treatment with various stimuli in glucose-free KRBH is described in the figure legends. Phospho-Akt immunoreactivity was detected as described above, except that cells were kept prior to stimulation for 12 h in medium containing 8.3 mM glucose and 0.1% serum.
Immunohistochemistry was performed on 4% (wt/vol) paraformaldehyde-fixed, cryopreserved sections (10 μm) of pancreata from B6C3F1 mice as described previously (43). For each age, 3 to 5 islets on 4 different pancreatic slices from 3 pancreata were investigated. Human pancreas was obtained from Cambridge Bioscience Ltd. (Cambridge, United Kingdom).
Antibodies are listed in Table S3 in the supplemental material. The polyclonal Rasgrf1 antiserum was raised in rabbits. Preabsorpt Zac1 and Rasgrf1 antisera failed to detect specific signals in pancreatic islets (data not shown).
ChIP.
The Magna chromatin immunoprecipitation (ChIP) kit (Millipore, Schwalbach, Germany) was used as described previously (42). In brief, DNA-protein complexes were cross-linked with 1% (vol/vol) formaldehyde. Chromatin was sheared with a Biorupter sonicator (Diagenode, Liege, Belgien) to an average length of 0.2 to 0.5 kb and immunoprecipitated with the indicated antibodies. Rabbit IgG or Zac1 preimmune serum served as a control for nonspecific binding. Purified DNA was analyzed by qPCR using Absolute Blue QPCR SYBR green master mix (Abgene, Thermo Fisher Scientific, Epsom, United Kingdom) and the MJ Mini Opticon Light Cycler (Bio-Rad, München, Germany). Data are diagramed as fold enrichment compared to control sera. qPCR primers are listed in Table S2 in the supplemental material.
Transplantation studies and glucose measurements.
Male B6C3F1 mice (Charles River, Sulzfeld, Germany) were housed under standard conditions with a 12-h light/dark cycle.
Streptozotocin (STZ; 200 mg/kg body weight; Sigma, München, Germany) or vehicle was injected once intraperitoneally. One week later, diabetic mice were intraperitoneally administered either R7T1 or R7-Z β cells (5 × 106 cells resuspended in 0.2 ml of 0.9% NaCl) or vehicle. Nondiabetic controls received vehicle alone. Drinking water was supplemented with doxycycline (1 mg/ml; Clontech, St-Germain-en-Laye, France) during the first day after transplantation.
In a second study group, half of the mice transplanted with R7T1 or R7-Z β cells were additionally administered liraglutide (0.2 mg/kg; Bachem, Weil am Rhein, Germany) or vehicle subcutaneously twice a day.
Blood glucose was measured every fourth day from mice fasted overnight with the Accu Check system (Roche, Mannheim, Germany), except for the liraglutide study, in which blood glucose was measured from nonfasting mice on each second day. Pancreata were dissected at the end of the experiment, weighed, and immediately frozen on dry ice.
Experiments were approved by the Regierung of Oberbayern (55.2-1-54-2531-163-09) in accordance with European Union Directive 86/609/EEC.
Insulin ELISA.
Total pancreatic insulin was extracted (37) and determined by enzyme-linked immunosorbent assay (ELISA) (DRG-Diagnostics, Marburg, Germany).
For cellular insulin secretion, cells were kept as indicated in the figure legends, subsequently preincubated for 2 h in glucose-free KRBH, and stimulated with various secretagogues for 2 h. The amount of insulin in the buffer and cell extract was measured.
Pancreatic islets.
Pancreata from 6 to 8 littermates (postnatal day 4 [P4]) were pooled, and islets were isolated (46) and cultured overnight in RPMI 1640 medium containing 11 mM glucose and 10% fetal calf serum (FCS). Transfections were performed using Lipofectamine (Life Technologies GmbH, Darmstadt, Germany). Insulin secretion was measured 36 h later following preincubation for 2 h in KRBH containing 2 mM glucose and stimulation for 2 h with 20 mM glucose or 50 mM KCl.
Statistical analysis.
Results represent the means and standard deviations (SD) from at least three independent experiments. Numerical data were analyzed by Student's t test. For animal experiments, statistical significance was assessed by one-way analysis of variance (ANOVA) followed by a Newman-Keuls post hoc test. The threshold for significance was set at P < 0.05.
RESULTS
Zac1 and Rasgrf1 are coexpressed in pancreatic β cells.
We previously observed in neuronal progenitor cells (in which we had overexpressed Zac1) that the expression of Rasgrf1 was repressed 2-fold at early (3 h) but less at late (6 h and 9 h) time points (5) (data not shown).
Initially described as having restricted expression in postnatal mice brain (21), Rasgrf1 expression has been reported in adult pancreatic islets as well (12). To determine whether and at which developmental stage Zac1 and Rasgrf1 are coexpressed, we analyzed their expression pattern in fetal, perinatal, and early adult mice pancreata.
We detected strong nuclear Zac1 immunoreactivity at fetal (embryonic day 18 [E18]) and neonatal (P1) stages in the endocrine pancreas (insulin-positive and -negative cells) and, to a lower degree, scattered throughout the exocrine pancreas (Fig. 1A and data not shown). Postnatally, Zac1 expression declined within a few days in both the endocrine and exocrine pancreas (compare P4 to P12). In early adult mice (P42), Zac1 expression was weaker in insulin-positive cells and was barely detectable in the parenchyma (Fig. 1A). Antisera raised against the Zac1 C terminus (Zac-C; rabbit) and the central part of Zac1 (Zac-LPR; guinea pig) gave similar results on pancreata sections (data not shown).
Fig 1.

Zac1 and Rasgrf1 expression by immunohistochemistry in mouse pancreas. (A) Strong nuclear Zac1 (red) immunostaining is detected in the endocrine and less is detected in the exocrine pancreas of E18 embryos and neonatal (P1) mice and declines postnatally (P4, P12). Low Zac1 expression is maintained in islets in early adulthood (P42) and colocalizes to insulin-positive (green) β cells. Sparse Zac1 immunoreactivity is detected in the exocrine pancreas. Cell nuclei are stained by DAPI (blue). (B) Rasgrf1 (red) is weakly expressed in the endocrine pancreas of E18 embryos and increases peri- and postnatally (P1, P4, and P12). High cytoplasmic expression is maintained in early adulthood (P42) in insulin-positive (green) β cells and slightly in exocrine cells. (C) Zac1 (red) and Rasgrf1 (green) are inversely expressed in pancreatic β cells. Strong nuclear Zac1 immunoreactivity decreases perinatally, in contrast to increased cytoplasmic Rasgrf1 expression. Size bar, 100 μm. Sections with higher magnifications are included to depict nuclear Zac1 and cytoplasmic Rasgrf1 staining. Arrows indicate Zac1-positive cells in the exocrine pancreas. (D) Zac1 and Rasgrf1 expression in whole pancreas determined by qRT-PCR at the indicated ages. Data represent means ± SD from three animals per time point. *, P < 0.05; **, P < 0.01.
In contrast, Rasgrf1 was faintly expressed in the endocrine pancreas at E18 and was hardly detectable in the parenchyma (Fig. 1B). Postnatally, Rasgrf1 expression increased within a few days in the cytoplasm of insulin-positive cells (Fig. 1B, compare P4 to P12). Both Zac1 and Rasgrf1 are coexpressed in insulin-positive β cells (Fig. 1C), a pattern that is well conserved in humans (see Fig. S1 in the supplemental material).
In accord with the results from immunohistochemistry, real-time PCR from total pancreas showed a strong postnatal decline in Zac1 expression in parallel to an inverted increase in Rasgrf1 expression (Fig. 1D). This inverse relationship was most prominent between P4 and P12.
Zac1 represses Rasgrf1 and insulin secretion in β cells.
The inverse expression of Zac1 and Rasgrf1 in pancreatic β cells is consistent with the results from expression profiling and raises the question of whether Rasgrf1 is a direct Zac1 target gene that contributes to Zac1's potential effect on insulin secretion. To test this hypothesis, we examined Zac1 and Rasgrf1 expression and the effects from Zac1 overexpression on Rasgrf1 and insulin secretion in different pancreatic β-cell lines and primary cultures of neonate islets.
The two mouse β-cell lines Min6 (41) and R7T1 (40) showed Zac1 and Rasgrf1 expression similar to that of neonate islets from postnatal day 4, while the rat β-cell line INS-1 (4) displayed slightly lower expression of both genes (Fig. 2A). Following the transfection of increasing doses of Zac1, Rasgrf1 expression steadily decreased in all three β-cell lines (Fig. 2B). A higher Zac1 dose was necessary to repress Rasgrf1 in neonate islets given the low transfection efficiency.
Fig 2.

Zac1 and Rasgrf1 expression in β-cell lines and ex vivo islets. (A) RT-PCR analysis of Zac1 and Rasgrf1 expression in INS-1, Min6, and R7T1 β-cell lines and ex vivo islets isolated at postnatal day 4. (B) Transfected Zac1 dose dependently (100, 250, and 500 ng) inhibits Rasgrf1 expression in β-cell lines and ex vivo islets. Rasgrf1 expression is measured by qRT-PCR analysis and normalized to mock transfection, which is set to 100%. Ex vivo islets were transfected with the highest Zac1 or mock dose. (C and D) Zac1 overexpression reduces insulin secretion in β-cell lines and ex vivo islets. Following Zac1 transfection (500 ng), cells were stimulated after 36 h for 2 h with 20 mM glucose (C) or 50 mM potassium chloride (D). Secreted insulin was normalized on intracellular insulin and expressed as percentages of basal insulin secretion. (A to D) R7T1 cells were kept in the presence of tetracycline. Data represent means ± SD from three independent experiments performed in duplicate. *, P < 0.05; **, P < 0.01.
Interestingly, Zac1 overexpression also impaired glucose- and KCl-stimulated insulin secretion in a dose-dependent manner in each cell line, giving rise to a 30 to 40% reduction at the highest doses tested (Fig. 2C and D and data not shown). This effect was recapitulated in primary islets, although to a lower degree.
Together, these findings indicate that Zac1 overexpression leads to the repression of Rasgrf1 and impaired insulin secretion.
Zac1 and Rasgrf1 expression during advanced differentiation of R7T1 cells.
To study further the Zac1-dependent regulation of Rasgrf1, we chose the reversibly immortalized β-cell line R7T1. These cells have been derived from heritably developing insulinomas in transgenic mice that harbor the simian virus 40 (SV40) large T antigen under the control of a β-cell-specific tetracycline regulatory expression system (40).
R7T1 β cells were negative for the progenitor cell state marker neurogenin 3 (Ngn3) but expressed early factors involved in the specification into mature endocrine cells (Pax4, Nkx2.2, Nkx6.1, and MafB) and the maturation of committed endocrine cells (Pax6, Isl1, Pdx1, Hlxb9, and MafA) (8) (data not shown). Following the withdrawal of tetracycline, R7T1 β cells ceased proliferation, flattened and spread, and produced large amounts of insulin, which led to a parallel increase in insulin secretion (Fig. 3A to E).
Fig 3.

Advanced differentiation elicits Zac1 downregulation and increased Rasgrf1 expression in R7T1 β cells. (A) Cell proliferation ceases in the absence of tetracycline (−Tc). (B) Bright-light microscopy. Multilayered clusters (top) segregate and flatten following 2 weeks of Tc withdrawal (bottom). Size bar, 200 μm. (C) Insulin immunoreactivity (green) increases following 2 weeks of Tc withdrawal. Cell nuclei were stained with DAPI (blue). Size bar, 100 μm. (D and E) Glucose-dependent insulin secretion. Insulin content and secretion concurrently increase during 2 weeks of advanced differentiation. The amount of insulin in the lysate and medium was determined by ELISA and normalized to cellular protein content. (F) Relative mRNA levels (note the logarithmic scale) of the islet hormones insulin 1 (Ins1), insulin 2 (Ins2), pancreatic polypeptide (PP), somatostatin (Sst), glucagon (Glu), and ghrelin (Ghr) in predifferentiated R7T1 β cells as measured by qRT-PCR analysis. Glucagon expression is not detectable (nd). (G) Hormone expression in β cells grown for 2 weeks in the presence and absence of tetracycline as evidenced by qRT-PCR analysis. (H and I) Zac1 and Rasgrf1 expression for the indicated period of advanced differentiation. (H) mRNAs and (I) whole-cell extracts (WCE; 75 μg) were analyzed by qRT-PCR and the indicated antibodies, respectively. Data represent means ± SD from four independent experiments. *, P < 0.05; **, P < 0.01.
The analysis of relative mRNA levels of pancreatic islet hormones revealed large amounts of insulin, small amounts of pancreatic polypeptide, somatostatin, and ghrelin, and undetectable amounts of glucagon in the predifferentiated state (Fig. 3F). Advanced differentiation caused an increase in the expression of insulin and pancreatic polypeptide, while the expression levels of somatostatin and ghrelin remained unaltered (Fig. 3G).
In accord with the data from mice and human pancreata, Zac1 and Rasgrf1 proteins were coexpressed in the nuclear and cytoplasmic compartments of differentiated R7T1 β cells, respectively (see Fig. S2 in the supplemental material). While Zac1 showed strong nuclear immunoreactivity, Rasgrf1 expression was weakly detectable in predifferentiated R7T1 β cells. Interestingly, the expression of Zac1 and Rasgrf1 correlated inversely with each other at the mRNA and protein levels upon advanced differentiation (Fig. 3H and I). This pattern recapitulates well the expression of Zac1 and Rasgrf1 in the developing pancreas and makes this cell line a suitable tool for studying the relationship between Zac1 and Rasgrf1 in perinatal β-cell function.
Zac1 occupancy at the Rasgrf1 locus confers gene repression.
To corroborate the role of Rasgrf1 as a direct Zac1 target gene, we carried out transfection studies in predifferentiated R7T1 β cells with a set of Zac1 zinc finger (ZF) constructs selectively impaired in DNA binding (15). Immunoblot analysis confirmed similar expression levels for these constructs (Fig. 4A). While Zac1 and a construct lacking the first zinc finger (ΔZF1) potently repressed Rasgrf1, the absence of the first two zinc fingers (ΔZF1-2) abolished repression (Fig. 4B). ZF2 is necessary for Zac1 binding to half-site (HS) and direct-repeat DNA elements (DR), whereas it is dispensable for binding to the palindrome (PAL). To distinguish between these cases, we transfected Zac1 harboring the mutation ZF7R201, which abolishes half-site binding but largely maintains direct-repeat binding (16). This mutation strongly diminished the repression of Rasgrf1. Finally, a broken ZF7 (ZF7mt) blocking any DNA binding did not alter Rasgrf1 expression (Fig. 4B). Together, these results suggest that Zac1 controls Rasgrf1 gene expression following binding to half-site DNA elements. Consistently with these findings, the knockdown of Zac1 by siRNA treatment efficiently enhanced Rasgrf1 expression (Fig. 4C and D).
Fig 4.
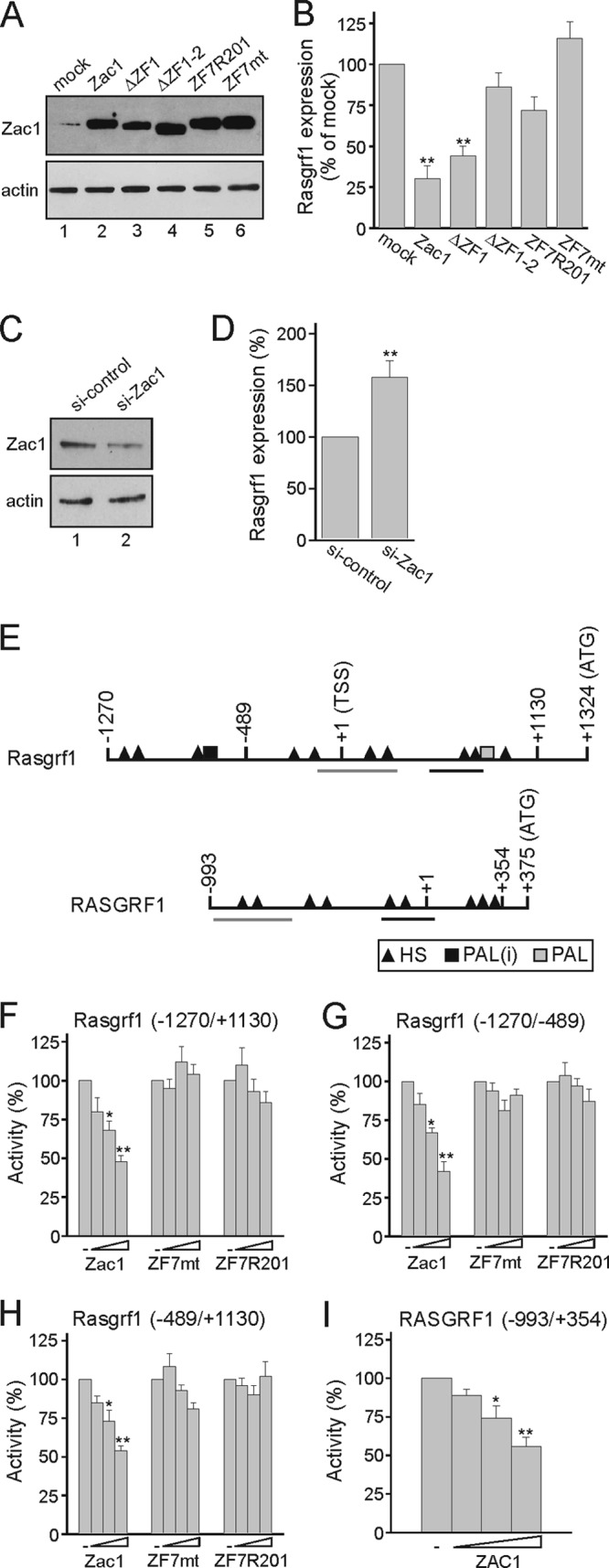
Zac1 inhibits Rasgrf1 gene expression. (A) Immunoblot analysis of Zac1 constructs transfected into predifferentiated R7T1 β cells. WCE (25 μg) were tested with the indicated antibodies. (B) Zac1 represses Rasgrf1 expression in predifferentiated R7T1 β cells. Following the transfection of Zac1 constructs, Rasgrf1 expression was determined by qRT-PCR analysis and normalized to the result from mock transfection, which was set to 100%. (C and D) Knockdown of Zac1 derepresses Rasgrf1. Control or Zac1 siRNA was transfected in predifferentiated R7T1 β cells, which were harvested 2 days later. (C) Immunoblot (50 μg WCE) shows Zac1 expression in control or Zac1 siRNA-transfected R7T1 cells. (D) Derepression of Rasgrf1 was investigated by qRT-PCR analysis; the expression of Rasgrf1 under control siRNA treatment was set to 100%. (E) Scheme of the mouse and human Rasgrf1 promoter. Transcriptional start site (TSS), translation start codon (ATG), and potential Zac1 DNA-binding sites are shown; abbreviations are half-site (HS), imperfect palindrome [PAL(i) (G3C4)], and palindrome [PAL (G5C4)]. In contrast to mice, the human RASGRF1 promoter contains only half-site repressor DNA elements. Highly conserved regions (>85%) between mouse and human Rasgrf1 promoters are underlined. (F to H) Zac1, Zac-ZF7mt, or Zac-ZF7R201 was transfected (100, 250, and 500 ng each) in predifferentiated R7T1 β cells together with the indicated Rasgrf1 promoter reporter constructs (2 μg). (I) Cotransfection of ZAC1 (50, 100, and 250 ng) in predifferentiated R7T1 β cells leads to the repression of a human RASGRF1 promoter reporter construct (2 μg). Data represent means ± SD from three (B and D) or five (F to I) independent experiments performed in duplicate. *, P < 0.05; **, P < 0.01.
Computational analysis of the Rasgrf1 gene revealed the presence of multiple half-sites across the promoter in addition to one imperfect and one perfect palindromic site (Fig. 4E). In agreement with this prediction, Zac1, but not the constructs ZF7mt and ZF7R201, potently repressed reporter constructs containing either the entire or separated distal and proximal promoter regions (Fig. 4F to H). Moreover, human ZAC1 potently repressed a RASGRF1 promoter construct which harbors solely multiple half-site DNA elements (Fig. 4I). These sites are highly conserved in part between human and mouse RASGRF1 genes (Fig. 4E).
We sought to verify the predicted binding sites by chromatin immunoprecipitation (ChIP) assays in R7T1 β cells in the presence and absence of tetracycline with a Zac1 antibody and primer pairs bracketing each of the potential binding regions (labeled I to V) (Fig. 5A). We detected decreased Zac1 occupancy upon advanced differentiation at each region, whereby region III showed low occupancy under either state. While regions I and II were located close to each other, making it difficult to distinguish between Zac1 occupancies, they behaved clearly distinctly from region III under the proliferative condition. Under advanced differentiation, residual Zac1 occupancy might reflect differences in DNA binding activities in proliferative versus quiescent R7T1 β cells or weak expression still detected by immunocytochemistry compared to less sensitive immunoblotting (compare Fig. 3I to Fig. S2 in the supplemental material).
Fig 5.
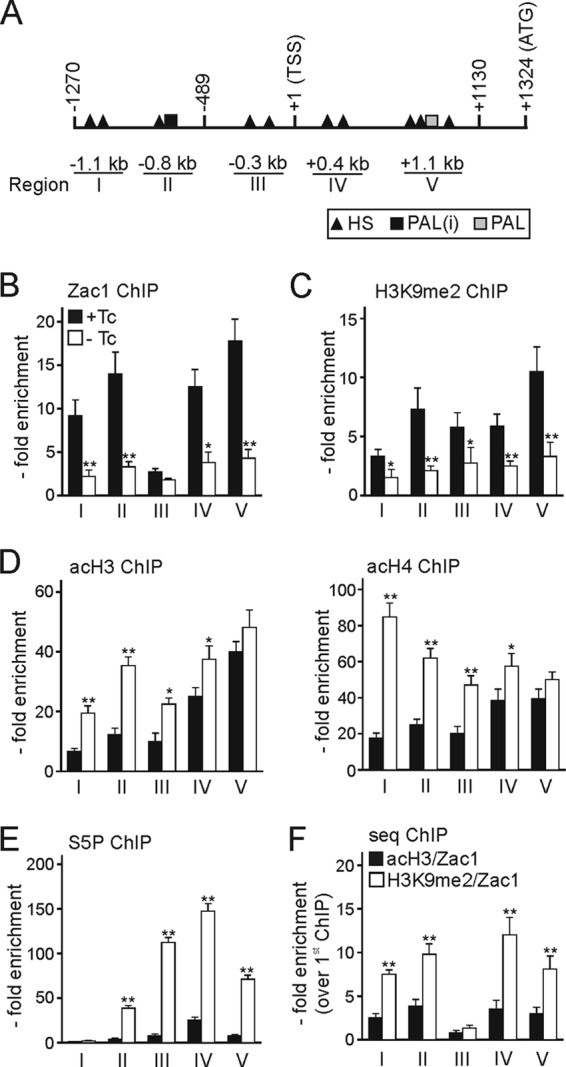
Zac1 binds to the Rasgrf1 promoter. (A) Scheme of the Rasgrf1 promoter. Abbreviations are the same as those for Fig. 4. Amplified ChIP regions are labeled I to V. (B to E) ChIP assays with R7T1 β cells grown in the presence (+Tc) or absence (−Tc) of tetracycline for 14 days using antibodies against Zac1 (B), H3K9me2 (C), acH3 and acH4 (D), and the active phosphoserine 5 form of RNA polymerase II (S5P) (E). (F) Zac1 associates with repressive chromatin marks at the Rasgrf1 promoter in predifferentiated R7T1 β cells. The first ChIP was done with an antibody against acH3 or against H3K9me2. Subsequently, the second ChIP was done with an antibody against Zac1. Zac1 constructs were transfected at 0.5 μg each. Data represent means ± SD from four independent experiments. *, P < 0.05; **, P < 0.01.
Consistently with a repressor function, Zac1 occupancy associated with the presence of the repressive chromatin mark H3K9me2 (dimethylated lysine 9 of histone 3) across the promoter (Fig. 5C). Conversely, active chromatin marks (panacetylated histone H3 and H4, respectively) strongly increased at the distal promoter region in the absence of Zac1 and concurred with the high occupancy of the active, serine 5-phosphorylated form of polymerase II (Fig. 5D and E).
To corroborate the role of Zac1 in the repression of Rasgrf1, we performed sequential ChIP experiments in predifferentiated R7T1 β cells by using in the primary ChIP antibodies against H3K9me2 or panacetyl-histone H3. Subsequently, the secondary ChIP was done with an anti-Zac1 antibody. In these experiments, Rasgrf1 promoter DNA was efficiently recovered from chromatin initially immunoprecipitated by antibodies against H3K9me2 but less so by antibodies against panacetyl-histone H3 (Fig. 5F).
Collectively, these results demonstrate that Zac1 preferentially associates with repressive marks at the Rasgrf1 gene that are compatible with its repressor function.
A cellular model for TNDM.
Considering the role of Rasgrf1 in controlling Ras's and Rac's impact on β-cell proliferation and insulin secretion (27, 28, 60), we hypothesized that repression due to biallelic Zac1 expression pertains to TNDM. To simulate biallelic Zac1 expression, we stably expressed Flag-tagged Zac1 in R7T1 β cells. Because single recombinant cell clones can misrepresent the properties of the parent cells, we pooled and amplified different transfected populations and determined transgene expression by qRT-PCR. One representative pool, designated R7-Z, was chosen for further analysis. The doubling of Zac1 expression halved Rasgrf1 expression, while insulin expression was unaltered (Fig. 6A). This increase in Zac1 expression reflected the presence of exogenous Zac1, as evidenced by RT-PCR and immunoblotting experiments (Fig. 6B).
Fig 6.
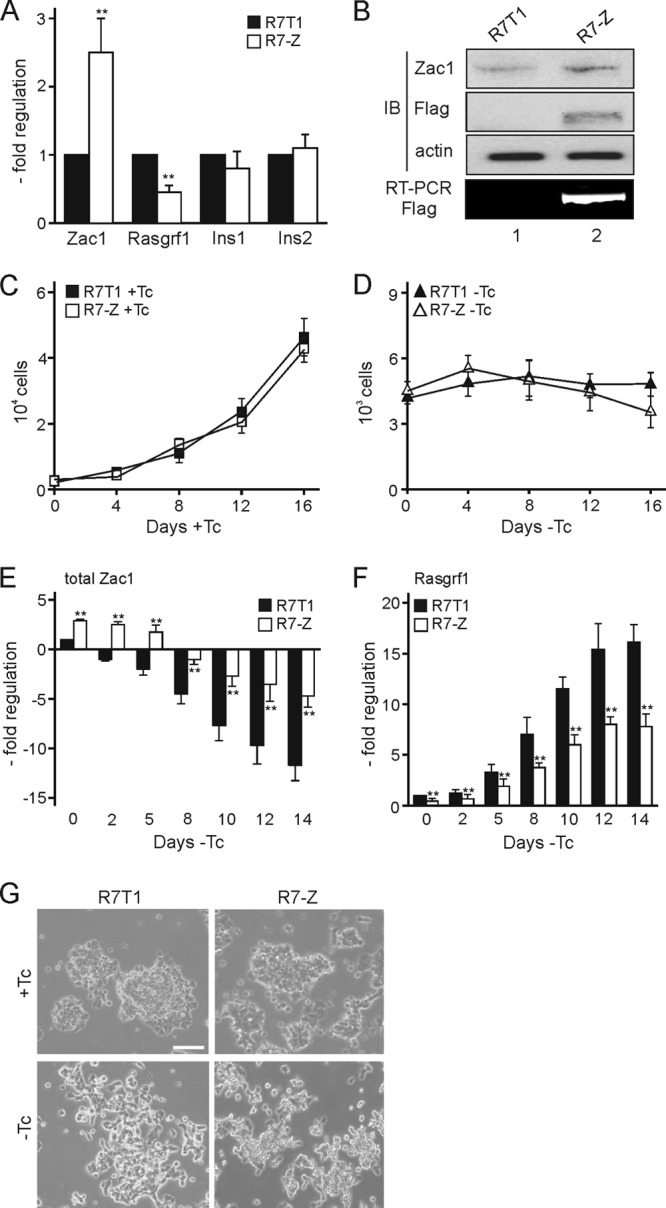
Zac1 overexpression reduces Rasgrf1 expression. (A) Zac1, Rasgrf1, and insulin (Ins1 and Ins2) expression was measured by qRT-PCR in predifferentiated R7T1 and R7-Z β cells. Data from R7T1 β cells were set to 1. (B) Immunoblot (IB) and RT-PCR analysis. WCE (75 μg) from predifferentiated R7T1 and R7-Z β cells were analyzed with Zac1 and Flag antibodies to detect endogenous and exogenous Zac1 proteins. The equal loading of cell extracts was verified by actin antibody. RT-PCR analysis was done with primers specific to Flag-tagged Zac1. (C and D) Maintained cell proliferation and survival of R7-Z β cells. Parent R7T1 and modified R7-Z β cells were grown in the presence (+Tc) (C) or absence (−Tc) (D) of tetracycline, and cell numbers were measured every fourth day. (E and F) Expression of total Zac1 (E) and Rasgrf1 (F) mRNA in R7T1 and R7-Z β cells was measured by qRT-PCR following the withdrawal of tetracycline for the indicated number of days. (G) Delayed differentiation of R7-Z β cells grown for 2 weeks in the absence of Tc. Representative bright-light microscopy; scale bar, 200 μm. Data represent means ± SD from four (A) or three (C to F) independent experiments. **, P < 0.01.
Compared to parent R7T1 β cells, doubling Zac1 expression did not influence proliferation or survival during 2 weeks in the presence or absence of tetracycline, as evidenced by cell count measurements (Fig. 6C and (D).
In the absence of tetracycline, endogenous Zac1 expression declined similarly in R7T1 and R7-Z β cells. Moreover, advanced differentiation led to progressive silencing of Zac1 transgenes (data not shown). Thus, the ratio between endogenous and exogenous Zac1 was largely maintained over time (Fig. 6E) and gave rise to a proportional decrease in Rasgrf1 expression (Fig. 6F).
Elevated levels of total Zac1 maintained cell proliferation despite decreased Rasgrf1 expression; however, they delayed slightly the flattening and separation of the densely packed cell clusters typical for proliferating R7T1 cells upon advanced differentiation (Fig. 6G).
Zac1 impairs glucose-dependent insulin secretion.
Glucose is the principal stimulus and regulator of β-cell function, also exerting a permissive action on the effectiveness of the vast majority of other modulators of insulin secretion. Here, we asked whether during advanced differentiation Rasgrf1 can integrate glucose-induced Ca2+ signaling by regulating the activity of Ras (27) and the downstream mediators mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K). These effectors coordinate a host of cellular functions, ranging from transcription to translation and from morphology to endo- and exocytosis (27, 28, 60).
To examine the consequences of Zac1 overexpression, we tested the activation of the MAPK and PI3K pathways in response to glucose-, insulin-, and potassium-induced depolarization. Following glucose stimulation, Erk1/2 phosphoimmunoreactivity (p-Erk1/2) peaked at 15 min and declined during 30 to 60 min (Fig. 7A). In contrast, additional amounts of Zac1 in R7T1 β cells largely prevented Erk1/2 activation. Consistently with these findings, the transient transfection of Zac1 into INS-1 and Min6 β cells caused comparable decreases in glucose-dependent Erk1/2 activation (see Fig. S3 in the supplemental materials).
Fig 7.

Zac1 inhibits regulated insulin secretion. (A to C) Immunoblots show Erk1/2 activation following treatment of parent and Zac1-overexpressing R7-Z β cells with 20 mM glucose (A), 100 mM insulin (B), or 50 mM potassium chloride (KCl) (C) for the indicated periods of time. WCE (25 μg) were analyzed by phosphospecific and total Erk1/2 antibodies. Graphs show quantified Erk1/2 phosphorylation normalized to total Erk1/2 (both bands were assessed) and the control (R7T1 at 0 min), which was set to 1. (D to F) Immunoblots showing Akt activation. R7T1 and R7-Z β cells were treated as indicated. WCE (100 μg) were analyzed by phosphospecific and total Akt antibodies. Graphs show quantified Akt phosphorylation normalized to total Akt and control values (R7T1 at 0 min). (A to F) R7T1 and R7-Z cells were grown in the absence of tetracycline for 5 days before stimulation. (G) Insulin secretion of R7T1 and R7-Z β cells grown for 5 days in the absence of Tc following 2 h of incubation with 20 mM glucose (Gluc) or 50 mM potassium chloride (KCl). Secreted insulin was normalized on intracellular insulin and expressed as a percentage of basal insulin secretion. (H) Reconstituted Rasgrf1 expression in R7-Z-R β cells. Total and HA-tagged Rasgrf1 expression was detected in predifferentiated R7T1 β cells with specific primer pairs. Total Rasgrf1 expression in R7T1 β cells was set to 1. (I) Reconstituting Rasgrf1 expression restores insulin secretion in R7-Z β cells. Insulin secretion following 2 h of incubation with 20 mM glucose (Gluc) or 50 mM potassium chloride (KCl) of R7-Z and R7-Z-R β cells grown for 5 days in the absence of Tc. Secreted insulin was normalized to intracellular insulin and expressed as a percentage of basal insulin secretion. Data represent means ± SD from four (A to F, H) or three (G and I) independent experiments. *, P < 0.05; **, P < 0.01.
A similar difference between R7T1 and R7-Z β cells, albeit at earlier time points, emerged for insulin- and depolarization-induced Erk1/2 phosphoimmunoreactivity (Fig. 7B and C).
The activation of the PI3K pathway in response to glucose, insulin, and depolarization was analyzed by the phosphorylation status of the downstream mediator Akt (p-Akt). In contrast to the parent cells, additional amounts of Zac1 largely abolished the increase in p-Akt immunoreactivity (Fig. 7D to F). In agreement, the transient transfection of Zac1 into INS-1 and Min6 β cells elicited similar decreases in glucose-dependent Akt activation (see Fig. S3 in the supplemental material).
Importantly, Zac1 overexpression strongly reduced the glucose- or depolarization-induced insulin secretion of β cells during advanced differentiation (Fig. 7G), suggesting that the repression of Rasgrf1 translates at least in part via diminished Erk1/2 and Akt activation into reduced insulin secretion.
Because Zac1 might regulate insulin secretion through additional targets unrelated to Rasgrf1, we asked whether normalizing Rasgrf1 expression reinstates insulin secretion in R7-Z β cells. The analysis of pooled cell populations was performed as described above, and one representative pool (R7-Z-R) containing largely restored total levels of Rasgrf1 mRNA was chosen for further experiments (Fig. 7H). In contrast to R7-Z cells, normalized levels of Rasgrf1 strongly increased p-Erk1/2 and p-Akt immunoreactivity in response to glucose (see Fig. S4 in the supplemental material) and largely reinstated glucose- and depolarization-induced insulin secretion (Fig. 7I). Together, these results strengthen a role for Rasgrf1 in Zac1-controlled insulin secretion in β cells.
Simulating biallelic Zac1 expression sustains hyperglycemia in diabetic mice.
We sought to investigate the effects from doubling Zac1 expression under conditions of continuous demand by transplantation studies in streptozotocin (STZ)-induced diabetic mice. The intraperitoneal administration of R7T1 β cells to hyperglycemic mice normalized the elevated blood glucose levels in accord with previous reports (40). In contrast, the transplantation of R7-Z β cells did not reinstate euglycemia across the entire study period and showed high glucose levels, similarly to STZ-treated, nontransplanted mice (Fig. 8A).
Fig 8.

Zac1 overexpression sustains hyperglycemia in transplanted diabetic mice. (A) Streptozotocin (STZ)-treated diabetic mice (10 weeks old) were intraperitoneally administered either parent or Zac1-overexpressing R7T1 β cells (n = 9 mice per condition). Fasting blood glucose levels were measured every fourth day using blood from the tip vein. The STZ/R7T1 group differed significantly from the STZ/R7-Z group as assessed by one-way ANOVA followed by Newman-Keuls post hoc test (P < 0.05). (B) Pancreatic insulin content from control groups or STZ-treated diabetic mice transplanted with R7T1 or R7-Z β cells was measured on day 40 after STZ treatment. (C) Glucagon-like peptide 1 or forskolin rescues insulin secretion. R7T1 and R7-Z β cells were grown in the absence of tetracycline for 5 days and subsequently treated with 20 mM glucose, 100 nM glucagon-like peptide 1 (GLP-1), or 10 μM forskolin (Forsk) in the presence and absence of glucose. (D and E) GLP-1 restores Erk1/2 and Akt activity in R7-Z cells. R7T1 and R7-Z cells were grown in the absence of tetracycline for 5 days before stimulation with 100 nM GLP-1 in the presence of 20 mM glucose for the indicated periods of time. (D) Quantification of Erk1/2 phosphorylation normalized to total Erk1/2 (both bands were assessed) and control (R7T1 at 0 min) set to 1. (E) Quantification of Akt phosphorylation normalized to total Akt and control (R7T1 at 0 min). (F) Liraglutide (Lira) rescues insulin secretion. R7T1 and R7-Z cells were grown in the absence of tetracycline for 5 days and subsequently treated with 20 mM glucose or increasing doses of liraglutide in the presence of glucose. (G) Liraglutide improves hyperglycemia. Experimental diabetic mice transplanted with R7T1 or R7-Z β cells were additionally administered liraglutide or vehicle (n = 9 mice per condition). Glucose was monitored as described above. The R7-Z/vehicle group differed significantly from the other three groups as assessed by one-way ANOVA followed by Newman-Keuls post hoc test (P < 0.05). (C to F) Data represent means ± SD from three independent experiments. *, P < 0.05; **, P < 0.01.
Pancreata from all groups were analyzed at the end of the experiment for total pancreatic insulin content, which was strongly and comparably reduced in all STZ-treated groups (Fig. 8B), arguing against differences in the recovery rates of endogenous β cells as a cause of altered insulin secretion among STZ-treated mice.
Zac1 spares GPCR-stimulated insulin secretion.
The insulinotropic effect of glucose is modulated by other factors that optimize insulin secretion, many of which act through the activation of G-protein-coupled receptors (GPCR) and subsequent increases in intracellular cyclic AMP (cAMP) (2).
Here, we chose the Gs-coupled glucagon-like peptide 1 receptor (GLP-1R) because of its well-known effect on Ras-independent activation of Erk1/2 (14) and insulin secretion.
While R7T1 and R7-Z β cells did not differ in the expression of GLP-1R (data not shown), GLP-1 at pharmacological doses strongly enhanced glucose-stimulated insulin secretion in both cell models to a similar degree under advanced differentiation (Fig. 8C). Moreover, treatment with forskolin, a direct activator of adenylate cyclase, strongly improved glucose-dependent insulin secretion in R7-Z cells (Fig. 8C).
Consistently with these findings, GLP-1, in the presence of glucose, increased Erk1/2 and Akt phosphorylation to a similar degree in both cell lines (Fig. 8D and E). Lastly, the clinically used GLP-1 analog liraglutide dose dependently increased glucose-stimulated insulin secretion in parent R7T1 and R7-Z cells undergoing advanced differentiation (Fig. 8F).
Therefore, we further studied the effects of liraglutide under conditions of sustained demand on β cells harboring elevated Zac1 levels. For this purpose, we transplanted experimental diabetic mice with R7T1 and R7-Z β cells and additionally administered them liraglutide or vehicle (Fig. 8G). While there were only minor differences between R7T1-transplanted vehicle and liraglutide-treated mice, liraglutide, but not vehicle, strongly reduced hyperglycemia in recipients of R7-Z β cells.
We measured across 2 weeks of liraglutide treatment, and no significant effect on body weight under any condition was found (data not shown).
In sum, these results show that Zac1 represses Rasgrf1-dependent, but not GPCR-dependent, insulin secretion under acute and continuous demand and raise the prospect of GLP-1 analogs in the treatment of TNDM.
DISCUSSION
In this study, we suggest that ZAC1-mediated repression of RASGRF1 contributes to insufficient perinatal insulin secretion in TNDM patients.
We detected prenatally high nuclear Zac1 immunoreactivity in insulin-positive β cells and, to a lower degree, in the exocrine pancreas. Postnatally, Zac1 expression declined rapidly, in accord with previous reports (11, 37). On the contrary, cytoplasmic Rasgrf1 was weakly expressed prenatally in insulin-positive and exocrine cells and strongly increased in insulin-positive cells within a few days following birth. Subsequently to this perinatal inversion in Zac1 and Rasgrf1 expression, we detected low nuclear Zac1 and high cytoplasmic Rasgf1 expression in adult β cells, a pattern well conserved in humans. Predifferentiated R7T1 β cells recapitulated this dynamic transition upon advanced differentiation with corresponding changes in Zac1 and Rasgrf1 immunoreactivities.
Zac1 expression in R7T1 β cells matches that from ex vivo neonate islets, and simulating biallelic expression interferes with advanced differentiation rather than antiproliferation. The effects of ZAC1 misexpression on pancreas development in mice have been reported to vary over time, whereby delayed embryonic growth contrasts with overcompensated postnatal increases in β-cell numbers (37). However, it should be noted that the effect on proliferation was never formally demonstrated, and the change in β-cell mass observed may have been caused by differentiation defects in Zac1-overexpressing mice.
Alternatively, doubling Zac1 expression might not interfere with R7T1 β-cell proliferation due to immortalization by the SV40 large T antigen, which inactivates the cell cycle regulators p53 and Rb. In fact, levels of enhanced Zac1 expression established in R7T1 β cells are far from those inducing cycle arrest and apoptosis following forced expression (7, 22, 50, 54).
Reporter assays and ChIP experiments revealed Zac1 binding to multiple half-site elements across the Rasgrf1 promoter which conferred transcriptional repression (16). Zac1 overexpression in R7T1 β cells inhibited Rasgrf1 expression and regulated p-Erk1/2 and p-Akt immunoreactivity and insulin secretion during advanced differentiation. Similar results were obtained for Zac1 transfection into unrelated INS-1 or Min6 β cells and ex vivo neonate islets, strengthening the relevance of these findings. It should be noted that a modulatory effect from positive- or negative-feedback actions of secreted insulin on β-cell function cannot be excluded presently (33).
Notwithstanding our findings, neonate islets from transgenic ZAC1 mice showed less impairment in stimulated insulin secretion (37). Although these mice were considered to simulate biallelic Zac1 expression due to similar levels of Zac1/ZAC1 mRNAs, the weak expression of ZAC1 protein compared to that of Zac1 questions this assumption (54). Moreover, differences in the extent of stimulation (for instance, 25 min versus 2 h in this study) could explain the differences in insulin secretion between the two studies.
Rasgrf1 is stimulated by Ca2+-calmodulin and serves as a bifunctional guanine nucleotide exchange factor (GEF) that catalyzes the activation of the small GTP-binding proteins Ras and Rac.
Ras is a central node in various signaling pathways; one of the best-characterized downstream effectors is the MAPK cascade, mainly Erk1/2, via activation of c-Raf or b-Raf (32, 58). Another well-known effector of Ras proteins is phosphoinositide 3-kinase (PI3K), which requires activation by Ras and Rap1 and drives the activation of Akt and the Rho family of small proteins (44, 45, 49). GTP-bound Ras can also turn on the GEF function of Tiam1, enabling the activation of Rac (31, 48, 59) with a regulatory role in glucose-stimulated insulin secretion (28, 34, 56). Since Rasgrf1 also has direct catalytic activity with Rac (20, 26), Zac1 effects on insulin secretion might operate through converging pathways. Compatibly with this view, Ras can stimulate RalGEF (25, 51, 57), which in turn activates Ral, a small G protein involved in different steps of insulin exocytosis (35, 36).
Our findings seem to conflict with an intact insulin response of ex vivo islets from adult Rasgrf1 null mice (12) which display a reduced body size, blood metabolomic profiles resembling calorie-restricted mice, increased life span, and Sirt1 expression (10). Sirt1 can promote glucose-dependent insulin secretion (9) and might have masked any deficits from impaired Rasgrf1 signaling in adult mice. Alternatively, the critical role of Rasgrf1 in insulin secretion might be confined to the perinatal time window, in accord with the original idea of an important early postnatal function (21) and a critical role in newborn TNDM patients.
In further support of our findings, previous reports have shown that β-cell-specific ablation of the insulin receptor (IR) or insulin-like growth factor 1 receptor (IGF-1R) abolishes glucose-stimulated insulin secretion, especially in the early phase (29, 30). Moreover, the ablation of the downstream effectors class IA PI3K and Akt revealed their role in insulin secretion at the level of the exocytosis machinery (6, 24).
Taken together, Zac1-dependent control of Rasgrf1 seems to translate through multiple routes in altered insulin secretion. Because ZAC1 is additionally expressed in various tissues participating in insulin and glucose homeostasis (i.e., liver, kidney, and hypothalamus), misexpression at these sites might further affect the complex phenotype of TNDM.
Clinical studies on TNDM agree that the transient manifestation of the disease probably reflects a permanent β-cell defect with variations during growth and development and a poor control of insulin release rather than an actual inability to produce insulin (47, 52). Our finding that simulating biallelic Zac1 expression causes Rasgrf1 repression but not insulin gene repression and impairs stimulated insulin secretion matches the latter assumption well.
Interestingly, relapsed TNDM patients show a subnormal insulin response to both the oral and intravenous glucose tolerance test, whereas glucagon-stimulated insulin secretion is maintained (52). These data indicate that in chromosome 6-associated TNDM, the β cell is preserved and able to secrete insulin through the stimulatory G-protein pathway while exhibiting a specific defect of insulin secretion after glucose stimulation. Compatibly with this concept, treatment with GLP-1 or the direct activation of adenylate cyclase reinstated regulated insulin secretion in R7T1 β cells, mimicking biallelic Zac1 expression. Moreover, treatment with liraglutide, a GLP-1R agonist, improved hyperglycemia in diabetic mice transplanted with Zac1-overexpressing β cells.
The activation of GLP-1R has been explored for many years as a potential therapy for type 2 diabetes (2) and may offer a promising target in neonate and relapsed adolescent TNDM patients.
Supplementary Material
ACKNOWLEDGMENTS
We are thankful to Alexandre Patchev, Albin Varga, Guillaume Daniel, and Udo Schmidt for help in animal experiments and discussions. INS-1, Min6, and R7T1 β cells were kindly provided by Claes Wollheim (University of Geneva, Geneva, Switzerland), Sabine Senkel (University Duisburg-Essen, Essen, Germany), Jun-ichi Miyazaki (Osaka University, Osaka, Japan), Annette Schuermann (German Institute of Human Nutrition, Nuthetal, Germany), and Norman Fleischer (Albert Einstein College of Medicine, New York, New York). The Rasgrf1 promoter and cDNA were kindly provided by Paul Soloway (Cornell University, Ithaca, New York) and Renata Zippel (University of Milan, Milan, Italy).
This study was funded by the Deutsche Forschungsgemeinschaft (SP 386/5-1) to D.S.
Footnotes
Published ahead of print 30 April 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Aguilar-Bryan L, Bryan J. 2008. Neonatal diabetes mellitus. Endocr. Rev. 29:265–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahrén B. 2009. Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat. Rev. Drug Discov. 8:369–385 [DOI] [PubMed] [Google Scholar]
- 3. Arima T, Drewell RA, Oshimura M, Wake N, Surani MA. 2000. A novel imprinted gene, HYMAI, is located within an imprinted domain on human chromosome 6 containing ZAC. Genomics 67:248–255 [DOI] [PubMed] [Google Scholar]
- 4. Asfari M, et al. 1992. Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology 130:167–178 [DOI] [PubMed] [Google Scholar]
- 5. Barz T, Hoffmann A, Panhuysen M, Spengler D. 2006. Peroxisome proliferator-activated receptor gamma is a Zac target gene mediating Zac antiproliferation. Cancer Res. 66:11975–11982 [DOI] [PubMed] [Google Scholar]
- 6. Bernal-Mizrachi E, et al. 2004. Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. J. Clin. Investig. 114:928–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bilanges B, et al. 2001. Alternative splicing of the imprinted candidate tumor suppressor gene ZAC regulates its antiproliferative and DNA binding activities. Oncogene 20:1246–1253 [DOI] [PubMed] [Google Scholar]
- 8. Bonal C, Herrera PL. 2008. Genes controlling pancreas ontogeny. Int. J. Dev. Biol. 52:823–835 [DOI] [PubMed] [Google Scholar]
- 9. Bordone L, Guarente L. 2007. Sirtuins and beta-cell function. Diabetes Obes. Metab. 9(Suppl. 2):23–27 [DOI] [PubMed] [Google Scholar]
- 10. Borrás C, et al. 2011. RasGrf1 deficiency delays aging in mice. Aging 3:262–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Du X, et al. 2011. Differential expression pattern of ZAC in developing mouse and human pancreas. J. Mol. Histol. 42:129–136 [DOI] [PubMed] [Google Scholar]
- 12. Font de Mora J, et al. 2003. Ras-GRF1 signaling is required for normal beta-cell development and glucose homeostasis. EMBO J. 22:3039–3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gardner RJ, et al. 2000. An imprinted locus associated with transient neonatal diabetes mellitus. Hum. Mol. Genet. 9:589–596 [DOI] [PubMed] [Google Scholar]
- 14. Gomez E, Pritchard C, Herbert TP. 2002. cAMP-dependent protein kinase and Ca2+ influx through L-type voltage-gated calcium channels mediate Raf-independent activation of extracellular regulated kinase in response to glucagon-like peptide-1 in pancreatic beta-cells. J. Biol. Chem. 277:48146–48151 [DOI] [PubMed] [Google Scholar]
- 15. Hoffmann A, Barz T, Spengler D. 2006. Multitasking C2H2 zinc fingers link Zac DNA binding to coordinated regulation of p300-histone acetyltransferase activity. Mol. Cell. Biol. 26:5544–5557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hoffmann A, et al. 2003. Transcriptional activities of the zinc finger protein Zac are differentially controlled by DNA binding. Mol. Cell. Biol. 23:988–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hoffmann A, Spengler D. 2008. A new coactivator function for Zac1's C2H2 zinc finger DNA-binding domain in selectively controlling PCAF activity. Mol. Cell. Biol. 28:6078–6093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang SM, Schönthal AH, Stallcup MR. 2001. Enhancement of p53-dependent gene activation by the transcriptional coactivator Zac1. Oncogene 20:2134–2143 [DOI] [PubMed] [Google Scholar]
- 19. Huang SM, Stallcup MR. 2000. Mouse Zac1, a transcriptional coactivator and repressor for nuclear receptors. Mol. Cell. Biol. 20:1855–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Innocenti M, Zippel R, Brambilla R, Sturani E. 1999. CDC25(Mm)/Ras-GRF1 regulates both Ras and Rac signaling pathways. FEBS Lett. 460:357–362 [DOI] [PubMed] [Google Scholar]
- 21. Itier JM, et al. 1998. Imprinted gene in postnatal growth role. Nature 393:125–126 [DOI] [PubMed] [Google Scholar]
- 22. Kamikihara T, et al. 2005. Epigenetic silencing of the imprinted gene ZAC by DNA methylation is an early event in the progression of human ovarian cancer. Int. J. Cancer 115:690–700 [DOI] [PubMed] [Google Scholar]
- 23. Kamiya M, et al. 2000. The cell cycle control gene ZAC/PLAGL1 is imprinted–a strong candidate gene for transient neonatal diabetes. Hum. Mol. Genet. 9:453–460 [DOI] [PubMed] [Google Scholar]
- 24. Kaneko K, et al. 2010. Class IA phosphatidylinositol 3-kinase in pancreatic β cells controls insulin secretion by multiple mechanisms. Cell Metab. 12:619–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kikuchi A, Williams LT. 1996. Regulation of interaction of ras p21 with RalGDS and Raf-1 by cyclic AMP-dependent protein kinase. J. Biol. Chem. 271:588–594 [DOI] [PubMed] [Google Scholar]
- 26. Kiyono M, Satoh T, Kaziro Y. 1999. G protein beta gamma subunit-dependent Rac-guanine nucleotide exchange activity of Ras-GRF1/CDC25(Mm). Proc. Natl. Acad. Sci. U. S. A. 96:4826–4831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kowluru A. 2010. Small G proteins in islet beta-cell function. Endocr. Rev. 31:52–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kowluru A. 2011. Friendly, and not so friendly, roles of Rac1 in islet β-cell function: lessons learnt from pharmacological and molecular biological approaches. Biochem. Pharmacol. 81:965–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kulkarni RN, et al. 1999. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 96:329–339 [DOI] [PubMed] [Google Scholar]
- 30. Kulkarni RN, et al. 2002. Beta-cell-specific deletion of the Igf1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter beta-cell mass. Nat. Genet. 31:111–115 [DOI] [PubMed] [Google Scholar]
- 31. Lambert JM, et al. 2002. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat. Cell Biol. 4:621–625 [DOI] [PubMed] [Google Scholar]
- 32. Lange-Carter CA, Johnson GL. 1994. Ras-dependent growth factor regulation of MEK kinase in PC12 cells. Science 265:1458–1461 [DOI] [PubMed] [Google Scholar]
- 33. Leibiger IB, Leibiger B, Berggren P-O. 2008. Insulin signaling in the pancreatic beta-cell. Annu. Rev. Nutr. 28:233–251 [DOI] [PubMed] [Google Scholar]
- 34. Li J, Luo R, Kowluru A, Li G. 2004. Novel regulation by Rac1 of glucose- and forskolin-induced insulin secretion in INS-1 beta-cells. Am. J. Physiol. Endocrinol. Metab. 286:E818–E827 [DOI] [PubMed] [Google Scholar]
- 35. Ljubicic S, Bezzi P, Vitale N, Regazzi R. 2009. The GTPase RalA regulates different steps of the secretory process in pancreatic beta-cells. PLoS One 4:e7770 doi:10.1371/journal.pone.0007770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lopez JA, et al. 2008. The RalA GTPase is a central regulator of insulin exocytosis from pancreatic islet beta cells. J. Biol. Chem. 283:17939–17945 [DOI] [PubMed] [Google Scholar]
- 37. Ma D, et al. 2004. Impaired glucose homeostasis in transgenic mice expressing the human transient neonatal diabetes mellitus locus, TNDM. J. Clin. Investig. 114:339–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mackay DJG, et al. 2002. Relaxation of imprinted expression of ZAC and HYMAI in a patient with transient neonatal diabetes mellitus. Hum. Genet. 110:139–144 [DOI] [PubMed] [Google Scholar]
- 39. Merglen A, et al. 2004. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology 145:667–678 [DOI] [PubMed] [Google Scholar]
- 40. Milo-Landesman D, et al. 2001. Correction of hyperglycemia in diabetic mice transplanted with reversibly immortalized pancreatic beta cells controlled by the tet-on regulatory system. Cell Transplant. 10:645–650 [PubMed] [Google Scholar]
- 41. Miyazaki J, et al. 1990. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 127:126–132 [DOI] [PubMed] [Google Scholar]
- 42. Murgatroyd C, Hoffmann A, Spengler D. 2012. In vivo ChIP for the analysis of microdissected tissue samples. Methods Mol. Biol. 809:135–148 [DOI] [PubMed] [Google Scholar]
- 43. Murgatroyd C, et al. 2009. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat. Neurosci. 12:1559–1566 [DOI] [PubMed] [Google Scholar]
- 44. Rodriguez-Viciana P, et al. 1994. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 370:527–532 [DOI] [PubMed] [Google Scholar]
- 45. Rodriguez-Viciana P, Warne PH, Vanhaesebroeck B, Waterfield MD, Downward J. 1996. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J. 15:2442–2451 [PMC free article] [PubMed] [Google Scholar]
- 46. Salvalaggio PRO, et al. 2002. Islet filtration: a simple and rapid new purification procedure that avoids Ficoll and improves islet mass and function. Transplantation 74:877–879 [DOI] [PubMed] [Google Scholar]
- 47. Shield JPH, et al. 2004. An assessment of pancreatic endocrine function and insulin sensitivity in patients with transient neonatal diabetes in remission. Arch. Dis. Child. Fetal Neonatal 89:F341–F343 doi:10.1136/adc.2003.030502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shirazi Fard S, Kele J, Vilar M, Paratcha G, Ledda F. 2010. Tiam1 as a signaling mediator of nerve growth factor-dependent neurite outgrowth. PLoS One 5:e9647 doi:10.1371/journal.pone.0009647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sjölander A, Yamamoto K, Huber BE, Lapetina EG. 1991. Association of p21ras with phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. U. S. A. 88:7908–7912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Spengler D, et al. 1997. Regulation of apoptosis and cell cycle arrest by Zac1, a novel zinc finger protein expressed in the pituitary gland and the brain. EMBO J. 16:2814–2825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Urano T, Emkey R, Feig LA. 1996. Ral-GTPases mediate a distinct downstream signaling pathway from Ras that facilitates cellular transformation. EMBO J. 15:810–816 [PMC free article] [PubMed] [Google Scholar]
- 52. Valerio G, et al. 2004. Beta-cell dysfunction in classic transient neonatal diabetes is characterized by impaired insulin response to glucose but normal response to glucagon. Diabetes Care 27:2405–2408 [DOI] [PubMed] [Google Scholar]
- 53. Varrault A, et al. 2001. Characterization of the methylation-sensitive promoter of the imprinted ZAC gene supports its role in transient neonatal diabetes mellitus. J. Biol. Chem. 276:18653–18656 [DOI] [PubMed] [Google Scholar]
- 54. Varrault A, et al. 1998. hZAC encodes a zinc finger protein with antiproliferative properties and maps to a chromosomal region frequently lost in cancer. Proc. Natl. Acad. Sci. U. S. A. 95:8835–8840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Varrault A, et al. 2006. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev. Cell 11:711–722 [DOI] [PubMed] [Google Scholar]
- 56. Veluthakal R, Madathilparambil SV, McDonald P, Olson LK, Kowluru A. 2009. Regulatory roles for Tiam1, a guanine nucleotide exchange factor for Rac1, in glucose-stimulated insulin secretion in pancreatic beta-cells. Biochem. Pharmacol. 77:101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. White MA, Vale T, Camonis JH, Schaefer E, Wigler MH. 1996. A role for the Ral guanine nucleotide dissociation stimulator in mediating Ras-induced transformation. J. Biol. Chem. 271:16439–16442 [DOI] [PubMed] [Google Scholar]
- 58. Wood KW, Sarnecki C, Roberts TM, Blenis J. 1992. ras mediates nerve growth factor receptor modulation of three signal-transducing protein kinases: MAP kinase, Raf-1, and RSK. Cell 68:1041–1050 [DOI] [PubMed] [Google Scholar]
- 59. Yamauchi J, Miyamoto Y, Tanoue A, Shooter EM, Chan JR. 2005. Ras activation of a Rac1 exchange factor, Tiam1, mediates neurotrophin-3-induced Schwann cell migration. Proc. Natl. Acad. Sci. U. S. A. 102:14889–14894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ye X, Carew TJ. 2010. Small G protein signaling in neuronal plasticity and memory formation: the specific role of ras family proteins. Neuron 68:340–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yuasa S, et al. 2010. Zac1 is an essential transcription factor for cardiac morphogenesis. Circ. Res. 106:1083–1091 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.