

Abstract
Constitutive NF-κB activation by proinflammatory cytokines plays a major role in cancer progression. However, the underlying mechanism is still unclear. We report here that histone methyltransferase NSD2 (also known as MMSET or WHSC1), a target of bromodomain protein ANCCA/ATAD2, acts as a strong coactivator of NF-κB by directly interacting with NF-κB for activation of target genes, including those for interleukin-6 (IL-6), IL-8, vascular endothelial growth factor A (VEGFA), cyclin D, Bcl-2, and survivin, in castration-resistant prostate cancer (CRPC) cells. NSD2 is recruited to the target gene promoters upon induction and mediates NF-κB activation-associated elevation of histone H3K36me2 and H3K36me3 marks at the promoter, which involves its methylase activity. Interestingly, we found that NSD2 is also critical for cytokine-induced recruitment of NF-κB and acetyltransferase p300 and histone hyperacetylation. Importantly, NSD2 is overexpressed in prostate cancer tumors, and its overexpression correlates with NF-κB activation. Furthermore, NSD2 expression is strongly induced by tumor necrosis factor alpha (TNF-α) and IL-6 via NF-κB and plays a crucial role in tumor growth. These results identify NSD2 to be a key chromatin regulator of NF-κB and mediator of the cytokine autocrine loop for constitutive NF-κB activation and emphasize the important roles played by NSD2 in cancer cell proliferation and survival and tumor growth.
INTRODUCTION
Progression to an advanced stage of malignancy involves hyperactivated or constitutive signaling pathways. One of them is mediated by the NF-κB transcription factor (5, 37). Activated NF-κB is found to play important roles in cancer cell proliferation, survival, invasion, and chemoresistance as well as tumor angiogenesis. NF-κB, with RelA/p65 and NF-κB1/p50 being the prototypical heterodimers, can be activated by multiple stimuli in tumors, including proinflammatory cytokines and cellular stress signals. The canonical NF-κB activation mechanism involves phosphorylation of IκB proteins by the IκB kinases, dissociation of NF-κB from IκB in the cytoplasm, and its subsequent translocation to the nucleus. In cancer cells, NF-κB can activate the transcription of a slew of genes crucial for proliferation (e.g., the genes for cyclin D1 and c-Myc), antiapoptosis (e.g., the genes for Bcl-2 and Birc5/survivin), proangiogenesis (e.g., the genes for interleukin-8 [IL-8] and vascular endothelial growth factors [VEGFs]), and proinflammation (e.g., the genes for IL-1, IL-6, and tumor necrosis factor alpha [TNF-α]). However, how NF-κB is persistently activated to stimulate gene expression is not well understood.
The molecular mechanisms underlying prostate cancer progression to castration therapy-resistant prostate cancer (CRPC) are still unclear, which hampers the development of rationale-based therapeutics for the advanced disease. Recent studies suggest that multiple mechanisms, including the aberrant function of androgen receptor (AR) and other transcriptional regulators, likely play important roles in the progression (18, 33, 34). Overexpression and/or hyperactivation of NF-κB is found in prostate cancer and correlates with tumor stages and metastasis (21, 45, 50, 60). CRPC cells often display constitutive NF-κB activation and impaired response to proapoptotic stimuli. Activated NF-κB may directly regulate multiple gene programs that are critical for prostate cancer progression (6, 9, 14, 29, 30, 55, 60).
Activation of target gene transcription by NF-κB likely involves layers of regulatory mechanisms (47). The well-characterized ones include NF-κB association with p300/CBP, which not only mediates histone hyperacetylation at NF-κB target gene promoters but also modulates NF-κB DNA binding activity or its association with other proteins such as bromodomain protein Brd4 and IκBα, through acetylation of specific lysines of NF-κB subunits (12, 38). NF-κB can also interact with the nuclear receptor coactivators (46, 54). Its transcriptional activity can also be modulated by other posttranslational modifications, including phosphorylation and methylation at specific residues of NF-κB proteins and by the local chromatin structure (7, 31, 35, 57, 61). Emerging evidence primarily from lipopolysaccharide (LPS)-stimulated immune cells indicates that NF-κB activation of inflammatory genes involves dynamic changes in histone methylations on H3K4, H3K9, and H3K27, likely to remove the repressive chromatin barrier (2, 10, 11, 48). However, how NF-κB deregulates gene expression in cancer cells is much less understood.
Recent studies indicate that epigenetic regulators with histone-modifying or -demodifying activity or chromatin-remodeling function (e.g., SRC/p160 nuclear receptor coactivators, subunits of the SWI/SNF complex, and p300) are overexpressed or amplified in subsets of prostate cancer tumors (1, 62). The overexpressed coactivators can promote androgen deprivation-resistant cell proliferation and survival. High levels of a histone lysine methyltransferase (EZH2) and histone demethylases LSD1 and JMJD2C are detected in subsets of prostate cancers and play important roles in prostate cancer progression (49, 51, 59). We and others recently identified a chromatin coregulator, ANCCA (AAA+ nuclear coregulator cancer associated [also known as ATAD2]), endowed with a bromodomain and found that ANCCA is overexpressed in multiple types of cancers, including prostate cancer tumors with high Gleason scores (3, 8, 15, 40, 63, 64). Our further study indicated that ANCCA is a novel coactivator of AR and mediates expression of a specific group of AR target genes, such as those for IGF1R, IRS2, SGK1, and survivin/Birc5, for proliferation and survival of prostate cancer cells (63).
NSD2 (nuclear receptor-binding SET domain protein 2) is a member of a SET histone methyltransferase family that also includes NSD1 and NSD3. In a subtype of multiple myeloma (MM), the NSD2 gene is fused to the IgH locus via t(4;14) translocation, and its overexpression is likely responsible for the tumorigenic expansion of MM cells (19, 27). NSD2 has been shown to mediate di- and trimethylation of H3K36 and dimethylation of H4K20 in different systems and has been characterized as a transcriptional repressor interacting with histone deacetylase HDAC1 and histone demethylase LSD1 (17, 27, 28, 32, 36). Recent studies found that the NSD2 protein is highly overexpressed in several types of human cancers, including neuroblastoma, carcinomas of stomach and colon, small-cell lung cancers, and bladder cancers, and that its overexpression tends to associate with tumor aggressiveness (13). However, whether NSD2 is deregulated in other types of cancer and how the aberrant NSD2 plays a role in tumorigenesis have not been explored. In our further study of ANCCA function in prostate cancer, we found that ANCCA controls NSD2 expression and that the NSD2 protein is overexpressed in a subset of prostate cancer tumors with high Gleason scores. More importantly, we found that NSD2 serves as a key mediator of constitutive activation of NF-κB and an important component of an autocrine signaling loop by the proinflammatory cytokines to promote cancer cell proliferation and survival.
MATERIALS AND METHODS
Cell culture, siRNA transfection, lentivirus vectors, and assays for gene expression and apoptosis.
PC-3, CWR22Rv1, and LNCaP cells were obtained from the American Type Culture Collection (ATCC) and cultured in RPMI 1640 (Invitrogen) with 10% fetal bovine serum (FBS). C4-2B cells derived from LNCaP cells were obtained from UroCoR and cultured in T medium with 5% fetal bovine serum. For small interfering RNA (siRNA) transfection, cells were seeded at a density of 2 × 105 cells per well in 6-well plates and transfected with synthetic siRNAs (Dharmacon) targeting the NSD2 sequence, siNSD2-JL (5′-GCACGCTACAACACCAAGTTT-3′) (19) and siNSD2 (5′-AGGGATCGGAAGAGTCTTCAA-3′) (23); a control sequence targeting the luciferase (Luc) gene, siControl (5′-CTTACGCTGAGTACTTCGA-3′); or siRNAs targeting the ANCCA sequence, siANCCA#6 (5′-GCTACTGTTTACTATCAGGCT-3′) and siANCCA#7 (5′-CAAGCTGCTAAGCCTCCTATA-3′), using Lipofectamine 2000 (Invitrogen) at a 100 nM concentration. Cells and culture supernatants were then harvested at the indicated times for the different assays. For lentivirus vectors, full-length human NSD2 cDNA was inserted into pLenti4 (Invitrogen) with modifications to allow a Flag tag at the N terminus. Viruses were produced by transfecting 293T cells with packaging plasmids psPAX2 and pMD2.G and purified by ultracentrifugation on a 20% sucrose cushion. A mutant form of NSD2 was generated by using a site-directed mutagenesis kit (Stratagene). TNF-α was from Peprotech, and IL-6 was from R&D Systems.
Secreted IL-6, IL-8, and VEGFA proteins in the culture medium were measured using an IL-6 or IL-8 enzyme-linked immunosorbent assay (ELISA) kit (eBioscience) or a VEGF ELISA kit (Abcam). For quantitative reverse transcription-PCR (qRT-PCR), total RNA was isolated with TRIzol reagent (Invitrogen). The cDNA was prepared using 3 μg of total RNA. cDNA sequences were amplified in the presence of SYBR green fluorophore and detected using an iCycler real-time PCR instrument (Bio-Rad). The primers for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were included in each experiment as a control, and its threshold cycle (CT) value was used for normalization. After each elongation step, fluorescent values were collected and a melting curve analysis was performed at the end of the PCR. Fold difference was calculated as described previously (24). The PCR primers and antibodies used for Western blotting are listed in the supplemental material. Apoptotic cell death was measured using a cell death detection ELISA kit (Roche) by measuring DNA fragmentation following the manufacturer's protocol, and the results were normalized by the cell numbers. Reporter gene assays were performed by transfecting LNCaP cells with a reporter plasmid containing five NF-κB binding sequences (5× NF-κB–Luc), and expression plasmids pcDHCMV-HA-Flag-NSD2, pEBB-HA-p50, and pEBB-HA-p65/RelA (53). A pCMV β-galactosidase expression plasmid (pCMV–β-Gal) was included in each transfection for normalization. Twenty-four hours after the transfection, cells were stimulated with TNF-α overnight and harvested 24 h later for luciferase and β-Gal assays.
ChIP and re-ChIP assays.
Chromatin immunoprecipitation (ChIP) was performed essentially as described previously (63, 64) with the following modifications. The crude chromatin solutions of C4-2B and PC-3 cells were first cleared with protein A beads (Invitrogen) that had been precoated with preimmune serum for 2 h at 4°C. Then, the precleared chromatin solutions were incubated at 4°C overnight with the other antibodies listed in the supplemental material, prior to precipitation with protein A beads that had been preblocked with bovine serum albumin and sonicated salmon sperm DNA. For re-ChIP, the immunoprecipitated complexes from the first ChIP were eluted with 20 mM dithiothreitol at 37°C for 30 min with brief vortexing, diluted with the ChIP buffer 50 times, and cleared by centrifugation, before they were incubated with antibodies for secondary ChIP overnight at 4°C. The immunoprecipitated DNA was analyzed by real-time PCR with SYBR green on an iCycler instrument. Enrichment of genomic DNA was presented as the percent recovery relative to the input. The primers are listed in the supplemental material.
Co-IP and GST pulldown assay.
Nuclear extracts of PC-3 cells were prepared and diluted for coimmunoprecipitation (co-IP) with p65/RelA rabbit antibody or normal rabbit IgG as a control as previously described (63). NSD2 associated with NF-κB was detected by Western blotting with anti-NSD2 monoclonal antibody (Abcam). Alternatively, HEK293T cells were cotransfected with plasmids for expression of Flag-NSD2, NF-κB1/p50, p65/RelA, or a vector control, and whole-cell extracts were used for co-IP. For detection of a direct association between NSD2 and the NF-κB subunit p50 or RelA/p65, HEK293T cells were transfected with plasmid pEBG-p50 or pEBG-p65 (a gift from Colin Duckett) for expression of glutathione S-transferase (GST)–p50 or GST-p65 fusion protein, and the transfected cells were treated with 10 ng/ml of TNF-α (BD Pharmingen) for 12 h prior to the lysis. The GST-p50 or GST-p65 fusion protein was purified with glutathione-agarose beads. The immobilized GST-p50 or GST-p65 was then incubated with purified Flag-NSD2 protein for the pulldown assay as described previously (63).
Xenograft tumor models.
PC-3 or CWR22Rv1 cells were infected with adenocarcinoma short hairpin RNA (adeno-shRNA)-NSD2 or adeno-shRNA control at multiplicities of infection of 50 and 20, respectively, as described previously (65). Cells were detached 48 h later, suspended with RPMI 1640–10% FBS, and mixed with an equal volume of prethawed Matrigel matrix (BD). The mixture (200 μl, 2 × 106 cells) was injected subcutaneously into the dorsal flanks of 6-week-old male athymic BALB/c nude mice (Harlan Laboratories), which were randomly segregated into two groups (8 mice per group) before injection. Tumors were measured using calipers, and tumor volume was calculated as previously described (65). Tumors were dissected and subjected to Western blotting.
IHC, patient tumor specimens, and statistics analysis.
Immunohistochemistry (IHC) was performed as previously described (15, 63). Tissue section slides were incubated with anti-NSD2 monoclonal antibody (29D1; Abcam) or anti-p65 antibody (sc-8008; Santa Cruz) at 1:50 and 1:100 dilutions, respectively, overnight at 4°C, followed by incubations with biotinylated secondary antibody and the ABC reagents in the Vectastain Elite kit. After rinsing, the contents of the slides were visualized by staining with diaminobenzidine chromogen solution, followed by counterstaining with hematoxylin. NSD2 IHC was performed on tissue microarrays (TMAs) containing specimens from 123 cases of informative prostate cancer or hyperplasia collected at the UC Davis Cancer Center. IHC analysis of the NSD2 and p65 correlation was performed on adjacent sections of a TMA containing specimens from 111 cases of informative prostate adenocarcinoma from U.S. Biomax, Inc. The tissue section was scored negative if <1% of the prostate epithelial cells (PrECs) displayed any staining and scored positive if >5% of the cells displayed staining in the nucleus with moderate to high intensity. Differences and correlations in immunostaining among groups were analyzed with the χ2 test. Other statistical analysis was performed as previously described (15).
RESULTS
NSD2, a target of ANCCA, is overexpressed in prostate cancer.
We recently demonstrated that ANCCA promotes cancer cell proliferation and survival through upregulation of oncogenic gene expression, including expression of histone methyltransferase gene EZH2 (15). Our further study showed that expression of histone methyltransferase gene NSD2 is strongly inhibited at the mRNA and protein levels in androgen-sensitive LNCaP prostate cancer cells by siRNA-mediated ANCCA silencing (Fig. 1A and B). Similar results were obtained in C4-2B CRPC cells (data not shown). Furthermore, ectopic ANCCA expression in LNCaP cells stimulated NSD2 expression (see Fig. S1 in the supplemental material). These data indicate that, like EZH2, NSD2 is also a downstream target of ANCCA. We thus examined the expression of NSD2 in a panel of normal and cancerous prostate epithelial cells and found that CRPC cells (C4-2B, CWR22Rv1, and PC-3) express much higher levels of NSD2 protein than the normal PrECs, the nonmalignant prostate epithelial cell line RWPE-1, or the androgen-dependent prostate cancer cell line LNCaP (Fig. 1C). Analysis of a microarray data set from a previous study (49) demonstrated that ANCCA and NSD2 expression was markedly elevated in metastatic prostate cancers compared to primary, localized tumors (4.29- and 3.38-fold, respectively) and that their overexpression was directly correlated (Pearson correlation coefficient r = 0.9160) (see Fig. S1 in the supplemental material). We thus examined whether the NSD2 protein is overexpressed in human prostate tumors by IHC analysis of prostate tissue specimens on two TMAs (specimens from a total of 234 cases, with 201 being tumors) and found that while normal or hyperplastic tissue displayed no or occasionally positive nuclear staining by the anti-NSD2 antibody, over 40% of tumors displayed NSD2 protein overexpression with nuclear staining in tumor epithelial cells. When tumors with low or high Gleason scores were compared, a significant correlation (P = 0.0002) between NSD2 protein overexpression and a high Gleason score was observed (Fig. 1D; see Fig. S2 in the supplemental material). Since tumors with high Gleason scores are generally more aggressive and have a worse prognosis, the above-described results suggest that NSD2 overexpression is associated with more advanced prostate cancer.
Fig 1.
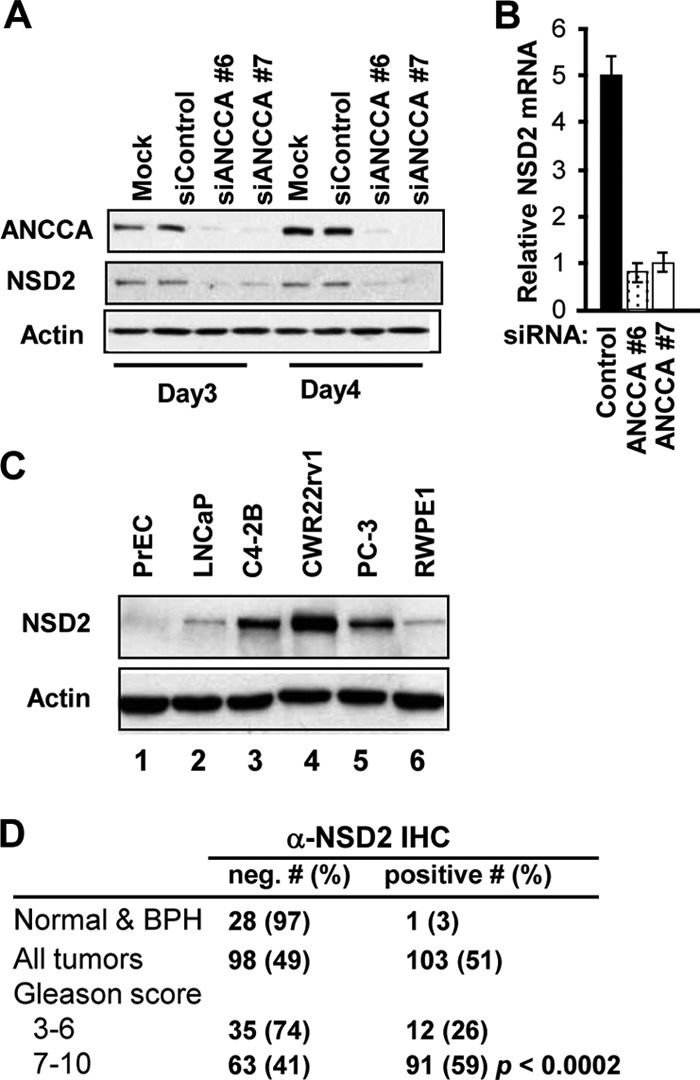
Expression of NSD2, a target of ANCCA, is highly elevated in prostate cancer cells and correlates with tumor Gleason scores. (A and B) Depletion of ANCCA with two siRNAs against different regions of ANCCA mRNA strongly inhibited NSD2 protein (A) and mRNA (B) expression in LNCaP cells. Relative transcript levels were obtained by normalization of expression units for NSD2 with those for GAPDH. (C) Western blot analysis of NSD2 expression in normal and cancerous prostate epithelial cells. (D) IHC analysis of NSD2 expression in normal, hyperplasia, or prostate cancer tissue with the indicated Gleason scores. Human prostate tissue on TMAs was subject to anti-NSD2 IHC analysis as described in Materials and Methods. BPH, benign prostatic hyperplasia. Differences and correlation in the immunostaining among groups were analyzed with the χ2 test.
NSD2 is required for proliferation and survival of CRPC cells and for tumor growth.
Given its high levels of expression in CRPC cells, we first examined the role of NSD2 in prostate cancer cell proliferation and survival. C4-2B, PC-3, and CWR22Rv1 cells were transfected with siRNAs targeting NSD2 or luciferase as a control. Cell numbers were counted at different days after the transfection. As shown in Fig. 2A and Fig. S3 and S4 in the supplemental material, NSD2-specific siRNA-mediated suppression of NSD2 (both the full-length NSD2, also known as MMSET II, and its isoform, MMSET I) dramatically inhibited proliferation of all three prostate cancer cell lines. To determine whether the decrease in cell growth was partly due to apoptotic cell death, cells were analyzed for DNA fragmentation, which was measured by a cell death detection ELISA. Indeed, NSD2 depletion resulted in a marked increase in cell death (>3-fold) in all three cell lines examined (Fig. 2B; see Fig. S3 and S4 in the supplemental material). Knockdown of NSD2 by a different siRNA yielded similar effects on cell proliferation and survival (see Fig. S4 in the supplemental material). To determine whether NSD2 plays a role in tumorigenesis, we suppressed NSD2 expression using adeno-shRNA-mediated silencing, injected the treated CWR22Rv1 or PC-3 cells into athymic male mice, and monitored the tumor growth rate. As expected, silencing of NSD2 strongly inhibited the tumor growth of PC-3 cells. Suppression of NSD2 in CWR22Rv1 cells also effectively blocked the tumor growth (Fig. 2C). Together, these data indicate that high levels of NSD2 are required for the proliferation and survival of CRPC cells and for tumor growth of prostate cancer cells.
Fig 2.
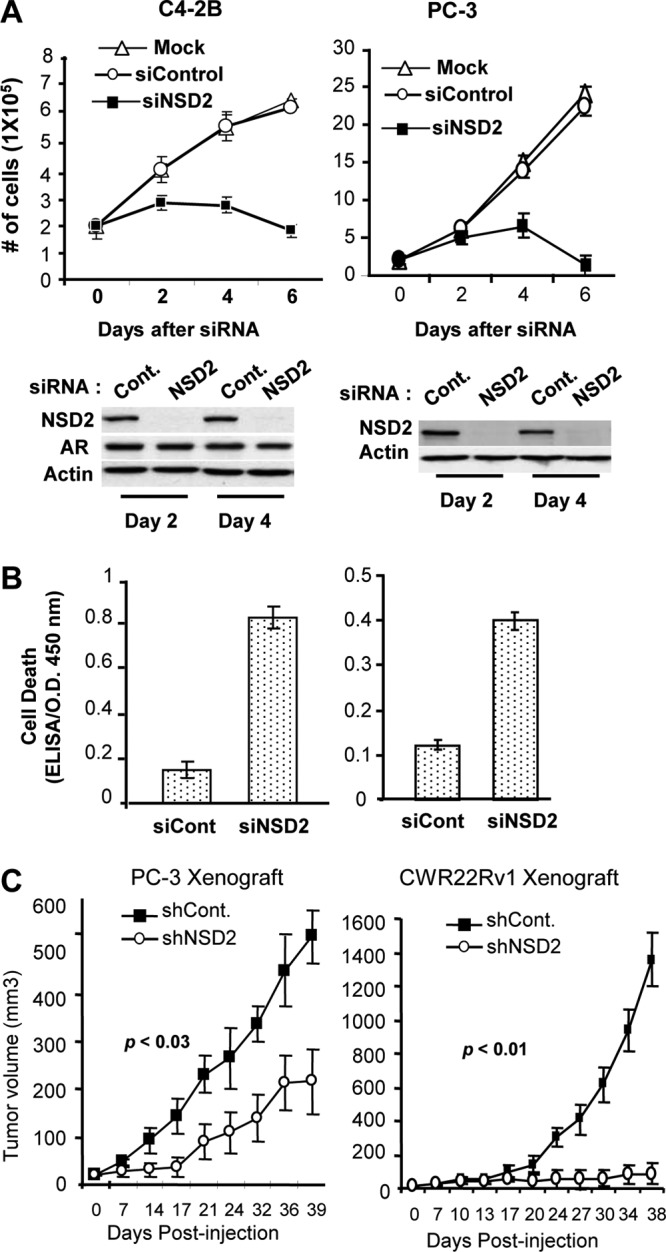
NSD2 is required for proliferation and survival of androgen-dependent and CRPC cells and for tumor growth. (A) Cells transfected with siRNA targeting NSD2 or control sequences were harvested for enumeration (top) or Western blot analysis (bottom) at the indicated times. (B) Three days after the transfection, cells were lysed and examined for apoptotic cell death by measuring DNA fragmentation using a cell death detection ELISA kit. O.D., optical density. (C) Adeno-shRNA-NSD2- or control-treated PC-3 or CWR22Rv1 cells were injected subcutaneously into the dorsal flanks of athymic nude mice (8 mice/group). Tumors were measured at the indicated times using calipers, and tumor volume was calculated as width × length × height × 0.5286.
High levels of NSD2 mediate expression of genes critical for cell proliferation and survival.
To examine the mechanisms of NSD2 function in prostate cancer, we assessed the effects of NSD2 knockdown on expression of genes that are well-known to play crucial roles in stimulation of prostate cancer cell proliferation and survival. As shown in Fig. 3A, knockdown of NSD2 in C4-2B cells showed a significant reduction (2- to 4-fold) in mRNA levels of genes controlling the cell cycle, such as those for cyclin A2, cyclin D2, and c-Myc. Consistent with its prominent role in cancer cell survival, NSD2 knockdown in C4-2B cells strongly inhibited the expression of genes with primary function in promoting cell survival, including genes for SGK1, Bcl2, survivin, and IL-6. Interestingly, silencing of NSD2 in AR-negative PC-3 cells also resulted in a strong inhibitory effect (3- to 9-fold) on a panel of genes with functions in cell proliferation, survival, inflammation, and angiogenesis, which included genes for cyclin D1, cyclin D3, cyclin E2, c-Myc, SGK-1, Bcl-2, Bcl-XL, survivin, IL-6, IL-8, VEGFA, and CXCL1. Western blotting demonstrated that the inhibitory effects can be observed at the protein level for most of the above-listed genes (Fig. 3C). ELISA detected significant decreases (2- to 3-fold) of secreted IL-6, IL-8, and VEGFA proteins in NSD2-depleted PC-3 cells (Fig. 3D). Knockdown of NSD2 in PC-3 xenograft tumors yielded similar effects on cell proliferation and survival genes (see Fig. S4 in the supplemental material). Together, these data suggest that high levels of NSD2 mediate the expression of genes that are critical for prostate cancer cell proliferation, survival, and angiogenesis.
Fig 3.

NSD2 controls the expression of NF-κB target genes that are important for cancer cell proliferation, survival, and angiogenesis. C4-2B or PC-3 cells were transfected with NSD2-specific or control siRNAs and harvested 48 h later for RNA extraction and real-time RT-PCR (A and B) or harvested at the indicated times for Western blotting (C). The fold reduction shown was obtained after normalization of expression with the expression levels of GAPDH. (D) The supernatants of PC-3 cells were collected 48 h after siRNA transfection and examined for secreted IL-6 or IL-8 or VEGF proteins by ELISA. Data for the RT-PCR and ELISA were obtained from triplicate experiments. *, P < 0.001.
NSD2 associates with NF-κB and acts as a novel coactivator for NF-κB.
Given that most of the genes affected by NSD2 depletion are known targets of NF-κB in prostate cancer and other types of cancer, we examined whether the function of NSD2 is through NF-κB. First, we performed a ChIP assay to determine whether NSD2 directly controls any of the endogenous NF-κB target genes in PC-3 cells where NF-κB is constitutively activated. Indeed, NSD2 occupies the promoter region of the genes for IL-6, IL-8, VEGFA, and survivin/Birc5, where NF-κB protein RelA/p65 also binds. In contrast, no significant occupancy was detected at the upstream region of the above-listed genes or at the promoter of GAPDH (Fig. 4A to C and data not shown). To examine whether NSD2 cooccupies the promoter region with NF-κB, ChIP–re-ChIP was performed. When NSD2-chromatin complexes enriched by the first ChIP with NSD2 antibody were subject to the second ChIP, a strong RelA association was detected by the RelA-specific antibody but not by control IgG at the IL-6, IL-8, and VEGFA gene promoters and not the promoter of the GAPDH control gene, indicating that NSD2 binds concurrently with RelA to the NF-κB target gene promoters (Fig. 4D). To determine whether the NSD2 occupancy is associated with NF-κB activation, ChIP was performed with PC-3 cells first deprived of serum to diminish NF-κB activation by undefined factors and then treated with TNF-α. As expected, the binding of NF-κB to its target sites was strongly stimulated by TNF-α (Fig. 4E). Remarkably, NSD2 recruitment to the NF-κB-targeted sites was also strongly induced by 1 h or 2 h of TNF-α treatment, indicating that NSD2 is recruited to NF-κB target genes upon NF-κB pathway activation.
Fig 4.
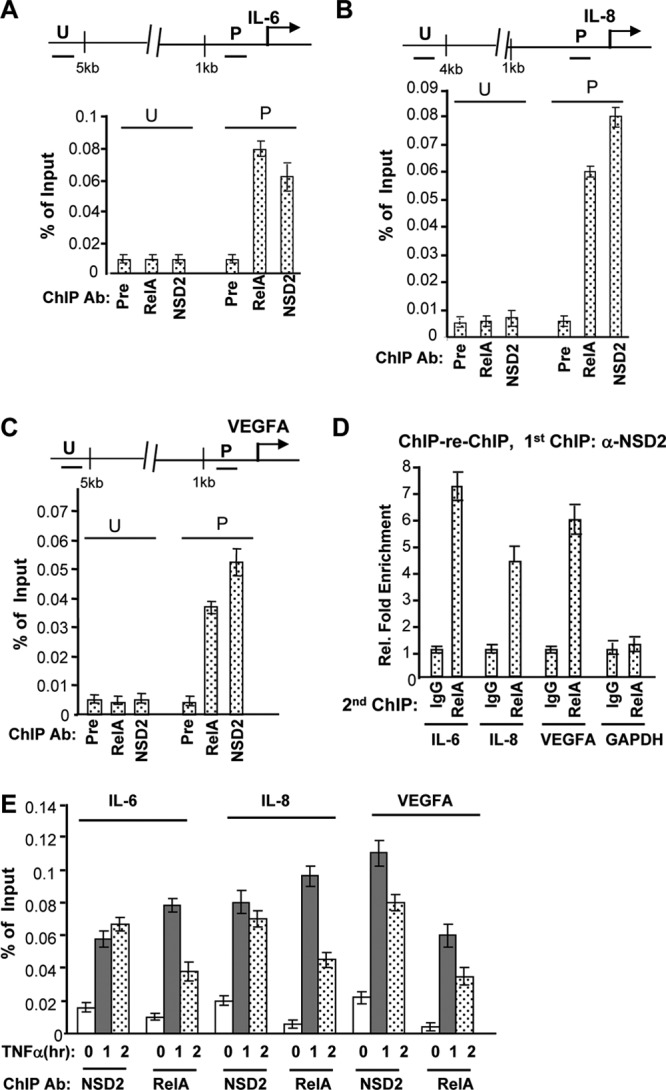
NSD2 associates with NF-κB at endogenous target gene promoters. (A to C) PC-3 cells were growing in regular growth medium and harvested for ChIP assays with preimmune serum or anti-NSD2 or anti-RelA/p65 antibodies (Ab). ChIP and input DNA was analyzed by real-time PCR with primers amplifying the indicated genomic regions of individual genes (U, upstream; P, promoter). ChIP data obtained from triplicate experiments were presented as a percentage of input signals. (D) The first ChIP was performed as described for panels A to C with NSD2 antibody. The immunoprecipitation complex was eluted and subject to the second ChIP procedures with control IgG or anti-RelA antibody. DNA from the second ChIP was analyzed by real-time PCR, as described for panels A to C. Data from control IgG were set as 1. (E) PC-3 cells were serum deprived for 16 h (to diminish NF-κB activation by undefined factors) and then treated with 10 nM TNF-α for the indicated times (hours) and harvested for ChIP assays with anti-NSD2 or anti-RelA/p65 antibodies, as described for panels A to C.
To further demonstrate NSD2 interaction with NF-κB, we performed co-IP. To facilitate the detection, we used cells ectopically expressing NF-κB subunit p50 or p65 and Flag-tagged NSD2. As shown in Fig. 5A, anti-Flag antibody effectively coprecipitated p50 and p65 proteins only in cells coexpressing Flag-NSD2 and p50 or p65 (lane 1, top and bottom), demonstrating that, indeed, NSD2 specifically formed complexes with both NF-κB proteins. To show that the association occurs with endogenous proteins, we performed co-IP with nuclear extracts of PC-3 cells where constitutive NF-κB activation is relatively high. Indeed, a portion of endogenous NSD2 protein was coprecipitated by anti-RelA/p65 antibody (Fig. 5B, lane 3) but not the control IgG, indicating the association of endogenous NSD2 with NF-κB. To further characterize the interaction, we performed GST pulldown experiments using purified Flag-NSD2 and GST-NF-κB proteins. Results in Fig. 5C clearly demonstrate that both GST-RelA/p65 and GST-p50 were able to pull down the NSD2 protein, whereas GST alone was not. Together, these results strongly suggest that NSD2 can directly interact with NF-κB proteins.
Fig 5.
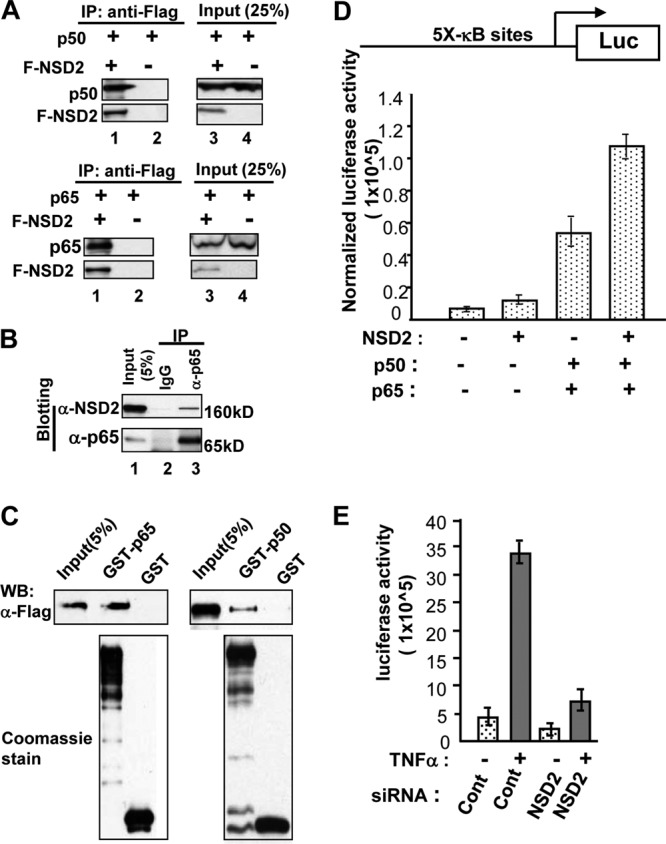
NSD2 directly associates with NF-κB. (A) Cell lysates from HEK293T cells transfected with plasmids expressing Flag-NSD2 (F-NSD2) and NF-κB1/p50 or RelA/p65 were incubated with M2 beads precoated with anti-Flag antibodies. The NF-κB proteins retained on the beads after extensive washing were detected by anti-p65 or -p50 antibody. (B) An equal amount of PC-3 cell nuclear extracts was incubated with anti-p65/RelA rabbit antibody or normal rabbit IgG. NSD2 associated with NF-κB was detected by Western blotting with anti-NSD2 monoclonal antibody. Input, 5% of cell lysates used for the co-IP. (C) GST, GST-p50, and GST-p65 were expressed in 293T cells by transfection with plasmid constructs, with transfected cells treated with 10 ng/ml of TNF-α. The GST and its fusion proteins were purified by glutathione agarose beads, separated by SDS-PAGE, and stained with Coomassie blue. The immobilized GST fusion proteins were incubated with purified Flag-NSD2 protein in the pulldown assay. NSD2 was detected by Western blotting (WB) using anti-Flag antibody. (D) LNCaP cells were transfected with a reporter plasmid containing five NF-κB binding sites (5×-κΒ–Luc) and with NSD2 or NF-κB (p50-p65) expression plasmids, as indicated. Twenty-four hours after the transfection, cells were stimulated with 2.5 nM TNF-α overnight and harvested 24 h later for luciferase and β-Gal assays. (E) HEK293T cells with chromosomally integrated 5 NF-κB sites–TATA–luciferase reporter were transfected with siRNAs and 48 h later maintained in serum-free medium for overnight before being treated with 2.5 nM TNF-α. Twenty-four hours later, cells were harvested for luciferase assay, with the protein concentration measured for normalization.
To determine whether NSD2 interaction with NF-κB affects the transcriptional regulation activity of NF-κB, we performed reporter gene assays. First, LNCaP cells that express very low levels of NF-κB were cotransfected with an NF-κB reporter construct containing five NF-κB enhancer elements and NSD2 and NF-κB expression plasmids. While expression of NF-κB heterodimer proteins alone resulted in about 7-fold activation, simultaneous expression of NF-κB and NSD2 yielded more than 16-fold induction (Fig. 5D). Next, a chromosomally integrated NF-κB binding site-luciferase reporter was used to further demonstrate the role of NSD2 in NF-κB-dependent activation. As expected, TNF-α robustly stimulated the reporter activity (about 7-fold; Fig. 5E). Remarkably, NSD2 depletion by siRNA resulted in almost complete blockade of the induction mediated by NF-κB. These results suggest that NSD2 can act as a potent coactivator of NF-κB.
NSD2 mediates NF-κB activation-associated elevation of H3K36 methylation and histone acetylation and the recruitment of NF-κB and coactivator p300.
We further examined the mechanism of NSD2 function in mediating NF-κB-dependent gene activation by assessing the involvement of its histone methylase activity. As described earlier, NSD2 can mediate di- and trimethylation of H3K36. We thus examined whether NF-κB-dependent gene activation is associated with changes in H3K36 methylation. PC-3 cells maintained under a regular growth condition displayed relatively high levels of constitutive NF-κB activation (58). ChIP assay demonstrated that in the absence of strong NF-κB activation (when cells were maintained in serum-free medium), the promoters of the IL-6, IL-8, and VEGFA genes were associated with relatively low levels of H3K36me2 and H3K36me3 mark. Upon NF-κB activation by TNF-α for 1 h or 3 h, levels of H3K36-me2 and -me3 increased significantly (Fig. 6A and B and data not shown). Importantly, NSD2 depletion significantly suppressed the increase of H3K36me2 and H3K36me3 induced by TNF-α at the NF-κB target promoters (Fig. 6A and B).
Fig 6.

NSD2 mediates NF-κB activation-associated histone H3K9K14 acetylation and H3K36 methylation and is required for recruitment of RelA and p300 at NF-κB target genes. (A and B) PC-3 cells were transfected with siRNAs and 48 h later maintained in serum-free medium for overnight. Cells were treated with 10 nM TNF-α for the indicated times (hours) before they were harvested for ChIP assay with H3K36me2 and H3K36me3 antibodies. ChIP DNA was analyzed as described in the legend to Fig. 4 with primers amplifying the indicated gene promoter regions with NF-κB sites. (C and D) PC-3 cells were transfected with siRNAs, treated with TNF-α for 1 h, and subjected to ChIP assay as described for panels A and B with anti-p300, anti-RelA, and anti-H3K9K14ac antibodies. ChIP data from cells treated with control siRNA and no TNF-α were set as 1.
NF-κB-mediated gene activation requires the function of coactivator p300/CBP-mediated histone acetylation. Strikingly, depletion of NSD2 strongly diminished H3K9K14 hyperacetylation induced by TNF-α at the IL-6 and IL-8 gene promoters (Fig. 6C and D), which is accompanied by a significant decrease in recruitment of HAT protein p300 (Fig. 6C and D; compare lane 6 with lane 8). Unexpectedly, when RelA recruitment was examined, it was found that NSD2 knockdown also resulted in a strong inhibition of RelA recruitment to the NF-κB target promoters (Fig. 6C and D; compare lane 10 with lane 12).
Given that NSD2 is required for H3K36 trimethylation at NF-κB target promoters, we asked whether the histone methylation function of NSD2 is critical for NF-κB-dependent gene activation. Substitution of a tyrosine to alanine at amino acid Y1179 in the SET domain was demonstrated to abolish the histone methylation activity of NSD2 in vitro and in cell culture systems (17). As shown in Fig. 7A, ectopic expression of NSD2 was readily achieved by lentivirus infection of C4-2B cells. Remarkably, wild-type NSD2 significantly increased the mRNA expression of IL-6, IL-8, and VEGFA and the secreted protein levels of IL-8 and VEGFA (Fig. 7B). In contrast, the NSD2 Y1179A mutant had much reduced activity to increase the NF-κB target gene expression. Moreover, although increased recruitment of both wild-type and SET mutant NSD2 was observed at the NF-κB target gene promoter, only wild-type NSD2 overexpression led to an increased level of H3K36me3 (Fig. 7C). Together with data in Fig. 6, these results suggest that NSD2 mediates NF-κB-dependent gene activation by promoting histone methylation at H3K36 and H3 acetylation through facilitating the occupancy of NF-κB and its coactivator p300 at the target gene promoters. The results also demonstrated that the methyltransferase activity of NSD2 is important for NSD2 to mediate NF-κB activation of gene expression.
Fig 7.
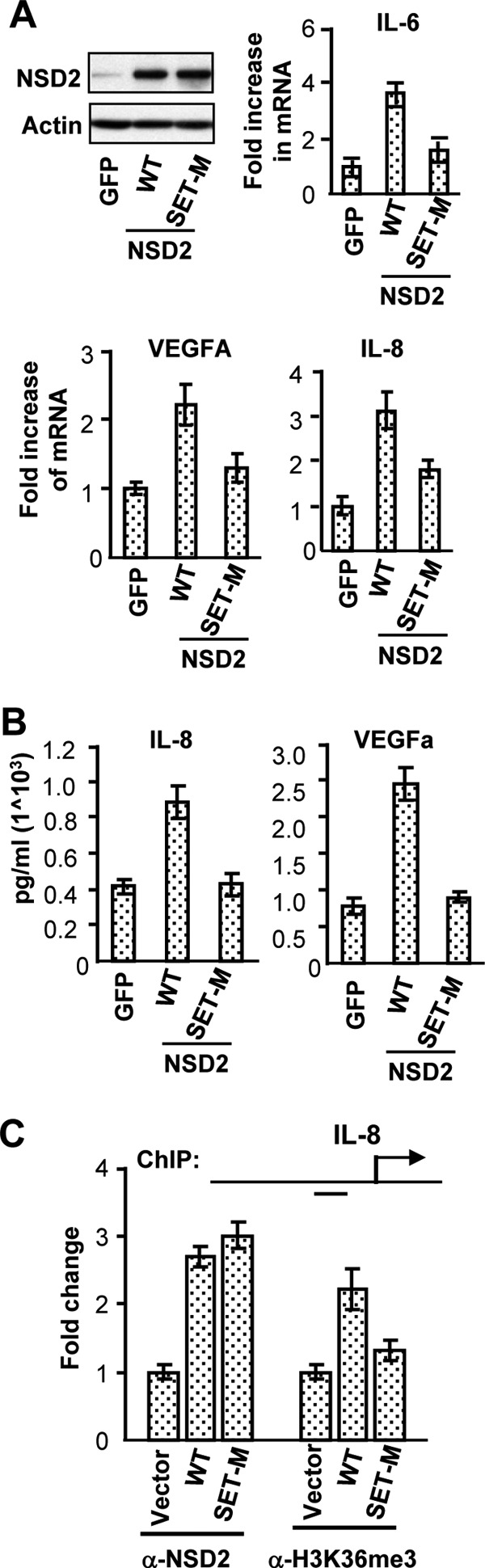
Overexpressed wild-type (WT) NSD2 and not its methylase mutant stimulates NF-κB target gene expression and histone methylation. (A and B) C4-2B cells were infected with lentivirus vectors expressing wild-type or a methylase-inactive mutant form of NSD2 or green fluorescent protein (GFP) as a control and harvested 3 days later for real-time RT-PCR and Western blotting (A) or ELISA (B) for analysis of expression of indicated genes. (C) C4-2B cells were treated as described for panel A and harvested for ChIP assays with the indicated antibodies. ChIP DNA was analyzed by real-time PCR for relative NSD2 occupancy and H3K36me3 mark change at the IL-8 NF-κB binding region, with signals from empty vector-treated cells set as 1.
NSD2 expression is stimulated by inflammatory cytokines via NF-κB, and its overexpression correlates with NF-κB activation in the tumors.
We recently found that the bromodomain protein ANCCA is a potent coactivator of E2Fs (40). Interestingly, ANCCA expression is regulated by E2Fs (8; our unpublished data). This prompted us to examine whether NSD2 is regulated by NF-κB. Thus, cells were treated with TNF-α to activate NF-κB. Remarkably, TNF-α treatment significantly induced NSD2 protein expression (>3-fold at 24 h) (Fig. 8A, top left). For comparison, cells were also treated with the other proinflammatory cytokine, IL-6. Surprisingly, IL-6 treatment also induced NSD2 expression (Fig. 8A, bottom left). Further study indicated that NSD2 induction by TNF-α or IL-6 was at the mRNA level (Fig. 8A, right). To establish that the induction by TNF-α is via NF-κB, cells were transfected with siRNA targeting RelA. Indeed, knockdown of RelA strongly diminished the NSD2 expression induced by TNF-α (Fig. 8B).
Fig 8.
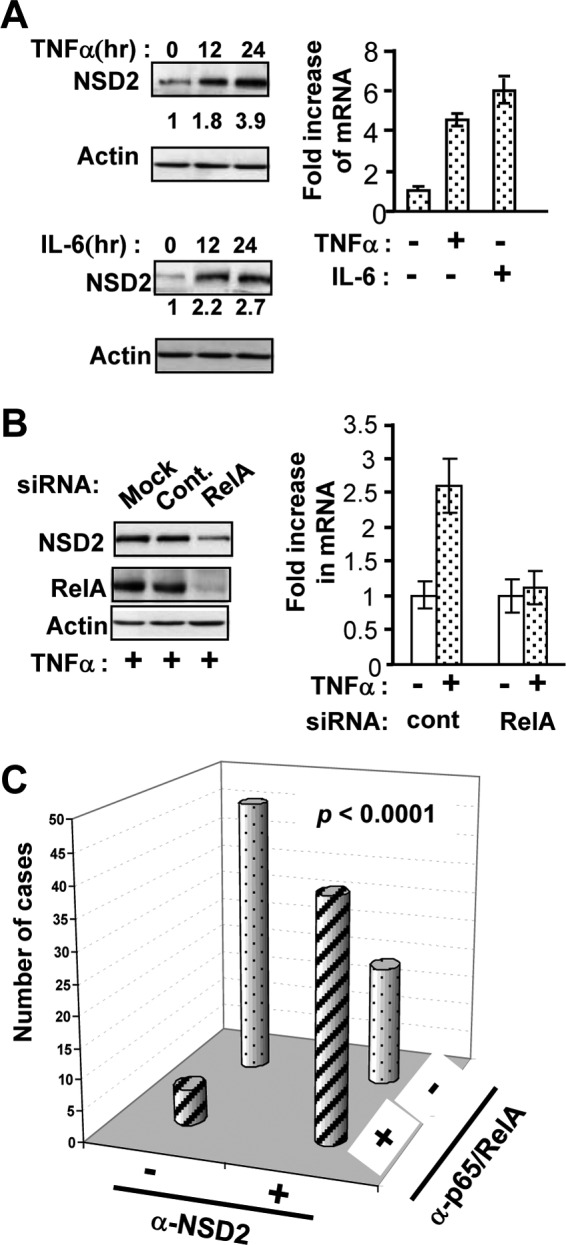
Proinflammatory cytokines stimulate NSD2 expression in advanced prostate cancer cells and tumors. (A) C4-2B cells were maintained in serum-free medium overnight and treated with TNF-α (10 ng/ml) or IL-6 (20 ng/ml) for 12 or 24 h before they were harvested for Western blotting or for 24 h for real-time RT-PCR analysis of NSD2 expression. The density of NSD2 (170-kDa) protein bands was normalized to the density of beta-actin. (B) C4-2B cells were transfected with siRNAs and 48 h later were treated as described for panel A with TNF-α for 24 h before they were harvested for Western blotting and real-time RT-PCR analysis of NSD2 expression. (C) IHC analysis of NSD2 and p65 was performed on adjacent sections of a TMA containing specimens from 111 cases of prostate adenocarcinoma. Pearson's χ2 test was used to determine the association between positive nuclear staining by anti-NSD2 and anti-RelA/p65 antibodies (P < 0.0001).
Next, we wondered if a similar regulation of NSD2 by NF-κB occurs in the tumors. We thus performed IHC analysis of NSD2 and p65 on adjacent sections of a TMA containing 111 prostate adenocarcinoma specimens. Consistent with previous studies (21, 42), we found that nuclear p65 can be detected in about 40% of the tumors (Fig. 8C). Interestingly, 35% of the tumors stained positive for both NSD2 and p65, while 41% stained negative for both. Statistical analysis indicated a strong correlation (P < 0.0001) between NSD2 overexpression and nuclear expression of NF-κB in the tumors. Together these results suggest that NF-κB activation may stimulate the expression of NSD2 in the tumor cells, which in turn promotes hyperactivation of NF-κB, thus forming a positive-feedback loop (Fig. 9).
Fig 9.
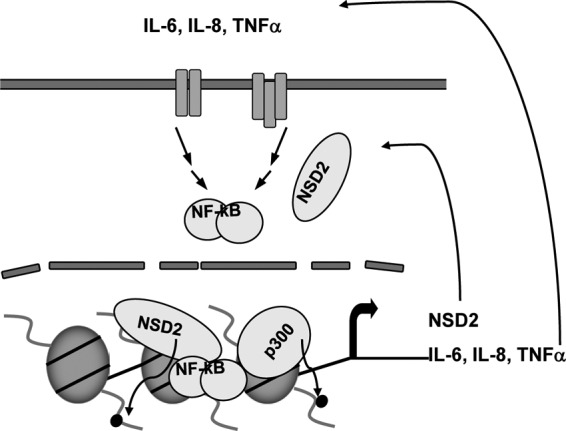
Proposed model for NSD2 functions in mediating constitutive NF-κB signaling. In addition to its histone methylation activity, NSD2 promotes the recruitment of NF-κB and p300 complexes for histone acetylation at the promoters of NF-κB target genes, which results in increased transcription of genes such as IL-6, IL-8, TNF-α, and NSD2 itself. Their elevation in turn further stimulates NF-κB signaling through the indicated positive feed-forward loops, leading to constitutive activation of NF-κB and its target gene expression. The upstream signaling events are omitted for clarity.
DISCUSSION
In this study, we provide evidence that NSD2 is overexpressed in tumors of more advanced prostate cancer and that elevated NSD2 plays a major role in prostate cancer cell proliferation, survival, and tumor angiogenesis. More importantly, we revealed that NSD2 acts as a novel transcriptional coactivator to mediate constitutive NF-κB signaling in castration therapy-resistant prostate cancer cells. Our study identified a number of NF-κB-controlled genes as direct targets of NSD2 in the cancer cells, which include the genes for IL-6, IL-8, survivin/Birc5, and VEGFA. Furthermore, we found that NSD2 expression is strongly stimulated by proinflammatory cytokines such as TNF-α and IL-6. Taken together, the results suggest that constitutive NF-κB activation involves a histone methyltransferase NSD2-mediated, positive feed-forward loop (Fig. 9), in which NSD2 serves as a major mediator of NF-κB-dependent signaling by its coactivator function and as a reinforcement of NF-κB constitutive activation via its own elevated expression and function stimulated by NF-κB signaling.
Cancer progression, such as the transition of androgen-responsive prostate cancer to CRPC, has been strongly linked to aberrant paracrine and sometimes autocrine signaling by the tumor cells, which are capable of both producing and responding to multiple inflammatory cytokines and chemokines, including TNF-α, IL-6, and IL-8 (20, 26, 41, 44). Importantly, overexpression and/or activation of canonical and noncanonical NF-κB pathways has also been found in high-grade prostate cancer and metastases and serves as the key mediator for autocrine signaling, thereby underscoring the critical role of NF-κB activation in driving disease progression. However, how NF-κB pathways are activated during the progression has been poorly understood. Results from this study provide strong evidence that the histone methyltransferase NSD2 plays a very crucial role in this regard. First, we demonstrate that NSD2 directly interacts with NF-κB and controls the expression of IL-6, IL-8, and CXCL1. Our ChIP assay demonstrated that NSD2 cooccupies with NF-κB at the cytokine gene promoters. We also found that depletion of NSD2 strongly inhibits the expression of Bcl2, Bcl-XL, survivin/Birc5, and VEGFA, which are key targets and mediators of NF-κB for its prosurvival signaling in cancer. Interestingly, we found that NSD2 overexpression in cells with otherwise low NF-κB activity can strongly promote the expression of NF-κB target genes. Our IHC analysis of clinical specimens of prostate cancer indicates that elevated levels of NSD2 strongly associate with NF-κB activation (indicated by nuclear localization) in the tumors. Thus, this study provides strong evidence that aberrant NSD2 is a key player in tumor autocrine signaling for progression of prostate cancer.
As a histone methyltransferase, NSD2 belongs to the SET2 family, which primarily methylates H3K36. However, its functional mechanism has been poorly understood. Saccharomyces cerevisiae SET2 protein appears to be important for suppressing spurious intragenic initiation of transcription via promoting deacetylation of chromatin at gene coding regions (4). Mammalian NSD2 was initially characterized as a transcriptional corepressor and found to associate with histone deacetylase complex proteins as well (16, 27, 32). Moreover, mouse ES cells with NSD2/WHSC1 gene knockouts displayed increased expression of genes regulated by NSD2 (32). Unexpectedly, in this study, we found that NSD2 acts as a strong transcriptional coactivator for NF-κB, which was evidenced by its potent coactivation activity in stimulation of endogenous NF-κB target genes and by a marked suppression (3- to 9-fold) of NF-κB target gene transcription upon its depletion. Consistent with the notion that NSD2 can act primarily as a coactivator, our unpublished genome-wide expression profiling of NSD2-knockdown prostate cancer cells indicated that among the genes with altered expression, over 70% of them were downregulated. Our ChIP assay demonstrated that NSD2 cooccupies with NF-κB at the NF-κB sites in cells with constitutive NF-κB activation and that blockage of NF-κB signaling inhibits the NSD2 occupancy, therefore suggesting that NSD2 is recruited by activated NF-κB to the gene regulatory region. Thus, this study provides new insights into the functional mechanism of NSD2 by showing that NSD2 can act primarily as a transcriptional coactivator and that NSD2 is targeted to chromatin by transcription factors such as NF-κB.
Recent studies suggest that transcriptional activation of target genes by NF-κB is regulated at several independent levels, including chromatin remodeling and switch of repressive chromatin modifications to active chromatin modifications (47). Intriguingly, NSD2 is found to be critical for TNF-α-induced histone hyperacetylation and the recruitment of histone acetyltransferase p300, suggesting that NSD2 is a mediator of the chromatin modification switch. Unexpectedly, NSD2 is also found to be important for NF-κB recruitment to target gene promoters. The latter observation is reminiscent of a recent study demonstrating that histone demethylase AOF1 is recruited through its interaction with c-Rel and in turn is needed for the recruitment of c-Rel and p65/RelA to target genes Mdc and IL-12 upon LPS stimulation (48). It also echoes our study on ANCCA, where ANCCA acts as an E2F coactivator and is crucial for E2F occupancy at promoters (40). Although understanding how such a functional loop initiates and operates requires more elaborate approaches, it is tempting to speculate that NSD2 may interact with other transcription factors and/or coregulators in addition to NF-κB at the target chromatin and that cooperative interactions among them allow strong induction of transcription upon NF-κB activation by the stimuli (39, 52). Alternatively, in the absence of stimulus, a low level of NSD2 may anchor itself at the local chromatin through its histone mark recognition modules such as its PHD domains, like ANCCA does through its bromodomain (40). Upon stimulation, NSD2 engages in recruitment of NF-κB through physical interaction. NF-κB activation leads to increased expression of NSD2 and more NSD2–NF-κB complex formation and occupancy at NF-κB sites. On the other hand, given the fact that nonhistone proteins are important substrates of histone-modifying enzymes, it is also possible that NSD2 may exert its coactivation activity via methylation of NF-κB. In this regard, several histone methylases and demethylases were recently demonstrated to modulate the function of NF-κB (22, 25, 56). NSD2 methylation of NF-κB at specific sites may facilitate NF-κB interaction with NSD2 and/or other coregulators to facilitate the process of transcriptional activation.
NSD2 may mediate NF-κB-dependent gene activation through histone H3K36 methylation. Both H3K36me2 and H3K36me3 have been found to antagonize H3K27 trimethylation by PRC2 (43). In this regard, a recent study demonstrated that purified NSD2 protein primarily converts H3K36me1 to H3K36me2 (17). Consistently, we found that NSD2 function in prostate cancer cells is associated with H3K36me2 at NF-κB target gene promoters, as NSD2 knockdown decreases the level of H3K36me2 and its ectopic expression increases the mark. Interestingly, we also observed similar results on H3K36me3 when we altered NSD2 expression in the prostate cancer cells, which is in agreement with other studies on NSD2 (28, 32). Whether NSD2 directly methylates H3K36me2 to H3K36me3 at the NF-κB targets is unclear at this point. It is conceivable, however, that the methylation activities of NSD2, including its substrate specificity, are regulated in a cell context- and chromatin locus-specific manner.
One important finding of this study is that about 40% of the primary prostate cancer tumors with high Gleason scores overexpress NSD2 protein. Remarkably, NSD2 overexpression is strongly associated with activation of NF-κB in the tumors, suggesting that one major mechanism of NSD2 deregulation is mediated by NF-κB. Interestingly, we also found that elevated levels of ANCCA may partly be responsible for NSD2 overexpression, therefore implying that there might be a coregulator network that drives prostate cancer progression. Given that aberrant NF-κB signaling is prevalent in many types of human malignancies and that excessive inhibition of NF-κB can result in unwanted effects such as immunosuppression (37), targeting of NF-κB coactivators such as NSD2 or its regulators such as ANCCA can be a new strategy for effective therapeutic intervention in the progression of prostate cancer and other types of cancer with aberrant expression and function of NSD2 and ANCCA.
Supplementary Material
ACKNOWLEDGMENTS
We thank C. Duckett for NF-κB expression plasmids. We appreciate the technical help from A. Gemo, F. He, Junjian Wang, E. Kalashnikova, and N. Nadiminty.
This work was supported by grants from the U.S. Department of Defense (DoD) (W81XWH-07-1-0312) to J. X. Zou, the NIH (R01CA134766, R01DK060019), and the DoD (W81XWH-08-1-0432) to H.-W. Chen.
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 29 May 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Agoulnik IU, et al. 2005. Role of SRC-1 in the promotion of prostate cancer cell growth and tumor progression. Cancer Res. 65:7959–7967 [DOI] [PubMed] [Google Scholar]
- 2. Barish GD, et al. 2010. Bcl-6 and NF-kappaB cistromes mediate opposing regulation of the innate immune response. Genes Dev. 24:2760–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Caron C, et al. 2010. Functional characterization of ATAD2 as a new cancer/testis factor and a predictor of poor prognosis in breast and lung cancers. Oncogene 29:5171–5181 [DOI] [PubMed] [Google Scholar]
- 4. Carrozza MJ, et al. 2005. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123:581–592 [DOI] [PubMed] [Google Scholar]
- 5. Chaturvedi MM, Sung B, Yadav VR, Kannappan R, Aggarwal BB. 2011. NF-kappaB addiction and its role in cancer: ‘one size does not fit all. ’ Oncogene 30:1615–1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen CD, Sawyers CL. 2002. NF-kappa B activates prostate-specific antigen expression and is upregulated in androgen-independent prostate cancer. Mol. Cell. Biol. 22:2862–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen LF, et al. 2005. NF-kappaB RelA phosphorylation regulates RelA acetylation. Mol. Cell. Biol. 25:7966–7975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ciro M, et al. 2009. ATAD2 is a novel cofactor for MYC, overexpressed and amplified in aggressive tumors. Cancer Res. 69:8491–8498 [DOI] [PubMed] [Google Scholar]
- 9. Dan HC, et al. 2008. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 22:1490–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Santa F, et al. 2007. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 130:1083–1094 [DOI] [PubMed] [Google Scholar]
- 11. Heinz S, et al. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38:576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang B, Yang XD, Zhou MM, Ozato K, Chen LF. 2009. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol. Cell. Biol. 29:1375–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hudlebusch HR, et al. 2011. The histone methyltransferase and putative oncoprotein MMSET is overexpressed in a large variety of human tumors. Clin. Cancer Res. 17:2919–2933 [DOI] [PubMed] [Google Scholar]
- 14. Jin RJ, et al. 2008. The nuclear factor-kappaB pathway controls the progression of prostate cancer to androgen-independent growth. Cancer Res. 68:6762–6769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kalashnikova EV, et al. 2010. ANCCA/ATAD2 overexpression identifies breast cancer patients with poor prognosis, acting to drive proliferation and survival of triple-negative cells through control of B-Myb and EZH2. Cancer Res. 70:9402–9412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim JY, et al. 2008. Multiple-myeloma-related WHSC1/MMSET isoform RE-IIBP is a histone methyltransferase with transcriptional repression activity. Mol. Cell. Biol. 28:2023–2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kuo AJ, et al. 2011. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell 44:609–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lamont KR, Tindall DJ. 2011. Minireview: alternative activation pathways for the androgen receptor in prostate cancer. Mol. Endocrinol. 25:897–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lauring J, et al. 2008. The multiple myeloma associated MMSET gene contributes to cellular adhesion, clonogenic growth, and tumorigenicity. Blood 111:856–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee LF, et al. 2004. Interleukin-8 confers androgen-independent growth and migration of LNCaP: differential effects of tyrosine kinases Src and FAK. Oncogene 23:2197–2205 [DOI] [PubMed] [Google Scholar]
- 21. Lessard L, et al. 2006. Nuclear localization of nuclear factor-kappaB p65 in primary prostate tumors is highly predictive of pelvic lymph node metastases. Clin. Cancer Res. 12:5741–5745 [DOI] [PubMed] [Google Scholar]
- 22. Li Y, et al. 2008. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes. Relevance to diabetes and inflammation. J. Biol. Chem. 283:26771–26781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li Y, et al. 2009. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J. Biol. Chem. 284:34283–34295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Louie MC, et al. 2003. Androgen-induced recruitment of RNA polymerase II to a nuclear receptor-p160 coactivator complex. Proc. Natl. Acad. Sci. U. S. A. 100:2226–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu T, et al. 2010. Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc. Natl. Acad. Sci. U. S. A. 107:46–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Malinowska K, et al. 2009. Interleukin-6 stimulation of growth of prostate cancer in vitro and in vivo through activation of the androgen receptor. Endocr. Relat. Cancer 16:155–169 [DOI] [PubMed] [Google Scholar]
- 27. Marango J, et al. 2008. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood 111:3145–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martinez-Garcia E, et al. 2011. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 117:211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Min J, et al. 2010. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat. Med. 16:286–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nadiminty N, et al. 2010. Aberrant activation of the androgen receptor by NF-kappaB2/p52 in prostate cancer cells. Cancer Res. 70:3309–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ndlovu MN, et al. 2009. Hyperactivated NF-{kappa}B and AP-1 transcription factors promote highly accessible chromatin and constitutive transcription across the interleukin-6 gene promoter in metastatic breast cancer cells. Mol. Cell. Biol. 29:5488–5504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nimura K, et al. 2009. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature 460:287–291 [DOI] [PubMed] [Google Scholar]
- 33. Niu Y, et al. 2010. Differential androgen receptor signals in different cells explain why androgen-deprivation therapy of prostate cancer fails. Oncogene 29:3593–3604 [DOI] [PubMed] [Google Scholar]
- 34. Norris JD, et al. 2009. The homeodomain protein HOXB13 regulates the cellular response to androgens. Mol. Cell 36:405–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nowak DE, et al. 2008. RelA Ser276 phosphorylation is required for activation of a subset of NF-kappaB-dependent genes by recruiting cyclin-dependent kinase 9/cyclin T1 complexes. Mol. Cell. Biol. 28:3623–3638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pei H, et al. 2011. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 470:124–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perkins ND. 2012. The diverse and complex roles of NF-kappaB subunits in cancer. Nat. Rev. Cancer 12:121–132 [DOI] [PubMed] [Google Scholar]
- 38. Pradhan M, Baumgarten SC, Bembinster LA, Frasor J. 2012. CBP mediates NF-kappaB-dependent histone acetylation and estrogen receptor recruitment to an estrogen response element in the BIRC3 promoter. Mol. Cell. Biol. 32:569–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ramadoss S, Li J, Ding X, Al Hezaimi K, Wang CY. 2011. Transducin beta-like protein 1 recruits nuclear factor kappaB to the target gene promoter for transcriptional activation. Mol. Cell. Biol. 31:924–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Revenko AS, Kalashnikova EV, Gemo AT, Zou JX, Chen HW. 2010. Chromatin loading of E2F-MLL complex by cancer-associated coregulator ANCCA via reading a specific histone mark. Mol. Cell. Biol. 30:5260–5272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rojas A, et al. 2011. IL-6 promotes prostate tumorigenesis and progression through autocrine cross-activation of IGF-IR. Oncogene 30:2345–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ross JS, et al. 2004. Expression of nuclear factor-kappa B and I kappa B alpha proteins in prostatic adenocarcinomas: correlation of nuclear factor-kappa B immunoreactivity with disease recurrence. Clin. Cancer Res. 10:2466–2472 [DOI] [PubMed] [Google Scholar]
- 43. Schmitges FW, et al. 2011. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell 42:330–341 [DOI] [PubMed] [Google Scholar]
- 44. Seaton A, et al. 2008. Interleukin-8 signaling promotes androgen-independent proliferation of prostate cancer cells via induction of androgen receptor expression and activation. Carcinogenesis 29:1148–1156 [DOI] [PubMed] [Google Scholar]
- 45. Setlur SR, et al. 2007. Integrative microarray analysis of pathways dysregulated in metastatic prostate cancer. Cancer Res. 67:10296–10303 [DOI] [PubMed] [Google Scholar]
- 46. Sheppard KA, et al. 1999. Transcriptional activation by NF-kappaB requires multiple coactivators. Mol. Cell. Biol. 19:6367–6378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smale ST. 2011. Hierarchies of NF-kappaB target-gene regulation. Nat. Immunol. 12:689–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van Essen D, Zhu Y, Saccani S. 2010. A feed-forward circuit controlling inducible NF-kappaB target gene activation by promoter histone demethylation. Mol. Cell 39:750–760 [DOI] [PubMed] [Google Scholar]
- 49. Varambally S, et al. 2005. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 8:393–406 [DOI] [PubMed] [Google Scholar]
- 50. Wang J, et al. 2011. Activation of NF-{kappa}B by TMPRSS2/ERG fusion isoforms through Toll-like receptor-4. Cancer Res. 71:1325–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wissmann M, et al. 2007. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 9:347–353 [DOI] [PubMed] [Google Scholar]
- 52. Wolter S, et al. 2008. c-Jun controls histone modifications, NF-kappaB recruitment, and RNA polymerase II function to activate the ccl2 gene. Mol. Cell. Biol. 28:4407–4423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wright CW, Duckett CS. 2009. The aryl hydrocarbon nuclear translocator alters CD30-mediated NF-kappaB-dependent transcription. Science 323:251–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu RC, et al. 2004. Selective phosphorylations of the SRC-3/AIB1 coactivator integrate genomic responses to multiple cellular signaling pathways. Mol. Cell 15:937–949 [DOI] [PubMed] [Google Scholar]
- 55. Xu Y, et al. 2009. RelB enhances prostate cancer growth: implications for the role of the nuclear factor-kappaB alternative pathway in tumorigenicity. Cancer Res. 69:3267–3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang XD, et al. 2009. Negative regulation of NF-kappaB action by Set9-mediated lysine methylation of the RelA subunit. EMBO J. 28:1055–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang XD, Tajkhorshid E, Chen LF. 2010. Functional interplay between acetylation and methylation of the RelA subunit of NF-kappaB. Mol. Cell. Biol. 30:2170–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yemelyanov A, et al. 2006. Effects of IKK inhibitor PS1145 on NF-kappaB function, proliferation, apoptosis and invasion activity in prostate carcinoma cells. Oncogene 25:387–398 [DOI] [PubMed] [Google Scholar]
- 59. Yu J, et al. 2010. The neuronal repellent SLIT2 is a target for repression by EZH2 in prostate cancer. Oncogene 29:5370–5380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang L, et al. 2009. NF-kappaB regulates androgen receptor expression and prostate cancer growth. Am. J. Pathol. 175:489–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhong H, May MJ, Jimi E, Ghosh S. 2002. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol. Cell 9:625–636 [DOI] [PubMed] [Google Scholar]
- 62. Zhou HJ, et al. 2005. SRC-3 is required for prostate cancer cell proliferation and survival. Cancer Res. 65:7976–7983 [DOI] [PubMed] [Google Scholar]
- 63. Zou JX, et al. 2009. Androgen-induced coactivator ANCCA mediates specific androgen receptor signaling in prostate cancer. Cancer Res. 69:3339–3346 [DOI] [PubMed] [Google Scholar]
- 64. Zou JX, Revenko AS, Li LB, Gemo AT, Chen HW. 2007. ANCCA, an estrogen-regulated AAA+ ATPase coactivator for ERalpha, is required for coregulator occupancy and chromatin modification. Proc. Natl. Acad. Sci. U. S. A. 104:18067–18072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zou JX, et al. 2006. ACTR/AIB1/SRC-3 and androgen receptor control prostate cancer cell proliferation and tumor growth through direct control of cell cycle genes. Prostate 66:1474–1486 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.