

Abstract
The formation of crossovers is a fundamental genetic process. The XPF-family endonuclease Mus81-Mms4 (Eme1) contributes significantly to crossing over in eukaryotes. A key question is whether Mus81-Mms4 can process Holliday junctions that contain four uninterrupted strands. Holliday junction cleavage requires the coordination of two active sites, necessitating the assembly of two Mus81-Mms4 heterodimers. Contrary to this expectation, we show that Saccharomyces cerevisiae Mus81-Mms4 exists as a single heterodimer both in solution and when bound to DNA substrates in vitro. Consistently, immunoprecipitation experiments demonstrate that Mus81-Mms4 does not multimerize in vivo. Moreover, chromatin-bound Mus81-Mms4 does not detectably form higher-order multimers. We show that Cdc5 kinase activates Mus81-Mms4 nuclease activity on 3′ flaps and Holliday junctions in vitro but that activation does not induce a preference for Holliday junctions and does not induce multimerization of the Mus81-Mms4 heterodimer. These data support a model in which Mus81-Mms4 cleaves nicked recombination intermediates such as displacement loops (D-loops), nicked Holliday junctions, or 3′ flaps but not intact Holliday junctions with four uninterrupted strands. We infer that Mus81-dependent crossing over occurs in a noncanonical manner that does not involve the coordinated cleavage of classic Holliday junctions.
INTRODUCTION
Robin Holliday first proposed a mechanism for crossover formation (29). Based on fungal tetrad data, he envisioned that nick-induced heteroduplex formation could result in a DNA intermediate composed of four intact strands after ligation of the strand interruptions. This intermediate was later termed the Holliday junction (HJ). Cleavage across the two alternative planes of this junction would result in crossover (CO) or noncrossover (NCO) products, depending on the orientation of cleavage. Biochemical analysis of the bacterial RuvC nuclease supports this model and provides a paradigm for a class of enzymes called Holliday junction resolvases. These nucleases form homodimeric complexes to deliver two coordinated and symmetric endonucleolytic cuts that generate DNA ends that can be directly ligated to form recombinant products (reviewed in reference 38). However, RuvC is not evolutionarily conserved in eukaryotes, and the specific mechanisms of crossover formation in eukaryotes are still undefined. Refinement and expansion of the original Holliday model have produced the current model of double-strand break repair, in which one subpathway is defined by the formation of a double Holliday junction (dHJ) (46). Physical analysis has demonstrated the existence of dHJs in meiotic and mitotic recombination (12, 48). However, these studies could not establish whether these junctions were truly dHJs, i.e., with each individual junction having four uninterrupted strands, or were nicked junctions in a dHJ population where each strand could be found to be full length (for more discussion, see reference 49). Hence, the importance of Holliday junctions and their cleavage in CO formation still remains to be demonstrated.
The structure-selective endonuclease Mus81-Mms4 (Mms4 is known as Eme1 in other organisms) contributes to CO formation in budding and fission yeast as well as in Arabidopsis and mice, suggesting a role in joint molecule processing (6, 20, 28, 30, 45, 52). Mus81 was identified through its physical interactions with the Saccharomyces cerevisiae recombination protein Rad54 and the Schizosaccharomyces pombe DNA damage response kinase Cds1, as well as in a genetic screen for genes required in the absence of the Sgs1 helicase (10, 31, 41). A fundamental question is, what are the DNA joint molecules targeted by Mus81-Mms4 in vivo? Biochemical analysis of purified and partially purified Mus81-Mms4 protein shows catalytic and robust cleavage of nicked, 3′-flap, and displacement loop (D-loop) substrates to be the preferential in vitro target for Mus81 (5, 9, 13, 14, 21, 23, 26, 45). Yet, it is the highly inefficient incision of intact, four-way HJs that has resulted in widely discussed models suggesting that intact HJs or dHJs are the physiological substrates for Mus81-Mms4 in CO formation (9, 14, 26, 38, 52, 57).
Sgs1, in conjunction with Top3 and Rmi1, provides an alternative mechanism to process dHJs termed dissolution (see Fig. 17B), a reaction discovered with the human BLM-TOPOIIIalpha-RMI1 complex (61). Dissolution describes the coordinate movement of both junctions in a dHJ toward each other by combined action of the Sgs1/BLM helicase and Top3/TOPOIIIalpha topoisomerase, resulting in a single hemicatenane, which is resolved by the type IA topoisomerase Top3/TOPOIIIalpha, stimulated by the Rmi1 specificity factor. Dissolution is the only biochemical mechanism demonstrated to process dHJs, but it always leads to NCO products, leaving the mechanism of CO formation in eukaryotes unaddressed.
Fig 17.

(A) Molecular models of recombinational DNA repair during replication fork support. Recombination supports replication fork (RF) restart by facilitating both break-induced replication (BIR) and gap repair. Potential substrates for Mus81-Mms4 and Yen cleavage based on their biochemical specificity are indicated. LOH, loss of heterozygosity; CO, crossover; NCO, noncrossover. (B) Model of processing of late recombination intermediates. After extension of the invading strand in the D-loop, D-loop reversal leads to a noncrossover outcome by synthesis-dependent strand annealing (SDSA). Alternatively, dual incision by Mus81-Mms4 on D-loops or nicked junction substrates (yellow triangles) leads to CO. Formation of a double Holliday junction (dHJ) and dissolution by Sgs1-Top3-Rmi1 leads to NCO. The activity of Yen1 on dHJ substrates has not yet been reported. Yen1 possibly cleaves an intact Holliday junction (dark triangles), resulting from branch migration of the D-loop, contributing to a minor pathway of CO formation (lighter arrow) in wild-type cells and to NCO or break-induced replication, depending on second end capture and D-loop cleavage (?), as proposed by Ho et al. (28). In the absence of Mus81-Mms4 (mus81Δ), the Yen1 pathway may act on Mus81-Mms4 substrates directly or their processing products, consistent with recent results analyzing the regulation of Mus81-Mms4 and Yen1 (40).
In budding yeast and Drosophila, the genetic requirement for Mus81 (or Mms4/Eme1) in the absence of Sgs1 is completely dependent on the key recombination protein Rad51, suggesting that both the Sgs1-Top3-Rmi1 complex and the Mus81-Mms4 heterodimer function in the late stages of recombination to resolve Rad51-dependent DNA joint molecules (5, 22, 58). Physical analysis of meiotic DNA products has shown that loss of both Mus81 and Sgs1 during meiosis results in meiotic catastrophe and a failure to segregate chromosomes, consistent with the inability to resolve DNA joint molecules (33, 43). These data provide compelling support for the model that Mus81-Mms4 cleaves late recombination intermediates to form COs but leaves open the question of the physical nature of these intermediates.
A long-term effort to identify the eukaryotic equivalent of the paradigmatic bacterial RuvC HJ resolvase resulted in the identification of S. cerevisiae Yen1/human GEN1 (32). An N-terminal fragment of Yen1 or GEN1 is capable of HJ resolution across the plane of the junction, resulting in religatable products, although Yen1/GEN1 and the full-length Drosophila GEN1 also cleave other substrates (32, 34, 47). Human GEN1 binds to the HJ substrate as a multimer, providing the subunit architecture for coordinated HJ cleavage by two active sites (47).
On the basis of the biochemical data, it was suggested that Yen1/GEN1 is the long-sought HJ resolvase responsible for CO formation in eukaryotes (32). However, genetic analysis failed to discover any phenotype in the budding yeast yen1 mutant or in human cells sensitized by a mutation in the BLM gene depleted of GEN1 by small interfering RNA (1, 8, 40, 56, 60). Fission yeast entirely lacks this gene, and CO formation is almost completely dependent on Mus81-Eme1 (52). Elegant genetic analysis of CO formation illuminated the enzymes required for CO formation in vegetative (somatic) budding yeast cells (28). Mutations in MUS81 reduced all CO classes, demonstrating the critical role of Mus81-Mms4 also in CO formation in vegetative yeast cells (28), after such a role was already documented for meiotic recombination (3, 19, 20, 43). Mutations in YEN1 had only a minor effect and reduced only COs that were associated with short conversion tracts (28). In the mus81 yen1 double mutants, somatic COs were essentially eliminated (28), suggesting that in the absence of Mus81-Mms4, Yen1 can cleave either the original Mus81-Mms4 substrate(s) or its processing products (see Discussion). These results, together with the biochemical substrate preferences for Mus81-Mms4 and Yen1, suggest that the majority of CO formation in wild-type vegetative budding yeast, and possibly all eukaryotes, does not involve classic HJs or dHJs but involves other types of recombination-dependent junctions.
Cdc5 (Polo) kinase regulates meiotic progression and crossover formation (16, 53), and Mus81-Mms4 controls about a third of the meiotic crossovers in budding yeast (3, 19, 20; reviewed in reference 49). Cdc5 expression is induced during meiotic prophase, which correlates with phosphorylation of Mms4 and activation of the Mus81-Mms4 nuclease activity on an oligonucleotide-based HJ substrate (40). The same Cdc5-dependent activation was demonstrated to occur during mitotic cell cycles at the onset of anaphase (40). Analysis of a phosphorylation-defective Mms4 mutant, in which 14 predicted or mapped phosphorylation sites were mutated to the nonphosphorylatable residue alanine, suggested that Cdc5-mediated phosphorylation of the Mms4 subunit activates Mus81-Mms4 nuclease activity (40). The mechanism by which phosphorylation activates Mus81-Mms4 nuclease activity and whether this involves tetramerization of the Mus81-Mms4 heterodimer remain to be determined.
We have previously demonstrated that budding yeast Mus81-Mms4 strongly prefers nicked junctions and D-loops as the substrates, whereas HJ cleavage was so weak that classic kinetic (Michaelis-Menten) analysis could not be performed (21; see also reference 23). To address whether Mus81-Mms4 might cleave HJs in vivo, we have determined the subunit composition of the Mus81-Mms4 complex in vitro and in vivo. The hydrodynamic properties of S. cerevisiae Mus81-Mms4 demonstrate that it exists as a single heterodimer in solution and when bound to 3′ Flap or HJ DNA substrates. Transmission electron microscopy (TEM) of individually cross-linked protein particles with subsequent volumetric analysis of particle classes supports a heterodimer model. In vivo coimmunoprecipitation (co-IP) of soluble protein failed to show any self-association of free or chromatin-associated subunits into larger oligomeric complexes. We demonstrate Cdc5-mediated activation with purified Mus81-Mms4 in vitro and show that this does not involve multimerization of the enzyme. These results also show that the catalytic form of Mus81-Mms4 in vivo contains a single nuclease active site for preferential cleavage of nicked substrates and D-loops, pointing to a mechanism of CO formation in eukaryotes that likely involves nicked junctions rather than the iconic single or double Holliday junctions with four intact strands.
MATERIALS AND METHODS
S. cerevisiae strains and DNA substrates.
A complete list of strains used in this study is found in Table 1. Tagging constructs were prepared as previously described (4), and correct integration was verified by PCR and immunoblot analysis. The functionality of tagged proteins is demonstrated in Fig. 9A. Oligonucleotide-based 5′-32P-labeled and nonlabeled substrates were produced as described previously (21).
Table 1.
Yeast strains
| Straina | Genotype |
|---|---|
| W303-RAD5b | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 RAD5 |
| W303-RAD5b | MATα ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 RAD5 |
| WDHY668 | MATa/α ura3-52/ura3-52 leu2Δ1/leu2Δ1 his3Δ200/his2Δ200 pep4::HIS3/pep4::HIS3 prb1Δ1.6R/prb1Δ1.6R + pWDH722 |
| WDHY1858 | W303-RAD5 MATa mus81::KANMX |
| WDHY1868 | W303-RAD5 MATa MUS81-9MYC::TRP1 |
| WDHY1872 | W303-RAD5 MATa MMS4-9MYC::TRP1 |
| WDHY2128 | W303-RAD5 MATα mms4::TRP1 |
| WDHY2420 | W303-RAD5 MATα MUS81-6HA::TRP1 |
| WDHY2423 | W303-RAD5 MATα MMS4-6HA::TRP1 |
| WDHY2491 | MATa/α ade2-1/ade2-1 can1-100/can1-100 his3-11,15/his3-11,15 leu2-3,112/leu2-3,112 trp1-1/trp1-1 ura3-1/ura3-1 RAD5/RAD5 |
| WDHY2455 | WDHY2491 MMS4-6HA::TRP1/MMS4-9MYC::TRP1 |
| WDHY2456 | WDHY2491 MUS81-6HA::TRP1/MUS81-9MYC::TRP1 |
| WDHY2492 | WDHY2491 MMS4/MMS4-6HA::TRP1 |
| WDHY2493 | WDHY2491 MUS81/MUS81-6HA::TRP1 |
| WDHY2494 | WDHY2491 MUS81/MUS81-9MYC::TRP1 |
| WDHY2495 | WDHY2491 MMS4/MMS4-9MYC::TRP1 |
| WDHY2612 | W303-RAD5 MATα mus81::KANMX mms4::TRP1 |
| WDHY2733 | W303-RAD5 MATa mus81::KANMX mms4::TRP1 |
| WDHY2783 | W303-RAD5 MATα SLX4-MYC13::KANMX |
| WDHY3267 | WDHY2491 hta1-htb1::LEU2/hta1-htb1::LEU2 MUS81-6HA::TRP1/MUS81-9MYC::TRP1 (hta2-htb2 status unknown), contains pZS145 (pFLAG-HTB1-HTA1::HIS3) |
| WDHY3268 | WDHY2491 hta1-htb1::LEU2/hta1-htb1::LEU2 MUS81/MUS81-6HA::TRP1 (hta2-htb2 status unknown), contains pZS145 (pFLAG-HTB1-HTA1::HIS3) |
| WDHY3269 | WDHY2491 hta1-htb1::LEU2/hta1-htb1::LEU2 MUS81/MUS81-9MYC::TRP1 (hta2-htb2 status unknown), contains pZS145 (pFLAG-HTB1-HTA1::HIS3) |
| WDHY3431 | W303-RAD5 mms4::TRP1 mus81::KANMX yen1::KANMX |
| SFY001c | SLX4-MYC13::KANMX MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 rad5-535 |
| MAO633d | W303 MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 hta1-htb1::LEU2 hta2-htb2 ura3-1::pRG145 UBI4-3HA::URA3 contains pZS145 (pFLAG-HTB1-HTA1::HIS3) |
All strains are W303 background with corrected RAD5 and constructed for this study unless otherwise noted.
Strains generously provided by Rodney Rothstein.
Strain generously provided by John Rouse.
Strain generously provided by Mary Ann Osley.
Fig 9.

HA- and MYC-tagged Mus81 and Mms4 are functional and (His10)Mus81-(GST)Mms4 complements a mus81 mms4 double deletion. (A) HA and Myc tags do not affect Mus81 or Mms4 protein function. Cell cultures containing HA- or Myc-tagged Mus81 (WDHY2420 and WDHY1868, respectively) or Mms4 (WDHY2423 and WDHY1872, respectively) were grown in YPD and diluted to an OD600 of 1, followed by six 5-fold serial dilutions. Cells were plated onto YPD containing 0.015% MMS, 15 μM CPT, 200 mM HU, or no addition. The cells were grown at 30°C for 2 days for MMS, CPT, and no-addition plates and 6 days for HU-containing plates. Images were taken daily to monitor cell growth. (B) (His10)Mus81-(GST)Mms4 retains biological function. The (His10)Mus81-(GST)Mms4 overexpression vector (pWDH722, pMUS81-MMS4) and the empty vector (pWDH403) were transformed into a mus81Δ mms4Δ double-deletion strain (WDHY2733). Three independent transformants were analyzed for growth and genotoxin sensitivity. Transformed strains were grown overnight in standard defined medium containing all required nutrients except uracil (SD-URA). Overnight cultures were diluted to an OD600 of 1, and six 5-fold serial dilutions were plated on YPD and SD-URA medium containing 0.015% MMS, 15 μM CPT, 200 mM HU, or no addition. Images were taken at day 3. On the YPD 15 µM CPT plate, two irrelevant contaminations occurred but are not visible in the figure.
Generation of plasmid pWDH722 for (His)10Mus81-(GST)Mms4 overexpression in S. cerevisiae.
The overexpression vector used to produce Mus81-Mms4 is essentially the same as the previously described (21) pWDH595 containing the GAL1-GAL10 divergent promoter on a 2μm-based overexpression vector. However, pWDH595 contains a tobacco etch virus (TEV) protease sequence between the (His)10 tag and MUS81 which was similar enough in amino acid sequence to the PreScission site on Mms4 to also be cleaved by PreScission protease when glutathione S-transferase (GST) was removed from Mms4. To remove the problematic TEV protease site, MUS81 was PCR amplified with primers introducing MluI sites and cloned into these sites in pWDH595, replacing the original TEV-MUS81 cassette C terminal to the (His)10 tag to result in pWDH722. The overexpression plasmid pWDH722 fully complemented the genotoxin sensitivities of the mus81Δ mms4Δ double mutant strain as well as the mus81Δ mms4Δ yen1Δ triple mutant strain (see Fig. 9B and 16).
Fig 16.

(His10)Mus81-(GST)Mms4 suppresses MMS sensitivity of a mus81 mms4 yen1 triple-deletion mutant. The (His10)Mus81-(GST)Mms4 overexpression vectors (pWDH722, pMUS81-MMS4; pWDH596, pmus81-dd-MMS4 [mus81-dd, mus81-D414A, D415A] [21]) were transformed into a mus81Δ mms4Δ yen1Δ triple-deletion strain (WDHY3431). Transformants were analyzed for growth and genotoxin sensitivity. Transformed strains were grown overnight in SD-URA. Overnight cultures were diluted to an OD600 of 1, and six 5-fold serial dilutions were plated on SD-URA medium containing 0.0075% MMS or no MMS. Images were taken at day 3.
Purification of GST-cleaved (His)10Mus81-Mms4.
(His)10Mus81-Mms4 was purified by two-column (glutathione-Sepharose 4b and nickel-nitrilotriacetic acid) affinity chromatography essentially as described previously (21), with the following modifications. The mass of cells (WDHY668/pWDH722) lysed was reduced to 60 g, and the volume of glutathione resin was increased to 12 ml. After binding GST-Mms4, the column was washed with three column volumes of 50 mM Tris-HCl, pH 7.5, 450 mM NaCl, 1 mM dithiothreitol (DTT), and 1 mM EDTA, after which 3 mg of PreScission protease was added and the resin bed was resuspended. The cleavage of the GST tag from Mms4 was allowed to proceed for 2.5 h at 4°C, and the (His)10Mus81-Mms4 heterodimer was then eluted with 25 ml of the same buffer. This material was applied to a 1-ml HisTrap FF column, and the purification was completed as described previously (21). Protein concentrations were determined by the Bradford assay using bovine serum albumin (BSA) as a standard. GST-cleaved Mms4 retains four additional amino acids, PRET, at the N terminus and is otherwise native, while Mus81 contains the N-terminal tag MRGS(His)10AS.
Gel filtration.
For gel filtration, 5 μg (His)10Mus81-Mms4 was applied to a Superdex 200 GL (GE Healthcare) gel filtration column preequilibrated with GF buffer (20 mM Tris-HCl, pH 7.5, 1 mM EDTA, 1 mM DTT, 10% glycerol, and 500 mM NaCl). The column was developed with GF buffer at 0.5 ml/min using an Åkta purifier system (GE Healthcare) collecting 0.1-ml fractions. Protein levels were determined by immunoblots of 2 μl/fraction using a rat anti-Mms4 antibody. Fractions were immediately assayed for nuclease activity on 3′-flap junctions and Holliday junctions with a fixed core (XO12) as described below.
Sucrose gradient sedimentation.
Sucrose gradients (4.5 ml, 5 to 20%) containing buffer GF with 500 or 150 mM NaCl was poured into 5-ml ultraclear tubes (13 by 51 mm; Beckman) using a plunger-type gradient maker (Jule, Inc.). For gradients containing 150 mM NaCl, Mus81-Mms4 samples (stored in 500 mM NaCl) were predialyzed against GF buffer containing 150 mM NaCl. The dialysis buffer ionic strength was gradually reduced by slowly mixing GF buffer without NaCl into GF buffer with 500 mM NaCl over the course of 4 h. The protein concentration was then redetermined by Bradford assay. Five micrograms of purified Mus81-Mms4 in 50 μl GF buffer was then carefully layered on top of the gradient. Standard protein stocks in GF buffer at 20 mg/ml were then mixed and layered on top of the gradients in a volume of 65 μl consisting of 5 μl BSA, 5 μl ovalbumin, 15 μl aldolase, 15 μl catalase, and 25 μl thyroglobulin. Gradients were centrifuged for 9 h at 55,000 rpm in an SW 55 Ti swinging-bucket rotor using a Beckman Optima LE ultracentrifuge. Gradients were then fractionated from the top down by carefully pipetting 175 μl solution into 26 fractions per gradient. Each gradient was calibrated by determining the position of standard proteins by 10% SDS-PAGE of fraction samples, followed by Coomassie blue staining and densitometry using ImageQuant software (GE) to determine the peak fraction. Peak fraction positions of standard proteins were then plotted against their known sedimentation coefficient (S) values to generate internal standard curves for each gradient. The Mms4 protein peak was determined by immunoblot analysis using rat anti-Mms4 antibody. To avoid further loss of the limited resolution of the technique, if the protein level in two fractions differed by less than 10%, the peak was taken to be between the two fractions (e.g., if fractions 7 and 8 differed by less than 10%, the peak was taken to be fraction 7.5). To avoid possible saturation of immunoblot signals on film, high and low exposures of the same blot were taken and monitored for consistency in the relative signal between fractions. The relative signals were normalized by taking the peak fraction to be 100%. Two exposures normalized in such a way were then averaged.
Nuclease assays.
For gel filtration samples, 2.5 μl of each fraction (in GF buffer containing 500 mM NaCl) was mixed with 7.5 μl of a cocktail to adjust the final reaction to 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 3 mM magnesium diacetate [Mg(OAc)2], 0.25 mg/ml BSA, 1 mM DTT. Reaction mixtures contained 50 nM 3′-flap (defined by unlabeled substrate concentration, containing 1,500 cpm labeled substrate per reaction mixture) or 1 nM molecules for XO12 reactions. Reactions were allowed to proceed for 15 min at 30°C and were then stopped with the addition of 2 μl stop mix (200 mM EDTA, 2.5% SDS, and 10 mg/ml proteinase K). For sedimentation fractions, a representative gradient containing 150 mM NaCl was assayed for cleavage of 3′-flap and XO12 structures in the same manner. The reactions were stopped after 10 min at 30°C with the addition of 2 μl stop mix. One-third of the reaction mixtures (500 cpm) was electrophoresed by 10- by 20-cm native 10% TBE (Tris-borate-EDTA)-PAGE at 100 V for 65 min. Gels were vacuum dried to Whatman paper at 80°C and then exposed overnight to a phosphorimager screen.
S. cerevisiae Slx1-Slx4 (courtesy of S. Brill) was titrated into 10-μl reaction volumes containing 5 nM Mus81-Mms4 and 50 nM substrate (nicked Holliday junction [nXO12], replication fork (RF)-like structure [RF-like], XO12, or Holliday junction with mobile core [X12]). Nuclease reactions were performed in 25 mM HEPES, pH 7.5, 100 mM NaCl, 3 mM Mg(OAc)2, 1 mM DTT, 100 μg/ml BSA, with the substrate concentration defined by that of the unlabeled substrate and with the mixture spiked with a negligible quantity of radiolabeled substrate to report on substrate turnover (the substrate preparation was described previously [21]). After 10 min incubation at 30°C, reactions were quenched at 40 mM EDTA, 0.5% SDS, 2 mg/ml proteinase K before electrophoresis by native 10% TBE-PAGE at 150 V for 1 h. Gels were dried for 1 h at 80°C and then processed for phosphorimaging.
In vitro phosphorylation of purified Mus81-Mms4 by Cdc5 kinase.
S. cerevisiae cells overexpressing 3× hemagglutinin (3HA)–Cdc5 wild-type or 3HA-Cdc5-K110A kinase-defective protein were grown and induced with galactose as described previously (59). Wild-type or kinase-defective Cdc5 was immunoprecipitated from 50 optical density (OD) units of cells, and kinase reactions were performed essentially as described previously (59) with purified Mus81-Mms4 as the substrate. For the experiment whose results are shown in Fig. 3A, 5 μg of Mus81-Mms4 was incubated with IP beads in 80 μl. After 15 min incubation at 30°C, the supernatant was removed from the beads. One-fourth of this material was treated with PP1 phosphatase (2.5 U) with or without prior heating or I2 PP1 inhibitor, as indicated. Reactions were stopped with Laemmli buffer for immunoblot analysis. For the experiments whose results are shown in Fig. 5B and C, the wild-type Cdc5 reaction was carried out in the same manner. As controls, purified Mus81-Mms4 was incubated with PP1 phosphatase or simply in kinase buffer for 15 min. For nuclease assays, heterodimer concentrations were redetermined (to account for loss to nonspecific binding) by anti-Mms4 Western blotting in comparison to purified protein standards.
Fig 3.

Sedimentation analysis of Mus81-Mms4. (A) Overlay of four sedimentation experiments in which the relative levels of heterodimer in the fractions were determined by anti-Mms4 immunoblots. Five to 10 μg (His)10Mus81-Mms4 was separated on 5 to 20% sucrose gradients. Fraction numbers were converted to sedimentation coefficient values (S value) by interpolation from standard curves generated by standard proteins present in the gradients. Each data set was calibrated using its own intragradient standard curve. Gradients were made with GF buffer containing 150 mM NaCl and no magnesium, except for the filled circles gradient, which contained 3 mM Mg(OAc)2, and the open squares gradient, which contained 500 mM NaCl. (B) Activity assays of a representative gradient fractionation (triangles in panel A). Two and a half microliters of each fraction was mixed with 7.5 μl of a cocktail to adjust the final mix to 150 mM NaCl, 3 mM Mg(OAc)2, and the indicated substrate concentrations. In the first lane, 2.5 μl of GF buffer was substituted as a negative control. The peak fraction contains 32 nM Mus81-Mms4, as determined by quantitative immunoblotting. HJ cleavage by Mus81-Mms4 requires excess protein over substrate, as previously found (2, 3). (C) Representative sedimentation standard curve used for S-value interpolation.
Fig 5.
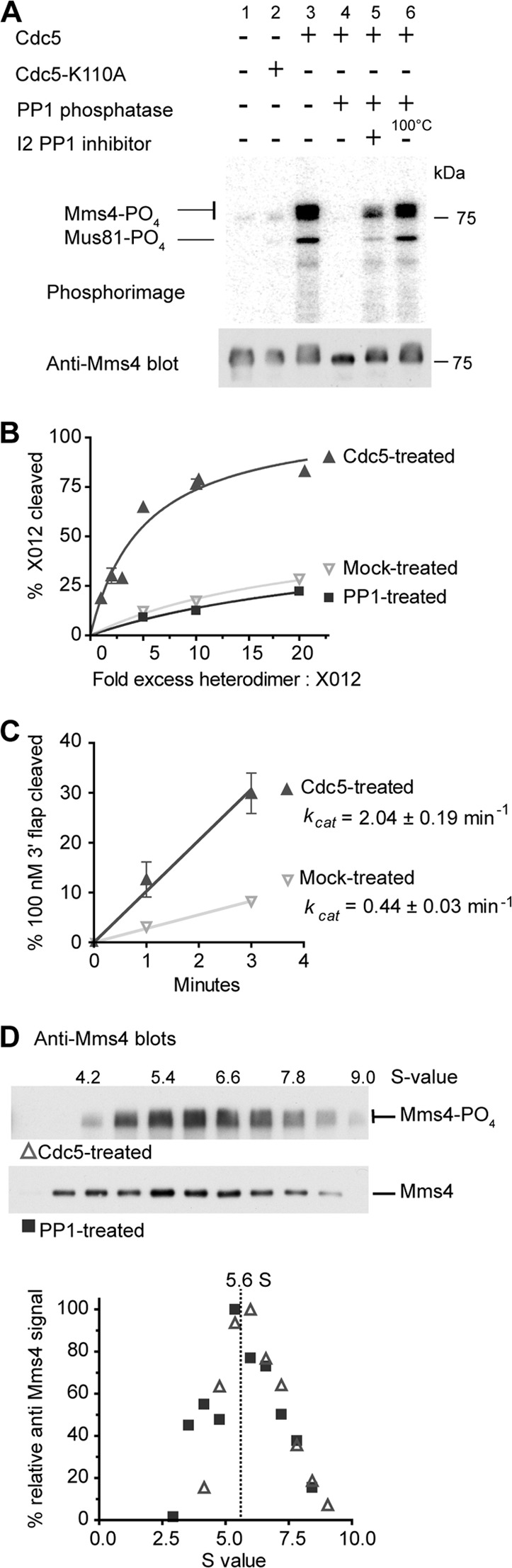
Cdc5 enhances the nuclease activity of Mus81-Mms4 without inducing dimerization of the Mus81-Mms4 heterodimer. (A) In vitro phosphorylation of Mus81-Mms4 by Cdc5. 3HA-Cdc5 or 3HA-Cdc5-K110A was immunoprecipitated and incubated in kinase buffer with purified Mus81-Mms4. A sample of this material was either loaded directly on an SDS-polyacrylamide gel or treated as indicated with PP1 phosphatase with or without I2 PP1 inhibitor. The PP1 added in lane 6 was first heat denatured. The gel was transferred to a PVDF membrane, quantified by phosphorimaging (upper), and then probed with anti-Mms4 antibody (lower). (B) XO12 cleavage is not catalytic, requiring excess protein to junction molecules. Percent XO12 cleavage is plotted as a function of fold excess Mus81-Mms4 and fitted to a hyperbolic curve. (C) Nuclease assays showing the initial rates of 5 nM Cdc5 or mock-treated Mus81-Mms4 on 100 nM 3′-flap substrate. Under these saturated substrate conditions ([substrate] ≫ Km), the initial velocity approximates the maximal velocity and, hence kcat (kcat = Vmax/[enzyme]). Enzyme concentrations were determined by densitometry of anti-Mms4 immunoblots with standards of purified protein. In panels B and C, for values without error bars, the error (±SEM, n = 3) was smaller than the plotting symbol. (D) Gradient sedimentation profiles of Cdc5-treated or PP1-treated purified Mus81-Mms4. Gradient fractions were calibrated to S values using a standard curve of fraction number versus S value of standard proteins present in the gradients.
Electron microscopy and image analysis.
Gradient fixation was performed essentially as described previously (35). Gradients were prepared as described above; however, 0 to 15% sucrose gradients were used in HB [25 mM HEPES, pH 7.9, 1 mM EDTA, 10% glycerol, 1 mM DTT, and 0.5 M or 1.0 M NaCl for (His)10Mus81-(GST)Mms4 and (His)10Mus81-Mms4, respectively, with or without 0.1% formaldehyde]. (His)10Mus81-Mms4 was slowly dialyzed overnight into a final HB solution containing 1.0 M NaCl. Five micrograms of protein was diluted in 200 μl HB with the corresponding salt concentrations and carefully applied to the gradient. Globular protein standards at 20 mg/ml were mixed in a volume of 40 μl (5 μl BSA, 5 μl ovalbumin, 15 μl aldolase, and 15 μl catalase) and applied to gradients without fixative to determine sedimentation values. Samples were centrifuged as described above and fractionated. Protein elution was determined by immuno-dot blot analysis using rabbit anti-Mus81. Protein-containing fractions were used for grid preparation and stained with 0.075% uranyl formate as described previously (44). Grids were visualized using a JEOL JEM-1230 microscope (JEOL Ltd., Japan) operated at an accelerating voltage of 120 keV. Images were acquired using minimum-dose procedures at a nominal magnification of 50,000× on a TVIPS TemCam-F224HD 2,048- by 2,048-pixel charge-coupled-device camera (TVIPS, Germany). Particle selection and classifications were completed using EMAN2 software, stable version 2.0RC3 (55).
Soluble protein immunoprecipitation.
Diploid cells were grown in yeast extract-peptone-dextrose (YPD) to an optical density at 600 nm (OD600) of 2.0, resuspended either in fresh YPD medium or in medium treated with either 0.015% methyl methanesulfonate (MMS), 15 μM camptothecin (CPT), or 200 mM hydroxyurea (HU), and incubated for 2 h. Cells were pelleted, washed in IP-A (20 mM Tris, pH 7.5, 100 mM NaCl, and 1 mM EDTA) containing 1 mM phenylmethylsulfonyl fluoride (PMSF), and flash frozen in liquid nitrogen. Immunoprecipitations of protein and immunoblotting of precipitates were completed as previously described (4) using anti-HA (MMS-101P; Covance, Princeton, NJ), anti-cMyc (Cell Signaling, Danvers, MA), rabbit anti-Mus81, or rabbit anti-Mms4 antibodies.
Chromatin immunoprecipitation (ChIP).
Cultures were grown and treated as described for soluble immunoprecipitation and were subsequently treated with 1% formaldehyde for 15 min, followed by incubation with 125 mM glycine for 5 min. Cells were washed with ice-cold 1× phosphate-buffered saline with 1 mM PMSF before pelleting and freezing. Fifty OD600 units of cells was resuspended in lysis buffer (50 mM HEPES, pH 7.5, 1 mM EDTA, 140 mM NaCl, 1% Triton X-100, 0.1% Na-deoxycholate, 1 mM DTT) with protease inhibitors (1 mM PMSF, 2 μM leupeptin, 1 μM benzamidine, 1.67 μM pepstatin), 1 ml glass beads was added (0.5 mm; Biospec Products, Inc., Bartlesville, OK), and cells were lysed using a Savant FastPrep FP120 device (Bio101) on setting 4 four times for 45 s each time with a 5-min incubation on ice after two cycles. Chromatin and debris were collected by centrifugation, washed two times with fresh lysis buffer, and finally, resuspended in lysis buffer. Bulk chromatin was sonicated to 250-bp fragments using a Diagenode Bioruptor sonicator (Denville, NJ). Debris and sheared chromatin were collected by centrifugation. For DNase assays, the chromatin-containing pellet was washed twice with DNase buffer (40 mM Tris-HCl, pH 7.4, 10 mM NaCl, 6 mM MgCl2, 10 mM CaCl2). Approximately 5.0 × 107 cells were resuspended in 95 μl DNase buffer, into which 5 units of DNase or DNase buffer was added. Samples were incubated at 4°C for 30 min, and the reactions were stopped with the addition of 10 μl 20 mM EGTA, pH 8.0. Cross-links were reversed with the addition of an equal volume of elution buffer (1% SDS in TE [Tris-EDTA], pH 8.0) and incubation overnight at 65°C, followed by the addition of 100 μl proteinase K for an additional 2 h incubation at 37°C. Phenol extraction and ethanol precipitation were used to further extract the DNA. RNA was degraded by the addition of DNase-free RNase and incubation at 37°C for 2 h.
Immunoprecipitation was carried out as previously described (36) with either anti-HA (Covance, Princeton, NJ), anti-Myc (Cell Signaling, Danvers, MA), 10 μl rabbit anti-Mus81 serum, or anti-histone H3 antibody (AbCam, Cambridge, MA). Immunoprecipitates were boiled for 10 min in 1× Laemmli buffer, separated on an SDS-polyacrylamide gel, transferred to nitrocellulose, and probed using rat anti-Mus81 and anti-Myc antibodies.
RESULTS
Hydrodynamic properties of Mus81-Mms4 identify a single heterodimer in vitro.
We originally purified Mus81-Mms4 from the cognate S. cerevisiae cells as a doubly tagged, (His)10Mus81-(GST)Mms4 heterodimer (21). Since GST is known to induce dimerization, which would interfere with our analysis, we proteolytically removed the GST tag and performed the analysis with the (His)10Mus81-Mms4 heterodimer (Fig. 1A). To establish the native molecular weight (Mr) of (His)10Mus81-Mms4, we determined its hydrodynamic properties. Together, the Stokes radius and sedimentation coefficient allow a very reliable estimation of the native Mr using the equation of Siegel and Monty (Fig. 1B, equation 1) (51).
Fig 1.

Mus81-Mms4 is a single heterodimer in solution. (A) Denville blue stain of (His)10Mus81-Mms4 heterodimer (1.5 μg). (B) Equations used to calculate the hydrodynamic properties of Mus81-Mms4 (51). M is the molecular mass (in daltons), N is Avogadro's number (6.022 × 1023), a is the Stokes radius (Mus81-Mms4 = 6.83 × 10−7 cm), S is the sedimentation coefficient (Mus81-Mms4 = 5.60 × 10−13 s), ν̄ is the partial specific volume of the protein (Mus81-Mms4 = 0.726 cm3/g), ρ is the density of water at 293 K (0.9982 g/cm3), and η is the viscosity of water at 293 K (0.01 g cm−1 s−1) f/fo is the frictional ratio (unitless). (C) Hydrodynamic properties of Mus81-Mms4. Errors are standard deviations of independent determinations (n = number of determinations). (D) Mus81-Mms4 elution profile from a Superdex 200 GL column (for raw data, see Fig. 2). Five micrograms (His)10Mus81-Mms4 was applied to the column, and fractions were assayed for activity on 3′-flap and XO12 junctions. (E) Sedimentation profile of Mus81-Mms4 on sucrose gradients (for raw data, see Fig. 3). Five micrograms (His)10Mus81-Mms4 was applied to the gradient, fractionated after ultracentrifugation, and assayed for activity on 3′-flap and XO12 junctions. Protein levels in each fraction were determined by anti-Mms4 immunoblots.
During gel filtration, (His)10Mus81-Mms4 elutes at a volume corresponding to a Stokes radius of 68 Å (Fig. 1C and D), which for a globular protein would correspond to 450 kDa. This value is in the range of values previously reported for S. cerevisiae Mus81-Mms4 and S. pombe Mus81-Eme1 (23, 26). However, to determine the native Mr, it is imperative to also determine the sedimentation coefficient, which yielded an S value of 5.6 (Fig. 1C and E). This is smaller than that expected for a single heterodimer. Using equation 1, these data result in a native molecular mass of (His)10Mus81-Mms4 of 158 ± 8 kDa, which is in excellent agreement with the predicted molecular mass of 153.6 kDa calculated from the amino acid sequence (Fig. 1C). Purified recombinant Mus81-Mms4 is therefore a single heterodimer in solution. Activity assays on the fractions from gel filtration and sucrose gradients revealed that the robust nuclease activity on 3′-Flap and the feeble activity on XO12 HJs coeluted with (His)10Mus81-Mms4 protein levels. There were no fractions containing enhanced activity on XO12 HJs compared to the 3′-flap nuclease activity (Fig. 1D and E, 2, and 3). The longer tails of the 3′-flap nuclease activity profile compared to the protein profile are related to the much higher sensitivity of the nuclease assay than the immunoblot (Fig. 1D and E). Robust cleavage of 50 nM 3′-flap was observed in fractions containing (His)10Mus81-Mms4, while XO12 cleavage could be visualized only in reactions with 1 nM substrate (Fig. 2 and 3). The relatively higher XO12 cleavage activity in the fractions from the sedimentation analysis (Fig. 1E) compared to the fractions from the gel filtration analysis (Fig. 1D) is due to the higher Mus81-Mms4 concentration in the fractions of the sedimentation analysis. Mus81-Mms4 cleaves HJs such as XO12 only when it is in significant excess of protein over substrate (see references 21 and 23 and references therein; Fig. 2 and 3; see also Fig. 5B). The sedimentation profiles were not changed in the presence or absence of 3 mM magnesium or by high (500 mM NaCl) versus optimal (150 mM) salt concentrations (21), indicating that these factors do not influence the oligomeric status of (His)10Mus81-Mms4 (Fig. 3).
Fig 2.

Endonuclease activity correlates with Mus81-Mms4 concentration for both 3′-flap and XO12 structures across the gel filtration elution profile. (A) Mus81-Mms4 elution profile from a Superdex 200 GL column eluted in the presence of 500 mM NaCl. Five micrograms (His)10Mus81-Mms4 was applied to the column and fractionated into 0.1-ml fractions, which were immediately assayed for activity on 3′-flap and XO12 junctions and for heterodimer protein level by anti-Mms4 immunoblots. Asterisks, 5′ 32P labels; double dagger, elution peak fraction; triangle, outlier not found in a second independent experiment, excluded in the quantification in Fig. 1C. (B) (Top) The peak gel filtration fractions spanning elution volumes (Ve) of 9.4 to 10.5 ml (fractions 20 to 30) were titrated against increasing concentrations of XO12 substrate. Vo, column void volume, measured by the elution volume of blue dextran (8.008 ml); Vt, total column volume (24 ml). Substrate concentration was defined by the amount of unlabeled XO12 junction present. The peak fraction contains 6 nM Mus81-Mms4, as determined by quantitative immunoblotting. HJ cleavage by Mus81-Mms4 requires excess protein over substrate, as previously found (2, 3). (Bottom) The top images with the contrast adjusted in ImageQuant (GE) to allow the low substrate cleavage to be visualized. (C) Standard curve used to interpolate the Stokes radius from the elution volume. Three hundred micrograms of standard proteins was applied to the column in separate runs and detected by UV absorbance at 280 nM. The average elution volume from 2 to 3 separate runs per standard was used to generate the standard curve. The equation shown defines Kav, which is the fraction of the gel phase of the column available to the protein, determined by the Stokes radius of that protein.
Mus81-Mms4 binds DNA joint molecules as a single heterodimer.
Since it is possible that Mus81-Mms4 multimerizes only when bound to DNA, we performed DNA binding experiments to test this possibility. Mus81-Mms4 (650 nM) was applied to 1 nM 5′ end 32P-labeled 3′-flaps or XO12 HJs, and the S value of these complexes was determined by sucrose gradient sedimentation. The shift in the DNA S value induced by Mus81-Mms4 binding in comparison to the values for gradients containing DNA alone was 4.4S and 4.8S for 3′-flap and XO12 HJ, respectively (Fig. 4A). This suggests that the same form of Mus81-Mms4 binds both structures. The magnitude of the DNA shifts was similar to the S value of the heterodimer in solution (5.6S) (Fig. 4B), strongly supporting the conclusion that a single heterodimer is also the species that binds DNA. It is noteworthy that even the high excess of Mus81-Mms4 (650-fold) to substrate did not lead to dimerization on the substrate.
Fig 4.
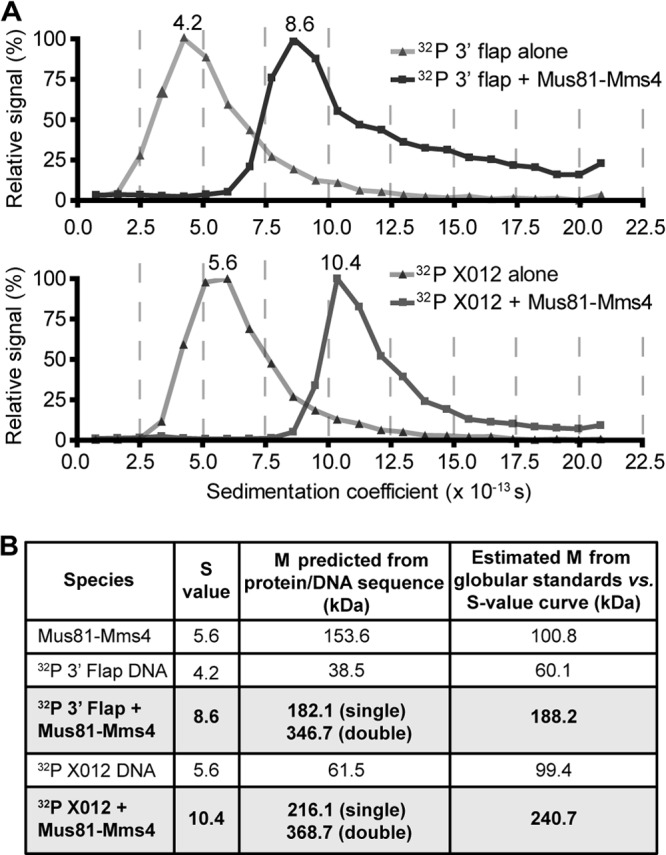
Mus81-Mms4 binds as a single heterodimer to 3′-flaps and Holliday junctions. (A) Five micrograms (650 nM) (His)10Mus81-Mms4 was added to limiting 32P-labeled 3′-flap or XO12 junctions (∼200,000 cpm, <1 nM) in 50 μl GF buffer and applied to sucrose gradients. DNA levels in each fraction were assessed by scintillation counting, and the data were normalized with the peak fraction, equal to a 100% signal. Values listed above the peaks are the S values interpolated from standard curves generated for each gradient. (B) Comparison of S values with predicted molecular mass (M) for a (His)10Mus81-Mms4 heterodimer or tetramer bound to DNA and estimates of molecular mass based on globular standards. While inaccurate in predicting the molecular mass of nonglobular proteins, the relative changes in S values shown are nonetheless informative.
Mus81-Mms4 nuclease activity is stimulated by Cdc5 in vitro without leading to Mus81-Mms4 multimerization.
Using immunoprecipitated Mus81-Mms4, Matos et al. (40) showed that Cdc5 kinase-dependent phosphorylation of Mms4 activated Mus81-Mms4 nuclease activity on oligonucleotide-based HJs. While the mechanism of activation remains to be determined, it is possible that Mms4 phosphorylation induces tetramerization of the Mus81-Mms4 heterodimer. To test this notion, we performed in vitro phosphorylation experiments using purified Mus81-Mms4 and Cdc5 kinase immunoprecipitated from budding yeast cells expressing an HA-tagged Cdc5 that was shown to be functional (59). Our Mus81-Mms4 is purified from budding yeast and known to be posttranslationally modified (21), consistent with the electrophoretic mobility of the Mms4 subunit shown in the lower panel of Fig. 5A. Incubation with catalytically active Cdc5 leads to additional phosphorylation, as indicated by the incorporation of radiolabeled ATP (Fig. 5A, lane 3). This additional phosphorylation is strongly reduced when using the catalytically defective Cdc5-K110A (lane 2), demonstrating the dependence of Mms4 in vitro phosphorylation on active Cdc5 kinase. Mms4 phosphorylation is abolished by phosphatase treatment, as shown by a lack of radiolabel and increased electrophoretic mobility (lane 4), but preserved when either phosphatase inhibitor (lane 5) or heat-inactivated phosphatase (lane 6) is added. In addition, we note that Mus81 is also phosphorylated by Cdc5 (Fig. 5A). As expected from the previous report (40), Cdc5 activates Mus81-Mms4 nuclease in vitro on an HJ substrate, while no enhancement of endonuclease activity is observed for phosphatase-treated or mock-treated controls (Fig. 5B). Cdc5 also activated the 3′-flap cleavage activity of Mus81-Mms4 (Fig. 5C), which is consistent with observations made with the immunoprecipitated Mus81-Mms4 (40; S. West, personal communication) that Cdc5 activation does not change Mus81-Mms4 substrate preference for nicked substrates over HJs. The kcat of Mus81-Mms4 for 3′-flaps was stimulated about 5-fold (Fig. 5C), whereas we could not determine the kcat for the HJ substrate because an excess of Mus81-Mms4 over substrate was required with either Cdc5-treated or untreated/phosphatase-treated Mus81-Mms4 (Fig. 5B). These results demonstrate that Cdc5 phosphorylates Mms4 and Mus81 in vitro, leading to a general activation of the Mus81-Mms4 nuclease activity.
To test whether activation is accompanied by tetramerization of the Mus81-Mms4 heterodimer, we determined the S value of Cdc5-treated and control phosphatase-treated Mus81-Mms4 by gradient centrifugation (Fig. 5D). The S value of 5.6 remained unchanged by phosphorylation (Fig. 5D; see also Fig. 1C). This result eliminates dimerization as a mechanism of Mus81-Mms4 activation, consistent with the observation that Cdc5-mediated activation does not change substrate specificity for substrates requiring one (3′ flaps) or two (HJ) active sites.
Visualization of single particles of Mus81-Mms4 shows predominately single heterodimers.
We used transmission electron microscopy (TEM) to visualize individual particles of (His)10Mus81-Mms4 (Fig. 6). Conditions for conventional negative-stain sample preparation were not conducive for the isolation of single particles due to the propensity of the protein to aggregate on carbon grids. Therefore, we slowly cross-linked the proteins by gradient fixation to stabilize individual protein complexes. The presence of cross-linker, necessary to visualize independent particles, resulted in a shift in the sedimentation coefficient value from 5.7 to 7.3 (Fig. 7A and B). This larger value corresponds to a globular protein with a molecular mass of 141 kDa. Visualization of the particles by TEM is consistent with this value, showing a globular single Mus81-Mms4 heterodimer (Fig. 6A and B). Volumetric analysis confirms the presence of a predominately single heterodimer population (Fig. 6C and D). A subset of particles (∼30%) showed larger volumes (Fig. 6B to D). Such particles are likely the result of random cross-linking, as indicated by the occurrence of some particles that correspond to trimers and the absence of a pattern to suggest specific tetramer formation. A complex of three heterodimers would result in cleavage products that appear to have little physiological relevance. The complete absence of assemblies larger than a heterodimer in the sedimentation and gel filtration analysis of non-cross-linked Mus81-Mms4 (Fig. 1D) excludes the possibility that cross-linking prevented multimerization.
Fig 6.
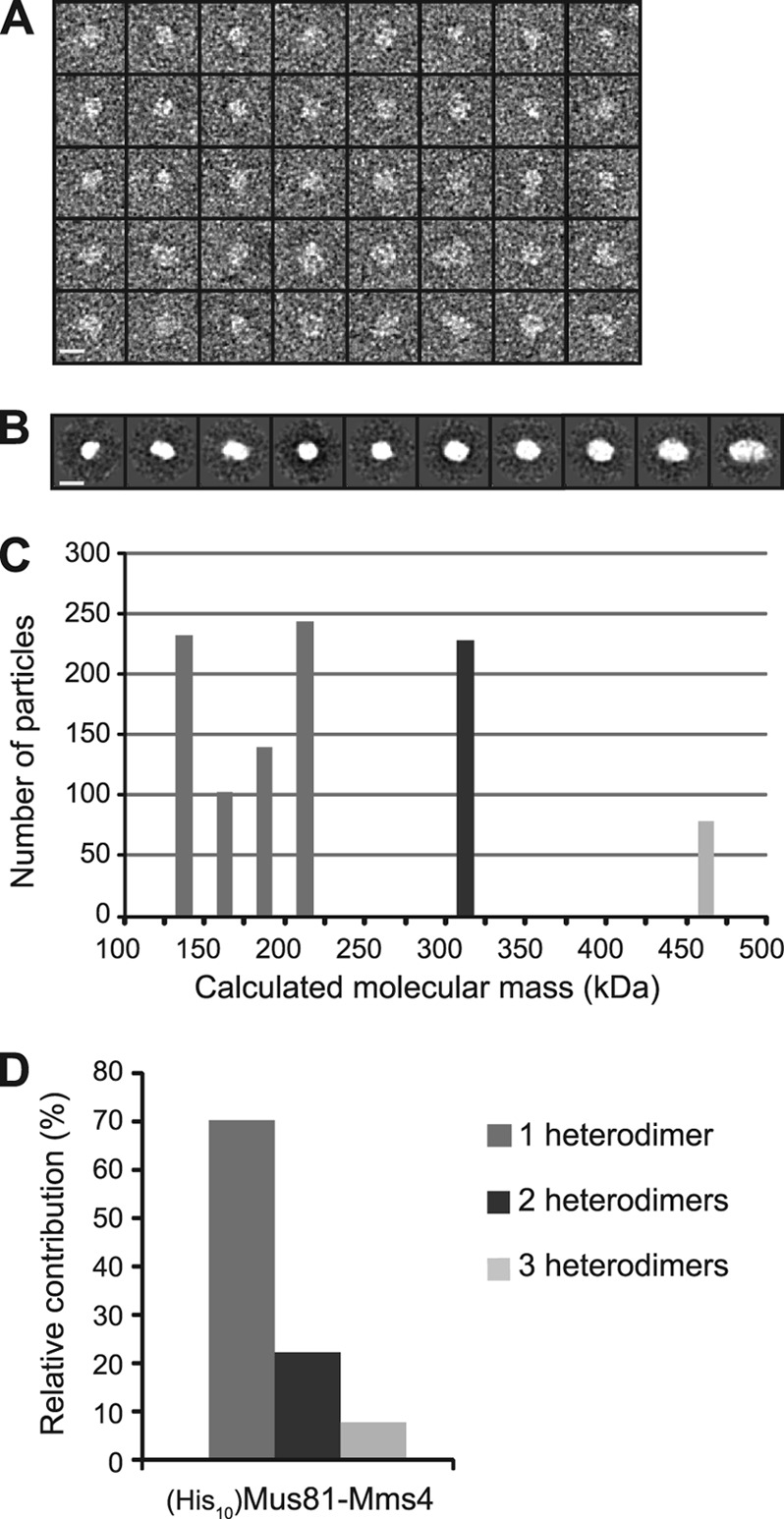
Structural analysis by transmission electron microscopy. (A) Using EMAN2 software, (His10)Mus81-Mms4 particles were selected for subsequent analysis. Results for a panel of 40 representative particles (n = 1,024 total) are shown. Bar, 10 nm. (B) Particles were grouped into 10 classes from which length measurements were made for each class average in the longest dimension and the perpendicular axis. Bar, 10 nm. (C) Volumetric analysis was conducted by assuming an ellipsoid shape and with a conversion factor of 0.82 Da/Å3 (2). The calculated molecular mass for each class was placed into bins plotted as a histogram relative to the number of particles within each class. (D) Expected molecular masses for the heterodimer, tetramer, and hexamer, as determined by amino acid sequence, are 153, 308, and 462 kDa, respectively. The relative contribution of each oligomeric class to the total number of particles is presented, as defined in panel B.
Fig 7.

Cross-linking results in a shift in sedimentation coefficient value and collapse of the protein complex into a globular form. (A) Protein sedimentation profile for (His10)Mus81-(GST)Mms4 in the presence and absence of formaldehyde cross-linker. (B) Table summary of sedimentation coefficient values (S-value) for both untagged (His10)Mus81-Mms4 and (His10)Mus81-(GST)Mms4-purified protein. Calculated Mrs are given as determined by the globular standard (indicated gMr), as are the predicted Mrs for the single heterodimer and dimer of heterodimers. (C) Eight randomly selected (His10)Mus81-(GST)Mms4 protein particles acquired with negative-stain transmission electron microscopy are shown along with their associated class averages. Bar, 10 nm. (D) Relative contribution of elongated particle classes between (His10)Mus81-Mms4 and (His10)Mus81-(GST)Mms4 to the total number of particles. Elongated particles were defined as having a calculated ratio greater than 1.5 when comparing measurements of the longest particle diameter against the corresponding perpendicular diameter.
To corroborate the conclusion that Mus81-Mms4 does not form a dimer of heterodimers, we performed single-particle analysis with the GST fusion protein (His)10Mus81-(GST)Mms4, which favors dimer formation through GST (Fig. 7B to D). As predicted, the S value upon fixation was two times that of the cross-linked untagged protein, and the fusion protein eluted with an S value of 15.1. This value is consistent with a globular molecular mass of 340 kDa, close to the predicted molecular mass of the heterotetramer at 356 kDa. Congruent with these findings, single-particle analysis showed that a majority of the particles with (His)10Mus81-(GST)Mms4 had an estimated volume consistent with a dimer of heterodimers (Fig. 7B). Control experiments to look at protein particles under tilted conditions (Fig. 8) excluded artifacts due to particle orientation or understaining. In sum, the TEM analysis independently confirms that Mus81-Mms4 primarily exists as a single heterodimer.
Fig 8.

Electron microscopy tilt analysis reveals predominantly globular protein population and sufficient z-axis information for volumetric studies. (A) Tilt analysis of negatively stained samples is critical for determining the reliability of z-axis information. From a top view, two particles can look identical but in reality have very different structures that are apparent only once the particle plane is tilted to an appreciable degree. Comparing the measured diameters of each particle both tilted and nontilted can determine if the stain is sufficiently thick to permit visualization of the entire particle and validate statistical analysis and classification. Additionally, tilt analysis can identify whether the purified sample exists as a heterogeneous population of different sizes or if each independent particle view is representative of an equivalent macromolecular complex. The tilt series images were taken at 0 and −47.6 degrees, and particle dimensions were measured using ImageJ software (1). θ = 47.6°. (B) Using micrographs collected for (His10)Mus81-Mms4, particles (n = 60) were selected and measured. Calculations of expected particle widths were determined assuming no z-axis information (ap) and compared to the measured x-axis lengths of the tilted images. (C) Two representative particles at 0° and −47.6°, one that had measurements compatible with a globular shape (top) and a second whose measurements predicted an elongated structure in the z axis (bottom).
Mus81-Mms4 is a single heterodimer in vivo.
There remains a possibility that posttranslational modifications, other than Cdc5-mediated phosphorylation, or accessory factors facilitate tetramer formation of Mus81-Mms4 in vivo. To determine the Mus81-Mms4 oligomeric state in vivo, we performed coimmunoprecipitation experiments in diploid cells containing either HA- and Myc-tagged Mus81 or HA- and Myc-tagged Mms4 expressed from their endogenous promoters. The tagged constructs were verified to be functional by wild-type levels of growth on genotoxin-containing medium (Fig. 9A). Immunoblot analysis of the fusion proteins showed expression levels similar to those of the native proteins (Fig. 10A and 11). No self-association into a larger oligomer was observed for either Mus81 or Mms4 (Fig. 10B and C). We also performed these experiments under conditions of genotoxic stress (HU, CPT, and MMS), which require Mus81-Mms4 for survival. Again, under no conditions did we obtain evidence for self-association of Mus81 or Mms4 (Fig. 10B and C). The detection limit of the antibodies was experimentally determined in reconstruction experiments to be 5 to 10%, depending on the antibody and protein (data not shown). This sensitivity ensures that we would have detected tetramers to this level and supports the conclusion from the EM experiments that the larger than heterodimeric assemblies are the result of nonspecific random cross-linking (Fig. 6). Using diploid control strains with either Myc- or HA-tagged Mus81, we demonstrated the specificity of the antibodies used for immunoprecipitation (Fig. 10B, first four lanes). In conclusion, S. cerevisiae Mus81-Mms4 exists as a single heterodimer in vivo.
Fig 10.
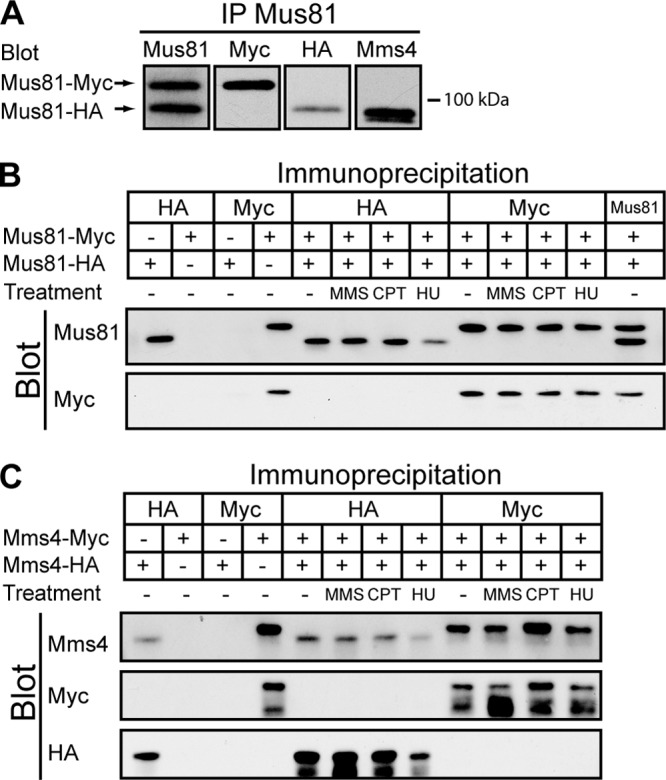
Neither Mus81 nor Mms4 self-associates into a larger oligomeric structure in vivo. (A) Mus81 was immunoprecipitated from WDHY2456 cell extracts containing endogenous HA- and Myc-tagged Mus81. Samples were separated by electrophoresis and subject to immunoblot analysis using anti-Mus81, anti-Myc, anti-HA, and anti-Mms4 antibodies. (B) Mus81-Myc and Mus81-HA proteins were immunoprecipitated from untreated or MMS-, CPT-, and HU-treated cell extracts with either anti-HA, anti-Myc, or anti-Mus81 antibodies. Immunoprecipitated products were analyzed by immunoblotting with anti-Mus81 and anti-Myc antibodies. Lane numbers represent unnumbered lanes from left to right, respectively, as follows: 1 and 3, WDHY2493; 2 and 4, WDHY2494 antibody specificity controls; 5 to 13, WDHY2456. (C) An analysis similar to that described for panel B was performed on strains containing HA- and Myc-tagged Mms4. Note that Mms4 is very sensitive to proteolysis, leading to truncated forms of the protein that are visible with the anti-Myc and anti-HA antibodies. Lane numbers represent unnumbered lanes from left to right, respectively, as follows: 1 and 3, WDHY2492; 2 and 4, WDHY2495 antibody specificity controls; 5 to 12, WDHY2455.
Fig 11.

HA and Myc tags do not severely affect Mus81 or Mms4 protein levels. (A) Strains containing HA- or Myc-tagged Mus81 (WDHY2420 and WDHY1868, respectively) or Mms4 (WDHY2423 and WDHY1872, respectively) were grown to an OD600 of 1, and protein was extracted by bead disruption and trichloroacetic acid precipitation. Fifty micrograms of total lysate was separated by electrophoresis, transferred to nitrocellulose, and subsequently immunoblotted with anti-Mus81, anti-Mms4, anti-HA, or anti-Myc antibodies to determine steady-state protein levels. Probing with anti-3-phosphoglycerate kinase (3-PGK) antibodies served as a loading control. (B) Quantification of Mus81 protein signal. (C) Quantification of Mms4 protein signal. For panels B and C, error bars represent the standard deviation for three independent experiments.
Mus81-Mms4 does not dimerize when bound to chromatin.
Next, we wanted to test the possibility that Mus81-Mms4 dimerizes when bound to chromatin. Using the same approach used in the solution co-IP experiments, we employed Mus81-HA and Mus81-Myc in chromatin IP (protein co-ChIP experiment [ Fig. 12A and B]) from diploid cells and found no evidence for self-association of Mus81 protein when bound to chromatin. Mus81-Mms4 cofractionated with histone H3, showing that chromatin-bound Mus81 was indeed analyzed (Fig. 12A). This conclusion was confirmed by additional control experiments that show that Mus81 and histone H3 can be solubilized from the chromatin-containing pellet by sonication, which fragments the DNA to a size of mostly 250 to 1,000 bp (Fig. 13). Also under conditions of relevant genotoxic stress (HU, CPT, and MMS), no Mus81 self-association could be detected by co-ChIP (Fig. 12B). Using diploid control strains with either Myc- or HA-tagged Mus81, we demonstrated the specificity of the antibodies used in the co-ChIP experiments (Fig. 12B, first four lanes). These results show that Mus81-Mms4 does not dimerize when bound to chromatin in vivo.
Fig 12.
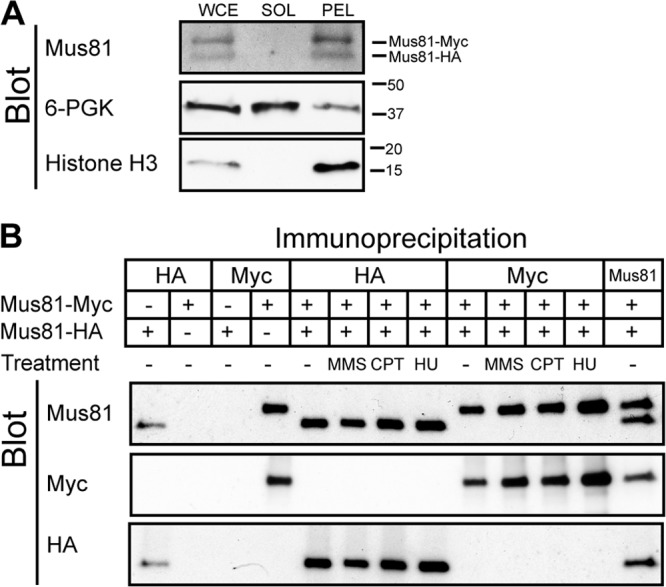
Mus81 does not self-associate when chromatin bound. (A) Formaldehyde-fixed cells (WDHY2456) expressing Myc- and HA-tagged Mus81 were lysed, and whole-cell extract (WCE) was isolated. Centrifugation separated soluble proteins (SOL) from the chromatin-containing pellet (PEL). Protein from each fraction was prepared by trichloroacetic acid precipitation and separated by SDS-PAGE prior to transfer to nitrocellulose. The membrane was probed using anti-Mus81, anti-3-phosphoglycerate kinase, and anti-histone H3 antibodies. (B) Co-chromatin immunoprecipitation analysis. Cells were treated as described for panel A, and the pellets were washed three times in fresh lysis buffer, sonicated, and cleared of precipitated proteins and debris prior to immunoprecipitation using anti-HA, anti-Myc, and anti-Mus81 antibodies. Immunoprecipitated samples were separated on an SDS-polyacrylamide gel, transferred to nitrocellulose membrane, and probed using anti-Mus81, anti-Myc, and anti-HA antibodies. Lane numbers represent unnumbered lanes from left to right, respectively, as follows: 1 and 3, WDHY3268; 2 and 4, WDHY3269 antibody specificity controls; 5 to 14 WDHY3267. For both panel A and panel B, membranes were stripped prior to each subsequent blot.
Fig 13.

Mus81 is solubilized from the chromatin pellet after sonication. (A) After cell lysis of cross-linked cells (WDHY2456; same results for WDHY3267 are not shown), whole-cell extract (WCE), soluble (SOL), and pellet (PEL) fractions were collected. The pellet fraction was washed three times, first in lysis buffer (W1) and then with an additional two washes in lysis buffer (W2S and W3S) or DNase buffer (W2D and W3D) for sonication and DNase experiments, respectively. The pellet fraction was then resuspended in either DNase or lysis buffer for the respective treatments with 5 units DNase, a single round of sonication, or four rounds of sonication (10 s at 15% impulse) or no treatment. Posttreatment, the samples were centrifuged into a soluble (Psol) and pellet (Ppel) fractions. All samples were precipitated with trichloroacetic acid, and protein concentrations were determined by Bradford analysis. Twenty-five micrograms of protein or the entire volume from each fraction was loaded on a 4 to 20% SDS-polyacrylamide gel. Proteins were transferred to nitrocellulose and blotted using anti-histone H3 and anti-Mus81 antibodies. (B) DNA was extracted from the samples for panel A at the step of posttreatment for the total pellet sample and then the soluble and pellet fractions to examine DNA distribution. (C) The total amount of DNA was calculated for each sample to analyze the DNA after the respective treatments.
S. cerevisiae Mus81-Mms4 does not interact with SLX4.
S. cerevisiae Slx1-Slx4 was identified together with Mus81-Mms4 in a screen for sgs1 synthetic lethals and also forms a heterodimeric endonuclease (24). Recent analyses in mammalian cells have suggested that Slx4 may be acting as an endonuclease scaffold to facilitate HJ cleavage (reviewed in reference 54). However, the presence of Slx4-Slx1 does not stimulate substrate cleavage or change the substrate specificity of purified Mus81-Mms4 in vitro (Fig. 14A and B). In congruence with these studies, in S. cerevisiae, no coprecipitation was observed between Slx4 and Mus81 or Mms4 (Fig. 14C). This suggests that budding yeast Mus81 is not interacting with Slx4 to facilitate coordinated cleavage of DNA junctions. The involvement of S. cerevisiae Slx1-Slx4, another nuclease proposed to cleave HJs, in processing recombination-mediated joint molecules is unlikely, because the synthetic lethality between mutations in Slx1-Slx4 and Sgs1 is not suppressed by a recombination defect (24). This shows that the synthetic lethality between Slx1-Slx4 and Sgs1 is not related to recombination but, rather, is probably related to DNA replication (24), and a similar conclusion was reached for fission yeast Slx1-Slx4 (17).
Fig 14.

S. cerevisiae Slx1-Slx4 and Mus81-Mms4 do not interact, are independently nucleolytically active, and do not modulate the nuclease activity of one another. (A) The Mus81-Mms4 concentration was fixed at 5 nM, and Slx1-Slx4 was titrated either in the absence or in the presence of Mus81-Mms4 on four substrates: X12 (Holliday junction with branch point migratable over a 12-bp distance), RF-like, XO12 (Holliday junction with immobile branch point), and nXO12 (nicked Holliday junction). Reaction mixtures were incubated for 10 min at 30°C. Asterisks on joint molecules represent radiolabeling on respective oligonucleotides or, in the case of lanes, label the addition of 5 μl Slx1-Slx4 storage buffer. Arrows indicate approximate cleavage sites of both enzymes: for the replication fork-like substrate, cleavage could occur on either strand (Mus81-Mms4 upper strand, Slx1-Slx4 lower strand). (B) Quantitation of the results shown in panel A. (C) Tagged Slx4-13Myc and native Mus81 and Mms4 were immunoprecipitated from both untreated and MMS-, CPT-, and HU-treated cell extracts using antibodies against raised against Myc, Mus81, or Mms4 (WDHY2783). Immunoprecipitation using anti-Myc antibodies in an untagged strain served as a negative control. Precipitated proteins were separated by electrophoresis and subjected to sequential immunoblot analysis, using anti-Myc,-Mus81, and -Mms4 antibodies. Antibodies were stripped from the membrane prior to each subsequent blot.
DISCUSSION
Key to understanding the biological function(s) of the Mus81-Mms4 structure-selective endonuclease is the nature of its in vivo substrates, specifically, whether it cleaves HJs with four uninterrupted strands or nicked junctions, D-loops, and flaps. HJ resolvases, such as bacterial RuvC and RusA, T4 endonuclease VII, archaeal Hje, or mitochondrial Cce1, exist as dimers, delivering coordinated cleavage across the junction using two active sites (37). Hence, it is critical to determine whether Mus81-Mms4 exists as a higher-order multimer.
The hydrodynamic data demonstrate that Mus81-Mms4 is a single heterodimer. Further analysis (Fig. 1B, equation 2) suggests a heterodimer with nonglobular dimensions, with hydrodynamic properties similar to those of Rad17-Rfc2, -Rfc3, -Rfc4, and -Rfc5, which adopt a washer-like structure (50) akin to the spiral formed by the classic Rfc1-5 clamp loader (11) (Fig. 15). The large Stokes radius of the native complex explains why previous studies had concluded that Mus81-Mms4 was either a dimer (26) or a trimer (23) of heterodimers solely on the basis of gel filtration results. While we hoped to reveal the native structure of Mus81-Mms4 by electron microscopic analysis, the necessary cross-linking procedure appeared to collapse Mus81-Mms4 into a more globular shape. However, the structural analysis by TEM supports the hydrodynamic data, identifying a predominant single heterodimer population. The larger oligomer classes likely result from nonspecific intermolecular cross-linking of individual protein particles to result in dimers and trimers of the heterodimers.
Fig 15.
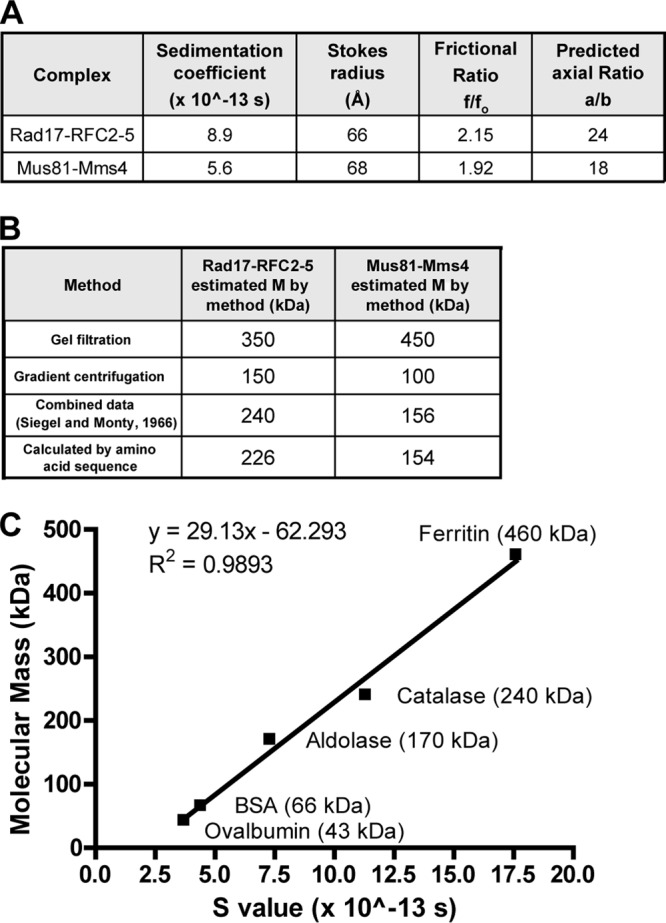
Hydrodynamic properties analysis. (A) Comparison of the hydrodynamic properties of Mus81-Mms4 to Rad17-RFC2-5; (B) comparison of the molecular mass (M) estimates by method of Mus81-Mms4 and Rad17-RFC; (C) curve relating known molecular masses to S value for a set of globular proteins, used to estimate molecular mass in panel B and in Fig. 4B.
Analysis of in vivo subunit associations under physiological protein concentrations shows no self-association of either Mus81 or Mms4 under normal growth conditions or in situations of replicative stress. Contrary to our findings, work in mammalian cells has identified coprecipitation of Mus81 subunits using an overexpression transfection system (7). Two possible explanations for the discrepancy are species-specific oligomerization or nonspecific homodimerization as a consequence of overexpression. Biochemical analysis showed that purified full-length human Mus81-Eme1 recombinant protein exhibits little activity on HJs (13), which does not support the notion of species-specific oligomerization. Another possibility is that the interactions observed under overexpressed conditions may not accurately reflect what occurs at physiological protein levels. Other XPF endonuclease family members have been shown to artificially associate when overexpressed or when their obligate partners are not present (15, 18, 25). Our analysis of the association of the Mus81 and Mms4 proteins at their endogenous levels eliminates any concern about artificial dimerization and provides a clearer depiction of what is occurring in vivo.
Four lines of evidence support the model that S. cerevisiae Mus81-Mms4 cleaves DNA junctions with at least one strand interruption but not single or double HJs (see Fig. 17). First, biochemical analyses demonstrate that HJs are an extremely poor substrate for Mus81-Mms4; in fact, they are so poor that even 5′-flaps, which are not considered a Mus81-Mms4 substrate, are cleaved catalytically, whereas HJs are not (21, 23). Second, the biochemical and in vivo analysis presented here demonstrates that Mus81-Mms4 does not exist in higher-order assemblies of the basic heterodimer structure in vitro or in vivo in solution or when bound to DNA. This implies that Mus81-Mms4 is inherently incapable of providing the requisite coordinate cleavage of HJs. Third, even when Mus81-Mms4 is forced into a tetrameric architecture by using a GST tag on Mms4, a fusion protein that is fully functional in vivo (Fig. 7B), the purified enzyme essentially does not cleave HJs (21). Fourth, elegant genetic analysis by Symington and coworkers (28) showed that Mus81-Mms4 largely controls CO formation in vegetative S. cerevisiae cells and that Yen1 primarily controls CO formation in the absence of Mus81-Mms4, such that COs are eliminated in the mus81 yen1 double mutant. Likewise, an additional mutation in YEN1 further sensitizes Mus81- or Mms4-deficient cells to genotoxic stress and exacerbates its defect in sister chromatid recombination (8, 39, 42, 56). To eliminate the possibility that the construct for Mus81-Mms4 purification specifically affects HJ cleavage over 3′-flap activity, we demonstrated that the expression construct shows no synthetic interaction with a yen1 mutation and fully suppresses the mus81 mms4 yen1 triple mutant (Fig. 16).
The virtual absence of a single mutant phenotype for Yen1-deficient cells suggests that in wild-type S. cerevisiae, most COs are controlled by Mus81-Mms4 processing of a nicked joint molecule intermediate with possibly a minor CO pathway through Yen1 involving classic HJs or dHJs (1, 8, 28, 56). Recent results concerning the regulation of Mus81-Mms4 and Yen1 activity in somatic and meiotic cells support this conclusion (40). Consistent with this study (40; S. West, personal communication), we find that Cdc5-mediated activation does not change the substrate specificity of Mus81-Mms4 and that activated Mus81-Mms4 still vastly prefers 3′ flaps (and, presumably, other nicked substrates) over HJs (Fig. 5B and C). In Mus81-Mms4-deficient cells, Yen1 may directly cleave either the Mus81-Mms4 substrates (nicked substrates or D-loops) or their ligated processing products (single or double HJs), although dHJ cleavage has not yet been demonstrated for Yen1. This model is highly consistent with the regulation pattern of Mus81-Mms4 and Yen1, showing that Yen1 is temporarily activated after Mus81-Mms4 (40).
In summary, kinetic (21, 23), structural/biochemical (this study), and genetic (28) data can be synthesized in a highly consistent manner into a model in which Mus81-Mms4 in S. cerevisiae, and likely other eukaryotes, functions to cleave nicked junctions and D-loops to form COs, while Sgs1-Top3-Rmi1 dissolves dHJs into NCO products. This model suggests that the Mus81 pathway of CO formation does not involve classical HJs or dHJs, i.e., junctions with four uninterrupted strands. Nicked-junction cleavage by Mus81-Mms4 provides an efficient pathway for CO formation in vegetative and meiotic cells (Fig. 17) (27, 28, 45). The question of the mechanism of the formation of meiotic COs that are associated with interference is still open, as Mus81-dependent COs lack interference (6, 20, 28, 30, 45, 52).
ACKNOWLEDGMENTS
We thank Mary Ann Osley, Rodney Rothstein, and John Rouse for providing strains; Steve Brill for providing purified Slx1-Slx4; David Toczyski for the Cdc5 constructs; Steve West for his personal communication of unpublished results; Po Lin Chiu, Lenin Dominguez-Ramirez, and David Carlson for sharing their electron microscopy expertise; and Stephen Kowalczykowski, Neil Hunter, Rinti Mukherjee, Clare Fasching, Ryan Janke, Xiao-Ping Zhang, and Jie Liu for critical comments on the manuscript.
This work was supported by grants from the U.S. National Institutes of Health (GM58015 to W.-D.H., U54GM74929 to H.S., and SystemsX.ch to H.S.). E.K.S. was supported by a fellowship from the HHMI-IMBS training grant at UC Davis, and W.D.W. was supported a by TRDRP predoctoral fellowship (17DT-0178).
Footnotes
Published ahead of print 29 May 2012
REFERENCES
- 1. Agmon N, Yovel M, Harari Y, Liefshitz B, Kupiec M. 2011. The role of Holliday junction resolvases in the repair of spontaneous and induced DNA damage. Nucleic Acids Res. 39:7009–7019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Andersson KM, Hovmöller S. 1998. The average Atomic volume and density of proteins. Z. Kristallogr. 213:369–373 [Google Scholar]
- 3. Argueso JL, Wanat J, Gemici Z, Alani E. 2004. Competing crossover pathways act during meiosis in Saccharomyces cerevisiae. Genetics 168:1805–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bashkirov VI, King JS, Bashkirova EV, Schmuckli-Maurer J, Heyer WD. 2000. DNA repair protein Rad55 is a terminal substrate of the DNA damage checkpoints. Mol. Cell. Biol. 20:4393–4404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bastin-Shanower SA, Fricke WM, Mullen JR, Brill SJ. 2003. The mechanism of Mus81-Mms4 cleavage site selection distinguishes it from the homologous endonuclease Rad1-Rad10. Mol. Cell. Biol. 23:3487–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berchowitz LE, Francis KE, Bey AL, Copenhaver GP. 2007. The role of AtMUS81 in interference-insensitive crossovers in A. thaliana. PLoS Genet. 3:e132 doi:10.1371/journal.pgen.0030132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blais V, et al. 2004. RNA interference inhibition of Mus81 reduces mitotic recombination in human cells. Mol. Biol. Cell 15:552–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blanco MG, Matos J, Rass U, Ip SC, West SC. 2010. Functional overlap between the structure-specific nucleases Yen1 and Mus81-Mms4 for DNA-damage repair in S. cerevisiae. DNA Repair 9:394–402 [DOI] [PubMed] [Google Scholar]
- 9. Boddy MN, et al. 2001. Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 107:537–548 [DOI] [PubMed] [Google Scholar]
- 10. Boddy MN, et al. 2000. Damage tolerance protein Mus81 associates with the FHA1 domain of checkpoint kinase Cds1. Mol. Cell. Biol. 20:8758–8766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bowman GD, O'Donnell M, Kuriyan J. 2004. Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature 429:724–730 [DOI] [PubMed] [Google Scholar]
- 12. Bzymek M, Thayer NH, Oh SD, Kleckner N, Hunter N. 2010. Double Holliday junctions are intermediates of DNA break repair. Nature 464:937–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chang JH, Kim JJ, Choi JM, Lee JH, Cho Y. 2008. Crystal structure of the Mus81-Eme1 complex. Genes Dev. 22:1093–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen X-B, et al. 2001. Human Mus81-associated endonuclease cleaves Holliday junctions in vitro. Mol. Cell 8:1117–1127 [DOI] [PubMed] [Google Scholar]
- 15. Choi YJ, et al. 2005. Biophysical characterization of the interaction domains and mapping of the contact residues in the XPF-ERCC1 complex. J. Biol. Chem. 280:28644–28652 [DOI] [PubMed] [Google Scholar]
- 16. Clyne RK, et al. 2003. Polo-like kinase Cdc5 promotes chiasmata formation and cosegregation of sister centromeres at meiosis I. Nat. Cell Biol. 5:480–485 [DOI] [PubMed] [Google Scholar]
- 17. Coulon S, et al. 2004. Slx1-Slx4 are subunits of a structure-specific endonuclease that maintains ribosomal DNA in fission yeast. Mol. Biol. Cell 15:71–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Das D, et al. 2008. The HhH domain of the human DNA repair protein XPF forms stable homodimers. Proteins 70:1551–1563 [DOI] [PubMed] [Google Scholar]
- 19. de los Santos T, et al. 2003. The Mus81/Mms4 endonuclease acts independently of double-Holliday junction resolution to promote a distinct subset of crossovers during meiosis in budding yeast. Genetics 164:81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de los Santos T, Loidl J, Larkin B, Hollingsworth N. 2001. A role for MMS4 in the processing of recombination intermediates during meiosis in Saccharomyces cerevisiae. Genetics 159:1511–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ehmsen KT, Heyer WD. 2008. Saccharomyces cerevisiae Mus81-Mms4 is a catalytic structure-selective endonuclease. Nucleic Acids Res. 36:2182–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fabre F, Chan A, Heyer WD, Gangloff S. 2002. Alternate pathways involving Sgs1/Top3, Mus81/Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc. Natl. Acad. Sci. U. S. A. 99:16887–16892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fricke WM, Bastin-Shanower SA, Brill SJ. 2005. Substrate specificity of the Saccharomyces cerevisiae Mus81-Mms4 endonuclease. DNA Repair 4:243–251 [DOI] [PubMed] [Google Scholar]
- 24. Fricke WM, Brill SJ. 2003. Slx1-Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1-Top3. Genes Dev. 17:1768–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gaillard PH, Wood RD. 2001. Activity of individual ERCC1 and XPF subunits in DNA nucleotide excision repair. Nucleic Acids Res. 29:872–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gaskell LJ, Osman F, Gilbert RJ, Whitby MC. 2007. Mus81 cleavage of Holliday junctions: a failsafe for processing meiotic recombination intermediates? EMBO J. 26:1891–1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heyer WD, Ehmsen KT, Solinger JA. 2003. Holliday junctions in the eukaryotic nucleus: resolution in sight? Trends Biochem. Sci. 10:548–557 [DOI] [PubMed] [Google Scholar]
- 28. Ho CK, Mazón G, Lam AF, Symington LS. 2010. Mus81 and Yen1 promote reciprocal exchange during mitotic recombination to maintain genome integrity in budding yeast. Mol. Cell 40:988–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Holliday R. 1964. A mechanism for gene conversion in fungi. Genet. Res. 5:282–304 [DOI] [PubMed] [Google Scholar]
- 30. Holloway JK, Booth J, Edelmann W, McGowan CH, Cohen PE. 2008. MUS81 generates a subset of MLH1-MLH3-independent crossovers in mammalian meiosis. PLoS Genet. 4:e1000186 doi:10.1371/journal.pgen.1000186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Interthal H, Heyer WD. 2000. MUS81 encodes a novel helix-hairpin-helix protein involved in the response to UV- and methylation-induced DNA damage in Saccharomyces cerevisiae. Mol. Gen. Genet. 263:812–827 [DOI] [PubMed] [Google Scholar]
- 32. Ip SCY, et al. 2008. Identification of Holliday junction resolvases from humans and yeast. Nature 456:357–361 [DOI] [PubMed] [Google Scholar]
- 33. Jessop L, Lichten M. 2008. Mus81/Mms4 endonuclease and sgs1 helicase collaborate to ensure proper recombination intermediate metabolism during meiosis. Mol. Cell 31:313–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kanai Y, et al. 2007. DmGEN shows a flap endonuclease activity, cleaving the blocked-flap structure and model replication fork. FEBS J. 274:3914–3927 [DOI] [PubMed] [Google Scholar]
- 35. Kastner B, et al. 2008. GraFix: sample preparation for single-particle electron cryomicroscopy. Nat. Methods 5:53–55 [DOI] [PubMed] [Google Scholar]
- 36. Kuo MH, Allis CD. 1999. In vivo cross-linking and immunoprecipitation for studying dynamic protein:DNA associations in a chromatin environment. Methods 19:425–433 [DOI] [PubMed] [Google Scholar]
- 37. Lilley DMJ, White MF. 2001. The junction-resolving enzymes. Nat. Cell Biol. 2:433–443 [DOI] [PubMed] [Google Scholar]
- 38. Liu YL, West SC. 2004. Timeline—happy Hollidays: 40th anniversary of the Holliday junction. Nat. Rev. Mol. Cell Biol. 5:937–944 [DOI] [PubMed] [Google Scholar]
- 39. Lorenz A, West SC, Whitby MC. 2010. The human Holliday junction resolvase GEN1 rescues the meiotic phenotype of a Schizosaccharomyces pombe mus81 mutant. Nucleic Acids Res. 38:1866–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matos J, Blanco MG, Maslen S, Skehel JM, West SC. 2011. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 147:158–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mullen JR, Kaliraman V, Ibrahim SS, Brill SJ. 2001. Requirement for three novel protein complexes in the absence of the sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics 157:103–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Munoz-Galvan S, et al. 2012. Distinct roles of mus81, yen1, slx1-slx4, and rad1 nucleases in the repair of replication-born double-strand breaks by sister chromatid exchange. Mol. Cell. Biol. 32:1592–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oh SD, Lao JP, Taylor AF, Smith GR, Hunter N. 2008. RecQ helicase, Sgs1, and XPF family endonuclease, Mus81-Mms4, resolve aberrant joint molecules during meiotic recombination. Mol. Cell 31:324–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ohi M, Li Y, Cheng Y, Walz T. 2004. Negative staining and image classification-powerful tools in modern electron microscopy. Biol. Proced. Online 6:23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Osman F, Dixon J, Doe CL, Whitby MC. 2003. Generating crossovers by resolution of nicked Holliday junctions: a role for Mus81-Eme1 in meiosis. Mol. Cell 12:761–774 [DOI] [PubMed] [Google Scholar]
- 46. Pâques F, Haber JE. 1999. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 63:349–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rass U, et al. 2010. Mechanism of Holliday junction resolution by the human GEN1 protein. Genes Dev. 24:1559–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schwacha A, Kleckner N. 1995. Identification of double Holliday junctions as intermediates in meiotic recombination. Cell 83:783–791 [DOI] [PubMed] [Google Scholar]
- 49. Schwartz EK, Heyer WD. 2011. Processing of joint molecule intermediates by structure-selective endonucleases during homologous recombination in eukaryotes. Chromosoma 120:109–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shiomi Y, et al. 2002. Clamp and clamp loader structures of the human checkpoint protein complexes, Rad9-1-1 and Rad17-RFC. Genes Cells 7:861–868 [DOI] [PubMed] [Google Scholar]
- 51. Siegel LM, Monty KJ. 1966. Determination of molecular weights and frictional ratios of protein in impure systems by use of gel filtration and density gradient centrifugation. Application to crude preparations of sulfite and hydroxylamine reductases. Biochim. Biophys. Acta 112:346–362 [DOI] [PubMed] [Google Scholar]
- 52. Smith GR, Boddy MN, Shanahan P, Russell P. 2003. Fission yeast Mus81 · Eme1 Holliday junction resolvase is required for meiotic crossing over but not for gene conversion. Genetics 165:2289–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sourirajan A, Lichten M. 2008. Polo-like kinase Cdc5 drives exit from pachytene during budding yeast meiosis. Genes Dev. 22:2627–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Svendsen JM, Harper JW. 2010. GEN1/Yen1 and the SLX4 complex: solutions to the problem of Holliday junction resolution. Genes Dev. 24:521–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tang G, et al. 2007. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157:38–46 [DOI] [PubMed] [Google Scholar]
- 56. Tay YD, Wu L. 2010. Overlapping roles for Yen1 and Mus81 in cellular Holliday junction processing. J. Biol. Chem. 285:11427–11432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Taylor ER, McGowan CH. 2008. Cleavage mechanism of human Mus81-Eme1 acting on Holliday-junction structures. Proc. Natl. Acad. Sci. U. S. A. 105:3757–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Trowbridge K, McKim K, Brill SJ, Sekelsky J. 2007. Synthetic lethality of Drosophila in the absence of the MUS81 endonuclease and the DmBlm helicase is associated with elevated apoptosis. Genetics 176:1993–2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vidanes GM, et al. 2010. CDC5 inhibits the hyperphosphorylation of the checkpoint kinase Rad53, leading to checkpoint adaptation. PLoS Biol 8:e1000286 doi:10.1371/journal.pbio.1000286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wechsler T, Newman S, West SC. 2011. Aberrant chromosome morphology in human cells defective for Holliday junction resolution. Nature 471:642–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wu LJ, Hickson ID. 2003. The Bloom's syndrome helicase suppresses crossing-over during homologous recombination. Nature 426:870–874 [DOI] [PubMed] [Google Scholar]