

Abstract
The E6AP ubiquitin ligase catalyzes the high-risk human papillomaviruses' E6-mediated ubiquitylation of p53, contributing to the neoplastic progression of cells infected by these viruses. Defects in the activity and the dosage of E6AP are linked to Angelman syndrome and to autism spectrum disorders, respectively, highlighting the need for precise control of the enzyme. With the exception of HERC2, which modulates the ubiquitin ligase activity of E6AP, little is known about the regulation or function of E6AP normally. Using a proteomic approach, we have identified and validated several new E6AP-interacting proteins, including HIF1AN, NEURL4, and mitogen-activated protein kinase 6 (MAPK6). E6AP exists as part of several different protein complexes, including the proteasome and an independent high-molecular-weight complex containing HERC2, NEURL4, and MAPK6. In examining the functional consequence of its interaction with the proteasome, we found that UBE3C (another proteasome-associated ubiquitin ligase), but not E6AP, contributes to proteasomal processivity in mammalian cells. We also found that E6 associates with the HERC2-containing high-molecular-weight complex through its binding to E6AP. These proteomic studies reveal a level of complexity for E6AP that has not been previously appreciated and identify a number of new cellular proteins through which E6AP may be regulated or functioning.
INTRODUCTION
E6AP was originally discovered as the cellular protein that mediates the binding of the E6 protein of the high-risk human papillomaviruses (HPVs) to the tumor suppressor p53 (35). Subsequently, E6AP was shown to catalyze the E6-dependent transfer of ubiquitin to p53, targeting it for degradation by the 26S proteasome. As such, E6AP was the first mammalian ubiquitin (Ub) ligase to be described (36, 37, 80).
E6AP is a 100-kDa protein containing a conserved carboxy-terminal domain spanning 350 amino acids, referred to as the HECT domain (homologous to E6AP carboxy terminus), that defines a family of E3 Ub ligases (34). This domain participates directly in the transfer of ubiquitin from an E2 ubiquitin-conjugating enzyme to a conserved cysteine residue through a thiolester bond (34). E6AP then directs the transfer of ubiquitin from this active-site cysteine to a lysine residue of an associated substrate. Replacement of the active-site cysteine with alanine renders E6AP unable to accept ubiquitin and therefore unable to catalyze the transfer of ubiquitin to the target protein. This C/A substitution mutant is inactive and functions in a dominant-negative (DN) manner (88). Three different isoforms of E6AP that are generated by differential splicing from the same gene have been described (98). These three isoforms differ in their amino-terminal sequences, and it is not yet known whether they differ in function or substrate specificity.
The E6AP protein is encoded by the UBE3A gene located in an imprinted region on chromosome 15q11-q13. As a consequence of the imprinting, only the maternal UBE3A allele is expressed in certain areas of the brain (75, 93). This region of chromosome 15 is implicated in the pathogenesis of Angelman syndrome (AS) (1, 43, 62), a neurogenetic disorder characterized by severe mental retardation, ataxia, loss of speech, seizures, and other abnormalities (3, 57, 96, 97). Four different mechanisms causing loss-of-function mutations or deletions in the maternally inherited allele of UBE3A can result in AS. They include deletions (58, 59), paternal uniparental disomy (24, 46), mutations of the UBE3A gene (43, 62), and defects in imprinting (10, 11). Loss of the paternal allele of this same 15q11-q13 region causes Prader-Willi syndrome (22, 90), a disorder closely related to AS. In addition, increased gene dosage of UBE3A has been implicated in some forms of autism spectrum disorders (31, 68, 84).
Since its discovery, a variety of cellular proteins have been shown to interact with E6AP, but the physiological relevance of most has not yet been established. E6AP is expressed in all cell types, but its normal cellular substrates and the pathways in which E6AP participates have not been fully elucidated. Some of the E6AP protein interactors and potential substrates are RAD23A (49), BLK (69), MCM7 (47), UBQLN1 and -2 (44, 45), the estrogen receptor (54), TSC2 (102), annexin A1 (82), PML (56), peroxiredoxin 1 (66), Arc (29), and Ring1B (99). In order to further understand the biological processes in which E6AP is involved, a comprehensive analysis of E6AP-interacting proteins (substrates and regulators) in the context of the whole proteome is required. To begin to define an E6AP interactome in a more comprehensive manner, we have employed a proteomic approach consisting of immunoprecipitations of the HA-tagged E6AP isoforms (wild type [wt] and DN), followed by the identification of associated proteins by liquid chromatography-tandem mass spectrometry (LC–MS-MS). These MS data were then analyzed with CompPASS (Comparative Proteomic Analysis Software Suite), which relies on an unbiased comparative approach for identifying high-confidence candidate-interacting proteins (HCIPs) (85). This technology has been successfully used to identify protein interactors among the hundreds of proteins typically identified in immunoprecipitation-MS experiments (5, 12, 72, 73, 95).
Using this approach, we report here the identification of several new E6AP interactors, including HIF1AN, mitogen-activated protein kinase 6 (MAPK6), and NEURL4. HIF1AN (also known as FIH) is an asparagine hydroxylase that negatively regulates the transcriptional activity of hypoxia-inducible factor 1α (HIF1α) (33, 50, 60), MAPK6 (or extracellular signal-regulated kinase 3 [ERK3]) is a serine/threonine kinase that has been associated with cell cycle control, cellular differentiation, and regulation of neuronal morphogenesis (9, 32, 55, 81, 86), and NEURL4, a protein that has recently been linked to regulation of the centrosome (2, 53), consists of a tandem array of six neuralized homology repeat (NHR) domains, which have been reported to mediate protein-protein interactions and Ub ligase substrate recognition (15, 26). Additionally, using size exclusion chromatography, we found that E6AP is present in several protein complexes of different molecular weights (MW) and compositions. E6AP is part of a high-molecular-mass complex of approximately 2 MDa, containing the known interactor HERC2 (48), as well as NEURL4 and MAPK6. HERC2 has been reported to enhance the Ub ligase activity of E6AP toward RING1B (48); however, we found that HERC2 does not contribute to the E6-mediated degradation of p53 in HeLa cells. We also confirmed that E6AP is found associated with the proteasome (6, 45, 78, 87, 94) in a complex that is distinct from the very high-molecular-weight complex containing HERC2 and NEURL4. Several Ub ligases have been associated with the mammalian proteasome, including UBE3C, a HECT domain E3 Ub ligase whose yeast ortholog, HUL5, has been shown to contribute to proteasomal processivity. We found that UBE3C, but not E6AP, contributes to general proteasomal processivity, establishing that this function of HUL5/UBE3C has been conserved throughout evolution. Finally, we found that most of E6AP is in lower-molecular-mass complexes ranging from 100 to 200 kDa. HIF1AN was found to associate with E6AP in this lower-molecular-mass range. This study establishes the association of E6AP with different protein complexes and reveals additional complexities regarding E6AP function and, possibly, regulation.
MATERIALS AND METHODS
Cloning and plasmids.
To generate vectors that express the hemagglutinin (HA)-tagged proteins (wt and DN) for each of the three isoforms of E6AP, the different E6AP cDNAs were amplified by PCR using the forward primers 5′-CACCATGAAGCGAGCAGCTGCAAAGC-3′, 5′-CACCATGGAGAAGCTGCACCAGTGTTATTG-3′, and 5′-CACCATGGCCACAGCTTGTAAAAGATCAGG-3′ for isoforms I, II, and III, respectively, and the reverse primer 5′-GGATCCTTACAGCATGCCAAATCCTTTGGC-3′. To obtain the HA-tagged NEDD4 proteins, the NEDD4 open reading frames were amplified from shorter cDNAs present in the laboratory using the primers 5′-CACCATGGCAACTTGCGCGGTGGAGGTGTTCGGGCTCCTGGAGGACGAGGAAAATTCACGAATTGTGAGAGTAAGAGTTATAGCCGGAATAGG-3′, which provided the missing N terminus, and 5′-GGATCCCTAATCAACTCCATCAAAGCCCTGGGTG-3′. The amplified DNAs were cloned into the pENTR/D-TOPO vector (Invitrogen) and then recombined into the vector pHAGE-P CMVt N-HA GAW, where GAW represents the GATEWAY cassette. This lentiviral vector was derived from an HIV-1-based backbone named pHAGE (65), which was modified by standard molecular biology techniques to work as a destination vector for the GATEWAY system (Invitrogen) and to express an N-terminally tagged protein under the control of a tetracycline-regulatable promoter (TetON). It also contains a puromycin resistance gene for selection (pHAGE-based vectors expressing HA-tagged proteins: E6AP I (p6895), E6AP I C820A (p6898), E6AP II (p6896), E6AP II C843A (p6899), E6AP III (p6897), E6AP III C840A (p6900), NEDD4 (p6270), NEDD4 C867A (p6271), and enhanced green fluorescent protein (EGFP) (p6901). To obtain a vector that expresses EGFP-Myc, the Myc cDNA was amplified by PCR using the primers 5′-GGGGACAACTTTGTACAAAAAAGTTGGCACCATGCCCCTCAACGTTAGCTTCACC-3′ and 5′-GGGGACAACTTTGTACAAGAAAGTTGGGTTACGCACAAGAGTTCCGTAGCTGTTC-3′, and the amplified fragment was recombined into the pDONR223 vector. From this vector, the Myc open reading frame was transferred into a pHAGE-based destination vector that expresses the recombined protein N-terminally fused to EGFP. This vector also contains a blasticidin resistance gene for selection (p6903). The murine stem cell virus (MSCV) and LXSH (a murine Moloney leukemia retrovirus-based vector) vectors (Clontech) vectors used to express the human papillomavirus type 16 (HPV16) and HPV8 E6 proteins have been described previously (89, 92). Plasmids generated in the Howley laboratory for this study have been deposited with Addgene.
Cell culture.
All cells lines used in this study were maintained in Dulbecco's modified Eagle's medium (DMEM)-10% fetal bovine serum (FBS) at 37°C and 5% CO2. T-REx 293 cells (Invitrogen), which stably express the Tet repressor (TetON), were maintained in 5 μg/ml blasticidin (InvivoGen). After infection with lentiviruses carrying pHAGE-P CMVt N-HA GAW-derived vectors, transduced cells were selected with 1 μg/ml puromycin (Sigma). To induce the expression of the HA-tagged proteins, the cells were kept for 24 h in medium without antibiotics and then induced with 1 μg/ml doxycycline (Sigma) for 24 h. In the case of 293T cells infected with lentiviruses carrying the pHAGE EGFP-Myc vector, transduced cells were selected with 5 μg/ml blasticidin. 293T cells infected with MSCV-based vectors for expression of the HPV8 and HPV16 E6 proteins were selected with puromycin as described above. For proteasome inhibition, cells were incubated for 3 h with 20 μM MG132 (Calbiochem). For the cycloheximide chase experiment, the cells were incubated with 100 μg/ml cycloheximide (Sigma) for the times indicated. C33A cells expressing HPV16 E6 and SiHa cells expressing HA-tagged HPV16 E6 protein have been previously described (92).
Immunoprecipitations and MS.
HA-tagged proteins were immunopurified, processed, and analyzed by MS as previously described (85). Several modifications to the purification protocol were introduced in order to speed it up to increase the likelihood of capturing transient interactions. Briefly, T-REx cells from five 15-cm culture dishes (approximately 80 to 90% confluent) expressing HA-tagged proteins were washed once with phosphate-buffered saline (PBS) at room temperature and then lysed on the plates with 5 ml ice-cold lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% NP-40, 12.5 mM NaF, 1 mM Na3VO4, 12.5 mM β-glycerophosphate, Complete protease inhibitor cocktail [Roche]). Cleared lysates were filtered through a 20-μm SFCA-PF syringe filter (Corning) and immunoprecipitated with 50 μl (settled) anti-HA–agarose beads (Sigma) for 2 h at 4°C. The beads were then washed twice with 10 ml lysis buffer and twice with 10 ml PBS, transferred with 1 ml PBS into a microcentrifuge tube, and then eluted with three 50-μl washes of 500 μg/ml HA-peptide (Sigma) in PBS.
MS data analysis using CompPASS.
The immunprecipitation (IP)-MS data from two biological repeats for each bait were analyzed by CompPASS as previously described (5, 85). We used a stats table with 106 pulldowns from 293 cells generated by the Harper laboratory. The weighted D score (WD) score threshold was calculated so that 95% of the data fell below it. The normalized WD (NWD) score results from dividing each WD score by the calculated threshold. We considered those proteins that had NWD scores equal to or higher than 1 (WD score equal to or higher than the calculated threshold) HCIPs. Only proteins that were considered HCIPs in both biological repeats were considered for further analysis. A complete set of data is presented in Tables S1 and S2 in the supplemental material. The heat map showing the NWD scores of the different HCIPs was generated using MultiExperiment Viewer software (76, 77). HCIPs were arranged by using a Pearson correlation hierarchical-clustering analysis.
Western blots.
Protein extracts were prepared using the lysis buffer described above. Immunoprecipitations from T-REx 293 cells analyzed by Western blotting were prepared using the same protocol used for pulldowns for MS, but the proteins were eluted from the agarose beads by boiling in loading buffer. One-fourth of the immunoprecipitated material was used for each analysis. Immunoprecipitations from T-REx cells for the characterization of the 2-MDa complex and from SiHa cells were done on a smaller scale, using the same protocol with only 20 μl of settled HA-agarose beads for the pulldowns. The samples were washed six times with 1 ml lysis buffer before elution. The proteins were resolved on NuPAGE gels (Invitrogen) and transferred to polyvinylidene difluoride (PVDF) membranes. The blots were blocked with 5% nonfat dry milk in TNET (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2.5 mM EDTA, and 0.1% Tween 20) and then incubated with the following primary antibodies: HA conjugated to horseradish peroxidase (HRP) (clone 3F10; Roche), E6AP (clone E6AP-300; Sigma), HERC2 (BD Transduction Laboratories), HIF1AN (EPR3658; Abcam), MAPK6 (ERK3; Cell Signaling), NEURL4 (ProteinExpress), PSMD4 (S5a-18; Enzo), PSMA4 (PAI-1962; Thermo), UBE3C (GeneTex), Myc (Ab-1; Calbiochem), EGFP (Cell Signaling), p53 (DO-1; Oncogene), actin (Millipore), and tubulin (Santa Cruz). Except when the HA-HRP antibody was used, the membranes were washed in TNET and then incubated with HRP-conjugated anti-mouse or anti-rabbit antibodies (GE Healthcare). Signals were generated using Western Lightning chemiluminescent substrate (Perkin Elmer) or West Femto (Pierce) and recorded on BioMax MR films (Kodak).
siRNA transfections.
Small interfering RNAs (siRNAs) were transfected into 293T cells using RNAiMax (Invitrogen) or into SiHa and HeLa cells using DharmaFect 1 and 4, respectively (Dharmacon/Thermo Scientific). In both cases, the manufacturers' instructions were followed, and the final concentration of the siRNA duplexes was 20 nM. All siRNA duplexes were obtained from Dharmacon/Thermo Scientific. Two of the siRNAs against E6AP, siE6AP-A and -B, used in this study have been previously described (42). The other two siRNAs against E6AP used in this study are siE6AP-C, whose target sequence is 5′-AGACAAAGAUGAAGAUGAA-3′, and siE6AP-04 (UBE3A; D-005137-04). Other siRNAs used in this work were against HERC2 (siHERC2-01 [J-007180-09], siHERC2-02 [J-007180-10], siHERC2-03 [J-007180-11], and siHERC2-04 [J-007180-12]); against NEURL4 (siNEURL4-01 [J-032714-08], siNEURL4-02 [J-032714-07], siNEURL4-03 [J-032714-06], and siNEURL4-04 [J-032714-05]); against MAPK6 (siMAPK6-01 [J-003594-11], siMAPK6-02 [J-003594-14], siMAPK6-03 [J-003594-13], and siMAPK6-04 [J-003594-12]); and against UBE3C (siUBE3C-01 [J-007183-08], siUBE3C-02 [J-007183-07], siUBE3C-03 [J-007183-06], and siUBE3C-04 [J-007183-05]). As controls, we used nontargeting siRNAs no. 1 (D-001810-01-20) and no. 2 (D-001210-02) and siGLO Red (D-001630-02) to assess transfection efficiency. After 24 h, the medium was exchanged, and at 72 h, the cells were harvested for immunoprecipitation or Western blot analysis or further treated as indicated for the specific experiments.
Size exclusion chromatography.
Protein extracts were obtained using the lysis buffer described above. The cleared lysate that was used for immunoprecipitations after fractionation was filtered through a 20-μm SFCA-PF syringe filter (Corning) before being loaded on the column. To avoid losses in the residual volume of the filter due to the small sample size, the other cleared lysates were filtered through Ultrafree centrifugal devices (0.45-μm Durapore PVDF; Millipore). Four to eight milligrams of total cellular protein was then applied to a Superose 6 10/300 GL column and fractionated using an Äkta FPLC (GE Healthcare) in 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.5% NP-40 as a running buffer. We used a 500-μl injection loop and a volume of 650 μl. The flow rate was 0.5 ml/min, and 500-μl fractions were collected after 5 ml. Twenty microliters from the collected fractions was analyzed by SDS-PAGE, followed by Western blotting with the indicated antibodies. Alternatively, following elution, individual fractions were immunoprecipitated with 20 μl anti-HA–agarose beads (settled), washed 6 times with 1 ml running buffer, and eluted with two washes of 40 μl of 0.5 N NH4OH, 0.5 mM EDTA. Sixteen microliters of the eluted proteins was used for SDS-PAGE and Western blot analysis. To estimate the molecular sizes of the proteins of interest, we used the Gel Filtration Markers Kit for Protein Molecular Weights 29,000 to 700,000 Da (Sigma), consisting of blue dextran (2,000 kDa; void volume marker), thyroglobulin (669 kDa), apoferritin (443 kDa), β-amylase (200 kDa), alcohol dehydrogenase (150 kDa), albumin (66 kDa), and carbonic anhydrase (29 kDa). One microgram of each protein standard was loaded on the column.
RESULTS
Identification of E6AP-interacting proteins.
To systematically identify proteins that interact with E6AP, we performed coprecipitation experiments using HA-tagged E6AP as bait. We used the three isoforms of E6AP in their wt and DN forms, the latter to increase the possibility of identifying proteins that might be substrates of E6AP. The coprecipitated proteins were identified by LC–MS-MS, and the data were analyzed by CompPASS to define HCIPs (85) (see Table S1 in the supplemental material). In this analysis, we also used NEDD4 (both wt and DN forms) as a control for specificity. The identification of several known E6AP interactors, such as HERC2 (48), proteasomal subunits, and proteasome-associated proteins (6, 45, 78, 87, 94), provided validation of the approach. In addition, a number of previously unknown interactors, such as HIF1AN, MAPK6, and NEURL4, were also identified as HCIPs. As further validation of this strategy, several known NEDD4-interacting proteins (for instance, AMOTL1, WBP2, and NDFIP1 [38, 41]) were identified as HCIPs of NEDD4, but not of E6AP.
As depicted in Fig. 1A (see Table S1 in the supplemental material), the HCIPs for each bait protein were very reproducible, as well as highly specific. There were only a few differences noted between the proteins pulled down by the three different isoforms of E6AP. Isoform I more efficiently pulled down proteasome subunits, as evidenced by the number of scan counts for each of those proteins. Only isoform II brought down KTN1, HIST1H2BK, MAP7D1, RPS26, EIF3D, and POTEF. The inclusion of the DN forms of E6AP in these studies was to enhance the likelihood of identifying potential substrates of the enzyme. However, the pulldowns done with the DN forms of E6AP were very similar to those with the wt proteins, perhaps due to the transient nature of the E6AP-substrate complexes. This suggests that the approach of using the DN form of E6AP to identify its substrates may not have been very efficient. However, the numbers of scans for peptides from HCIPs and E6AP itself were higher with the DN forms, most likely due to the increased levels and stability of the dominant-negative forms compared to wt E6AP (88).
Fig 1.

HCIPs identified by CompPASS analysis. (A) Heat map of NWD scores of HCIPs identified by CompPASS from two independent pulldowns for each bait. HA-tagged E6AP (isoforms I, II, and III, wt and dominant-negative mutants) and NEDD4 (wt and a dominant-negative mutant) were immunoprecipitated from T-REx 293 cells using anti-HA–agarose beads. Coimmunoprecipitated proteins were identified by mass spectrometry and analyzed by CompPASS. Each tagged protein was used in two separate experiments. Only proteins that were considered HCIPs in both experiments are listed. Only NWD scores equal to or higher than 1 are shown. Proteasomal subunits (PS) from the 20S and 19S particles are indicated. (B, C, and D) Confirmation of interaction of E6AP with several HCIPs. HA-tagged EGFP, E6AP III, or E6AP III C840A was immunoprecipitated from T-REx 293 cells using anti-HA–agarose beads. Protein extracts and immunoprecipitates were analyzed by SDS-PAGE and Western blotting using antibodies against MAPK6 (A), HIF1AN (B), or NEURL4 (C). In all cases, the HA-tagged proteins were detected by an anti-HA epitope antibody. HA-EGFP was used as a negative control.
We confirmed the interactions of E6AP with HIF1AN, MAPK6, and NEURL4 by coprecipitation and Western blotting (Fig. 1B, C, and D), since interactions between each of these proteins and E6AP had not been previously described. Since there were no major differences in the HCIPs found for the different E6AP isoforms, we used only the HA-tagged isoform III for these and later experiments. Interactions of E6AP with HERC2 and with the proteasome were not validated in this set of experiments but was confirmed in later ones. The studies presented here focus on the interactions of E6AP with HERC2 and the proteasome.
E6AP is a component of several protein complexes.
Next, we proceeded to determine whether E6AP and its interactors are parts of a single or several protein complexes. We performed a series of gel filtration experiments with T-REx 293 cells to examine the recruitment of endogenous E6AP in supramolecular complexes discriminated by their masses. As shown in Fig. 2A, E6AP could be detected more prominently from fractions 20 to 22, corresponding to a mass ranging from approximately 100 to 200 kDa, but it also showed a wide distribution, extending to fractions 05 and 06 (∼2 MDa). The HA-tagged wt and DN forms of E6AP expressed in T-REx 293 cells had elution profiles similar to that of endogenous E6AP (Fig. 2B and C). To determine the proteasome distribution in these gel filtration experiments, we used antibodies against the PSMD4 (19S) and PSMA5 (20S) subunits of the proteasome. We also examined the elution profile of UBE3C, another HECT Ub ligase, which has been previously shown to be associated with the proteasome (19, 51, 94). The fraction distribution of the proteasome in these experiments is similar to that described in the literature (7, 28, 70, 74) and overlaps that of UBE3C (Fig. 2). Addition of 5 mM ATP, 5 mM MgCl2, and 1 mM dithiothreitol (DTT) to the buffer resulted in a similar elution profile for E6AP and only minor differences in the elution profile for the proteasomal subunits in these gel filtration experiments (data not shown).
Fig 2.
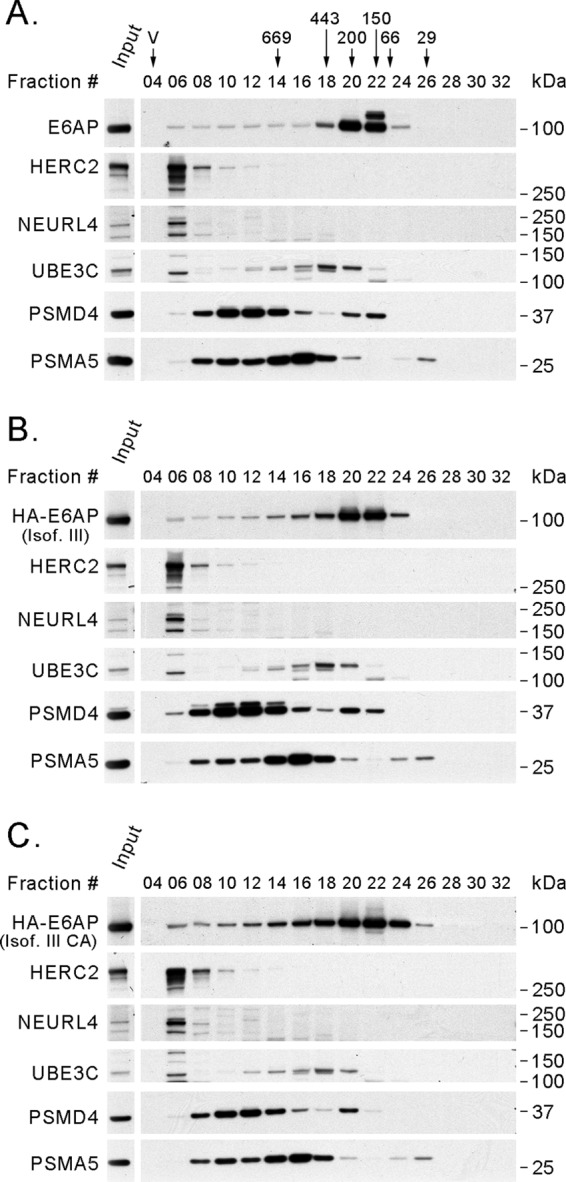
Endogenous E6AP and exogenously expressed HA-tagged E6AP have similar elution profiles in size exclusion chromatography experiments with T-REx 293 cell protein extracts. In each case, 4 mg of protein extract was loaded on a Superose 6 column for size exclusion chromatography. After fraction collection, 20 μl (4%) of even-numbered fractions was analyzed by SDS-PAGE and Western blotting using antibodies against E6AP, HERC2, NEURL4, UBE3C, PSMD4, and PSMA5 and against the HA tag in the case of tagged E6AP. (A) T-REx 293 cells. (B) T-REx 293 cells expressing HA-E6AP III. (C) T-REx 293 cells expressing HA-E6AP III C840A. The positions where the peaks of protein standards (669, 443, 200, 150, 66, and 29 kDa) eluted and the void volume (V) are indicated over the fraction numbers in panel A. Isof. III, isoform III; Isof. III CA, isoform III C840A; input, 20 μg of protein extract before fractionation.
To further investigate E6AP complexes with its associated interacting proteins, we performed HA pulldowns from individual fractions of T-REx 293 cells expressing the HA-tagged DN form of E6AP III subjected to size exclusion chromatography. Following the immunoprecipitations, the proteins were resolved by PAGE and detected by Western blotting (Fig. 3). The Western blots of the E6AP-coprecipitated proteins clearly demonstrate that E6AP forms several distinct complexes with its interacting proteins. E6AP associates with HERC2 in a high-molecular-mass complex of about 2 MDa, as previously described (48). NEURL4 and MAPK6 are also part of this complex, since NEURL4 and HERC2 interact with E6AP in the same fraction in a gel filtration experiment and since siRNA-mediated knockdown of HERC2 abolishes the interaction of E6AP with NEURL4 and MAPK6 (see below). E6AP also interacts with the proteasome subunit PSMD4 (also known as RPN10 or S5A) in two complexes with different masses of approximately 700 and 200 kDa. The largest complex probably corresponds to the 19S and/or the 26S particle, since E6AP pulled down proteasomal subunits of both the 19S and the 20S particles. The smallest complex, possibly containing non-proteasome-associated PSMD4, partially coelutes in fraction 20 with HIF1AN. These are probably independent protein complexes, since HIF1AN coprecipitates E6AP but no proteasomal subunits when used as the bait protein (data not shown). Altogether, these results establish that E6AP is engaged in different protein complexes of various MWs and compositions.
Fig 3.
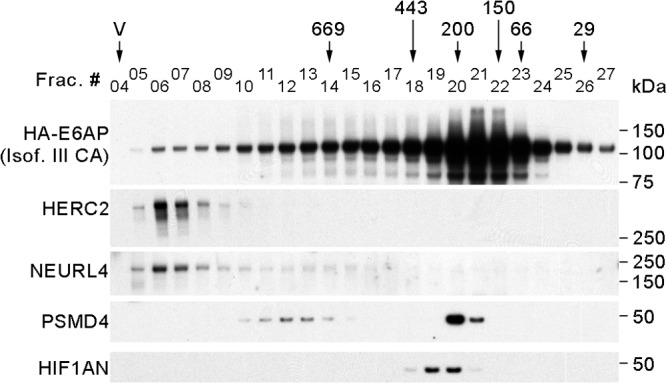
E6AP is a component of different protein complexes. Eight milligrams of protein extract from T-REx 293 cells expressing HA-tagged E6AP III C840A was loaded on a Superose 6 column for size exclusion chromatography. After fractionation, HA-tagged E6AP III C840A was immunoprecipitated from each fraction using HA-agarose beads, and the pulldowns were analyzed by SDS-PAGE and Western blotting using antibodies against the HA tag (for HA-tagged E6AP), HERC2, NEURL4, PSMD4, and HIF1AN. The positions where the peaks of protein standards eluted (669, 443, 200, 150, 66, and 29 kDa) and the void volume (V) are indicated over the fraction numbers.
Characterization of E6AP complexed with the proteasome.
We next examined the association of E6AP with the proteasome. The HECT Ub ligase HUL5, which associates with the proteasome, has been shown to contribute to proteasomal processivity in yeast (4). UBE3C is the closest ortholog of HUL5 in mammalian cells. We therefore tested whether E6AP might also contribute to the processivity of the proteasome in mammalian cells. We used a system similar to that employed by Aviram and Kornitzer (4), expressing a fusion protein containing an easily degradable domain (Myc) joined to a more stable domain (EGFP) that is difficult to degrade processively (61, 101) (Fig. 4A). We assayed the ability of the proteasome to proceed with the protein degradation from the Myc domain into the EGFP domain by measuring the levels of degradation intermediates. Expression of EGFP-Myc in 293T cells produced, in addition to the full-length protein, several bands of lower MW detectable with an anti-EGFP antibody (Fig. 4B). In Western blotting, shorter protein products could be the result either of protein degradation or of truncated synthesis. To identify which of the observed fragments were degradation intermediates generated by the proteolytic activity of the proteasome, we treated those cells with MG132 and identified bands whose intensity decreased with proteasome inhibition, indicating the need for proteasomal proteolytic activity for their generation. These bands correspond to the EGFP domain itself, because they were detected with the anti-EGFP antibody but not with an anti-Myc antibody and because their masses of about 30 kDa corresponded to that predicted for the EGFP domain (Fig. 4A). In addition, the siRNA-mediated knockdown of UBE3C stabilized these specific bands without affecting the level or stability of the full-length protein. The degree of 30-kDa EGFP domain stabilization correlated with the efficiency of the UBE3C knockdown (Fig. 4C and D). We therefore concluded that the 30-kDa bands are degradation intermediates of the EGFP-Myc protein generated by the proteasome.
Fig 4.

E6AP does not contribute to proteasomal processivity in 293T cells. (A) Schematic representation of the EGFP-Myc fusion protein indicating the positions of both domains and their predicted masses. (B) Western blot of protein extracts from cells expressing EGFP-Myc and control cells treated with MG132 to identify bands that correspond to degradation intermediates generated by the proteasome. (C) Western blot showing the effect of siRNA-mediated knockdown of UBE3C or E6AP in cells expressing EGFP-Myc on the level of the fusion protein and its degradation intermediates. (D) Western blot of a cycloheximide (CHX) chase experiment showing the result of siRNA-mediated knockdown of UBE3C (siUBE3C-02) or E6AP (siE6AP-A) on the stability of EGFP-Myc and its degradation intermediates. The arrows indicate the positions of bands corresponding to EGFP-Myc degradation intermediates containing the EGFP domain. The asterisks show the bands that correspond to UBE3C. siCont., siRNA control no. 2; siE6AP, siRNAs against E6AP; siUBE3C, siRNAs against UBE3C.
To ask whether E6AP has a role in proteasome processivity, we examined the effect of siRNA-mediated knockdown of E6AP on the abundance and stability of the 30-kDa degradation intermediates (Fig. 4C and D). Neither the level nor the stability of the full-length protein nor those of its degradation intermediates changed in the absence of E6AP. We therefore concluded that E6AP does not contribute to general proteasomal processivity under the conditions used in this assay.
The binding of the HPV16 E6 protein to E6AP has profound effects on the behavior of this ubiquitin ligase, redirecting the enzymatic activity of E6AP toward the ubiquitylation of p53 (80). To test whether the expression of the HPV16 E6 protein could somehow enable E6AP proteasomal processivity, we expressed the HPV16 E6 in 293T cells already expressing EGFP-Myc and examined the levels of the 30-kDa degradation intermediates following siRNA-mediated knockdown of UBE3C or E6AP. 293T cells expressing the HPV8 E6 protein, which does not bind to E6AP (89), and cells transduced with an empty vector were used as negative controls. As shown in Fig. 5, E6AP did not contribute to general proteosomal processivity in any of the cell lines tested, indicating that HPV16 E6 does not enable E6AP to contribute to the processivity of the proteasome. However, these experiments do not exclude the possibility that E6AP might affect the proteasomal processivity of a subpopulation of proteasomes and/or of specific substrates.
Fig 5.
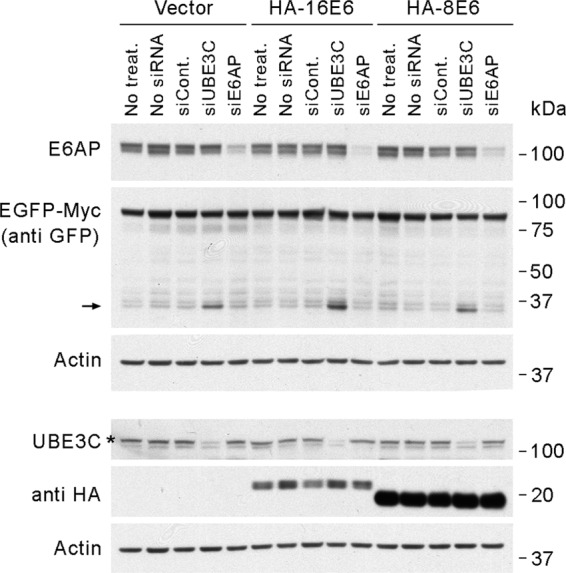
HPV16 E6 does not enable E6AP proteasomal processivity. The Western blot shows the effect of siRNA-mediated knockdown of UBE3C (siUBE3C-02) or E6AP (siE6AP-A) in cells expressing EGFP-Myc on the level of the 30-kDa EGFP domain degradation intermediates in 293T cells expressing empty vector and HA-tagged HPV16 E6 or HPV8 E6 protein. The EGFP-Myc fusion protein and the degradation intermediates were detected using an anti-GFP antibody. The HA-tagged E6 proteins were detected using an anti-HA epitope antibody. The arrow indicates the positions of bands corresponding to 30-kDa degradation intermediates containing the EGFP domain. The asterisk shows the band that corresponds to UBE3C. siCont., siRNA control no. 1; siE6AP, siRNA against E6AP; siUBE3C, siRNA against UBE3C; treat., treatment.
Characterization of the 2-MDa complex.
To further characterize the relationships between E6AP and the other components of the 2-MDa complex, we performed siRNA knockdowns of HERC2, NEURL4, and MAPK6 and assessed the ability of E6AP to coprecipitate the different components of the complex. As shown in Fig. 6, knockdown of HERC2 compromised the ability of E6AP to coprecipitate either NEURL4 or MAPK6, whereas reduced levels of NEURL4 affected only the binding of E6AP to MAPK6. The knockdown of MAPK6 had no effect on the coprecipitation of HERC2 and NEURL4 by E6AP. These data show that HERC2 binds to E6AP independently of NEURL4 and MAPK6 and thus most likely bridges the interaction between these two proteins and E6AP. It is possible, however, that NEURL4 could also independently interact with E6AP, since the knockdown of HERC2 results in a marked reduction in the levels of NEURL4, as has been recently reported (2). Our data do not discriminate whether the reduced levels of the coprecipitated NEURL4 are due to the lower levels of HERC2 bridging this interaction or a consequence of the reduced levels of NEURL4 in the cells. Since the levels of MAPK6 coprecipitated with E6AP, but not those of HERC2, were strongly reduced with the NEURL4 knockdown, and since MAPK6 knockdown did not affect the levels of HERC2 or NEURL4 pulled down by E6AP, we conclude that MAPK6 binding is mediated through its interaction with NEURL4. Further support for this conclusion comes from the observation that the levels of MAPK6 brought down by E6AP correlated with the levels of HERC2 and NEURL4 when they were knocked down. As expected, the levels of HIF1AN coprecipitated with E6AP did not correlate with the amount of coprecipitated HERC2, NEURL4, or MAPK6 but with the amount of precipitated E6AP, since HIF1AN binds to E6AP in a different protein complex (Fig. 3). Altogether, these data suggest that HERC2 bridges the interaction between E6AP and NEURL4 and that MAPK6 is recruited into this complex through its interaction with NEURL4 (Fig. 7).
Fig 6.

Characterization of the 2-MDa complex. T-REx 293 cells expressing HA-tagged E6AP III C840A were transfected with siRNA against HERC2, NEURL4, or MAPK6. Forty-eight hours after transfection, the expression of HA-tagged E6AP was induced by the addition of 1 μg/ml doxycycline. Seventy-two hours after transfection, the cells were lysed and HA-E6AP was immunoprecipitated using anti-HA-agarose beads. The pulldowns were resolved by SDS-PAGE and analyzed by Western blotting. siCont., siRNA control no 2; siHERC2, siRNAs against HERC2; siNEURL4, siRNAs against HERC2; siMAPK6, siRNAs against MAPK6.
Fig 7.
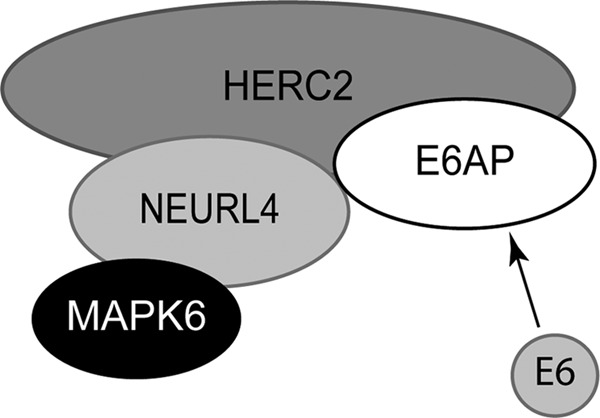
Model illustrating the relationships between the identified components of the 2-MDa complex. In high-risk HPV-infected cells, the HPV E6 protein is recruited to the 2-MDa complex trough its interaction with E6AP.
Expression of the HPV16 E6 protein does not significantly alter the recruitment of E6AP into the several protein complexes.
Next, we wondered whether the presence of the E6 protein could affect the recruitment of E6AP into the different protein complexes. To answer this question, we prepared C33A cell lines transduced with an empty retroviral vector or with a vector that expresses the HPV16 E6 protein. We fractionated protein extracts from those cells in a gel filtration experiment, and then we analyzed the elution profile of E6AP and several of its interacting proteins by Western blotting (Fig. 8). The expression of HPV16 E6 (Fig. 8B) did not produce changes in the elution profile of E6AP or of HERC2, NEURL4, or the proteasome (PSMD4 and PSMA5) compared to the fractionated protein extract from C33A cells transduced with the empty vector (Fig. 8A). Even with this experimental design, it would be difficult to see minor changes in the association of E6AP with the different protein complexes, but major changes (e.g., loss of interaction with the proteasome or the 2-MDa complex) should result in alterations in the E6AP elution profile. This was not observed in this experiment, allowing us to conclude that HPV16 E6 does not significantly change the association of E6AP with the different cellular protein complexes.
Fig 8.
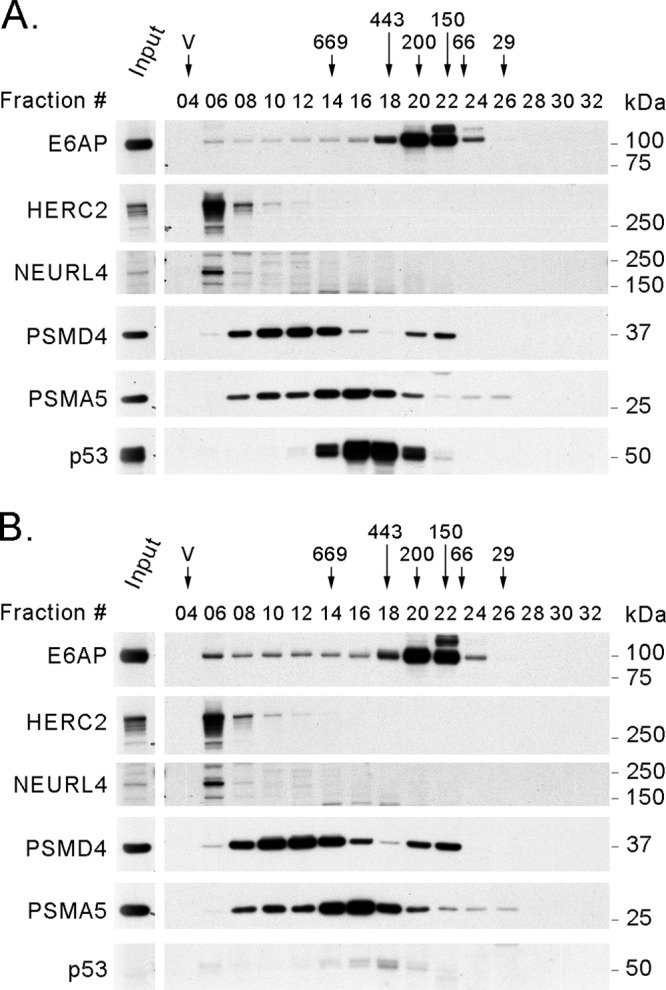
The elution profile of E6AP and several of its interacting proteins is not changed by the expression of HPV16 E6. Four milligrams of protein extract from C33A cells carrying an empty vector as a control or expressing HPV16 E6 was loaded on a Superose 6 column for size exclusion chromatography. After fraction collection, 20 μl (4%) from every second fraction was analyzed by SDS-PAGE and Western blotting using antibodies against E6AP, HERC2, NEURL4, PSMD4, PSMA5, and p53. (A) C33A control cells. (B) C33A cells expressing HPV16 E6. The positions where the peaks of protein standards eluted (669, 443, 200, 150, 66, and 29 kDa) and the void volume (V) are indicated above the fraction numbers. P53 was used as an indicator of E6 expression and function.
HERC2 does not contribute significantly to the E6-mediated degradation of p53.
The Ub ligase activity of E6AP mediates high-risk HPV E6 targeted degradation of p53 (80). It was previously shown that binding to HERC2 can stimulate the Ub ligase activity of E6AP (48). We therefore examined whether HERC2 contributes to the degradation of p53 in cells expressing high-risk HPV E6 proteins. We observed that knocking down E6AP, as expected, resulted in stabilization and increased levels of p53 in HeLa cells. However, knockdown of HERC2 had no measurable effect on p53 levels (Fig. 9). We therefore concluded that HERC2 does not contribute significantly to the E6/E6AP-mediated degradation of p53 in HeLa cells.
Fig 9.
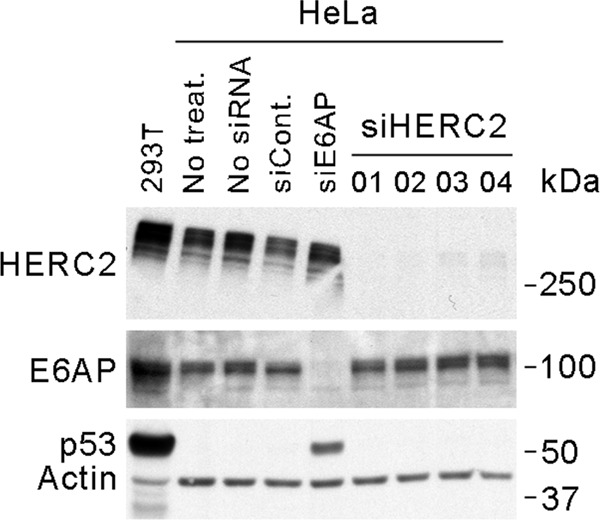
HERC2 does not contribute to the E6-E6AP-mediated degradation of p53 in HeLa cells. Western blot analysis shows the effect of siRNA-mediated knockdown of HERC2 or E6AP on the level of p53 in HeLa cells. siCont., siRNA control number 2; siE6AP, siRNA number 04 against E6AP; siHERC2, siRNAs against HERC2. Protein extract from 293T cells was used as a positive control for p53 detection.
HPV16 E6 is recruited to the high-molecular-weight complex through E6AP.
It has been previously observed that HA-tagged HPV16 E6 can coprecipitate HERC2 (92). That study, however, did not determine whether the interaction was direct or mediated by another protein. We therefore performed a gel filtration experiment using SiHa cells expressing HA-tagged HPV16 E6 (Fig. 10A) and found that E6, in addition to a peak in fraction 26 (about 30 kDa) that probably corresponds to the monomeric and/or dimeric form of the protein (100), had an elution profile that mirrored that of E6AP. A small amount of E6 that coelutes with the 2-MDa complex containing E6AP and HERC2 was present in fraction 6 (Fig. 3 and 10A). Because E6AP can also bind HERC2 in the absence of E6 (48), we examined whether the interaction of E6 with HERC2 was direct or mediated by E6AP. By siRNA-mediated knockdown of E6AP, we found that E6 could no longer pull down HERC2 (Fig. 10B). Furthermore, the ability of E6 to pull down HERC2 was inversely correlated with the efficiency of the E6AP knockdown. These results indicate that E6AP recruits E6 into a high-MW complex containing HERC2 by bridging their interaction.
Fig 10.
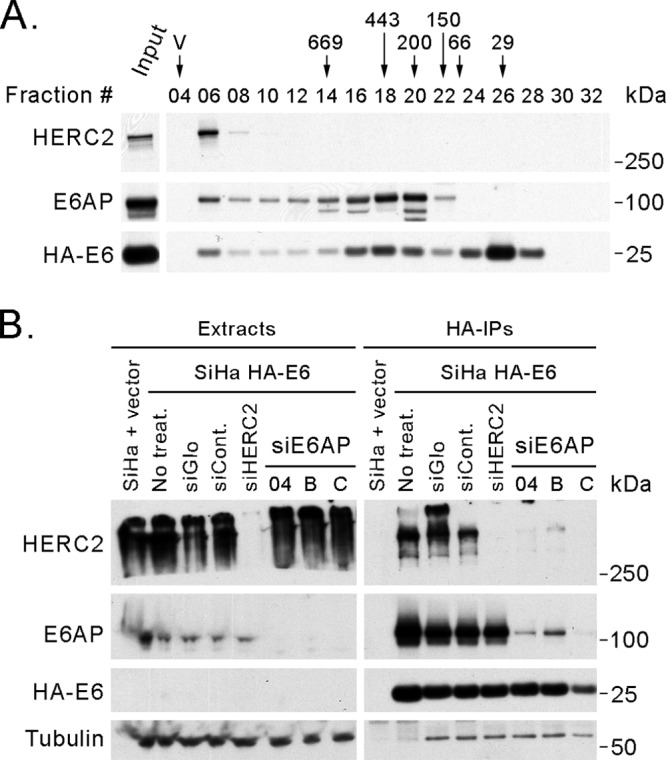
E6AP bridges the interaction between E6 and HERC2. (A) E6AP and HPV16 E6 have similar elution profiles in a gel filtration experiment. Eight milligrams of protein extract from SiHa cells expressing HA-tagged HPV16 E6 was loaded on a Superose 6 column for size exclusion chromatography. After fractionation, 20 μl (4%) from even-numbered fractions was analyzed by SDS-PAGE and Western blotting using antibodies against E6AP and HERC2 and against the HA tag for detection of the tagged E6. The positions where the peaks of protein standards (669, 443, 200, 150, 66, and 29 kDa) eluted and the void volume (V) are indicated above the fraction numbers. Input, 20 μg of protein extract before fractionation. (B) E6AP is necessary for the interaction between E6 and HERC2. SiHa cells expressing HA-tagged HPV16 E6 were transfected with siRNAs against E6AP or HERC2 and then immunoprecipitated using anti-HA-agarose beads. The pulldowns were resolved by SDS-PAGE and analyzed by Western blotting. SiHa HA-E6, SiHa cells expressing HA-tagged HPV16 E6; siCont., siRNA control; siE6AP, siRNAs against E6AP; siHERC2, siRNA against HERC2 number 04.
DISCUSSION
Proteins assemble into complexes to exert their functions at precise times and locations within the cell and, in the case of enzymes, with the correct substrates. Understanding the composition of those complexes and the nature of the relationship between their components is fundamental to comprehending the roles played by individual proteins in cell biology and the impact of their absence or malfunction in cells and organisms. In this study, we have identified several new HCIPs of E6AP, and by gel filtration experiments, we have shown that E6AP is a component of several distinct protein complexes.
We found only a few differences between the proteins pulled down by the three different isoforms of E6AP. According to the MS data, E6AP isoform II specifically coprecipitated KTN1, HIST1H2BK, MAP7D1, RPS26, EIF3D, and POTEF, but these potential interactions have not yet been further validated. Regardless, most of the HCIPs were the same for the three E6AP isoforms, arguing that there must be a high degree of redundancy between the proteins in terms of their abilities to associate with proteins and protein complexes in the cell. However, these data do not rule out the possibility that there are physiologically important differences among the E6AP isoforms related to specific interacting proteins (as might be the case for isoform II), subcellular localization, tissue-specific expression patterns, or developmentally specific expression patterns.
For the newly identified E6AP HCIPs, we validated interactions with HIF1AN, MAPK6, and NEURL4, however, the functional consequences of the interaction of each of these proteins with E6AP is still open. These interactors could be (i) substrates of E6AP, (ii) modifiers of E6AP when those interactors have enzymatic activity (e.g., HIF1AN and MAPK6), (iii) both E6AP and the interacting enzyme could modify each other, or (iv) none of these, as components of a complex that function on other proteins. From the Western blot analysis shown in Fig. 1C, HIF1AN would be a candidate for a ubiquitylation substrate of E6AP, since the amount of HIF1AN pulled down by the wt E6AP is much smaller (relative to E6AP) than that pulled down by the DN form of the ligase. However, we have not found any difference in the steady-state levels of HIF1AN following siRNA-mediated knockdown of E6AP (data not shown). These observations could be explained if only a small fraction of HIF1AN protein in the cell were the target of E6AP or if redundant Ub ligase activities regulated HIF1AN. Several proteins containing ankyrin repeat domains have been shown to interact with HIF1AN, and it has been postulated that through such binding, ankyrin repeat proteins might regulate the HIF1AN activity on HIF1α by sequestering HIF1AN in the cell (13, 14). Similarly, these ankyrin repeat proteins might limit the amount of HIF1AN available as the substrate for E6AP. Thus, this interaction between E6AP and HIF1AN could potentially be quite interesting in that E6AP might then contribute to the regulation of HIF1α transcriptional activity by controlling the levels of HIF1AN and/or in that E6AP activity itself might be regulated by oxygen levels in the cell through hydroxylation by HIF1AN.
In agreement with previous observations, we found that some E6AP is associated with the proteasome (6, 45, 78, 87, 94). The functional consequence of the association of E6AP with the proteasome is an open question. There is no E6AP ortholog in yeast. Therefore, perhaps the association of E6AP with the proteasome reflects a more complex regulation of the mammalian proteasome compared to the yeast proteasome in order to deal with more complex sets of inputs and responses. The dissociation of E6AP from proteasomes in neurons treated with N-methyl-d-aspartic acid (NMDA) (87) suggests an important contribution of the enzyme to the proteosomal function, since its association with the proteasome can be regulated in response to specific stimuli, perhaps, as suggested by Tai et al. (87), modulating the proteasome activity to produce the correct response. Another HECT Ub ligase, HUL5, has also been shown to associate with the proteasome in yeast, where it contributes to proteasomal processivity (4). We therefore tested whether E6AP could fulfill a similar function in mammalian cells. As shown in Fig. 4, we found that UBE3C, the mammalian ortholog of HUL5, contributes to proteasomal processivity in human cells, indicating that this role of HUL5/UBE3C has been conserved throughout evolution. In contrast, we did not find any general effect of E6AP on proteasomal processivity. Parkin, a RING finger Ub ligase, has also been shown to bind the proteasome and to modulate its activity (91). However, neither UBE3C nor E6AP seemed to affect the proteasome activity itself, since siRNA knockdown of either did not alter the level of the full-length EGFP-Myc protein (Fig. 4). These experiments do not rule out the possibility that E6AP could have a role in proteasomal activity or processivity for a specific subpopulation of proteasomes, for specific substrates, under certain conditions, or a combination of those factors. HUL5 has recently been shown to play a major role in the ubiquitylation and turnover of cytosolic misfolded proteins as part of the heat shock stress response, whereas in unstressed cells it is involved in the ubiquitylation of low-solubility proteins but without affecting the overall ubiquitylation levels in the cells (25). Since E6AP has also been associated with the degradation of misfolded proteins (20, 63, 64), perhaps a contribution of E6AP to the proteasome function becomes more significant under conditions that produce proteotoxic stress. Another possibility is that E6AP might affect the subcellular distribution of associated proteasomes, perhaps in a manner similar to that of CaMKIIα, which can act as a scaffold in neurons to recruit proteasomes to dendritic spines (8).
E6AP associates with HERC2, NEURL4 and MAPK6 in a large complex of about 2 MDa through its interaction with HERC2, since the E6AP-HERC2 interaction seems to be independent of other proteins in the complex, and the knockdown of HERC2 releases E6AP from this high-MW complex by gel filtration (48) and its interaction with NEURL4 and MAPK6 (Fig. 6). In this complex, HERC2 binds to NEURL4, which in turn interacts with MAPK6 (Fig. 7). MAPK6 is a Ser/Thr protein kinase closely related to mitogen-activated protein kinases and is regulated by protein stability (17, 18). It will be of interest to determine what roles, if any, E6AP, HERC2, or some other ubiquitin ligase in this 2-MDa complex may have in regulating MAPK6 stability.
MAPK6 is a binding partner of MK5 (also known as MAPK-activated protein kinase 5 [MAPKAPK5] or PRAK) and directs the phosphorylation and activation of MK5 (81). MK5 is also likely part of this 2-MDa complex, since it was present in several of the E6AP pulldowns (see Tables S1 and S2 in the supplemental material). The MAPK6/MK5 signaling module has been recently shown to play a role in neural morphogenesis through the regulation of neuronal cytoskeleton and dendritic-spine formation (9). Interestingly, a mouse model for AS has been shown to have abnormally shaped dendritic spines, reduced in size and density, suggesting that E6AP may be involved in the regulation of dendritic-spine development and synaptic plasticity (21). This potential functional overlap, added to the fact that E6AP and MAPK6/MK5 are associated, suggests that these proteins may work together in a pathway to regulate dendritic-spine formation and synaptic plasticity.
NEURL4, in contrast to HIF1AN and MAPK6, does not contain any defined catalytic domain but does contain six neuralized homology repeat (NHR) domains that have been implicated in mediating protein-protein interactions. As such, NEURL4 might serve as a platform on which a multiprotein complex assembles. The Western blot in Fig. 1D suggests that NEURL4 is not actively targeted for degradation by E6AP, since the wt and DN forms pulled down similar amounts of NEURL4. A search using the Conserved Domain Architecture Retrieval Tool (CDART) (27) revealed that the NHR domain often appears in proteins that also contain either a RING finger domain, which defines a large family of Ub ligases (40), or a SOCS box domain, which participates in the formation of cullin ring Ub ligase complexes (71). Indeed, an NHR domain of the Drosophila RING finger Ub ligase Neuralized has been reported to be responsible for substrate recognition (15). In the context of the 2-MDa complex, NEURL4 could function as an adaptor, bringing substrates to the E6AP and HERC2 ubiquitin ligases. Such a model has recently been proposed by Al-Hakim et al. for NEURL4 and HERC2 (2). NEURL4 has been shown to promote the ubiquitylation and degradation of the centrosomal protein CP110 (53). Perhaps NEURL4 mediates the recruitment of CP110 to E6AP, HERC2, or some other Ub ligase.
AS results from the lack of E6AP Ub ligase activity in the imprinted regions of the brain (16, 67). Interestingly, some of the symptoms described for mice bearing spontaneous mutations of HERC2 (rjs or jdf2 phenotypes) include tremors and jerky gait (39, 52), which are also present in AS (3, 30). The partial overlap of symptoms between AS and mice lacking HERC2 suggests that some of the symptoms observed in AS could be the result of the absence of E6AP ligase activity associated with the 2-MDa complex, since the absence of HERC2 also eliminates E6AP from the complex.
Previously, our laboratory reported that the HPV16 E6 protein coprecipitates with HERC2 (92). Subsequently, Kühnle et al. showed that HERC2 can bind to E6AP in the absence of E6 and that upon binding, it enhances E6AP Ub ligase activity toward RING1B (48). We found in this study that E6AP bridges the interaction between HPV16 E6 and HERC2 and that HERC2 does not contribute significantly to E6/E6AP-mediated degradation of p53 in HeLa cells. A gel filtration experiment using extracts from SiHa cells expressing HA-tagged HPV16 E6 showed that the elution profile of E6 mirrors that of E6AP (Fig. 10A). Moreover, a similar experiment using C33A cells plus or minus HPV16 E6 expression revealed that E6 does not produce any major alteration in the elution profile of E6AP or the HCIPs tested in this experiment, namely, HERC2, NEURL4, PSMD4, and PSMA5 (Fig. 8). This suggests that E6 does not significantly affect the binding of E6AP to the complexes that it normally associates with and that E6 itself associates with these various complexes through its binding to E6AP.
Both E6AP and a NEURL4/HERC2 complex have been reported to localize and regulate centrosomes (2, 83). The fact that E6AP interacts with NEURL4 and HERC2 only in the 2-MDa complex (Fig. 3) suggests that these proteins might contribute to the regulation of the centrosomes through this complex. Interestingly, the high-risk HPV E6 proteins have been reported to cause centrosomal aberrations (23, 79). Our results indicate that E6 binds to the 2-MDa complex through its interaction with E6AP, since it associates with HERC2 through E6AP and cofractionates with the complex in gel filtration experiments (Fig. 10). Therefore, the effects of E6 on centrosomes could be a consequence of E6 binding to the complex, perhaps affecting its normal function(s). Extending this line of thinking and given the extensive association between E6 and E6AP, it might also be possible that the recruitment of E6 to different protein complexes through binding to E6AP contributes to a number of the cellular phenotypes that have been attributed to the high-risk HPV E6 proteins but are not yet mechanistically well understood.
In this study, we have identified several new E6AP interactors and have shown that this Ub ligase is part of several different protein complexes. Our study reveals more complexity than previously appreciated for E6AP. We were able to assign several E6AP interactors to different protein complexes and to begin to characterize a 2-MDa complex that contains MAPK6, NEURL4, HERC2, and E6AP, whose functions may have relevance for AS and for some of the activities that have been ascribed to HPV E6. Studies are now under way to further characterize this high-MW complex and the other E6AP associations identified in this paper.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Howley and Harper laboratories for helpful discussions and suggestions.
This work was supported by a grant from the Angelman Syndrome Foundation (to P.M.H.), as well as National Institutes of Health grants P01CA50661 (to P.M.H.) and R01GM054137 and R01GM070565 (to J.W.H).
Footnotes
Published ahead of print 29 May 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Albrecht U, et al. 1997. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat. Genet. 17:75–78. [DOI] [PubMed] [Google Scholar]
- 2. Al-Hakim AK, Bashkurov M, Gingras AC, Durocher D, Pelletier L. 2012. Interaction proteomics identify NEURL4 and the HECT E3 ligase HERC2 as novel modulators of centrosome architecture. Mol. Cell. Proteomics 11:M111.014233 doi:10.1074/mcp.M111.014233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Angelman H. 1965. ‘Puppet’ children: a report on three cases. Dev. Med. Child Neurol. 7:681–688 [DOI] [PubMed] [Google Scholar]
- 4. Aviram S, Kornitzer D. 2010. The ubiquitin ligase Hul5 promotes proteasomal processivity. Mol. Cell. Biol. 30:985–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Behrends C, Sowa ME, Gygi SP, Harper JW. 2010. Network organization of the human autophagy system. Nature 466:68–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Besche HC, Haas W, Gygi SP, Goldberg AL. 2009. Isolation of mammalian 26S proteasomes and p97/VCP complexes using the ubiquitin-like domain from HHR23B reveals novel proteasome-associated proteins. Biochemistry 48:2538–2549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beyette J, Mason GG, Murray RZ, Cohen GM, Rivett AJ. 1998. Proteasome activities decrease during dexamethasone-induced apoptosis of thymocytes. Biochem. J. 332:315–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bingol B, et al. 2010. Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell 140:567–578 [DOI] [PubMed] [Google Scholar]
- 9. Brand F, et al. 2012. The ERK3 (MAPK6)-MAPKAP kinase 5 signalling complex regulates septin function and dendrite morphology. Mol. Cell. Biol., 32:2467–2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buiting K, et al. 2001. Disruption of the bipartite imprinting center in a family with Angelman syndrome. Am. J. Hum. Genet. 68:1290–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Buiting K, et al. 1998. Sporadic imprinting defects in Prader-Willi syndrome and Angelman syndrome: implications for imprint-switch models, genetic counseling, and prenatal diagnosis. Am. J. Hum. Genet. 63:170–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Christianson JC, et al. 2012. Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol. 14:93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cockman ME, Webb JD, Kramer HB, Kessler BM, Ratcliffe PJ. 2009. Proteomics-based identification of novel factor inhibiting hypoxia-inducible factor (FIH) substrates indicates widespread asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Mol. Cell. Proteomics 8:535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cockman ME, Webb JD, Ratcliffe PJ. 2009. FIH-dependent asparaginyl hydroxylation of ankyrin repeat domain-containing proteins. Ann. N. Y. Acad. Sci. 1177:9–18 [DOI] [PubMed] [Google Scholar]
- 15. Commisso C, Boulianne GL. 2007. The NHR1 domain of Neuralized binds Delta and mediates Delta trafficking and Notch signaling. Mol. Biol. Cell 18:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cooper EM, Hudson AW, Amos J, Wagstaff J, Howley PM. 2004. Biochemical analysis of Angelman syndrome-associated mutations in the E3 ubiquitin ligase E6-associated protein. J. Biol. Chem. 279:41208–41217 [DOI] [PubMed] [Google Scholar]
- 17. Coulombe P, Rodier G, Bonneil E, Thibault P, Meloche S. 2004. N-Terminal ubiquitination of extracellular signal-regulated kinase 3 and p21 directs their degradation by the proteasome. Mol. Cell. Biol. 24:6140–6150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Coulombe P, Rodier G, Pelletier S, Pellerin J, Meloche S. 2003. Rapid turnover of extracellular signal-regulated kinase 3 by the ubiquitin-proteasome pathway defines a novel paradigm of mitogen-activated protein kinase regulation during cellular differentiation. Mol. Cell. Biol. 23:4542–4558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crosas B, et al. 2006. Ubiquitin chains are remodeled at the proteasome by opposing ubiquitin ligase and deubiquitinating activities. Cell 127:1401–1413 [DOI] [PubMed] [Google Scholar]
- 20. Cummings CJ, et al. 1999. Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron 24:879–892 [DOI] [PubMed] [Google Scholar]
- 21. Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL. 2008. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 17:111–118 [DOI] [PubMed] [Google Scholar]
- 22. Driscoll DJ, et al. 1992. A DNA methylation imprint, determined by the sex of the parent, distinguishes the Angelman and Prader-Willi syndromes. Genomics 13:917–924 [DOI] [PubMed] [Google Scholar]
- 23. Duensing S, Munger K. 2002. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 62:7075–7082 [PubMed] [Google Scholar]
- 24. Engel E. 1997. Uniparental disomy (UPD). Genomic imprinting and a case for new genetics (prenatal and clinical implications: the “Likon” concept). Ann. Genet. 40:24–34 [PubMed] [Google Scholar]
- 25. Fang NN, Ng AH, Measday V, Mayor T. 2011. Hul5 HECT ubiquitin ligase plays a major role in the ubiquitylation and turnover of cytosolic misfolded proteins. Nat. Cell Biol. 13:1344–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fontana JR, Posakony JW. 2009. Both inhibition and activation of Notch signaling rely on a conserved Neuralized-binding motif in Bearded proteins and the Notch ligand Delta. Dev. Biol. 333:373–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Geer LY, Domrachev M, Lipman DJ, Bryant SH. 2002. CDART: protein homology by domain architecture. Genome Res. 12:1619–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Glas R, Bogyo M, McMaster JS, Gaczynska M, Ploegh HL. 1998. A proteolytic system that compensates for loss of proteasome function. Nature 392:618–622 [DOI] [PubMed] [Google Scholar]
- 29. Greer PL, et al. 2010. The Angelman syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 140:704–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guerrini R, Carrozzo R, Rinaldi R, Bonanni P. 2003. Angelman syndrome: etiology, clinical features, diagnosis, and management of symptoms. Paediatr. Drugs. 5:647–661 [DOI] [PubMed] [Google Scholar]
- 31. Guffanti G, et al. 2011. Role of UBE3A and ATP10A genes in autism susceptibility region 15q11-q13 in an Italian population: a positive replication for UBE3A. Psychiatry Res. 185:33–38 [DOI] [PubMed] [Google Scholar]
- 32. Hansen CA, Bartek J, Jensen S. 2008. A functional link between the human cell cycle-regulatory phosphatase Cdc14A and the atypical mitogen-activated kinase Erk3. Cell Cycle 7:325–334 [DOI] [PubMed] [Google Scholar]
- 33. Hewitson KS, et al. 2002. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J. Biol. Chem. 277:26351–26355 [DOI] [PubMed] [Google Scholar]
- 34. Huibregtse JM, Scheffner M, Beaudenon S, Howley PM. 1995. A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. U. S. A. 92:5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huibregtse JM, Scheffner M, Howley PM. 1991. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 10:4129–4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huibregtse JM, Scheffner M, Howley PM. 1993. Cloning and expression of the cDNA for E6-AP, a protein that mediates the interaction of the human papillomavirus E6 oncoprotein with p53. Mol. Cell. Biol. 13:775–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huibregtse JM, Scheffner M, Howley PM. 1994. E6-AP directs the HPV E6-dependent inactivation of p53 and is representative of a family of structurally and functionally related proteins. Cold Spring Harbor Symp. Quant. Biol. 59:237–245 [DOI] [PubMed] [Google Scholar]
- 38. Ingham RJ, et al. 2005. WW domains provide a platform for the assembly of multiprotein networks. Mol. Cell. Biol. 25:7092–7106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ji Y, et al. 1999. The ancestral gene for transcribed, low-copy repeats in the Prader-Willi/Angelman region encodes a large protein implicated in protein trafficking, which is deficient in mice with neuromuscular and spermiogenic abnormalities. Hum. Mol. Genet. 8:533–542 [DOI] [PubMed] [Google Scholar]
- 40. Joazeiro CA, Weissman AM. 2000. RING finger proteins: mediators of ubiquitin ligase activity. Cell 102:549–552 [DOI] [PubMed] [Google Scholar]
- 41. Jolliffe CN, Harvey KF, Haines BP, Parasivam G, Kumar S. 2000. Identification of multiple proteins expressed in murine embryos as binding partners for the WW domains of the ubiquitin-protein ligase Nedd4. Biochem. J. 351:557–565 [PMC free article] [PubMed] [Google Scholar]
- 42. Kelley ML, Keiger KE, Lee CJ, Huibregtse JM. 2005. The global transcriptional effects of the human papillomavirus E6 protein in cervical carcinoma cell lines are mediated by the E6AP ubiquitin ligase. J. Virol. 79:3737–3747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kishino T, Lalande M, Wagstaff J. 1997. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 15:70–73 [DOI] [PubMed] [Google Scholar]
- 44. Kleijnen MF, Alarcon RM, Howley PM. 2003. The ubiquitin-associated domain of hPLIC-2 interacts with the proteasome. Mol. Biol. Cell 14:3868–3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kleijnen MF, et al. 2000. The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol. Cell 6:409–419 [DOI] [PubMed] [Google Scholar]
- 46. Knoll JH, Nicholls RD, Lalande M. 1989. On the parental origin of the deletion in Angelman syndrome. Hum. Genet. 83:205–207 [DOI] [PubMed] [Google Scholar]
- 47. Kuhne C, Banks L. 1998. E3-ubiquitin ligase/E6-AP links multicopy maintenance protein 7 to the ubiquitination pathway by a novel motif, the L2G box. J. Biol. Chem. 273:34302–34309 [DOI] [PubMed] [Google Scholar]
- 48. Kuhnle S, et al. 2011. Physical and functional interaction of the HECT ubiquitin-protein ligases E6AP and HERC2. J. Biol. Chem. 286:19410–19416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kumar S, Talis AL, Howley PM. 1999. Identification of HHR23A as a substrate for E6-associated protein-mediated ubiquitination. J. Biol. Chem. 274:18785–18792 [DOI] [PubMed] [Google Scholar]
- 50. Lando D, et al. 2002. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 16:1466–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Leggett DS, et al. 2002. Multiple associated proteins regulate proteasome structure and function. Mol. Cell 10:495–507 [DOI] [PubMed] [Google Scholar]
- 52. Lehman AL, et al. 1998. A very large protein with diverse functional motifs is deficient in rjs (runty, jerky, sterile) mice. Proc. Natl. Acad. Sci. U. S. A. 95:9436–9441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li J, et al. 2012. Neurl4, a novel daughter centriole protein, prevents formation of ectopic microtubule organizing centres. EMBO Rep. 13:547–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li L, Li Z, Howley PM, Sacks DB. 2006. E6AP and calmodulin reciprocally regulate estrogen receptor stability. J. Biol. Chem. 281:1978–1985 [DOI] [PubMed] [Google Scholar]
- 55. Li S, et al. 2010. ERK3 is required for metaphase-anaphase transition in mouse oocyte meiosis. PLoS One 5:e13074 doi:10.1371/journal.pone.0013074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Louria-Hayon I, et al. 2009. E6AP promotes the degradation of the PML tumor suppressor. Cell Death Differ. 16:1156–1166 [DOI] [PubMed] [Google Scholar]
- 57. Mabb AM, Judson MC, Zylka MJ, Philpot BD. 2011. Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 34:293–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Magenis RE, Brown MG, Lacy DA, Budden S, LaFranchi S. 1987. Is Angelman syndrome an alternate result of del(15)(q11q13)? Am. J. Med. Genet. 28:829–838 [DOI] [PubMed] [Google Scholar]
- 59. Magenis RE, et al. 1990. Comparison of the 15q deletions in Prader-Willi and Angelman syndromes: specific regions, extent of deletions, parental origin, and clinical consequences. Am. J. Med. Genet. 35:333–349 [DOI] [PubMed] [Google Scholar]
- 60. Mahon PC, Hirota K, Semenza GL. 2001. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 15:2675–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Martin A, Baker TA, Sauer RT. 2008. Protein unfolding by a AAA+ protease is dependent on ATP-hydrolysis rates and substrate energy landscapes. Nat. Struct. Mol. Biol. 15:139–145 [DOI] [PubMed] [Google Scholar]
- 62. Matsuura T, et al. 1997. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 15:74–77 [DOI] [PubMed] [Google Scholar]
- 63. Mishra A, et al. 2008. E6-AP promotes misfolded polyglutamine proteins for proteasomal degradation and suppresses polyglutamine protein aggregation and toxicity. J. Biol. Chem. 283:7648–7656 [DOI] [PubMed] [Google Scholar]
- 64. Mishra A, Godavarthi SK, Maheshwari M, Goswami A, Jana NR. 2009. The ubiquitin ligase E6-AP is induced and recruited to aggresomes in response to proteasome inhibition and may be involved in the ubiquitination of Hsp70-bound misfolded proteins. J. Biol. Chem. 284:10537–10545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Murphy GJ, Mostoslavsky G, Kotton DN, Mulligan RC. 2006. Exogenous control of mammalian gene expression via modulation of translational termination. Nat. Med. 12:1093–1099 [DOI] [PubMed] [Google Scholar]
- 66. Nasu J, et al. 2010. E6AP ubiquitin ligase mediates ubiquitin-dependent degradation of peroxiredoxin 1. J. Cell Biochem. 111:676–685 [DOI] [PubMed] [Google Scholar]
- 67. Nawaz Z, et al. 1999. The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol. Cell. Biol. 19:1182–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nurmi EL, et al. 2001. Linkage disequilibrium at the Angelman syndrome gene UBE3A in autism families. Genomics 77:105–113 [DOI] [PubMed] [Google Scholar]
- 69. Oda H, Kumar S, Howley PM. 1999. Regulation of the Src family tyrosine kinase Blk through E6AP-mediated ubiquitination. Proc. Natl. Acad. Sci. U. S. A. 96:9557–9562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Papa FR, Amerik AY, Hochstrasser M. 1999. Interaction of the Doa4 deubiquitinating enzyme with the yeast 26S proteasome. Mol. Biol. Cell 10:741–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Piessevaux J, Lavens D, Peelman F, Tavernier J. 2008. The many faces of the SOCS box. Cytokine Growth Factor Rev. 19:371–381 [DOI] [PubMed] [Google Scholar]
- 72. Powell ML, et al. 2010. NCoR1 mediates papillomavirus E8:E2C transcriptional repression. J. Virol. 84:4451–4460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rahman S, et al. 2011. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell. Biol. 31:2641–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rivett AJ, et al. 2002. Assays of proteasome activity in relation to aging. Exp. Gerontol. 37:1217–1222 [DOI] [PubMed] [Google Scholar]
- 75. Rougeulle C, Glatt H, Lalande M. 1997. The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat. Genet. 17:14–15 [DOI] [PubMed] [Google Scholar]
- 76. Saeed AI, et al. 2006. TM4 microarray software suite. Methods Enzymol. 411:134–193 [DOI] [PubMed] [Google Scholar]
- 77. Saeed AI, et al. 2003. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34:374–378 [DOI] [PubMed] [Google Scholar]
- 78. Scanlon TC, et al. 2009. Isolation of human proteasomes and putative proteasome-interacting proteins using a novel affinity chromatography method. Exp. Cell Res. 315:176–189 [DOI] [PubMed] [Google Scholar]
- 79. Schaeffer AJ, et al. 2004. E6 and E7 oncoproteins induce distinct patterns of chromosomal aneuploidy in skin tumors from transgenic mice. Cancer Res. 64:538–546 [DOI] [PubMed] [Google Scholar]
- 80. Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. 1993. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75:495–505 [DOI] [PubMed] [Google Scholar]
- 81. Seternes OM, et al. 2004. Activation of MK5/PRAK by the atypical MAP kinase ERK3 defines a novel signal transduction pathway. EMBO J. 23:4780–4791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shimoji T, et al. 2009. Identification of annexin A1 as a novel substrate for E6AP-mediated ubiquitylation. J. Cell Biochem. 106:1123–1135 [DOI] [PubMed] [Google Scholar]
- 83. Singhmar P, Kumar A. 2011. Angelman syndrome protein UBE3A interacts with primary microcephaly protein ASPM, localizes to centrosomes and regulates chromosome segregation. PLoS One 6:e20397 doi:10.1371/journal.pone.0020397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Smith SE, et al. 2011. Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci. Transl. Med. 3:103ra97 doi:10.1126/scitranslmed.3002627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Sowa ME, Bennett EJ, Gygi SP, Harper JW. 2009. Defining the human deubiquitinating enzyme interaction landscape. Cell 138:389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sun M, et al. 2006. Identification of extracellular signal-regulated kinase 3 as a new interaction partner of cyclin D3. Biochem. Biophys. Res. Commun. 340:209–214 [DOI] [PubMed] [Google Scholar]
- 87. Tai HC, Besche H, Goldberg AL, Schuman EM. 2010. Characterization of the brain 26S proteasome and its interacting proteins. Front. Mol. Neurosci. 3:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Talis AL, Huibregtse JM, Howley PM. 1998. The role of E6AP in the regulation of p53 protein levels in human papillomavirus (HPV)-positive and HPV-negative cells. J. Biol. Chem. 273:6439–6445 [DOI] [PubMed] [Google Scholar]
- 89. Tan MJA, et al. 2012. Cutaneous beta-human papillomavirus E6 proteins bind Mastermind-like coactivators and repress Notch signaling. Proc. Natl. Acad. Sci. U. S. A., 109:E1473–E1480 doi:10.1073/pnas.1205991109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tsai TF, Jiang YH, Bressler J, Armstrong D, Beaudet AL. 1999. Paternal deletion from Snrpn to Ube3a in the mouse causes hypotonia, growth retardation and partial lethality and provides evidence for a gene contributing to Prader-Willi syndrome. Hum. Mol. Genet. 8:1357–1364 [DOI] [PubMed] [Google Scholar]
- 91. Um JW, et al. 2010. Parkin directly modulates 26S proteasome activity. J. Neurosci. 30:11805–11814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Vos RM, Altreuter J, White EA, Howley PM. 2009. The ubiquitin-specific peptidase USP15 regulates human papillomavirus type 16 E6 protein stability. J. Virol. 83:8885–8892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Vu TH, Hoffman AR. 1997. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat. Genet. 17:12–13 [DOI] [PubMed] [Google Scholar]
- 94. Wang X, et al. 2007. Mass spectrometric characterization of the affinity-purified human 26S proteasome complex. Biochemistry 46:3553–3565 [DOI] [PubMed] [Google Scholar]
- 95. White EA, et al. 2012. Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proc. Natl. Acad. Sci. U. S. A. 109:E260–E267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Williams CA. 2010. The behavioral phenotype of the Angelman syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 154C:432–437 [DOI] [PubMed] [Google Scholar]
- 97. Williams CA, Driscoll DJ, Dagli AI. 2010. Clinical and genetic aspects of Angelman syndrome. Genet. Med. 12:385–395 [DOI] [PubMed] [Google Scholar]
- 98. Yamamoto Y, Huibregtse JM, Howley PM. 1997. The human E6-AP gene (UBE3A) encodes three potential protein isoforms generated by differential splicing. Genomics 41:263–266 [DOI] [PubMed] [Google Scholar]
- 99. Zaaroor-Regev D, et al. 2010. Regulation of the polycomb protein Ring1B by self-ubiquitination or by E6-AP may have implications to the pathogenesis of Angelman syndrome. Proc. Natl. Acad. Sci. U. S. A. 107:6788–6793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zanier K, et al. 2010. E6 proteins from diverse papillomaviruses self-associate both in vitro and in vivo. J. Mol. Biol. 396:90–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhang M, Coffino P. 2004. Repeat sequence of Epstein-Barr virus-encoded nuclear antigen 1 protein interrupts proteasome substrate processing. J. Biol. Chem. 279:8635–8641 [DOI] [PubMed] [Google Scholar]
- 102. Zheng L, et al. 2008. E3 ubiquitin ligase E6AP-mediated TSC2 turnover in the presence and absence of HPV16 E6. Genes Cells 13:285–294 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.