

Abstract
The Wiskott-Aldrich syndrome protein (WASp) is a key regulator of actin dynamics during cell motility and adhesion, and mutations in its gene are responsible for Wiskott-Aldrich syndrome (WAS). Here, we demonstrate that WASp is ubiquitylated following T-cell antigen receptor (TCR) activation. WASp phosphorylation at tyrosine 291 results in recruitment of the E3 ligase Cbl-b, which, together with c-Cbl, carries out WASp ubiquitylation. Lysine residues 76 and 81, located at the WASp WH1 domain, which contains the vast majority of WASp gene mutations, serve as the ubiquitylation sites. Disruption of WASp ubiquitylation causes WASp accumulation and alters actin dynamics and the formation of actin-dependent structures. Our data suggest that regulated degradation of activated WASp might be an efficient strategy by which the duration and localization of actin rearrangement and the intensity of T-cell activation are controlled.
INTRODUCTION
The Wiskott-Aldrich syndrome protein (WASp) is an actin filament nucleation protein. WASp not only is involved in actin cytoskeletal reorganization but also regulates transcriptional activity, cytokine production, cell proliferation, and motility (35, 38, 42). Previously, we showed that following initial T-cell activation, WASp is recruited to the T-cell antigen receptor (TCR) site, subsequently driving nucleation of filamentous actin (F-actin) networks (5, 34). Much of the F-actin is found in narrow protrusive filopodial or flat lamellipodial structures, thereby enabling processes critical for maintaining a functional immune response, including T-cell spreading, adhesion, and formation and maintenance of the immunological synapse essential for the recognition of foreign antigens by antigen-presenting cells (APCs) (11). These changes in actin filaments were found to be highly dynamic and are regulated by signaling molecules, including the linker for the activation of T cells (LAT), SLP-76, Nck, WASp, and others (5, 10, 26, 27). WASp binds and activates the Arp2/3 complex and promotes actin polymerization and its recruitment to the TCR site (3, 5, 30). Late in the activation process, vesicles containing both SLP-76 and WASp are endocytosed via a lipid raft-dependent pathway (5, 7).
The involvement of WASp in actin filament formation depends on its functional activation and recruitment to the T cell-APC contact site. WASp adopts an autoinhibited conformation in which its basic region, located at the N terminus of WASp and flanked by the Wiskott homology 1 (WH1) domain and the GTPase-binding domain (GBD), forms an intramolecular interaction with the verprolin homology, central hydrophobic region, and acidic region (VCA) domains located at its C terminus. The Rho family GTPase, Cdc42, when activated by the guanine nucleotide exchange factor (GEF) VAV1, binds to the WASp GBD. This binding, together with phosphorylation of WASp on tyrosine 291, induces a dramatic conformational change (1). The hydrophobic core is disrupted, releasing the VCA domain and enabling its interaction with the Arp2/3 complex, thereby promoting actin polymerization (14). WASp associates with multiple proteins through several of its domains, including the association between the WASp WH1 domain and the WASp-interacting protein (WIP) (15, 29) and the association of the WASp proline-rich domain (PRD) with SH3 domain adaptors such as Nck, which recruits WASp to the TCR site (5, 31).
The essential contribution of WASp to the lymphocyte-mediated immune response is reflected by Wiskott-Aldrich syndrome (WAS), a severe X-linked immunodeficiency disease that is caused by WASp gene deletions or mutations, leading to WASp deficiency or its reduced expression (15, 16, 21, 28, 41).
Recent studies suggested that WASp, and specifically its homologue, neural WASp (N-WASp), might be a target of proteasomal degradation (20); however, the molecular mechanism that mediates this degradation process and its functional consequences is unknown. In the present study, we demonstrate that WASp is ubiquitylated on lysine residues 76 and 81, encoded by exon 2 in the WH1 domain. This process is mediated by the E3 ligases c-Cbl and Cbl-b. WASp ubiquitylation depends on its phosphorylation at the tyrosine 291 site, which associates with the tyrosine kinase-binding (TKB) domain of Cbl-b. The expression of specific WASp mutants in the WH1 domain resulted in WASp accumulation, impairment of WASp dynamics, and aberrant actin rearrangement. In addition, upregulation of nuclear factor of activated T cell (NFAT) transcription factor activity and an increase in the intracellular calcium concentration were detected in the nonubiquitylated WASp mutants. Our findings demonstrate that regulation of the WASp degradation process plays an important role in WASp localization and activity and directly controls TCR signaling and actin-dependent processes.
MATERIALS AND METHODS
Reagents.
Antibodies and their sources were as follows. Antibodies for imaging were mouse anti-CD3ε (UCHT or HIT3a) and anti-CD28 (Becton, Dickinson [BD] Biosciences) and Alexa-conjugated isotype-specific secondary antibodies (Molecular Probes). Rabbit antiphosphotyrosine antibody was a kind gift of Lawrence E. Samelson. Primary antibodies for immunoprecipitation and immunoblotting were mouse anti-GFP (Roche), mouse anti-GAPDH (Biodesign), rabbit anti-WASp H-250, mouse anti-WASp D1, mouse anti-ubiquitin P4D1, mouse anti-Cbl-b, and rabbit anti-c-Cbl (Santa Cruz), and rat anti-HA–peroxidase 3F10 (Roche). Secondary antibodies were goat anti-mouse (Sigma-Aldrich) and goat anti-rabbit (Santa Cruz) antibodies. Antibodies for T cell stimulation were anti-CD3ε (OKT3 ascites) or mouse IgM anti-T-cell antigen receptor (clone C3O5).
For knockdown of gene expression, pools of independent specific small interfering RNA (siRNA) oligonucleotides (Smartpools) were purchased from Dharmacon.
The c-Cbl siRNA sequences were CCAGCAGAUUGAUAGCUGUACGUAU, GCGGAGAAUCAACUCUGAACGGAAA, and CCUACCAGGACAUCCAGAAAGCUUU.
The Cbl-b siRNA sequences were UCAUCCCACCCUGUUUCCCUGAAUU, GGUCCAUCUUCAGAGAAGAAAUCAA, and CAUGGGAGAGGGUUAUGCCUUUGAA.
Expression vectors and plasmids.
Human WASp cDNA was kindly provided by D. Nelson (National Cancer Institute, NIH, Bethesda, MD). The cDNA was cloned into the expression vectors pEYFP-N1, pEYFP-C1, and pECFP-C1 from Clontech to obtain the cyan fluorescent protein (CFP)- or yellow fluorescent protein (YFP)-tagged WASp. Green fluorescent protein (GFP) fusions were rendered monomeric by an A206K substitution. For untagged protein expression, WASp was cloned into pcDNA3.1+Hygro vector from Invitrogen. The c-Cbl cDNAs from the pSX c-Cbl wild type (wt) and the pSX 70z/3 c-Cbl, which has a 17-amino acid (aa) deletion (residues 366 to 382), were cloned into the pcDNA3.1+Hygro vector. The hemagglutinin (HA)-tagged ubiquitin plasmid was previously described (4). The pCEFL Cbl-b cDNA was cloned into pcDNA3.1+Hygro and pEYFP-N1 vectors. Deletion of the N-terminal WH1 domain of WASp (ΔWH1; deletion of residues 1 to 164) was performed by PCR amplification of WASp beyond WH1 using the primer pair 5′ GACAAGCTTCCATGGCCAATGAAGAGAGAAGAGG, which includes a HindIII site and ATG start codon, and 5′ ATGCGGCCGCTAGTCATCCCATTCATCATCTTC, which includes a NotI site and a TAG stop codon. The amplification product was cloned into pEYFP-C1. WASp point mutations and WASp deletion mutants, WASpΔexon2 (aa 56 to 102) and WASpΔexon4 (aa 131 to 166), were introduced into the indicated constructs using the QuikChange II XL site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. All constructs were verified using DNA sequencing.
Cell culture and transfection of 293T cells.
Human embryonic kidney 293T cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 50 μg/ml penicillin, and 50 μg/ml streptomycin. The 293T cells were transiently transfected by DNA-calcium phosphate coprecipitation or by using Metafectene reagent according to the manufacturer's instructions (Biontex).
Transfection of Jurkat T cells and fluorescence-activated cell sorter (FACS) analysis.
Jurkat T cells were transfected with an Amaxa electroporator using Amaxa solution T and protocol H-10. Transiently transfected cells were used after 48 h. Stable clones were derived from transiently transfected cells by a combination of drug selection and cell sorting. Cells transiently expressing chimeric proteins were selected in either neomycin or hygromycin. Fluorescence analysis and cell sorting were performed using FACSVantage (BD Biosciences). Single-cell suspensions were analyzed by standard flow cytometry. Flow cytometry was performed using FACSCalibur and FlowJo software. Fluorochrome-conjugated antibodies were purchased from BD Biosciences.
PBL isolation and stimulation.
Human primary peripheral blood lymphocytes (PBLs) were isolated from whole blood of healthy donors as previously described (5). The cells were activated with anti-CD3ε (OKT3, 10 μg/ml) and anti-CD28 (10 μg/ml) for 30 min on ice. The cells were then warmed to 37°C for 10 min and stimulated with anti-mouse IgG (50 μg/ml) for 2 min.
siRNA treatment.
siRNA to human c-Cbl and Cbl-b and a control nontargeting siRNA pool were purchased from Dharmacon Inc. Jurkat cells were transfected with control siRNA or siRNA specific for Cbl proteins using an Amaxa electroporator, Amaxa solution, and protocol H-10.
Spreading assays and molecular imaging.
Spreading assays were performed as previously described by us (5). Dynamic fluorescent and interference reflection microscopy images were collected on a Zeiss LSM510 Meta confocal microscope. All images were collected with a 63 Plan-Apochromat objective (Carl Zeiss).
FRET analysis.
Fluorescence resonance energy transfer (FRET) was measured by the donor-sensitized acceptor fluorescence technique. Three sets of filters were used: one optimized for donor fluorescence (excitation, 468 nm; emission, 475 to 505 nm), a second for acceptor fluorescence (excitation, 514 nm; emission, 530 nm long pass [LP]), and a third for FRET (excitation, 468 nm; emission, 530 nm LP). FRET was corrected, and its efficiency was determined as described in detail in the supplemental Materials and Methods.
Actin shape index.
A quantitative estimate of the actin shape changes was obtained as previously described (17) with some modifications: actin shape index was defined as P2/4πS, where P and S are the perimeter and the surface of the cell, respectively. These values were calculated from a semiautomatic definition of the outline of the polymerized actin (as discerned from the phalloidin staining) obtained using IPLab imaging software. A perfectly circular shape of the polymerized actin yields a shape index of 1; departure from a circle yields a shape index larger than 1.
Colocalization analysis.
Pearson's colocalization correlation coefficient analysis was performed as previously described (33). Imaris 7.0 software (Bitplane AG, Zurich, Switzerland) was used to distinguish between actual colocalization and random association between signals from two fluorescent labels over the entire three-dimensional (3D) image. Pearson's colocalization coefficient varies from −1 to 1, where 1 represents perfect colocalization and −1 denotes complete exclusion.
Cluster determination.
To quantify the percentage of cells with WASp clusters out of the total number of cells, Imaris 7.0 was used to produce tilted views of 3D projections of confocal z series. Each field was examined from several angles to view each cell relative to the phosphotyrosine clusters on the bottom of the cell. Only activated cells exhibiting phosphotyrosine clusters were evaluated (8).
Immunoprecipitation and immunoblotting.
Jurkat cells were either stimulated with anti-CD3ε (OKT3, 10 μg/ml) or anti-TCR (C305, 10 μg/ml) for 2 min at 37°C or left untreated. PBLs were stimulated and lysed, as previously described (10). Transfected 293T cells were lysed 48 h after transfection in ice-cold NP-40 lysis buffer containing 50 mM Tris-HCl (pH 7.6), 150 mM NaCl, 5 mM EDTA, 5 mM EGTA, 50 mM NaF, 1% NP-40, and complete protease inhibitor tablets (Roche). Proteasome activity was blocked by addition of MG132 (Sigma-Aldrich) to Jurkat and 293T cell medium at final concentrations of 50 μM and 10 μM, respectively, for 3 h before the cells were harvested. Immunoprecipitation and immunoblotting were performed as previously described (10). Denaturation of immunoprecipitated complexes and reprecipitation of the WASp protein were performed as previously described (4). Briefly, the precipitated complex was resuspended and boiled for 5 min in 100 μl of denaturation buffer (20 mM Tris-HCl [pH 8], 50 mM NaCl, 5 mM dithiothreitol [DTT], 1% SDS, and 1 mM sodium orthovanadate), the denatured sample was then diluted to 1 ml with cell lysis buffer, and a second immunoprecipitation was performed as described above. Densitometry was performed using ImageJ software with final results normalized using GAPDH (glyceraldehyde-3-phosphate dehydrogenase) as a loading control.
Measurement of intracellular calcium concentrations.
Cells were incubated with 5 μM indo-1-acetoxymethylester (Indo-1-AM; Teflabs) and 0.5 mM probenecid (MPB) in RPMI 1640 medium at 37°C for 45 min. The cells were washed once, resuspended in RPMI 1640 without phenol red containing 10 mM HEPES and 0.5 mM probenecid, and maintained at room temperature for 20 min. The cells were incubated at 37°C for 5 min before measurements and then stimulated with 1 μg/ml OKT3 antibody, and the Ca2+ influx was measured by spectrofluorometer using the Synergy 4 microplate reader (Bio Tek).
Luciferase assay.
Jurkat T cells were transiently cotransfected with 15 μg of an NFAT luciferase reporter plasmid along with various forms of YFP-WASp, as indicated in the figure legends, and 5 μg of a Renilla-luciferase plasmid to correct for variations in transfection efficiency (dual-luciferase reporter assay; Promega). After 48 h, cells were aliquoted in triplicate into a 96-well cell culture dish at 2 × 105 cells/well and left unstimulated, stimulated with plate-bound anti-TCR (2 μg/ml), or stimulated with phorbol 12-myristate 13-acetate (PMA) (50 ng/ml) and ionomycin (1 μg/ml). Cultures were incubated at 37°C for 6 h. Cells were harvested, lysed, and assayed for luciferase activity. The luminescent signal was quantitated using the Synergy 4 microplate reader (Bio Tek). NFAT luciferase activity obtained upon anti-TCR stimulation was normalized to an internal Renilla-luciferase control and expressed as a fraction of the maximal stimulation (PMA and ionomycin).
Statistical analyses.
Standard errors were calculated with the use of Microsoft Excel. Student's t test was used to evaluate significance. In all cases, the threshold P value required for significance was 0.05.
RESULTS
WASp is modified by ubiquitylation.
Previously, we showed that following recruitment to TCR-based clusters, WASp is internalized with phosphorylated SLP-76 vesicles along microtubules (5). Clusters containing SLP-76 rapidly internalize in a raft- and ubiquitin-dependent manner, followed by loss of the clusters (7). This endocytic trafficking of signaling molecules (5, 7) suggested a possible mechanism for their downregulation and raised the question of whether activated WASp molecules might undergo a similar process. Since ubiquitin carries an endocytosis signal and can target the degradation of proteins, we tested whether WASp is ubiquitylated.
First, WASp and hemagglutinin (HA)-tagged ubiquitin (HA-Ub) were expressed in 293T cells. WASp ubiquitylation was demonstrated by coimmunoprecipitation and Western blot and appeared as a smear of anti-HA reactive bands above the 75-kDa marker (Fig. 1A). Furthermore, inhibition of the proteasome using MG132 led to accumulation of ubiquitylated WASp proteins, showing that WASp degradation is proteasome dependent, as was previously suggested (15).
Fig 1.
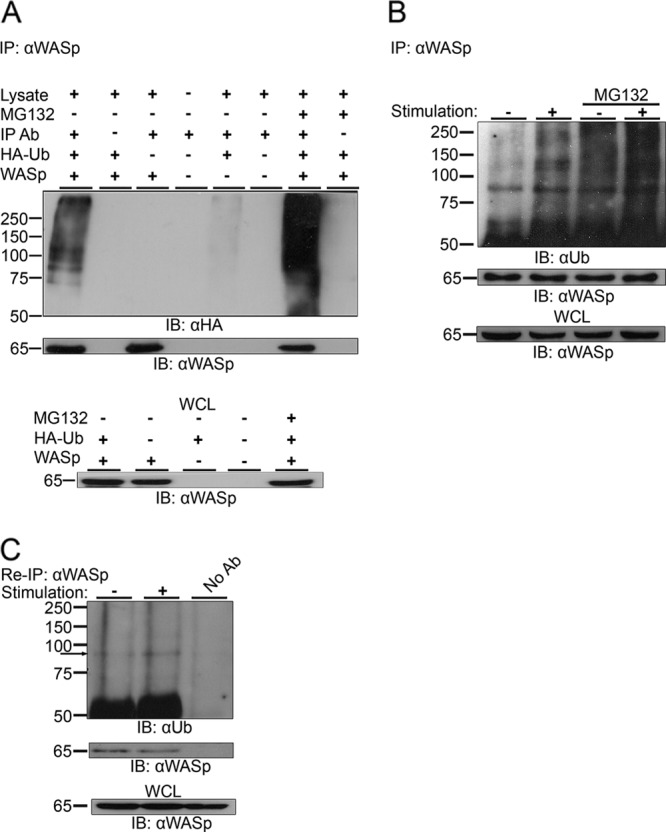
Ubiquitylation of WASp. (A) WASp was immunoprecipitated (IP: αWASp) from 293T cells transiently expressing WASp and HA-tagged ubiquitin (HA-Ub), WASp alone, or HA-Ub alone. Cells coexpressing WASp and HA-Ub were left untreated or incubated in the presence of MG132. The immunoprecipitates were resolved by SDS-PAGE and immunoblotted for ubiquitin using the anti-HA antibody (IB: αHA) and for WASp using anti-WASp antibody. Ubiquitylated WASp bands appear as a smear of bands above the molecular mass of ∼81 kDa. Whole-cell lysates (WCL) of the indicated samples were also blotted for WASp (lower panel). (B) Jurkat T cells were left unstimulated (−) or stimulated with anti-CD3 antibody (+). MG132-treated cells were also left unstimulated (−) or stimulated (+). Cell lysates were immunoprecipitated using anti-WASp antibody and immunoblotted for ubiquitin. Ubiquitylated endogenous WASp bands appear as a smear of bands around the molecular mass of ∼81 kDa. WCL of the indicated samples were also blotted for WASp (lower panel). Data shown are representative of five independent experiments. (C) Jurkat T cells were left unstimulated (−) or stimulated with anti-CD3 antibody (+). Cell lysates were immunoprecipitated using anti-WASp antibody, followed by boiling in a denaturing buffer to separate WASp-associated proteins. After dilution in cell lysis buffer, WASp was reimmunoprecipitated (Re-IP: αWASp). The precipitates from the denatured/renatured samples were immunoblotted for ubiquitin and WASp (IB: αUb and IB: αWASp, respectively). Ubiquitylated endogenous WASp bands are indicated with an arrow. WCL of the indicated samples were also blotted for WASp (lower panel). Data shown are representative of two independent experiments.
Ubiquitylation of endogenous WASp is induced by TCR activation.
To further establish WASp modification, we tested if this protein is ubiquitylated in T lymphocytes expressing endogenous levels of WASp and, furthermore, whether this ubiquitylation is dependent upon TCR stimulation. Untreated Jurkat T cells or MG132-pretreated cells were either left unstimulated or stimulated by anti-CD3 antibody. Consistent with the observations in the 293T cells, immunoprecipitation of endogenous WASp from the T-cell line resulted in the coprecipitation of ubiquitylated proteins, seen as a smear at around ∼81 kDa (Fig. 1B). This ubiquitylation was induced upon TCR stimulation and was further enhanced by the incubation of the cells with MG132. These results were confirmed by a reciprocal immunoprecipitation demonstrating a strong precipitate at ∼81 kDa and above (see Fig. S1A in the supplemental material).
In order to confirm that the ubiquitylated bands detected by WASp immunoprecipitation consisted of modified WASp and not WASp-associated proteins, we immunoprecipitated WASp from the T cell line and denatured the immunoprecipitated proteins, allowed them to renature, and then reimmunoprecipitated WASp. As seen in Fig. 1C, ubiquitylated bands of ∼81 kDa were detected in both the first (Fig. 1B) and second (Fig. 1C) WASp immunoprecipitations, indicating that the bands detected by the Ub antibody represent directly ubiquitylated WASp.
We further characterized WASp ubiquitylation in nontransformed, primary human peripheral blood lymphocytes (PBLs). WASp ubiquitylation, seen as a smear above the ∼81-kDa marker, was upregulated in PBLs upon activation (see Fig. S1B in the supplemental material).
Phosphorylation of WASp Y291 is required for its ubiquitylation.
Since WASp ubiquitylation is dependent on its activation, we explored the possibility that WASp phosphorylation is required to enable this modification. WASp contains seven tyrosines; however, only tyrosine 291 was identified as the major site of TCR-induced WASp phosphorylation, activating WASp by disrupting its autoinhibited conformation (1, 40). Replacing tyrosine 291 with phenylalanine (WASp-CFP Y291F) abrogated the ubiquitylation process, as evidenced by the fainter Ub-reactive smear of bands around the size of ∼100 kDa (Fig. 2A), suggesting that WASp activation, i.e., phosphorylation by protein tyrosine kinases (PTK), is required for its ubiquitylation and, potentially, for degradation. These data provide a rational mechanism for the downregulation of WASp, in which activated WASp molecules, able to recruit the Arp2/3 complex and the actin machinery, are ubiquitylated and ultimately degraded.
Fig 2.
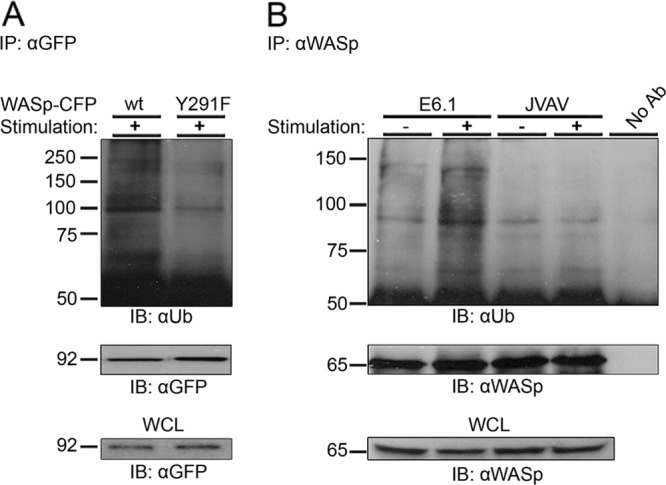
TCR-induced WASp phosphorylation on the tyrosine 291 site is required for WASp ubiquitylation. (A) Jurkat T cells transiently expressing WASp wt-CFP or the Y291F-CFP mutant form of WASp were stimulated (+), and WASp-CFP was immunoprecipitated using anti-GFP antibody. Immunoblotting analysis for ubiquitin and for WASp-CFP with antiubiquitin and anti-GFP antibodies, respectively, was performed. Ubiquitylated WASp-CFP bands appear as a smear of bands around the molecular mass of ∼100 kDa. WCL of the indicated samples were also blotted for WASp-CFP (lower panel). Data shown are representative of three independent experiments. (B) VAV1, a guanine nucleotide exchange factor, is required for WASp ubiquitylation. WASp ubiquitylation was examined in Jurkat E6.1 and in VAV1-deficient T cells (JVAV). WASp was immunoprecipitated with anti-WASp antibody from unstimulated or TCR-stimulated cell lysates. WASp precipitates were analyzed by immunoblotting with anti-Ub antibody. Precipitates were also analyzed by WASp immunoblotting. Ubiquitylated endogenous WASp bands appear as a smear of bands around the molecular mass of ∼81 kDa. WCL of the indicated samples were also blotted for WASp (lower panel). Data shown are representative of three independent experiments.
Functional activity of VAV1 is essential for WASp ubiquitylation.
Phosphorylation of WASp on tyrosine 291 by PTK was shown to enhance the sensitivity of WASp to the small G protein Cdc42 (39, 40). To further establish the necessity of the open nonautoinhibited conformation of WASp for its regulation, we tested the role of VAV1, which functions as a GEF for Cdc42. Cdc42-GTP then competitively binds to WASp GBD, releasing it from its autoinhibited conformation. To study the role of VAV1 in the regulation of WASp ubiquitylation, the VAV1-deficient cell line, JVAV, was used in comparison to E6.1 Jurkat cells expressing endogenous VAV1. WASp ubiquitylation was analyzed and found to be almost totally eliminated in VAV1-deficient T cells following TCR activation (Fig. 2B). Strikingly, WASp ubiquitylation was reconstituted by expression of either VAV1 wt or the VAV1 R694K SH2 mutant (abrogating the potential interaction of VAV1 with ubiquitin ligases, such as Cbls) (see Fig. S1C in the supplemental material). Together, our data suggest that the functional activity of VAV1 as a GEF, rather than its role as an adaptor protein, is essential for WASp ubiquitylation.
c-Cbl and Cbl-b physically associate with WASp and serve as its E3 ligase.
The c-Cbl and Cbl-b E3 ligases have emerged as key negative regulators of the immune response by regulating the activation thresholds of signaling pathways downstream of immune receptors (18, 37). Loss of both c-Cbl and Cbl-b proteins is embryonic lethal, suggesting that these proteins have necessary and overlapping functions (25). We next focused on the molecular mechanism that can mediate the ubiquitylation of WASp. Since the ubiquitin ligase c-Cbl is involved in the downregulation of several proximal TCR proteins, including the TCR itself (4, 24, 37), we examined whether WASp ubiquitylation is affected by the expression of a c-Cbl RING mutant protein lacking ligase activity (70Z). Our results indicated no differences in WASp ubiquitylation between cells expressing the c-Cbl 70Z mutant and those expressing the wt form (see Fig. S1D in the supplemental material), indicating that c-Cbl does not uniquely mediate the ubiquitylation of WASp. We therefore examined the possible involvement of the c-Cbl homologue, Cbl-b, in this system. Previously, it was shown that similar to c-Cbl, the Cbl-b E3 ligase leads to downregulation of TCR expression (25) in addition to the induction of clonal anergy and reduction of actin polymerization (18, 22). Thus, we first investigated whether Cbl-b associates with WASp, since its structural features seem to suggest a molecular interaction. WASp was precipitated from T-cell lysates, and the precipitates were analyzed for the presence of Cbl-b and c-Cbl (Fig. 3A). Our results show a molecular association between endogenous WASp and both c-Cbl and Cbl-b, mainly in TCR-stimulated cells.
Fig 3.

The E3 ligases, Cbl-b and c-Cbl, physically associate with endogenous WASp in T cells. (A) E6.1 T cells were left unstimulated or were stimulated using anti-TCR antibody (C3O5). Cells were lysed, followed by immunoprecipitation of WASp (IP: αWASp). The immunoprecipitates and the WCL were resolved on SDS-PAGE and immunoblotted for Cbl-b (upper panel), c-Cbl (middle panel), and WASp (lower panel). Data shown are representative of six independent experiments. (B) Gene silencing of both Cbl-b and c-Cbl reduces WASp ubiquitylation. E6.1 T cells were transfected with siRNA specific to human Cbl-b, c-Cbl, or a mixture of both as well as with a nontargeting siRNA control (nonspecific). After 48 h, cell lysates were prepared and analyzed for Cbl protein levels. The gene-silencing efficiencies were calculated by ImageJ densitometry results for Cbl-b and c-Cbl after normalization against the GAPDH values and comparison to the values of the negative controls (lower panel). Endogenous WASp was immunoprecipitated by anti-WASp, and the membranes were blotted with anti-Ub and anti-WASp. Ubiquitylated endogenous WASp bands appear as a smear of bands around the molecular mass of ∼81 kDa. Data shown are representative of six independent experiments. (C) Phosphorylation of WASp on the tyrosine 291 site is required for its interaction with Cbl-b. E6.1 T cells stably expressing WASp (wt-CFP) or a mutant form of WASp (Y291F-CFP) were stimulated (+) and immunoprecipitated for WASp-CFP using anti-GFP antibody. Immunoblotting with anti-Cbl-b, -GFP, and -phosphotyrosine (pTy) was performed. WCL of the indicated samples were also blotted for WASp (lower panel). Data shown are representative of three independent experiments.
In view of the results described above, we asked if the Cbls serve as the WASp E3 ligases in T cells and whether they act exclusively. Since c-Cbl and Cbl-b can have equal E3 ubiquitin ligase activity toward a similar range of substrates (37), we examined their individual and combined effects on WASp ubiquitylation. T cells were transfected with siRNA pools targeting Cbl-b, c-Cbl, or both, followed by WASp immunoprecipitation and anti-ubiquitin immunoblotting (Fig. 3B). A control containing a nonspecific scrambled siRNA pool was used as a negative control. Cbl-b expression was reduced by >97%, whereas c-Cbl expression was reduced by >80%, as measured by densitometry analysis (Fig. 3B, lower panels). As shown in Fig. 3B, individual knockdown of c-Cbl or Cbl-b did not cause a substantial reduction in WASp ubiquitylation. However, silencing both Cbl-b and c-Cbl caused a marked decrease in the ubiquitylation levels. A similar pattern was detected in PBLs (see Fig. S2A in the supplemental material). These results suggest that both Cbl-b and c-Cbl can serve as the E3 ligase of WASp, together regulating its expression.
Phosphorylation of WASp on tyrosine 291 is essential for the interaction of WASp with Cbl-b.
In the next experiments, we determined the site of interaction of WASp with Cbl-b (Fig. 3C; see also Fig. S2B in the supplemental material). Since WASp association with Cbl-b is induced upon TCR activation, it is possible that the Cbl-b TKB domain associates with phosphorylated tyrosines on WASp. As the WASp tyrosine 291 site is heavily phosphorylated following TCR activation, we determined whether this residue serves as the interaction site. To this end, we analyzed precipitates of WASp with Cbl-b in unstimulated and stimulated T-cell lysates stably expressing the Y291F WASp mutant. Binding of Cbl-b to WASp was greatly reduced in cells expressing WASp Y291F following TCR activation (Fig. 3C, upper panel). Immunoblotting with antiphosphotyrosine showed, as expected, a weaker band of activated WASp in the lane of cells expressing WASp Y291F compared to that in the lane of cells expressing WASp wt. Consistent with these observations, an SH2 loss-of-function mutant form of Cbl-b (Cbl-b G298E) showed reduced ability to interact with WASp following TCR activation (see Fig. S2B in the supplemental material).
Mapping of the ubiquitylation sites on WASp.
To further characterize the ubiquitylation sites on WASp, we examined the sequences in WASp that are important for its ubiquitylation. WASp contains several domains that are critical for its activity; however, only the WH1 domain is important for maintaining its stability (21) (Fig. 4Ai). Furthermore, this domain contains most of the spontaneous mutations of WASp (19, 29). When introduced into 293T cells, deletion of the WH1 domain completely abolished WASp ubiquitylation, while YFP-WASp wt was normally ubiquitylated (Fig. 4B). This result indicated that the WH1 domain is the site of WASp ubiquitylation and allowed us to examine the WASp WH1 domain in greater detail. As shown in the scheme in Fig. 4Ai, the WH1 domain contains six lysines and is encoded by exons 1 to 4 of the WASp gene; within this region, exon 2 contains the largest number of WAS missense mutations and the most common ones (19, 23), including three lysines. Based on this lysine mapping and on the clinical analysis of WASp mutations, we prepared additional WASp mutant forms (Fig. 4Aii), including WASp Δexon2 (Fig. 4B), WASp Δexon4, and WASp K42R (see Fig. S3A in the supplemental material).
Fig 4.

Identification of WASp domains and lysine residues required for its ubiquitylation. (Ai) Schematic illustration of WASp structural domains. WH1, WASp homology 1 domain; B, basic region; GBD, GTPase-binding domain; PRD, proline-rich domain; VCA, verprolin homology/central region/acidic region. The WASp WH1 domain consists of four exons containing six lysines. (Aii) Schemes of WASp wt and WASp mutant forms: WH1-deleted WASp (ΔWH1), exon 2-deleted WASp (Δexon2), exon 4-deleted WASp (Δexon4), and point-mutated WASp with the indicated lysine residues replaced by arginine. (B) 293T cells were cotransfected with constructs encoding HA-tagged ubiquitin together with YFP-WASp wt or with YFP-WASp mutant forms described above (Aii). YFP-WASp was immunoprecipitated using anti-GFP antibody; complexes were resolved by SDS-PAGE and immunoblotted for ubiquitin using the anti-HA and anti-GFP antibodies. Ubiquitylated YFP-WASp bands appear as a smear of bands around the molecular mass of ∼100 kDa. WCL of the indicated samples were also blotted for YFP-WASp (lower panel). Data shown are representative of five independent experiments. (C) Accumulation of WASp ΔWH1, WASp Δexon2, and WASp 2KR mutant forms. Jurkat T cells expressing YFP-WASp wt or the YFP-WASp mutant forms ΔWH1, Δexon2, and 2KR were sorted for GFP-positive cells. Anti-CD3 antibody-stimulated cells were lysed and analyzed for WASp protein levels by immunoblotting with anti-GFP antibody or anti-GAPDH as a loading control. Densitometric analysis of the bands presented was performed by using ImageJ and normalized by the GAPDH densitometry values. Data shown are representative of five independent experiments.
YFP-WASp mutant forms were expressed and tested for ubiquitylation by immunoprecipitation. No reduction in WASp ubiquitylation following replacement of lysine 42 with arginine or deletion of exon 4 was detected (see Fig. S3A, right and left panel, respectively, in the supplemental material). However, the deletion of exon 2 led to a substantial decrease in ubiquitylation (Fig. 4B). These results suggest that the lysines expressed in exon 2 of the WH1 domain are crucial for WASp ubiquitylation. Based on these findings, we individually mutated each of the lysines located on exon 2. Strikingly, while mutation of lysine 66 encoded by exon 2 had no effect on ubiquitylation levels (Fig. 4B), the WASp K76R and K81R mutant immunoprecipitates showed decreased anti-HA reactivity (Fig. 4B). Not surprisingly, the WASp 2KR mutant, in which both WASp K76 and K81 were mutated, showed greatly decreased ubiquitylation. The WASp K76, 81R (WASp 2KR) mutant was expressed at a level equal to that of the WASp wt construct (Fig. 4B). These results indicate that WASp is ubiquitylated predominantly on two sites located within the WH1 domain, K76 and K81.
WASp ΔWH1, WASp Δexon2, and WASp 2KR mutant forms accumulate within the cell and induce aberrant spreading and WASp cluster formation.
Next, we examined how the expression of WASp ΔWH1, WASp Δexon2, and WASp 2KR affects WASp abundance, distribution, and dynamics in T cells.
To quantify the expression levels of YFP-WASp ΔWH1, WASp Δexon2, and WASp 2KR in comparison to YFP-WASp wt proteins, cell lysates were prepared from YFP-positive sorted cells following TCR stimulation. The results confirmed a substantial accumulation of the nonubiquitylated forms of WASp, WASp ΔWH1, WASp Δexon2, and WASp 2KR compared to the full-length WASp wt (Fig. 4C). These data were verified by FACS analysis, indicating that YFP-WASp levels were increased by more than ∼12-, 6-, and 4-fold in cells expressing WASp ΔWH1, WASp Δexon2, and WASp 2KR, respectively, compared to cells expressing YFP-WASp wt (see Fig. S3B in the supplemental material). These data also correlate WASp ubiquitylation with WASp degradation.
Real-time imaging of YFP-WASp wt was next performed in comparison to YFP-tagged WASp mutant forms (see Movies S1 to S4 and Fig. S4 in the supplemental material) during the T-cell activation process. In addition, we performed spreading assays, as previously described by us (27). As previously shown (5) and as demonstrated in the left panel of Fig. 5A and B and in Movie S1 in the supplemental material, YFP-WASp wt microclusters were formed at the TCR activation sites. Along with cell spreading, the clusters moved toward the cell periphery, where a complete lamellipodium was formed (Fig. 5A, left panel; see also Movie S1 in the supplemental material). WASp ΔWH1 (see Movie S2), similarly to WASp Δexon2 (see Movie S3) and WASp 2KR (see Movie S4), was accumulated in the cells and was not degraded, as observed by the enhanced fluorescence. These nonubiquitylated WASp mutants, i.e., ΔWH1, Δexon2, and 2KR, were diffusely distributed over the cell and formed substantially reduced microclusters.
Fig 5.

Accumulation of WASp ΔWH1, WASp Δexon2, and WASp 2KR mutant forms causes aberrant WASp cluster formation and T-cell spreading. (A) The distribution of WASp wt was compared to those of the YFP-WASp mutant forms ΔWH1, Δexon2, and 2KR in activated T cells. E6.1 T cells expressing YFP-WASp wt or mutants (green) were plated on a stimulatory coverslip and then fixed and stained with antiphosphotyrosine (red). Right panels display collected images at 2 and 5 min into the spreading process. The images of YFP-WASp wt were taken under the same conditions as those of the YFP-WASp ΔWH1, WASp Δexon2, and WASp 2KR proteins. Left panels represent the DIC channel. Images on the bottom show the entire field of cells. (B) The percentage of cells that produced WASp clusters in cells expressing YFP-WASp wt was compared to that in cells expressing the WASp mutant forms at 2 and 5 min into the spreading process. A t test analysis between WASp wt and the WASp mutants was performed at 2 (*) and 5 (**) min (P < 0.0001 for both time points). (C) Colocalization analysis based on Pearson's colocalization coefficients. Image analysis was performed on more than 50 cells for each experimental group. Data shown are representative of three independent experiments.
We then more specifically documented the requirement for the WASp WH1 domain for WASp cluster formation and distribution (Fig. 5A and B). As shown by the percentages presented in Fig. 5B, after 2 min of activation, YFP-WASp ΔWH1 cluster formation was significantly decreased (14.7%, versus 68.4% for YFP-WASp wt) and colocalization between WASp mutants and phosphotyrosine-containing clusters was significantly reduced, as determined by Pearson's correlation coefficient (Fig. 5C). After several minutes of activation, WASp ΔWH1 was not translocated to the periphery as the wt molecules were. Cluster distributions similar to those for WASp wt were observed for YFP-WASp K66R and, to a lesser extent, for the single-mutated YFP-WASp K76R- or K81R-expressing cells (see Fig. S5A and B in the supplemental material). In contrast, WASp 2KR cluster formation was significantly reduced (P < 0.0001) (see Fig. S5B in the supplemental material).
Furthermore, WASp mutant cells, i.e., WASp ΔWH1, WASp Δexon2, and WASp 2KR cells, did not spread normally, as demonstrated by their actin shape indexes (Fig. 6) and as demonstrated by the tiny filopodium spikes protruding from the cell surface (Fig. 6; see also Movies S2 to S4, respectively, and Fig. S4 in the supplemental material); furthermore, they did not succeed in forming a complete lamellipodium structure (Fig. 6, differential interference contrast [DIC]) and were significantly (P ≤ 0.03) smaller than the WASp wt cells. Unlike the nonubiquitylated WASp mutants, the single WASp mutants K66R, K76R, and K81R successfully formed complete lamellipodium structures and spread normally after 5 min of activation, as analyzed by their actin shape indexes (see Fig. S6 in the supplemental material).
Fig 6.

WASp ΔWH1-, WASp Δexon2-, or WASp 2KR-expressing cells form mainly membrane spike structures and fewer lamellipodia. E6.1 T cells expressing YFP-WASp wt, WASp ΔWH1, WASp Δexon2, or WASp 2KR were sorted for YFP-positive cells, plated, fixed after 5 min of activation, and stained with phalloidin. Cells producing lamellipodia were counted, and their percentage of the total cells was calculated. Actin shape index was analyzed by IPLab software as described in Materials and Methods. The images of at least 50 cells (for each cell type) were analyzed. A t test analysis between WASp wt and the WASp mutants was performed with a P value of <0.05, as indicated within the figure.
The results described above demonstrate that WASp is degraded through the WH1 domain and that this domain plays a pivotal role in the dynamics of WASp and its recruitment to the cell membrane.
Ubiquitylation of active WASp leads to its degradation.
So far, we have shown that WASp ubiquitylation is dependent on its phosphorylation on tyrosine 291 following TCR activation. Since the activated form of WASp comprises only a fraction of the total WASp, this might explain why a global decrease of WASp levels could not be systematically detected in the blots for WASp. However, since the active phosphorylated WASp is the functional protein that serves as a key player in the regulation of the actin machinery, our next experiments aimed to monitor the levels of the active WASp form over time following TCR activation and to compare YFP-WASp wt and YFP-WASp 2KR (nonubiquitylated)-expressing cells. In order to distinguish active WASp levels from total WASp levels, YFP-WASp was immunoprecipitated and immunoblotted with antiphosphotyrosine (see Fig. S7 in the supplemental material). Following T-cell stimulation, almost no phosphorylated YFP-WASp wt was precipitated, in contrast to substantial and persistent precipitation of the phosphorylated YFP-WASp 2KR mutant form (see Fig. S7 in the supplemental material). These results suggest that only the activated pool of WASp is ubiquitylated and subsequently degraded.
Mutations at the WASp ubiquitylation sites (lysine 76 and 81) alter the WASp-WIP interaction.
Regulation of WASp expression is critical for membrane structure formation, i.e., lamellipodia, and in the absence of the WH1 domain, the cells do not spread normally. One of the critical binding partners of WASp is WIP, a widely expressed protein that is known to bind the WH1 domain of WASp through its C-terminal end (aa 451 to 485) and, in addition, also binds F-actin. The WASp-WIP interaction depends on residues throughout the N terminus of WASp (15, 21). Therefore, we wanted to examine whether WASp ubiquitylation sites, which are located at the WH1 domain and are critical for WASp stability, are essential for WIP binding. To this end, we transfected CFP-WIP into T cells stably expressing YFP-WASp wt or YFP-WASp 2KR (nonubiquitylated) and determined their binding by measuring the fluorescence resonance energy transfer (FRET) efficiency between YFP-WASp and CFP-WIP, as previously described by us (5). Interestingly, a significant reduction of the FRET efficiency between WIP and WASp 2KR from that between WIP and WASp wt was detected (24.64% ± 5.48% versus 39.22% ± 6.78%, respectively; P ≤ 0.02) (see Fig. S8 in the supplemental material). These results provide direct evidence that mutations in WASp ubiquitylation sites alter the interaction with WIP.
Abrogation of WASp degradation enhances T-cell activation, as detected by NFAT activity and intracellular calcium levels.
To assess the effect of the WASp WH1 domain on T-cell activation, we transfected T cells with YFP-WASp, wt WASp ΔWH1, WASp Δexon2, and WASp 2KR. Cells were activated with anti-TCR and evaluated for NFAT activity and for increases in intracellular calcium concentrations. Intracellular calcium levels (Fig. 7A) and NFAT activity (Fig. 7B) were upregulated in cells expressing WASp ΔWH1, WASp Δexon2, and WASp 2KR compared to the levels in cells expressing WASp wt. In order to exclude the possibility that these effects were caused only by overexpression of the WASp VCA domain, which might result in enhanced actin polymerization, we performed spreading assays with WASp, wt WASp ΔWH1, WASp Δexon2, and WASp 2KR. The integrated fluorescence intensity was analyzed and compared as a measure of actin polymerization levels, as previously described by us (6). Our analysis showed no significant differences in the actin polymerization levels between cells expressing WASp wt and those expressing WASp mutants (see Fig. S9 in the supplemental material). Thus, our data show that abrogation of WASp degradation upregulates T-cell activation and NFAT activity. These experiments indicate that the intracellular level of WASp affects TCR activation.
Fig 7.
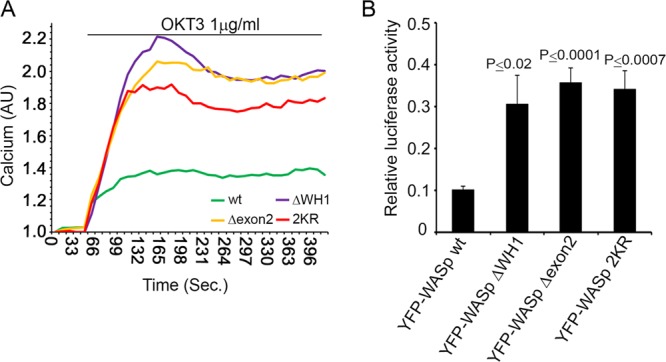
Abrogation of WASp negative regulation enhances T-cell activation. (A) Jurkat T cells were transfected with YFP-WASp wt or mutant forms. YFP-WASp-positive cells were sorted and loaded with 5 μg calcium-sensitive Indo-1-AM as described in Materials and Methods. Cells were then analyzed for intracellular calcium levels by spectrofluorometry following stimulation with anti-CD3 (OKT3, 1 μg/ml) antibody. Data shown are representative of four independent experiments. (B) Jurkat T cells were cotransfected with YFP-WASp wt or its mutant forms, ΔWH1, Δexon2, or 2KR, and the NFAT luciferase reporter plasmid. Cells were lysed and assayed for luciferase activity. NFAT luciferase activity obtained upon anti-TCR stimulation (2 μg/ml) was normalized for transfection efficiency using values from the Renilla-luciferin reaction and expressed as a fraction of the maximal stimulation (PMA and ionomycin). A t test analysis between WASp wt and the WASp mutants is presented. Data shown are representative of at least five independent experiments.
DISCUSSION
WASp is a key regulator of the actin cytoskeleton that plays a pivotal role in controlling vital cellular processes, including T-cell motility and proliferation. Importantly, alterations in dynamic actin cytoskeletal rearrangement result in T-cell unresponsiveness and are linked to pathological conditions, such as WAS.
Our previous studies demonstrated that following TCR activation, actin polymerization is driven by WASp and is dependent on its dynamic localization (5). Late in the activation process, vesicles containing WASp leave the periphery and move along microtubules to a central structure, where they are targeted to internalization and degradation (4, 5, 7). WASp was initially suggested to be degraded by a calpain-dependent degradation pathway (32); however, a calpain inhibitor could only mitigate but not abolish WASp degradation (15), indicating that additional factors contribute to this process. Indeed, in the current study, we identified the molecular mechanisms governing the degradation of the active WASp molecular complex.
We demonstrated that WASp ubiquitylation is phosphorylation dependent and occurs through the WASp WH1 domain, which is the region containing the vast majority (85%) of WASp mutations (28). Biochemical analysis suggested that Cbl-b directly interacts with WASp and that this interaction is stabilized by the binding of the WASp Y291 residue to the TKB domain of Cbl-b. We found that both Cbl-b and c-Cbl act as common E3 ligases of WASp and that they act in a synergistic manner.
Our results indicate that WASp activation is a prerequisite event for its ubiquitylation. Among WASp phosphotyrosines, tyrosine 291 (Y291) of WASp was previously shown to be the major site of WASp phosphorylation and to be critical for the activation of WASp effector function in T cells, i.e., actin polymerization (1, 9). The necessity of WASp phosphorylation on Y291 for its ubiquitylation was demonstrated by biochemical approaches.
Furthermore, since VAV1 is the GEF of Cdc42-GTP and is required for the activation of WASp, we tested its requirement for WASp ubiquitylation by using VAV1-deficient T cells (JVAV). In these cells, WASp ubiquitylation was abrogated, suggesting that VAV1 is required for this process. Moreover, we show that VAV1 plays a role in regulating ubiquitylation through the activation of Cdc42 rather than through its function as an adaptor protein. Thus, our data indicate that WASp ubiquitylation is triggered by the activation of WASp and its phosphorylation at the Y291 site.
Interestingly, while VAV1 is required for WASp ubiquitylation, SLP-76 is not necessary for this process. Using SLP-76-deficient T cells (J14) and J14 reconstituted with SLP-76-YFP (J14/SLP-76-YFP), no change in WASp ubiquitylation between the presence and the absence of SLP-76 was found, suggesting that this process is independent of SLP-76 (see Fig. S10 in the supplemental material). However, these data do not rule out the possibility that SLP-76 and WASp are simultaneously internalized by endocytic vesicles and degraded.
Ubiquitylation of WASp occurs on lysines 76 and 81, located on the WASp WH1 domain. Strikingly, mutations at these sites prevented the degradation of active WASp following TCR stimulation and led to WASp accumulation, thus supporting the role of ubiquitylation as a mechanism for negative WASp regulation. Our data indicate that the phosphorylated, active WASp undergoes ubiquitylation and degradation. We show that impairment of WASp degradation via deletions or point mutations at the WH1 domain causes upregulation of T-cell activation. In addition, impairment of WASp dynamics and aberrant actin rearrangement following TCR activation are observed, most likely due to disruption of the interaction of WASp with WIP, which plays a role in actin polymerization.
Given the essential role of WASp in the immune cell response and cytoskeletal remodeling, the regulated degradation of activated WASp signaling complexes might be an efficient strategy by which WASp regulates the homeostasis of actin polymerization and T-cell activation. Deletion of the WASp WH1 domain or exon 2-encoded fragment causes a dramatic accumulation of WASp in the cells, as detected by both biochemical and FACS analyses. Expression of the WASp 2KR mutant causes a lesser accumulation. These data indicate that both WASp ubiquitylation motifs, i.e., lysine residues 76 and 81, and calpain degradation sites located on the N-terminal WH1 domain of WASp are important for the regulation of the WASp expression level. FRET analysis showed that mutations at both WASp ubiquitylation sites, i.e., lysine residues 76 and 81, significantly reduced but did not eliminate the WASp-WIP interaction. Thus, our data suggest that WIP masks and protects WASp ubiquitylation sites. Upon TCR activation, a shift occurs in the WASp-WIP interaction that exposes the WASp WH1 domain and, specifically, these lysine residues to ubiquitylation by the Cbls, thereby promoting WASp degradation (Fig. 8). Interestingly, the mutation in codon 86 (located at exon 2 and very close to the WASp ubiquitylation sites) is the most common missense mutation seen in WAS and can produce severe or mild disease (36).
Fig 8.

Schematic representation of the molecular mechanism of WASp ubiquitylation. In naive T cells, WASp tightly associates with WIP via its N terminus and exon 2-encoded fragment (aa 56 to 102). WIP masks lysine residues 76 and 81 and inhibits their modification via ubiquitin, and thus, WASp accumulates within the T cell. Upon cellular activation, the Rho family GEF VAV1 activates Cdc42, which binds to the WASp GBD domain, thereby releasing WASp from its autoinhibitory conformation and exposing the VCA domain. The VCA domain of WASp binds to the Arp2/3 complex, promoting local actin polymerization. Phosphorylation of WASp on Y291 by PTK increases the sensitivity of WASp to Cdc42. WASp-WIP interactions are altered following activation. Phosphorylation of WASp on Y291 mediates its interaction with the E3 ligases. This event leads to the ubiquitylation of WASp on lysine residues 76 and 81 and to its degradation.
Identification of the sites at which WASp is ubiquitylated can shed light on some of the clinical manifestations of WAS. We showed that deletion of the WASp WH1 domain, which contains most WASp mutations (21, 28, 36), abolishes WASp ubiquitylation. A more detailed examination of this domain revealed that the lysines encoded by exon 2, K76, and K81 are essential for WASp degradation. Several point mutations, such as E31K, E131K, and E133K, that are located on WASp WH1 of WAS patients and that replace glutamic acid with lysines were identified. These mutations cause a very severe phenotype correlated with the absence of WASp expression (19, 23). Thus, it is possible that these mutations enhance WASp susceptibility to ubiquitylation and degradation. Further characterization of this class of mutations might assist in the diagnosis and classification of WAS disease subtypes.
Our study indicates that abrogation of WASp ubiquitylation resulting in WASp accumulation enhances T-cell activation, as detected by elevation of intracellular calcium concentrations and NFAT activity. Indeed, T cells from WAS patients and WASp-deficient T cells show reduced levels of calcium influx (12) and of NFAT transcriptional activity (2, 13), demonstrating the crucial role of WASp regulation.
In this study, we demonstrated that the regulation of WASp degradation is critical for its functional activity, i.e., actin remodeling and T-cell activation. We show that the instability of WASp and its degradation via ubiquitylation result in the regulation of T-cell signaling and support the homeostasis of actin polymerization.
Supplementary Material
ACKNOWLEDGMENTS
We thank Elad Noy for technical assistance.
This research was funded by the Israel Cancer Association through the estate of the late Alexander Smidoda, the Israeli Ministry of Health through the Office of the Chief Scientist, and by the Israel Science Foundation (grant numbers 1659/08, 971/08, and 491/10).
Footnotes
Published ahead of print 4 June 2012
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Badour K, et al. 2004. Fyn and PTP-PEST-mediated regulation of Wiskott-Aldrich syndrome protein (WASp) tyrosine phosphorylation is required for coupling T cell antigen receptor engagement to WASp effector function and T cell activation. J. Exp. Med. 199:99–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Badour K, Zhang J, Siminovitch KA. 2004. Involvement of the Wiskott-Aldrich syndrome protein and other actin regulatory adaptors in T cell activation. Semin. Immunol. 16:395–407 [DOI] [PubMed] [Google Scholar]
- 3. Badour K, Zhang J, Siminovitch KA. 2003. The Wiskott-Aldrich syndrome protein: forging the link between actin and cell activation. Immunol. Rev. 192:98–112 [DOI] [PubMed] [Google Scholar]
- 4. Balagopalan L, et al. 2007. c-Cbl-mediated regulation of LAT-nucleated signaling complexes. Mol. Cell Biol. 27:8622–8636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barda-Saad M, et al. 2005. Dynamic molecular interactions linking the T cell antigen receptor to the actin cytoskeleton. Nat. Immunol. 6:80–89 [DOI] [PubMed] [Google Scholar]
- 6. Barda-Saad M, et al. 2010. Cooperative interactions at the SLP-76 complex are critical for actin polymerization. EMBO J. 29:2315–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barr VA, et al. 2006. T-cell antigen receptor-induced signaling complexes: internalization via a cholesterol-dependent endocytic pathway. Traffic 7:1143–1162 [DOI] [PubMed] [Google Scholar]
- 8. Barr VA, et al. 2008. Dynamic movement of the calcium sensor STIM1 and the calcium channel Orai1 in activated T-cells: puncta and distal caps. Mol. Biol. Cell 19:2802–2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blundell MP, et al. 2009. Phosphorylation of WASp is a key regulator of activity and stability in vivo. Proc. Natl. Acad. Sci. U. S. A. 106:15738–15743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Braiman A, Barda-Saad M, Sommers CL, Samelson LE. 2006. Recruitment and activation of PLCγ1 in T cells: a new insight into old domains. EMBO J. 25:774–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bunnell SC, Kapoor V, Trible RP, Zhang W, Samelson LE. 2001. Dynamic actin polymerization drives T cell receptor-induced spreading: a role for the signal transduction adaptor LAT. Immunity 14:315–329 [DOI] [PubMed] [Google Scholar]
- 12. Calvez R, et al. 2011. The Wiskott-Aldrich syndrome protein permits the assembly of a focused immunological synapse enabling sustained TCR signalling. Haematologica 96:1415–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cannon JL, Burkhardt JK. 2004. Differential roles for Wiskott-Aldrich syndrome protein in immune synapse formation and IL-2 production. J. Immunol. 173:1658–1662 [DOI] [PubMed] [Google Scholar]
- 14. Cannon JL, et al. 2001. Wasp recruitment to the T cell:APC contact site occurs independently of Cdc42 activation. Immunity 15:249–259 [DOI] [PubMed] [Google Scholar]
- 15. de la Fuente MA, et al. 2007. WIP is a chaperone for Wiskott-Aldrich syndrome protein (WASP). Proc. Natl. Acad. Sci. U. S. A. 104:926–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dong X, Patino-Lopez G, Candotti F, Shaw S. 2007. Structure-function analysis of the WIP role in T cell receptor-stimulated NFAT activation: evidence that WIP-WASP dissociation is not required and that the WIP NH2 terminus is inhibitory. J. Biol. Chem. 282:30303–30310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Donnadieu E, Bismuth G, Trautmann A. 1994. Antigen recognition by helper T cells elicits a sequence of distinct changes of their shape and intracellular calcium. Curr. Biol. 4:584–595 [DOI] [PubMed] [Google Scholar]
- 18. Duan L, Reddi AL, Ghosh A, Dimri M, Band H. 2004. The Cbl family and other ubiquitin ligases: destructive forces in control of antigen receptor signaling. Immunity 21:7–17 [DOI] [PubMed] [Google Scholar]
- 19. Jin Y, et al. 2004. Mutations of the Wiskott-Aldrich syndrome protein (WASP): hotspots, effect on transcription, and translation and phenotype/genotype correlation. Blood 104:4010–4019 [DOI] [PubMed] [Google Scholar]
- 20. King SJ, et al. 2011. β1 integrins regulate fibroblast chemotaxis through control of N-WASP stability. EMBO J. 30:1705–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Konno A, Kirby M, Anderson SA, Schwartzberg PL, Candotti F. 2007. The expression of Wiskott-Aldrich syndrome protein (WASP) is dependent on WASP-interacting protein (WIP). Int. Immunol. 19:185–192 [DOI] [PubMed] [Google Scholar]
- 22. Krawczyk C, et al. 2000. Cbl-b is a negative regulator of receptor clustering and raft aggregation in T cells. Immunity 13:463–473 [DOI] [PubMed] [Google Scholar]
- 23. Lutskiy MI, Rosen FS, Remold-O'Donnell E. 2005. Genotype-proteotype linkage in the Wiskott-Aldrich syndrome. J. Immunol. 175:1329–1336 [DOI] [PubMed] [Google Scholar]
- 24. Myers MD, et al. 2006. Src-like adaptor protein regulates TCR expression on thymocytes by linking the ubiquitin ligase c-Cbl to the TCR complex. Nat. Immunol. 7:57–66 [DOI] [PubMed] [Google Scholar]
- 25. Naramura M, et al. 2002. c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat. Immunol. 3:1192–1199 [DOI] [PubMed] [Google Scholar]
- 26. Pauker MH, Barda-Saad M. 2011. Studies of novel interactions between Nck and VAV SH3 domains. Commun. Integr. Biol. 4:175–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pauker MH, Reicher B, Fried S, Perl O, Barda-Saad M. 2011. Functional cooperation between the proteins Nck and ADAP is fundamental for actin reorganization. Mol. Cell Biol. 31:2653–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rajmohan R, Raodah A, Wong MH, Thanabalu T. 2009. Characterization of Wiskott-Aldrich syndrome (WAS) mutants using Saccharomyces cerevisiae. FEMS Yeast Res. 9:1226–1235 [DOI] [PubMed] [Google Scholar]
- 29. Ramesh N, Anton IM, Hartwig JH, Geha RS. 1997. WIP, a protein associated with Wiskott-Aldrich syndrome protein, induces actin polymerization and redistribution in lymphoid cells. Proc. Natl. Acad. Sci. U. S. A. 94:14671–14676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reicher B, Barda-Saad M. 2010. Multiple pathways leading from the T-cell antigen receptor to the actin cytoskeleton network. FEBS Lett. 584:4858–4864 [DOI] [PubMed] [Google Scholar]
- 31. Rivero-Lezcano OM, Marcilla A, Sameshima JH, Robbins KC. 1995. Wiskott-Aldrich syndrome protein physically associates with Nck through Src homology 3 domains. Mol. Cell Biol. 15:5725–5731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shcherbina A, et al. 2001. WASP and N-WASP in human platelets differ in sensitivity to protease calpain. Blood 98:2988–2991 [DOI] [PubMed] [Google Scholar]
- 33. Singh S, Plassmeyer M, Gaur D, Miller LH. 2007. Mononeme: a new secretory organelle in Plasmodium falciparum merozoites identified by localization of rhomboid-1 protease. Proc. Natl. Acad. Sci. U. S. A. 104:20043–20048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smith-Garvin JE, Koretzky GA, Jordan MS. 2009. T cell activation. Annu. Rev. Immunol. 27:591–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Snapper SB, et al. 1998. Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity 9:81–91 [DOI] [PubMed] [Google Scholar]
- 36. Stewart DM, Tian L, Nelson DL. 1999. Mutations that cause the Wiskott-Aldrich syndrome impair the interaction of Wiskott-Aldrich syndrome protein (WASP) with WASP interacting protein. J. Immunol. 162:5019–5024 [PubMed] [Google Scholar]
- 37. Thien CB, Langdon WY. 2005. c-Cbl and Cbl-b ubiquitin ligases: substrate diversity and the negative regulation of signalling responses. Biochem. J. 391:153–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thrasher AJ, Burns SO. 2010. WASP: a key immunological multitasker. Nat. Rev. Immunol. 10:182–192 [DOI] [PubMed] [Google Scholar]
- 39. Torres E, Rosen MK. 2003. Contingent phosphorylation/dephosphorylation provides a mechanism of molecular memory in WASP. Mol. Cell 11:1215–1227 [DOI] [PubMed] [Google Scholar]
- 40. Torres E, Rosen MK. 2006. Protein-tyrosine kinase and GTPase signals cooperate to phosphorylate and activate Wiskott-Aldrich syndrome protein (WASP)/neuronal WASP. J. Biol. Chem. 281:3513–3520 [DOI] [PubMed] [Google Scholar]
- 41. Westerberg LS, et al. 2010. Activating WASP mutations associated with X-linked neutropenia result in enhanced actin polymerization, altered cytoskeletal responses, and genomic instability in lymphocytes. J. Exp. Med. 207:1145–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang J, et al. 1999. Antigen receptor-induced activation and cytoskeletal rearrangement are impaired in Wiskott-Aldrich syndrome protein-deficient lymphocytes. J. Exp. Med. 190:1329–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.