

Abstract
Cross-linking of the IgE receptor (FcεRI) on mast cells plays a critical role in IgE-dependent allergy, including allergic rhinitis, asthma, anaphylaxis, and immediate-type hypersensitivity reactions. Previous studies have demonstrated that the K+ channel, KCa3.1, plays a critical role in IgE-stimulated Ca2+ entry and degranulation in both human and mouse mast cells. We now have shown that the class II phosphatidylinositol-3-kinase C2β (PI3KC2β) is necessary for FcεRI-stimulated activation of KCa3.1, Ca2+ influx, cytokine production, and degranulation of bone marrow-derived mast cells (BMMC). In addition, we found that the E3 ubiquitin ligase, tripartite motif containing protein 27 (TRIM27), negatively regulates FcεRI activation of KCa3.1 and downstream signaling by ubiquitinating and inhibiting PI3KC2β. TRIM27−/− mice are also more susceptible in vivo to acute anaphylaxis. These findings identify TRIM27 as an important negative regulator of mast cells in vivo and suggest that PI3KC2β is a potential new pharmacologic target to treat IgE-mediated disease.
INTRODUCTION
Mast cells play an important role in IgE-mediated allergic reactions, such as allergic rhinitis, anaphylaxis, and asthma. Binding of IgE to the high-affinity IgE receptor, FcεRI, on mast cells triggers receptor oligomerization and activation (3, 17). Activation then results in the immediate release of preformed mediators, including histamine, proteases, and a number of cytokines that are stored in cytoplasmic granules. In addition, activation of FcεRI results in the de novo synthesis of a number of proinflammatory cytokines and lipids.
Ca2+ functions as a critical second messenger to mediate both degranulation and the production of proinflammatory cytokines (12, 14, 18). Cross-linking of FcεRI activates phospholipase Cγ, resulting in the generation of inositol-1,4,5-trisphosphate (IP3), which binds its receptor on the endoplasmic reticulum, leading to the release of Ca2+ into the cytoplasm. This in turn results in the oligomerization of STIM1 and its subsequent association with and activation of the calcium-release-activated Ca2+ (CRAC) channels at the plasma membrane, which is the predominant mechanism for Ca2+ influx into mast cells and other immune cells (11, 19, 26, 27, 38). The important role for this pathway in mast cells is supported by the findings that FcεRI-stimulated degranulation is markedly defective in bone marrow-derived mast cells (BMMC) derived from Stim1−/− and CRAC1M−/− mice (2, 37). In addition, FcεRI-induced in vivo anaphylaxis is markedly inhibited in CRAC1M−/− mice (37).
The influx of Ca2+ into mast cells via CRAC channels is dependent on maintaining a negative membrane potential, which provides the electrical driving force for rapid Ca2+ influx. Previous studies have shown that the intermediate-conductance Ca2+-activated K+ channel, KCa3.1 (also known as SK4 and KCNN4), via the efflux of K+, is critical for maintaining a negative membrane potential and is required for maximal FcεRI-stimulated Ca2+ influx and degranulation in mast cells (8, 29). On the other hand, activation of the Ca2+-activated nonselective transient receptor potential melastatin 4 (TRPM4) channel functions to depolarize the membrane potential and limit FcεRI-stimulated Ca2+ influx (36).
KCa3.1 also plays a critical role in Ca2+ flux and cytokine production following T cell receptor (TCR) activation (13, 32). Previously, it has been shown that TCR stimulation results in the activation of phosphatidylinositol-3-kinase C2β (PI3KC2β), leading to the generation of phosphatidylinositol 3 phosphate (PI3P), which is required for the histidine kinase nucleoside diphosphate kinase B (NDPKB) to phosphorylate and activate KCa3.1 (31, 32). A critical role for PI3KC2β in KCa3.1 activation is supported by the findings that small interfering RNA (siRNA) knockdown of PI3KC2β results in decreased KCa3.1 channel activity and TCR-stimulated Ca2+ flux and cytokine production, while these same responses are all increased by T cells overexpressing PI3KC2β (31).
We have recently found that the tripartite motif containing protein TRIM27 functions as an E3 ligase to ubiquitinate and inhibit PI3KC2β's kinase activity and, via this effect, functions to negatively regulate KCa3.1 channel activity and TCR-stimulated Ca2+ influx and cytokine production in activated CD4 T cells (6). We now show that PI3KC2β is also essential for FcεRI activation of KCa3.1 and Ca2+ influx in mast cells and that TRIM27 plays a similar role to negatively regulate KCa3.1 channel activity and FcεRI activation of mast cells in vitro and in vivo.
MATERIALS AND METHODS
Cells and constructs.
Bone marrow-derived mast cells (BMMC) were generated from 6- to 8-week-old TRIM27+/+ and TRIM27−/− mice that were backcrossed 8 generations to C57BL/6 mice as previously described (29). TRIM27−/− mice were generated from the embryonic stem (ES) cell line 345D11(The Center for Disease Modeling at The University of Toronto), which contained the exon-trapping plasmid pUPA, located between exons 1 and 2 of TRIM27 on mouse chromosome 13, and has been previously described (6). Bone marrow cells were cultured for 6 to 8 weeks in RPMI supplemented with interleukin 3 (IL-3) (20 ng/ml), stem cell factor (100 ng/ml), and 10% fetal calf serum (FCS). Generation of a pure population of mast cells after 6 weeks of culture was verified by staining with phycoerythrin (PE)-labeled anti-FcεR1 antibody followed by fluorescence-activated cell sorter (FACS) analysis.
Antibodies.
Anti-TRIM27 antibodies were purchased from IBL America. Anti-PI3KC2β antibody 3E2 (Novus Biologicals, Littleton, CO) was used to immunoblot mouse PI3KC2β.
Whole-cell patch clamping.
Whole-cell patch clamping was performed with TRIM27+/+ and TRIM27−/− BMMCs that were first sensitized overnight with anti-2,4-dinitrophenol (DNP) IgE and then stimulated with DNP-human serum albumin (HSA) using conditions previously described (7). Briefly, the standard pipette solution contained 140 mM KCl, 2 mM MgCl2, 10 mM HEPES, 2 mM Na+-ATP, and 0.1 mM GTP, pH 7.3. The standard external solution contained 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, and 10 mM HEPES, pH 7.3. Whole-cell currents were recorded using an Axoclamp 200B amplifier (Axon Instruments, Foster City, CA), and currents were evoked by applying voltage commands to a range of potentials in 10-mV steps from a holding potential of −20 mV. For measuring the membrane potential, ruptured patch was used in current clamp mode as described in reference 36 with some modifications. The standard pipette solution contained 134 mM KCl, 1 mM CaCl2, 1.1 mM EGTA, 2 mM MgCl2, 10mM HEPES, and 5 mM Na+, pH 7.2.
To verify that PI3KC2β mediated activation of KCa3.1 via the generation of PI3P, PI3P (100 nM) was added to the pipette solution during patch clamping in BMMCs, in which PI3KC2β was knocked down using siRNA (34). PI(3)P diC16 [C41H45Na3O16P2 (C6)] was purchased from Echelon Biosciences and used according to specifications at a concentration of 100 nM in the pipette solution.
Intracellular Ca2+ activity.
BMMCs from TRIM27+/+ and TRIM27−/− mice were sensitized overnight with anti-DNP IgE (100 ng/ml) and subsequently loaded with 5 μM Fura-2 acetoxymethyl (AM) ester (Molecular Probes) in RPMI medium for 30 min at room temperature, washed, and then resuspended in RPMI. Cells were attached to poly(l)lysine-coated coverslips for 20 min in an RC-20 bath flow chamber (Warner Instrument Corp., Hamden, CT), and Fura-2 fluorescence was recorded (Delta Ram; PTI Inc., South Brunswick, NJ) at excitation wavelengths of 340 and 380 nm. Data are represented as the 340/380 ratio after background subtraction. Intracellular Ca2+ was measured before and after the perfusion of DNP-HSA in the Hanks balanced salt solution (HBSS) in the presence of 1 mM extracellular Ca2+.
β-Hexosaminidase release and cytokine production.
BMMCs were plated at 1 × 106 cells/96 well plate in medium supplemented with DNP-IgE antibody for 4 h. Cells were then washed and stimulated with various concentrations of DNP-HSA for 30 min in Tyrode's buffer (10 mM HEPES, pH 7.4, 130 mM NaCl, 5 mM KCl, 1.4 mM CaCl2, 1 mM MgCl2, 5.6 mM glucose, and 0.1% [wt/vol] bovine serum albumin (BSA). Cells were then spun at 1,200 rpm, and β-hexosaminidase was measured in the supernatant by incubating 30 μl of supernatant with 3.3 μl of p-nitrophenyl-N-acetyl-β-d-glucosamide (10 mM) diluted in 0.2 M citrate buffer, pH 4.5, for 1.5 h at 37°C. The reaction was then stopped by adding 135 μl of a 0.1 M Na2CO3–0.1 M NaHCO3 solution and then assayed on an ELISA plate reader at an optical density (OD) at 405 nm. β-Hexosaminidase was measured in the pellet following a similar protocol, with the exception that the cell pellet was lysed in Tyrode's buffer with 1% Triton.
To assay for cytokines, mast cells were stimulated as described above, and total RNA was isolated using TRIzol reagent and then reverse transcribed using random hexamer primers. Quantitative PCR was then assessed using SYBR green 1 with an iCycler iQ (Bio-Rad) system using cytokine-specific primers purchased from Qiagen.
siRNA knockdown of PI3KC2β.
BMMCs were transfected with 2 independent siRNAs to PI3KC2β using the Lipofectamine RNAiMAX reagent, and cells were studied 48 h after transfection. Silencing of PI3KC2β was confirmed by reverse transcription-PCR (RT-PCR) and immunoblotting. The siRNAs used were as follows: siRNA 1, 5′-CCAAGAUCUCUCAGCCUAATT-3′ (sense sequence) and 5′-UUAGGCUGAGAGAUCUUGGAG-3′ (antisense sequence); siRNA 2, 5′GGGUGGUCCAGUCUGUCAATT-3′ (sense sequence) and 5′-UUGACAGACUGGACCACCCTG-3′ (antisense sequence).
Passive systemic and cutaneous anaphylaxis.
To assess whether TRIM27−/− mice are also more sensitive to passive systemic anaphylaxis, TRIM27+/+ and TRIM27−/− mice were first sensitized with anti-DNP IgE (30 μg) administered by intraperitoneal injection. After 5 h, mice were challenged with either DNP-HSA (50 μg) or a phosphate-buffered saline (PBS) control, and body temperature was measured before and then at 5-min intervals following challenge using a rectal probe (40). Blood was also collected 30 min following challenge and assayed for histamine as described previously (2).
To assess passive cutaneous anaphylaxis, mice were sensitized intradermally with 100 ng of anti-DNP IgE and 24 h later were injected intravenously with 100 μg of DNP-HSA containing 0.5% Evans blue dye. Control mice were given dye in saline without DNP-HSA. Thirty minutes after dye injection, mice were sacrificed and tissue sections around the intradermal injection site were excised and weighed. Evans blue dye was then extracted from the tissue by incubation of biopsy specimens in 0.5 ml formamide at 55°C for 24 h and quantitated by measuring absorbance at 620 nm (1).
Immunoblotting.
BMMCs from TRIM27+/+ and TRIM27−/− mice were sensitized for 4 h with anti-DNP IgE (100 ng/ml) and then stimulated with DNP-HSA for various periods of time. Lysates were then immunoblotted with various antibodies as previously described (30, 34).
RESULTS
PI3KC2β activation is required for FcεRI-stimulated KCa3.1 channel activity, Ca2+ influx, β-hexosaminidase release, and cytokine production by BMMCs.
Previous studies have shown that activation of KCa3.1 in CD4 T cells is mediated via TCR-stimulated activation of PI3KC2β, which functions to generate the pool of PI3P required for KCa3.1 channel activation (31). To test whether PI3KC2β is also required for KCa3.1 activation by FcεRI, BMMCs were generated from TRIM27+/+ mice. We found that FcεRI-stimulated KCa3.1 activation was decreased in TRIM27+/+ BMMCs following siRNA knockdown of PI3KC2β using 2 independent siRNAs to PI3KC2β (Fig. 1A to C). This was due to decreased PI3P, because dialyzing siRNA knockdown cells with PI3P restored FcεRI-stimulated KCa3.1 channel activity (Fig. 1B and C), while other phosphoinositides, such as PI4P and PI(3,4,5)P3, failed to rescue (Fig. 1C). The decrease in KCa3.1 channel activity also led to decreased FcεRI-stimulated Ca2+ influx; both the acute rise as well as the sustained plateau phase of Ca2+ influx was decreased in PI3KC2β-knocked-down cells (Fig. 1D).
Fig 1.

PI3KC2β is necessary for FcεRI-stimulated activation of KCa3.1 and Ca2+ influx of BMMCs. (A) Real-time PCR of PI3KC2β in TRIM27+/+ BMMCs transfected with a control or siRNA to PI3KC2β. (B) TRIM27+/+ BMMCs transfected with an siRNA to PI3KC2β were sensitized with anti-DNP IgE, and whole-cell patch clamping was performed without (a) or with (b) stimulation of FcεRI with DNP-HSA. (C) Bar graph summary of whole-cell patch clamp experiments performed for panel B at +40 mV. Also shown is that a second siRNA to PI3KC2β (PI3KC2β2) inhibits KCa3.1 channel activity and that the decrease in KCa3.1 channel activity in PI3KC2β siRNA-transfected cells is rescued by the addition of PI3P (100 nM) but not PI(4)P and PI(3,4,5)P3 to the pipette solution during patch clamping. All experiments shown are representative of at least three experiments performed with cells isolated from three separate mice. ∗, P < 0.05 as compared to the current in TRIM27+/+ BMMC or as indicated. (D) Mast cells were loaded with Fura-2 AM (5 mM), and Ca2+ flux was determined after cross-linking with DNP-HSA as described for panel B. (E) TRIM27+/+ BMMCs (1 × 106) were plated in 96-well plates, sensitized with anti-DNP IgE, and then stimulated with various concentrations of DNP-HSA for 30 min. Shown is the release of β-hexosaminidase into the supernatants after correcting for spontaneous release. ∗, P < 0.05 as compared to the release measured for the wild type (WT) at the same concentration. (F) Cells were stimulated as for panel E for various periods of time, and induction of TNF-α (F), IL-6 (G), or IL-13 (H) mRNA was assessed by RT-PCR. ∗, P < 0.05 compared to the mRNA levels in the WT at the same time point.
FcεRI-mediated rapid degranulation and cytokine production are dependent upon Ca2+ entry into mast cells (3, 17). β-Hexosaminidase is stored in preformed granules in BMMCs and is released into the supernatant following FcεRI stimulation. While basal β-hexosaminidase release was similar between control and siRNA-transfected cells, FcεR1-stimulated β-hexosaminidase release was significantly decreased in PI3KC2β siRNA-transfected BMMCs (Fig. 1E). In addition, FcεRI-stimulated induction of mRNA for the cytokines tumor necrosis factor alpha (TNF-α), IL-6, and IL-13 was decreased in PI3KC2β siRNA-transfected BMMCs (Fig. 1F to H).
FcεRI-stimulated KCa3.1 channel activity and Ca2+ influx are increased in TRIM27−/− mast cells.
The finding that PI3KC2β is required for FcεRI-stimulated KCa3.1 channel activity and Ca2+ influx suggested that TRIM27, via inhibition of PI3KC2β, may also function to negatively regulate mast cells. BMMCs were generated from littermate control TRIM27+/+ and TRIM27−/− mice. TRIM27−/− BMMCs differentiated normally and expressed levels of FcεRI similar to those of TRIM27+/+ cells (Fig. 2A and B). While basal KCa3.1 channel activity was similar between TRIM27+/+ and TRIM27−/− BMMCs, FcεRI-stimulated KCa3.1 channel activity was increased about 50% in TRIM27−/− BMMCs (Fig. 3A and B). Consistent with the increase in KCa3.1 channel activity, TRIM27−/− BMMCs also had an increase in FcεRI-stimulated Ca2+ influx (Fig. 3C), as well as a more negative membrane potential (Fig. 3D). Both the acute rise and the sustained plateau phase of Ca2+ influx were increased in TRIM27−/− BMMCs as a result of the increased negative membrane potential, which would provide the electrical driving force for increased Ca2+ influx. The increase in KCa3.1 channel activity and Ca2+ influx was due to an increase in PI3KC2β activity leading to the generation of more PI3P, since siRNA knockdown of PI3KC2β decreased KCa3.1 channel activity and Ca2+ flux in TRIM27−/− BMMCs to levels similar to those found in TRIM27+/+ BMMCs (Fig. 3E and F). Furthermore, the decrease in KCa3.1 channel activity in PI3KC2β siRNA-transfected BMMCs was rescued by dialyzing cells with PI3P (Fig. 3E).
Fig 2.

Generation of BMMCs from TRIM27+/+ and TRIM27−/− mice. (A) Lysates of TRIM27+/+ and TRIM27−/− BMMCs immunoblotted with antibodies to TRIM27 and PI3KC2β. (B) FACS analysis demonstrating similar levels of expression of FcεR1 on TRIM27+/+ and TRIM27−/− BMMCs. SSC, side scatter.
Fig 3.

Increased KCa3.1 channel activity and FcεRI-stimulated Ca2+ influx in TRIM27−/− BMMCs. (A) TRIM27+/+ and TRIM27−/− BMMCs were sensitized with anti-DNP IgE, and KCa3.1 channel activity was assessed before (a) and after (b) stimulation with DNP-HSA. (B) Bar graph summary of results in panel A at +40 mV (n = 10 cells) is shown. ∗, P < 0.05 compared to the current in TRIM27+/+ BMMC. (C) Ca2+ influx was assessed in TRIM27+/+ and TRIM27−/− BMMCs as described for Fig. 2D. (D) TRIM27+/+ and TRIM27−/− BMMCs were activated as described above, and the change in membrane potential was determined. ∗, P < 0.05 compared to the membrane potential measured in TRIM27+/+ BMMCs. TRIM27−/− BMMCs were transfected with either a control siRNA or a siRNA to PI3KC2β, and whole-cell patch results (E) or Ca2+ influx (F) was assessed. Also shown in panel E is rescue of KCa3.1 channel activity in PI3KC2β siRNA-transfected cells by the addition of PI3P to the pipette solution. ∗, P < 0.05 compared to the current in TRIM27−/− BMMCs or as indicated.
FcεRI-stimulated β-hexosaminidase release and cytokine production are increased in TRIM27−/− BMMCs.
The amounts of basal β-hexosaminidase released were similar between TRIM27+/+ and TRIM27−/− BMMCs (Fig. 4A). However, a significant increase in β-hexosaminidase release in TRIM27−/− BMMCs was seen following FcεRI stimulation (Fig. 4A). In addition, FcεRI-stimulated induction of mRNA for the cytokines TNF-α, IL-6, and IL-13 was increased in TRIM27−/− BMMCs (Fig. 4Bi, ii, and iii). Thus, increased FcεRI-stimulated Ca2+ influx in TRIM27−/− BMMCs is associated with increased degranulation and production of inflammatory cytokines that mediate allergic responses.
Fig 4.
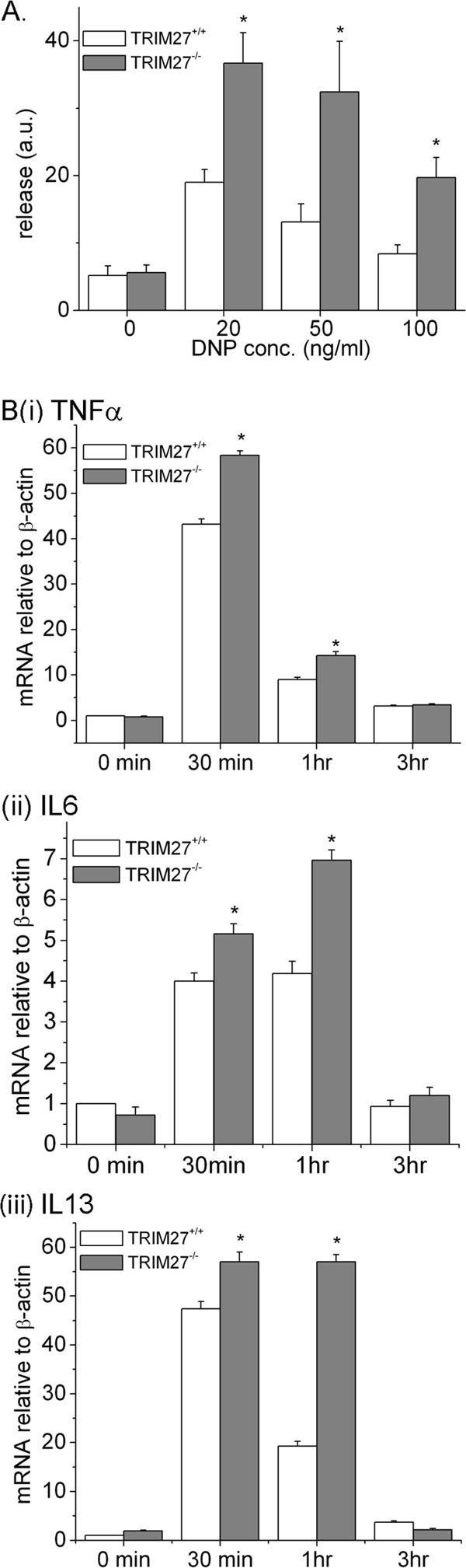
β-Hexosaminidase release and cytokine production are increased in TRIM27−/− BMMCs. (A) TRIM27+/+ and TRIM27−/− BMMCs (1 × 106) were plated in 96-well plates, sensitized with anti-DNP IgE, and then stimulated with various concentrations of DNP-HSA for 30 min, and β-hexosaminidase release was quantitated as described for Fig. 2E. ∗, P < 0.05 compared to the release measured for the WT at the same concentration. a.u., arbitrary units. (B) Cells were stimulated as for panel A for various periods of time, and induction of TNF-α, IL-6, and IL-13 mRNA was assessed by RT-PCR. ∗, P < 0.05 compared to the mRNA levels for the WT at the same time point.
TRIM27−/− mice are more susceptible to acute anaphylaxis.
To assess whether changes in BMMCs in vitro are also relevant in vivo in a model of passive systemic anaphylaxis, TRIM27−/− and TRIM27+/+ mice were sensitized intraperitoneally (i.p.) with anti-DNP IgE. After resting overnight, mice were then challenged i.p. with DNP-HSA or a saline control, and body temperature and serum histamine levels were assessed over time. The decrease in body temperature and histamine release at 30 min following treatment with antigen were significantly increased in TRIM27−/− mice in comparison to results for TRIM27+/+ mice (Fig. 5A and B).
Fig 5.

Systemic anaphylaxis in TRIM27+/+ and TRIM27−/− mice. (A) Mean decrease in body temperature (°C) of TRIM27+/+ and TRIM27−/− mice following induction of passive systemic anaphylaxis (n = 5 mice in each group). (B) Mean serum histamine levels 30 min after induction of anaphylaxis (n = 5 mice in each group). ∗, P < 0.05 as compared to results for the WT. (C) Passive cutaneous anaphylaxis in TRIM27+/+ and TRIM27−/− mice. Data are expressed as A620 per g of skin (n = 5 to 8 mice in each group). ∗, P < 0.05 compared to results for the WT or as indicated.
TRIM27−/− mice are also more sensitive to passive cutaneous anaphylaxis. TRIM27−/− and TRIM27+/+ mice were sensitized intradermally with anti-DNP IgE and 24 h later were injected intravenously with DNP-HSA containing 0.5% Evan's blue dye. The extravasation of Evan's blue dye was significantly increased in TRIM27−/− mice as quantitated by absorbance at 620 nm (Fig. 5C).Thus, these findings indicate that mast cells in vivo in TRIM27−/− mice are more sensitive to degranulation following antigen stimulation.
Activation of FcεR1-stimulated proximal and MAP kinase signaling pathways is similar in TRIM27+/+ and TRIM27−/− BMMCs.
If TRIM27 mediates the inhibition of FcεR1-stimulated KCa3.1 activation via inhibition of PI3KC2β, we would predict that tyrosine phosphorylation of proximal signaling molecules, such as PLCγ1 and Syk, should be similar between TRIM27+/+ and TRIM27−/− BMMCs. TRIM27−/− and TRIM27+/+ BMMCs were stimulated with DNP-HSA for various times following sensitization with DNP-IgE, and activation of signaling molecules was assessed by Western blotting with anti-phospho-specific antibodies. These studies demonstrated that tyrosine phosphorylation of PLCγ1 and Syk was similar between TRIM27+/+ and TRIM27−/− BMMCs (Fig. 6). In addition, activation of extracellular signal-regulated kinase (ERK) mitogen-activated protein (MAP) kinase was also similar between TRIM27−/− and TRIM27+/+ cells (Fig. 6).
Fig 6.
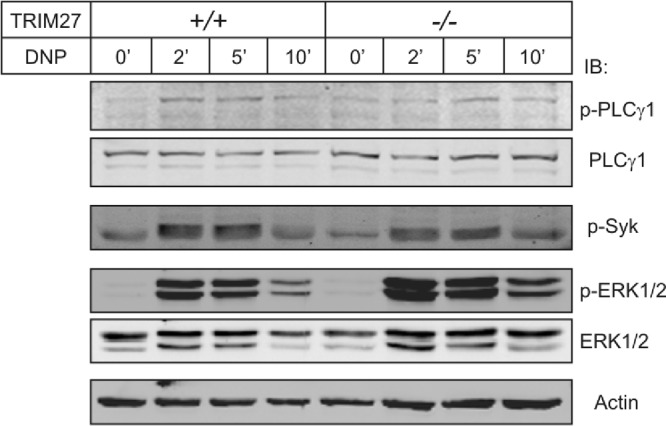
FcεRI-stimulated tyrosine phosphorylation of proximal signaling molecules and the ERK MAP kinase pathway are similar between TRIM27+/+ and TRIM27−/− BMMCs. Lysates from TRIM27+/+ and TRIM27−/− BMMCs were stimulated with DNP-HSA for various lengths of time following sensitization with DNP-IgE and then immunoblotted with anti-phospho-specific antibodies as indicated.
DISCUSSION
IgE-stimulated influx of extracellular Ca2+ via CRAC channels in mast cells is critical for FcεRI-stimulated degranulation and cytokine production (2, 37). CRAC channel-mediated influx of Ca2+ is also regulated by other channels that include KCa3.1 and TRPM4, which by regulating membrane potential play critical roles in modulating IgE-stimulated Ca2+ influx (29, 36). Thus, understanding the mechanisms whereby KCa3.1 and TRPM4 are regulated in mast cells will likely uncover important regulators of allergic responses and provide new therapeutic targets to treat allergic disease. We now demonstrate for the first time that both PI3KC2β and TRIM27 play critical but opposite roles in FcεR1-stimulated activation of KCa3.1, Ca2+ influx, degranulation, and cytokine production in BMMCs. Moreover, we have extended these findings in vivo by demonstrating increased IgE-mediated anaphylactic response in TRIM27−/− mice.
KCa3.1 is an intermediate-conductance Ca2+-activated K+ channel. By mediating the efflux of K+, KCa3.1 functions to maintain a negative membrane potential, which provides the electrical force to drive Ca2+ entry into mast cells and some subsets of CD4 T cells and B lymphocytes (4, 13, 39). It has been known for some time that binding of Ca2+ to calmodulin bound to the carboxy terminus (CT) of KCa3.1 is critical for KCa3.1 activation (10, 16, 22, 28). More recently, studies in CD4 T cells have identified a second signaling pathway that is required for KCa3.1 activation. These studies demonstrated that following TCR activation, PI3KC2β is recruited to the immunological synapse leading to the generation of PI3P, which is required for NDPKB to phosphorylate histidine 358 in the CT of KCa3.1, thereby providing the second signal for KCa3.1 channel activation (31, 33). We now demonstrate that PI3KC2β is also required for activation of KCa3.1 in BMMCs and, via KCa3.1 activation, is required for FcεR1-stimulated Ca2+ influx, degranulation, and cytokine production. This is supported by our finding that siRNA knockdown of PI3KC2β inhibits FcεR1-stimulated KCa3.1 channel activity and Ca2+ influx. Moreover, this inhibition is due to decreased levels of PI3P, because dialyzing PI3KC2β siRNA-transfected cells with PI3P but not other phosphoinositides during whole-cell patch clamping rescued KCa3.1 channel activity. Thus, these findings when taken together demonstrate that PI3KC2β functions downstream of FcεR1 and suggest that PI3KC2β may be a common mechanism for linking other antigen receptors, such the B cell antigen receptor, to KCa3.1 activation.
TRIM family members have been shown to regulate a plethora of biological responses, including innate and adaptive response to infection, cell proliferation, antiviral responses, and development by functioning as a novel family of E3 ligases (23–25). We recently found that TRIM27 downregulates TCR-stimulated activation of KCa3.1 and Ca2+ influx in CD4 T cells by ubiquitinating and inhibiting PI3KC2β enzyme activity (5). We now have shown that TRIM27 also functions as a negative regulator of mast cells. BMMCs derived from TRIM27−/− mice exhibit increased FcεR1-stimulated KCa3.1 channel activity, Ca2+ influx, degranulation, and production of inflammatory cytokines compared with findings for TRIM27+/+ BMMCs. Moreover, we have provided evidence in knockout mice that TRIM27 functions to regulate IgE-mediated degranulation of mast cells and the anaphylactic response in vivo. Consistent with TRIM27 mediating its effects via increased KCa3.1 channel activity, TRIM27−/− BMMCs had a more negative membrane potential following FcεRI stimulation, which would then provide a more favorable electrochemical driving force for Ca2+ influx. In addition, differences in tyrosine phosphorylation of proximal signaling pathways downstream of FcεR1 stimulation, such as phospholipase Cγ1 and Syk, were not detected in TRIM27−/− BMMCs as would be predicted if TRIM27 mediated its effects predominantly via the regulation of PI3KC2β and KCa3.1.
In comparison with the better-studied class I PI3Ks, much less is known about the class II PI3Ks (9, 35). Nevertheless, studies over the past several years have demonstrated critical roles for class II PI3Ks in a number of biological processes (20, 21, 31). The distinct biological roles for class II PI3Ks are likely mediated by their ability to generate a different lipid product in vivo, PI3P, which activates different intracellular signaling pathways than the class I PI3Ks, which generate predominately PI(3,4,5)P3 and PI(4,5)2 (9, 35). This model is consistent with our findings that PI3P is the only phosphatidylinositol generated downstream of PI3KC2β that is required for KCa3.1 activation (31, 34). Moreover, our finding reported here that PI3KC2β plays a critical role in FCεR1 and mast cell activation suggests that a specific pharmacological inhibitor of PI3KC2β may provide a unique opportunity to more surgically treat allergic disease, with a better safety profile than drugs being developed that target the better-studied class I PI3Ks, since PI3KC2β−/− mice do not display overt abnormalities (15).
ACKNOWLEDGMENTS
E.Y.S. is supported by grants R01GM084195 and R01AI052459.
We declare that we have no conflict of interest.
Footnotes
Published ahead of print 29 May 2012
REFERENCES
- 1. Ali K, et al. 2004. Essential role for the p110delta phosphoinositide 3-kinase in the allergic response. Nature 431:1007–1011 [DOI] [PubMed] [Google Scholar]
- 2. Baba Y, et al. 2008. Essential function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nat. Immunol. 9:81–88 [DOI] [PubMed] [Google Scholar]
- 3. Bischoff SC. 2007. Role of mast cells in allergic and non-allergic immune responses: comparison of human and murine data. Nat. Rev. Immunol. 7:93–104 [DOI] [PubMed] [Google Scholar]
- 4. Cahalan MD, Wulff H, Chandy KG. 2001. Molecular properties and physiological roles of ion channels in the immune system. J. Clin. Immunol. 21:235–252. [DOI] [PubMed] [Google Scholar]
- 5. Reference deleted.
- 6. Cai X, et al. 2011. Tripartite motif containing protein 27 negatively regulates CD4 T cells by ubiquitinating and inhibiting the class II PI3K-C2beta. Proc. Natl. Acad. Sci. U. S. A. 108:20072–20077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Duffy SM, Cruse G, Brightling CE, Bradding P. 2007. Adenosine closes the K+ channel KCa3.1 in human lung mast cells and inhibits their migration via the adenosine A2A receptor. Eur. J. Immunol. 37:1653–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Duffy MS, et al. 2004. The K+ channel iKCA1 potentiates Ca2+ influx and degranulation in human lung mast cells. J. Allergy Clin. Immunol. 114:66–72 [DOI] [PubMed] [Google Scholar]
- 9. Falasca M, et al. 2007. The role of phosphoinositide 3-kinase C2alpha in insulin signaling. J. Biol. Chem. 282:28226–28236 [DOI] [PubMed] [Google Scholar]
- 10. Fanger CM, et al. 1999. Calmodulin mediates calcium-dependent activation of the intermediate conductance KCa channel, IKCa1. J. Biol. Chem. 274:5746–5754 [DOI] [PubMed] [Google Scholar]
- 11. Feske S, et al. 2006. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441:179–185 [DOI] [PubMed] [Google Scholar]
- 12. Galli SJ, et al. 2005. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu. Rev. Immunol. 23:749–786 [DOI] [PubMed] [Google Scholar]
- 13. Ghanshani S, et al. 2000. Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J. Biol. Chem. 275:37137–37149 [DOI] [PubMed] [Google Scholar]
- 14. Gilfillan AM, Tkaczyk C. 2006. Integrated signalling pathways for mast-cell activation. Nat. Rev. Immunol. 6:218–230 [DOI] [PubMed] [Google Scholar]
- 15. Harada K, Truong AB, Cai T, Khavari PA. 2005. The class II phosphoinositide 3-kinase C2beta is not essential for epidermal differentiation. Mol. Cell. Biol. 25:11122–11130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Joiner WJ, Khanna R, Schlichter LC, Kaczmarek LK. 2001. Calmodulin regulates assembly and trafficking of SK4/IK1 Ca2+-activated K+ channels. J. Biol. Chem. 276:37980–37985 [DOI] [PubMed] [Google Scholar]
- 17. Kalesnikoff J, Galli SJ. 2008. New developments in mast cell biology. Nat. Immunol. 9:1215–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kraft S, Kinet JP. 2007. New developments in FcepsilonRI regulation, function and inhibition. Nat. Rev. Immunol. 7:365–378 [DOI] [PubMed] [Google Scholar]
- 19. Liou J, et al. 2005. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15:1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maffucci T, et al. 2005. Class II phosphoinositide 3-kinase defines a novel signaling pathway in cell migration. J. Cell Biol. 169:789–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maffucci T, Falasca M. 2007. Phosphoinositide 3-kinase-dependent regulation of phospholipase Cgamma. Biochem. Soc. Trans. 35:229–230 [DOI] [PubMed] [Google Scholar]
- 22. Maylie J, Bond CT, Herson PS, Lee WS, Adelman JP. 2004. Small conductance Ca2+-activated K+ channels and calmodulin. J. Physiol. 554:255–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meroni G, Diez-Roux G. 2005. TRIM/RBCC, a novel class of ‘single protein RING finger' E3 ubiquitin ligases. Bioessays 27:1147–1157 [DOI] [PubMed] [Google Scholar]
- 24. Napolitano LM, Jaffray EG, Hay RT, Meroni G. 2011. Functional interactions between ubiquitin E2 enzymes and TRIM proteins. Biochem. J. 434:309–319 [DOI] [PubMed] [Google Scholar]
- 25. Ozato K, Shin DM, Chang TH, Morse HC., III 2008. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 8:849–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Prakriya M, et al. 2006. Orai1 is an essential pore subunit of the CRAC channel. Nature 443:230–233 [DOI] [PubMed] [Google Scholar]
- 27. Roos J, et al. 2005. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 169:435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schumacher MA, Rivard AF, Bachinger HP, Adelman JP. 2001. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature 410:1120–1124 [DOI] [PubMed] [Google Scholar]
- 29. Shumilina E, et al. 2008. Blunted IgE-mediated activation of mast cells in mice lacking the Ca2+-activated K+ channel KCa3.1. J. Immunol. 180:8040–8047 [DOI] [PubMed] [Google Scholar]
- 30. Srivastava S, et al. 2006. Phosphatidylinositol 3-phosphate indirectly activates KCa3.1 via 14 amino acids in the carboxy terminus of KCa3.1. Mol. Biol. Cell 17:146–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Srivastava S, et al. 2009. The class II phosphatidylinositol 3 kinase C2beta is required for the activation of the K+ channel KCa3.1 and CD4 T-cells. Mol. Biol. Cell 20:3783–3791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Srivastava S, et al. 2006. Phosphatidylinositol-3 phosphatase myotubularin-related protein 6 negatively regulates CD4 T cells. Mol. Cell. Biol. 26:5595–5602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Srivastava S, et al. 2006. Histidine phosphorylation of the potassium channel KCa3.1 by nucleoside diphosphate kinase B is required for activation of KCa3.1 and CD4 T cells. Mol. Cell 24:665–675 [DOI] [PubMed] [Google Scholar]
- 34. Srivastava S, et al. 2005. The phosphatidylinositol 3-phosphate phosphatase myotubularin-related protein 6 (MTMR6) is a negative regulator of the Ca2+-activated K+ channel KCa3.1. Mol. Cell. Biol. 25:3630–3638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. 2010. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 11:329–341 [DOI] [PubMed] [Google Scholar]
- 36. Vennekens R, et al. 2007. Increased IgE-dependent mast cell activation and anaphylactic responses in mice lacking the calcium-activated nonselective cation channel TRPM4. Nat. Immunol. 8:312–320 [DOI] [PubMed] [Google Scholar]
- 37. Vig M, et al. 2008. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat. Immunol. 9:89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vig M, et al. 2006. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312:1220–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wulff H, Beeton C, Chandy KG. 2003. Potassium channels as therapeutic targets for autoimmune disorders. Curr. Opin. Drug Discov. Dev. 6:640–647 [PubMed] [Google Scholar]
- 40. Zemtsova IM, et al. 2010. Blunted IgE-mediated activation of mast cells in mice lacking the serum- and glucocorticoid-inducible kinase SGK3. Am. J. Physiol. Cell Physiol. 299:C1007–C1014 [DOI] [PMC free article] [PubMed] [Google Scholar]